

Abstract
After exocytosis, synaptic vesicle components are selectively retrieved by clathrin-mediated endocytosis and then re-used in future rounds of transmitter release. Under some conditions, synaptic terminals in addition perform bulk endocytosis of large membranous sacs. Bulk endocytosis is less selective than clathrin-mediated endocytosis and probably internalizes components normally targeted to the plasma membrane. Nonetheless, this process plays a major role in some tonic ribbon-type synapses, which release neurotransmitter for prolonged periods of time. We show here, that large endosomes formed after strong and prolonged stimulation undergo stimulated exocytosis in retinal bipolar neurons. The result suggests how cells might return erroneously internalized components to the plasma membrane, and also demonstrates that synaptic vesicles are not the only neuronal organelle that stains with styryl dyes and undergoes stimulated exocytosis.
Neurotransmitter is thought to be released from synaptic vesicles by two mechanisms: (1) through the transient opening of a fusion pore, which allows release of neurotransmitter without the loss of some or all of the vesicle membrane and/or protein; and (2) through the complete incorporation of the synaptic vesicle membrane into the plasmalemma (reviewed in An & Zenisek, 2004). While the relative significance of the first mechanism in synaptic terminals is still debated, there is general acceptance that the second mechanism, known as full-fusion, has an important role and the recycling of synaptic vesicle membrane and protein. Retrieval of membrane following full-fusion is thought to occur by at least two independent pathways. The first is believed to dominate following mild stimulation. In this pathway, exocytosed membrane and membrane proteins are collected at clathrin-coated pits in the perisynaptic regions. Clathrin-coated pits pinch off as small clathrin-coated vesicles, which either uncoat and become synaptic vesicles (Takei et al. 1996) or fuse with endosomes (Heuser & Reese, 1973). The clathrin-mediated pathway, although fast in neurons, may have limited capacity and may be unable to keep up with exocytosis under stronger or prolonged stimulation (Sankaranarayanan & Ryan, 2000).
The second process takes up membrane into large vacuoles or cisternae by bulk endocytosis (Heuser & Reese, 1973; Fried & Blaustein, 1978; Miller & Heuser, 1984; Takei et al. 1996; Gad et al. 1998; Marxen et al. 1999; Teng et al. 1999; Richards et al. 2000; Lenzi et al. 2002; de Lange et al. 2003; Holt et al. 2003; Paillart et al. 2003). Since large amounts of membrane are internalized at once without the benefit of sorting, this process almost certainly internalizes a mixture of plasma membrane and synaptic vesicle components (Miller & Heuser, 1984; Takei et al. 1996). Indeed, structures formed in this manner do not seem to concentrate or exclude intramembrane particles thought to be synaptic vesicle proteins. The sorting of synaptic vesicle components is thought to occur later by clathrin-coated vesicles budding from the cisternae (Miller & Heuser, 1984; Takei et al. 1996; Gad et al. 1998; Richards et al. 2000; Teng & Wilkinson, 2000). Once the synaptic vesicle components have been removed, what is the fate of the plasma membrane components left behind in the remnants of the cisternae? The predominant pathway in most cells may be retrograde transport and lysosomal degradation (LaVail & LaVail, 1974; Teichberg et al. 1975; Kristensson, 1977) but an interesting alternative suggestion (Takei et al. 1996) is that they return to the surface membrane by exocytosis. Indeed, exocytosis of lysosomes/endosomes has been demonstrated in non-neuronal cells as a mechanism for membrane repair (reviewed in Andrews, 2002; McNeil et al. 2003). However, there is as yet little direct evidence for the exocytosis of endosomes in neurons.
In the synaptic terminal of the retinal bipolar cell, a tonic ribbon-type synapse, bulk endocytosis appears to play a major role in membrane internalization (Holt et al. 2003, 2004; Paillart et al. 2003). Indeed, most membrane retrieval following exocytosis in these cells is thought to be retrieved in this manner, even under mild stimulus conditions (Paillart et al. 2003). Here we confirm that large endosomal compartments formed following stimulation in goldfish retinal bipolar neurons. To our surprise, we also found that such compartments fuse with the plasma membrane in a stimulation-dependent manner. We found that the properties of these fusion events differed from those of synaptic vesicles in their kinetics relative to the stimulation and in the time course of lipid mixing. Although the onset of fusion was delayed relative to synaptic vesicles, it occurred at physiologically relevant membrane potentials and in the absence of any external stimulus. Together our results suggest a role for regulated exocytosis of endosomes in nerve terminals. Since bulk endocytosis exists in many synapses, both such endocytosis and the exocytosis of endosomes may influence analyses of membrane recycling, whether they are carried out with styryl dyes or by capacitance measurements.
Methods
Cell preparation
All procedures for animal care were carried out according to Yale Animal Care and Use Committee (YACUC). Bipolar neurons were prepared as previously described (Zenisek et al. 2002). Briefly, adult goldfish (10–15 cm in length) were killed by decapitation followed immediately by pithing of the brain and spinal cord. Next, eyes were removed, hemisected and lenses removed from the eyecup. Eyecups were incubated for 20 min in hyaluronidase (Sigma type V; 1100 units ml−1) in a saline solution containing (mm): 120 NaCl, 0.5 CaCl2, 2.5 KCl, 1.0 MgCl2, 10 glucose and 10 Hepes (pH 7.4 with NaOH). After incubation, retinas were then removed from eyecups, cut into four to eight pieces each, and incubated for 30–35 min in a solution of 35 units ml−1 papain (lyophilized powder; Sigma), 0.5 mg ml−1 cysteine, and (mm): 120 NaCl, 0.5 CaCl2, 2.5 KCl, 1.0 MgCl2, 10 glucose and 10 Hepes (pH 7.4 with NaOH). After enzymatic treatment, tissue was washed in enzyme-free solution and stored at 12°C for up to 5 h. The pieces of retina were dissociated into single cells by mechanical trituration through a fire-polished Pasteur pipette. For imaging, cells were plated in recording solution containing (mm): 120 NaCl, 2.5 CaCl2, 2.5 KCl, 1.0 MgCl2, 10 glucose and 10 Hepes (pH 7.4 with NaOH) on special coverslips made from high refractive index glass (n488= 1.80; Plan Optik, Germany). Bipolar neurons were recognized by their large synaptic terminal and unique cell body morphology and all experiments were performed within 90 min of dissociation from the retina.
In some experiments, retina was dissociated into single cells by mechanical trituration and plated in the above recording solution with the addition of 20 μm latrunculin B. Cells remained in the latrunculin B solution for > 15 min before any experiments were begun.
FM1-43 labelling of bipolar neurons and quenching
We used two labelling protocols. A weak labelling–long wash protocol was used to label synaptic vesicles, as previously described (Zenisek et al. 2000). Briefly, a selected neuron was locally superfused for 10–15 s with a labelling solution containing 5 μm FM1-43 (Molecular Probes) and elevated [K+] and (mm): 2.5 CaCl2, 25 KCl, 95 NaCl, 1.0 MgCl2, 10 glucose and 10 Hepes (pH 7.4 with CsOH). Next, the neuron was washed by local superfusion for 30–60 min with a low calcium solution designed to stop exocytosis. It contained (mm): 120 NaCl, 0.5 CaCl2, 2.5 KCl, 1.0 MgCl2, 10 glucose, 10 Hepes and 1.25 mm EGTA (pH 7.4 with NaOH). A strong labelling–short wash protocol was used to label endosomal compartments that may form as a result of bulk endocytosis. It was carried out as above except that the labelling solution contained more KCl (50 mm) and less NaCl (70 mm), and was applied for longer (15–30 s) and washed for a briefer period (10–15 min).
After the wash, the superfusion was switched to the recording solution described above, and the cell bodies of bipolar neurons were whole-cell voltage clamped using an EPC-9 or EPC-10 amplifier (Heka), running Pulse (Instrutech) stimulus and acquisition software. The pipette solution contained (mm): 120 caesium glutamate, 4 Na2ATP, 0.5 GTP, 4 MgCl2 and 10 TEA-Cl (pH 7.4 with CsOH). Except when otherwise noted, exocytosis was triggered by voltage steps to 0 mV. Cells were deemed acceptable for analysis when voltage steps to 0 mV elicited inward calcium currents > 50 pA and resting leak currents at −60 mV were < 50 pA. A gravity-fed bath perfusion system perfused the entire chamber with recording solution throughout the experiment.
In some experiments, FM1-43 fluorescence was quenched with FM4-64 (Rouze & Schwartz, 1998) to assay whether organelles were open to the external space. For quenching experiments, we superfused bipolar neurons with 5 μm FM1-43 for 20–30 s in 50 mm K+ solution and then washed the neurons with an EGTA-buffered, low Ca2+ solution (0.5 mm Ca2+ and 0.75 mm EGTA). After a brief (5–20 min) wash, we superfused the neuron with 20 μm FM4-64, to quench FM1-43 fluorescence in structures with aqueous or lipidic access to the extracellular environment. To monitor for deleterious effects of photobleaching, at the end of each experiment where bipolar cells were not patch clamped, cells were monitored in bright field for changes in morphology.
Fluorescence microscopy
Bipolar neurons were imaged as previously described (Zenisek et al. 2002). Briefly, cells were observed through an inverted microscope (Zeiss Axiovert 135 or Olympus IX-70) modified for objective-type evanescent field illumination (Axelrod, 2001) using a 1.65 NA objective and an argon or 488 nm solid state laser (Coherent Inc.). The laser beam left the objective at an angle of 65.6–68.0 deg. Given the refractive indices of cytosol (n = 1.37) and glass (n = 1.80) and the 65.6–68.0 deg angle, the intensity of the evanescent field is expected to decline e-fold every 41–43 nm. Fluorescence images were captured using an intensified CCD camera (three different cameras were used in these studies: I-Pentamax, Cascade 512B, Cascade 650; all were from Roper Scientific). Images were recorded at 20–83.3 Hz in bursts lasting 1.5–3 s, except where otherwise indicated. Cells were rested for approximately 30 s between bursts. Illumination was controlled by opening and closing of a shutter (Uniblitz) placed in the beam path. The ‘shutter out’ port of the camera was used to trigger both shutter opening and to synchronize the timing of voltage pulses by the patch clamp amplifier (EPC-10; HEKA). A 505 nm long-pass dichroic mirror (Chroma) was used, as well as a 510 nm long pass filter except in quenching experiments, as follows.
For quenching experiments, bipolar neurons were imaged using a back-illuminated CCD camera with on-chip gain intensification (Cascade 512B; Roper Scientific; Tucson, AZ, USA). FM4-64 is itself fluorescent, but with a longer wavelength emission spectrum than FM1-43. To reject most light emitted from FM4-64, it was necessary to use a band-pass emission filter (HQ525/50; Chroma, Brattleboro, VT, USA). Cells were imaged using 500 ms exposures at 0.1 Hz for approximately 5 min, using intensifier and analog-to-digital gain settings of 3481 and 4, respectively. The exposures were longer in order to compensate for light lost through the band-pass emission filter.
Measuring stimulus strength
To determine the amount of membrane turnover in response to different stimulus conditions, we plated bipolar cells on coverslips and illuminated cells by directing laser light to the centre of the back focal plane of a high NA objective (NA = 1.65; Olympus) using an epifluorescence microscope (Olympus). Bipolar cells were first exposed to 0.1 μm FM1-43 in a 0 Ca2+ Ringer solution to determine the fluorescence of stained plasma membrane without membrane turnover. This fluorescence value served as a standard to estimate the amount of membrane turnover relative to the surface membrane. Next, cells were superfused with a solution containing either the 25 mm KCl stimulus solution or 50 mm KCl stimulus solution in the continued presence of 0.1 μm FM1-43. The fluorescence intensity after 15 s of depolarization was normalized to the fluorescence just before application of the depolarizing solution to determine the membrane turnover in response to the depolarizing solution.
Analysis
2D Gaussian fits
For analysis, we fitted images of organelles with 2D Gaussian functions on inclined planes. The function that was used for these purposes was:
where x and y are the x and y coordinates of the image and w is the ‘Gaussian width’ of the fit, and xcyc are the x and y coordinates of the centre of the object and
For these fits, the free parameters were A, B, C, D, xc, yc and w were allowed to be free parameters. Note that the function used here is a slightly different definition for width than in our previous work (Zenisek et al. 2002).
Measurement of fusing organelle surface area
The surface area of fusing organelles was calculated from their FM1-43 content, measured as previously described (Zenisek et al. 2002). Briefly, the images from 1.7 μm × 1.7 μm square regions centred on each fusing organelle were extracted from our image stacks to form ‘mini-stacks’ centred on exocytosing organelles. Next, the five images in these mini-stacks that immediately preceded exocytosis were averaged. The resultant image was best least-squares fitted to a 2D Gaussian function on an inclined plane. The light arising from the Gaussian function was taken to represent light from the organelle, whereas the remainder comprised our estimate of the local background. This local background was subtracted from all images before and after exocytosis in order to estimate the fluorescence arising solely from the vesicle at each time point. The fluorescence across the region was then integrated and taken to represent fluorescence of dye arising from the fusing vesicle. To estimate the surface area, we compared the fluorescence arising from the fusing organelle to the fluorescence of 1 μm2 of surface membrane stained to equilibrium with the same concentration of FM1 43 used during staining (5 μm).
For some experiments, we analysed organelles by plotting their pixel intensity as a function of distance from the centre of the object (radial sweeps). This was accomplished as previously described (Zenisek et al. 2002). Briefly, the centre of the object was determined to sub-pixel accuracy by fitting a 2D Gaussian fit on an inclined plane to its image. Next, circles were drawn around the centre in increments of 1 pixel unit (85 nm). For each circle, 633 evenly spaced points around the circle were measured and averaged using a MATLAB program to determine the fluorescence intensity as a function of distance from the centre of the object.
Counting organelles
For Fig. 2 and the estimation of percentage of endosomes fusing per stimulus, it was necessary to identify and count the number of endosomes. For the histograms in Fig. 2, time-lapse movies of bipolar cell terminals were taken and individual spots were visually identified for each frame during the movie and each identified spot was measured and used as a data point.
Figure 2.
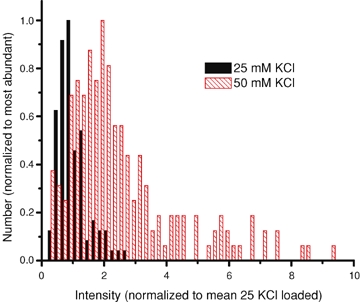
Organelles labelled using 50 mm KCl protocol are brighter than those loaded with 25 mm KCl Terminals were labelled at 25 and 50 mm[K+] for 15 s and then washed with dye-free solution for 10–15 min. The fluorescence intensity of organelles was measured and divided by the mean value observed after labelling at 25 mm[K+]. Histograms were scaled to a peak value of 1.0.
To estimate the percentage of endosomes fusing, the number of endosomes was visually determined in movies in which bipolar cells were depolarized for 0.5 s to 0 mV. The number of endosomes visible prior to the stimulus was determined by taking an average of the images in the 0.5 s just prior to the stimulus. Individual organelles were visually identified and counted and then compared against the number of organelles fusing in the same movies.
Electron microscopy
All of the EM reagents were purchased from Electron Microscopy Sciences (EMS; Hatfield, PA, USA). Dissociated retinal cells were prepared and plated on glass coverslips as described above. Once the cells attached to the coverslip, ∼20 min after plating, the cells were carefully rinsed in 2.5 mm Ca2+ recording media, and equilibrated for 10 min at room temperature (RT). The cells were fixed by rapidly exchanging the recording media with the following primary fixative: 1.5% glutaraldehyde, 1% paraformaldehyde, 0.06 m sodium cacodylate, pH 7.4, and they remained in fixative at RT for 30 min. Next, the cells were washed three times with 0.1 m sodium cacodylate, pH 7.4, and then post-fixed in 2% osmium tetroxide, 0.1 m sodium cacodylate, pH 7.4, to which 2% potassium ferrocyanide was added immediately before use. The cells incubated in the post-fix solution for 30 min at RT. Next, the cells were rinsed three times in 0.1 m sodium cacodylate, pH 7.4, and this was followed by two rinses in 0.04 m sodium maleate, pH 5.4, and then stained with 2% uranyl acetate in 0.04 m sodium maleate, pH 5.4, for 20 min in the dark at RT. Next, cells were rinsed twice with 0.04 m sodium maleate, pH 5.4, and then rinsed three times with doubly distilled (dd) H2O before dehydration. Serial graded dehydration consisted of 5 min incubations in the following EtOH–ddH20 mixtures: 50%, 70%, 90% and twice in 100% EtOH. Immediately after dehydration cells were transferred to a 50 : 50 mixture of 812 epoxy resin–EtOH for 1 h, followed by 70% epoxy for 1 h, and finally 100% epoxy for 16 h. Prior to heat curing the resin, the empty face of the coverslip was wiped clean of epoxy, and the face with cells was inverted onto a bead of fresh resin and left for 16 h at RT. Finally, the samples were cured in a 60°C oven for 24 h.
Once the resin cured, the glass was separated from the resin–cells by placing the glass face onto dry ice until the glass detached from the resin. Small portions of the resin–cell block were mounted for thin sectioning. Sections were made with a diamond blade that was positioned parallel to the surface of the sample. Thin sections that had an even, silver colour were deemed to be 60 nm in thickness (Sakai, 1980), and they were collected on carbon-coated Formvar slot grids (1 mm × 2 mm slots). Sections were counterstained in 2% uranyl acetate, followed by lead citrate. Sections were viewed on a Tecnai 12 Biotwin electron microscope (FEI Company; Hillsboro, OR, USA) that is maintained by the Center for Cell and Molecular Imaging at the Yale School of Medicine. Digital images were acquired from serial sections at 16 000 × magnification.
Measuring vesicle size
Vesicle diameter was estimated from vesicle area using ImageJ software (sponsored by the NIH, Bethesda, MD, USA; http://rsb.info.nih.gov/ij/). This was done with the aid of the circle tool in ImageJ, which was used to place a circle around the outer membrane of the vesicle. Diameter was calculated from the circle's area using the relationship: Diameter = 2(Area/π)0.5. Only vesicle profiles that were circular were included. The periphery of the cell was selected for analysis, as this region is most relevant to TIRF microscopy, and the periphery has fewer mitochondria and tubular structures, which can confound the vesicle size analysis. To estimate the size of large vesicles that were over > 60 nm in diameter (greater in diameter than the section thickness), large vesicular structures were tracked through multiple sections to determine whether the structure was a vesicle or some other organelle of no interest to the analysis. To do this, serial sections were aligned with Reconstruct software (available at http://synapses.bu.edu), and all circular vesicles > 60 nm were inspected, and the section with the vesicle's widest diameter was identified (also see Grabner et al. 2005). The density of large vesicles within 60 nm of the cell membrane was determined by dividing the number of large vesicles (diameter > 60 nm) identified from serial sections by the cell's surface area (vesicles per μm2). The cell's surface area was calculated as the product of the terminal's circumference and the height/thickness of the section. A density value was calculated per section, and this value was averaged across sections to generate the final average density. Results are presented as mean +/−s.e
Results
Labelling of endosomes in bipolar cell terminals
In order to investigate membrane recycling in retinal bipolar neurons, we imaged cells using evanescent field fluorescence microscopy (EFM). EFM uses an exponentially decaying layer of light to selectively excite fluorophores within 100 nm of the glass coverslip (see Methods). Using this technique, it is possible to image 30 nm diameter FM1-43-labelled vesicles in bipolar cell axon terminals (Zenisek et al. 2000, 2002). Terminals were labelled with the styryl dye FM1-43. FM1-43 reversibly intercalates into the outer leaflet of the plasma membrane, and is usually assumed not to cross the lipid bilayer; thus it labels membrane that is either surface exposed, or has been retrieved from the surface of the cell during endo-cytosis (Betz & Bewick, 1992). Under some conditions, however, FM1-43 has been shown to cross the membrane in the goldfish retinal bipolar cells (Rouze & Schwartz, 1998). Rouze & Schwartz (1998) found that FM1-43 permeability required divalent cations, but not calcium, and is concentration and exposure time dependent. To avoid membrane permeation, Rouze & Schwartz (1998) reduced FM1-43 concentration to 20 μm and exposure time to 30 s. In our experiments, to prevent FM1-43 from crossing the membrane, we loaded cells for less than 30 s and kept FM1-43 concentrations to 5 μm.
During the duration of these experiments, we used two loading protocols to label organelles with FM1-43. For protocol 1, we superfused cells with 25 mm KCl for 15 s in the presence of 4 μm FM1-43, which causes turnover of the equivalent of 22 ± 9% (n = 3) of the surface membrane of the terminal. For protocol 2, we superfused cells with 50 mm KCl for 15–30 s in the presence of 4 μm FM1-43. We found that a 15 s application of 50 mm KCl causes the equivalent of 71 ± 29% (n = 3) of the surface membrane of the terminal to turnover, similar to rates previously reported for the same preparation (Lagnado et al. 1996). The fluorescence pattern of labelled terminals varied strikingly with loading and washing conditions. After application of 50 mm KCl for 15–30 s and a 15 min wash, terminals fluoresced brightly, with punctate fluorescence that probably arose from membrane-bound organelles (Fig. 1A). On average, there were 5.4 ± 0.4 organelles visible per frame (n = 14 cells). Punctate structures were rare (0.2 ± 0.1 spots per frame; n = 4 cells) when calcium was omitted or 100 μm CdCl2 was added to the 50 mm KCl perfusate (0.5 ± 0.5 spots per frame; n = 4 cells), consistent with the idea that punctate structures arise as a consequence of cell activity. After 25 mm KCl and a 45 min wash, fluorescence was barely visible (Fig. 1B) at the contrast settings of Fig. 1A. Nonetheless, when the contrast is adjusted to image lower light levels, punctate fluorescence is clearly seen (Fig. 1C), and results from stained synaptic vesicles (Zenisek et al. 2000).
Figure 1.
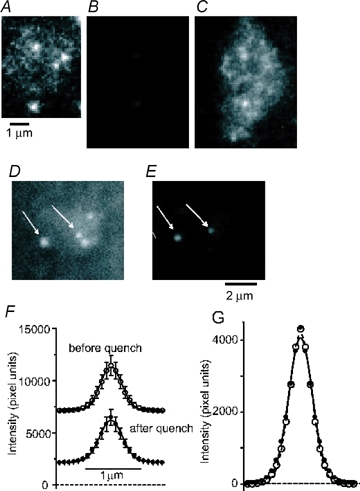
Endosomes and synaptic vesicles in retinal bipolar neurons A, footprint of the fluorescent terminal of a bipolar cell where it adhered closely to a coverslip. Terminal was labelled with FM1-43 using our stronger stimulus protocol (protocol 2; see text for details), and washed with dye-free solution for 15 min before imaging. B, as in A, but using protocol 1 and washing for > 40 min before imaging. Same contrast setting and illumination intensity as in A, note dimmer fluorescence. A was a single 30 ms exposure and B the average of five consecutive 30 ms exposures. C, image in B with contrast enhanced. D, another terminal as in A imaged after a 215 s wash with dye-free solution. E, same terminal as in D but 20 s later, after application of 20 μm FM4-64. Arrows indicate spots that were visible before and after application of FM4-64. F, average fluorescence profiles (radial sweeps) of a subset of vesicles (n = 11) that remained at the same site in images taken first without (○) and 10–60 s later with FM4-64 (•). Lines connecting the points represent best-fits of Gaussian functions. Error bars indicate standard errors of values after the asymptotes of the Gaussian functions were subtracted. G, data in F shifted vertically, setting the average values at a distance of 1.47 μm from the centre to zero. Error bars removed for clarity.
The stronger fluorescence of the organelles in Fig. 1A suggests that they were larger than synaptic vesicles. This would be consistent with results at the frog and lizard neuromuscular junctions, where strong stimulation generates large cisternal structures that are contiguous with the extracellular environment and retain FM1-43 over minutes (Richards et al. 2000; Teng & Wilkinson, 2000). To test whether the structures we observed were open to the external space, we used FM4-64 to quench the fluorescence of surface-exposed FM1-43 (Rouze & Schwartz, 1998). Figure 1D shows a terminal imaged approximately 4 min after strong loading. Application of 20 μm FM4-64 diminished the fluorescence in the entire image (Fig. 1E), both within the terminal (to 41 ± 5% in 11 terminals) and outside it (to 18 ± 2%). The result documents the presence of FM1-43 both on the coverslip and on the terminal surface, as well as its efficient quenching by FM4-64. However, organelles within the terminal remained clearly visible. Some moved or appeared during the solution change while others vanished. When spots remained in place sufficiently well to be recognizable both before and after quenching (11 of 23 spots in 8 cells; arrows in Fig. 1D and E), it appeared that they remained just as fluorescent in the presence of FM4-64 as they had been in its absence (Fig. 1F and G), provided the local background was subtracted. This can be shown by first fitting a small region surrounding each spot with the sum of a plane and a Gaussian function, and then plotting the fluorescence as a function of distance from the centre. Whereas FM4-64 lowered the plane nearly threefold (from 7382 ± 1591 to 2438 ± 432 pixel units, measured at the centre of the Gaussian), the amplitude of the Gaussian did not change (4720 ± 888 pixel units without and 4697 ± 1128 pixel units with FM4-64). At least these 11 organelles therefore were not quenched by FM4-64 and thus were inaccessible to the dye. Moreover, FM4-64 did not lower the number of fluorescent organelles. Prior to quenching, bipolar terminals exhibited 2.8 ± 0.8 spots frame, while after quenching terminals contained 4.1 ± 0.9 spots frame. The increase in the number of spots probably reflects the improvement of the signal-to-noise ratio observed after quenching background fluorescence. Even after 2 min in the continued presence of FM4-64, the number of visible spots (4.3 ± 0.5) had not changed significantly. Evidently, spots that disappeared during the application of FM4-64 were replaced by others. We conclude that the spots visualized here are inaccessible to FM4-64, and hence not connected to the external space.
To compare the loading conditions more directly, terminals were loaded at either 50 or 25 mm KCl and washed for 10–15 min and imaged during 100 ms exposures at intervals of 10 to 30 s. The brightness of organelles was measured in a 9 pixel diameter circle (pixel area of 63 pixel units) centred on each organelle, and the fluorescence in a surrounding annulus (14 pixel diameter annulus of 5 pixel width) subtracted as background. Figure 2 plots the organelle brightness as a histogram. At 50 mm KCl (335 spots in 4 cells), organelles, on average, took up 2.77 ± 0.11 times more dye than at 25 mm KCl (n = 66 spots in 3 cells). After a longer washing time (35–47 min) the multiple diminished to 2.00 ± 0.11 times (P < 0.01, two-sample t test; n = 251 spots in 5 cells). In an alternative analysis, the mean and standard deviation at 25 mm K+ were measured, and organelles exceeding the mean by 3 s.d.s were defined as bright. There were no bright organelles (0 of 66) after loading at 25 mm KCl. However, 131 of 335 spots (39%) were bright after loading at 50 mm KCl and 10–15 min washing. The percentage diminished (22%) after 35–47 min washing.
Lagnado and colleagues (Holt et al. 2003) demonstrated that bulk endocytosis of soluble dyes can be suppressed by application of actin depolymerizing drugs or inhibitors of PI-3 kinases. In particular, they showed that latrunculin B caused an approximately 40% decrease in the loading of dextrans via bulk endocytosis, with no effect on the loading of synaptic vesicles. We asked whether latrunculin inhibits the formation of bright organelles. Terminals were loaded at 50 mm KCl and again subjected to time lapse imaging. When 20 μm latrunculin was applied to the cells during the loading period, the percentage of bright organelles was reduced to 27% (47 of 171 spots) from 39% (see earlier). On average, organelles were 2.11 ± 0.14 times brighter than those loaded with 25 mm KCl, compared with 2.77 without drug (P < 0.01). Our results suggest a relationship between bright organelles and the bulk endocytosis described by Lagnado and colleagues.
Stimulated exocytosis of endosomes
Surprisingly, after returning strongly loaded cells to normal calcium concentration (2.5 mm), we twice observed endosomes whose fluorescence suddenly brightened and spread across the membrane, indicating exocytosis. Although these events were rare (2 events in 200 s of imaging), this finding motivated us to explore the possibility that endosomes could undergo stimulated exocytosis with the plasma membrane. Terminals were subjected to 50 mm KCl labelling with FM1-43, and neurons were whole-cell voltage clamped. In order to maximally open voltage-gated Ca2+ channels, cells were depolarized to 0 mV for 0.5 s. Upon depolarization, bipolar cells exhibited 96 ± 3.3 pA of calcium current (n = 54 depolarizations in 11 cells). Approximately 9% (32 of 364; n = 11 cells) of visible organelles exhibited fusion events during 0.5 s depolarizations (Fig. 3A and B and Supplemental movie 1). These events were observed at a rate of 1.19 s−1 (32 events in 27 s at 0 mV; n = 11 cells). By contrast, fusion events were much rarer during 2 s depolarizations to −25 mV (0.07 events s−1; 3 events; ICa= 22 ± 8 pA; n = 6 cells) and even rarer (1 event in 69 s; n = 11 cells) at −60 mV where calcium channels are closed (Heidelberger & Matthews, 1992; Tachibana et al. 1993).
Figure 3.
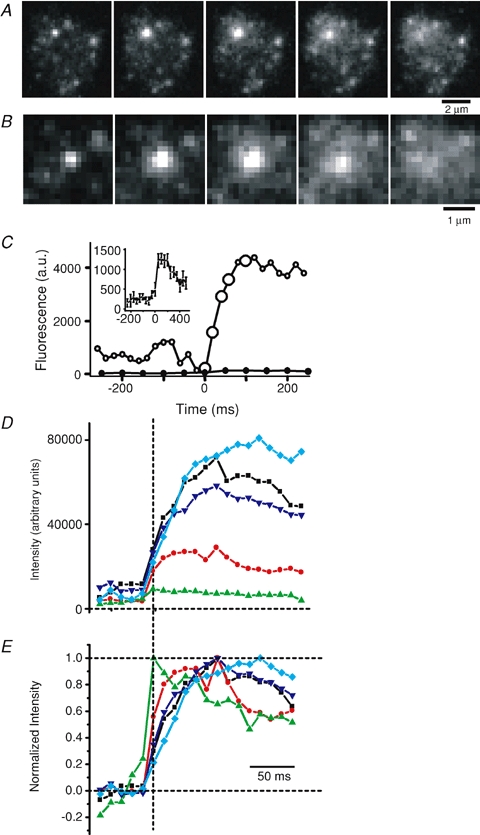
Stimulated exocytosis of endosomes A, sequence of images during exocytosis of an endosome in a terminal subjected to strong labelling. B, images in A magnified. C, total fluorescence (○) of a 1.7 μm × 1.7 μm square placed over the centre of the exocytic site in A, after subtracting a background image (see Methods). Origin is assigned to the first frame before exocytosis. Filled circles represent a similar analysis on 20 fusing organelles labelled using the vesicle labelling protocol. Inset shows these data on an expanded ordinate. D, total fluorescence of a 2.5 μm × 2.5 μm square placed over the centre of five exocytic events imaged at 83.3 Hz, after subtracting a background image (see Methods). Dashed vertical line indicates the first frame of exocytosis. Note that fluorescence peaked many frames after the initiation of exocytosis for some fusing organelles. E, same data as in D, normalized so that the average fluorescence during the five frames immediately preceding exocytosis before exocytosis is 0 and the peak fluorescence is 1. Same colours and symbols in D and E indicate the same exocytic event.
When fusing organelles were well separated both from the edge of the terminal and from other fusion events, it was possible to estimate the surface area of the fusing organelle. This was done by fitting a Gaussian function on an inclined plane to a region encompassing an endosome before exocytosis. The Gaussian function was taken to represent the fluorescence arising from the endosome and the residual after subtracting the Gaussian was taken as the local background. This local background was subtracted from each frame in the online supplemental movie of the endosome undergoing exocytosis and the integrated light after background subtraction was plotted against time as FM1-43 spilled from the endosome into the plasma membrane (Zenisek et al. 2002). The open symbols in Fig. 3C show the integrated light, whose value reaches a peak of about 45 000 pixel units. When plasma membrane was stained to equilibrium with the same concentration of FM1-43 that was also used for labelling endosomes (5 μm), the light from 1 nm2 of plasma membrane was 0.65 units. On this basis we estimate that the organelle in Fig. 3C had a surface area of 65 556 nm2. On average, organelles labelled with our strong labelling protocol had a surface area of 56 700 ± 12 100 nm2 if they fused within 10–20 min after labelling (n = 23 organelles in 8 cells). If spherical, the organelles had diameters of 134 nm. Organelles fusing more than 20 min after labelling had an average surface area of only 10 500 ± 170 nm2 (n = 7 organelles in 3 cells), corresponding to a diameter of only 57 nm. The decline with time is statistically significant (P < 0.05, t test). It is consistent with the idea that the organelles are broken down into smaller structures with time.
In parallel experiments, we measured the size of fusing organelles labelled with FM1-43 using our weak labelling protocol. A similar analysis (Fig. 3C, filled symbols and inset) yielded an average surface area of 3123 ± 408 nm2 (n = 22 vesicles, 5 cells). This value is similar to earlier results using the same loading protocol in a different population of synaptic terminals (2490 ± 300 nm2; Zenisek et al. 2000); and also similar to the size expected for a 30 nm spherical vesicle in terminals of bipolar cells (2827 nm2).
During exocytosis, fluorescence rises as FM1-43 escapes from the organelle into the plasma membrane, where it is better excited due to changes in its orientation and proximity to the coverslip, thus the time course of the fluorescence rise associated with exocytosis can be used to monitor the timing of dye escape (Zenisek et al. 2002). In the example in Fig. 3A–C, the fluorescence rise associated with exocytosis continued for ∼80 ms. Figure 3D shows the time course of fluorescence increase for five fusing organelles imaged at high rates (83.3 Hz) and aligned to the first frame of exocytosis determined visually (vertical dashed line). The same five fusion events are normalized to the peak fluorescence in Fig. 3E. Note that dye release from the organelles often continues for several frames. This is strikingly different from the time course of brightening observed following the weak load–long wash protocol, when organelles release their dye within 10 ms of exocytosis (Zenisek et al. 2002). In cells loaded using the strong stimulus–short wash protocol, we imaged 23 fusion events that were far away from the edge of the cell: of those, 14 reached maximum fluorescence within two images, and thus the time course of dye release was indistinguishable from instantaneous for these organelles. On average, these organelles had a surface area of 23 100 ± 4700 nm2, measured as described above. The remaining nine organelles released their dye more slowly and had an average surface of 125 500 ± 17 200 nm2, significantly larger than the fast releasing organelles (P < 0.0001). Evidently, the slow time course of dye release was more pronounced with the larger organelles.
Figure 4B shows the relationship between the timing of depolarization and exocytosis of all organelles labelled with our strong stimulus–short wash labelling protocol. The organelles observed to undergo exocytosis were 2.3- to 71-fold larger in surface area than expected for synaptic vesicles. Although all but one fusion event occurred during the depolarization, we found that endosomes underwent exocytosis only after a delay of > 80 ms. This result differs markedly from the exocytosis of small vesicles, which were labelled using the weak stimulus–long wash protocol (Fig. 4A). Small vesicles have the highest rate of exocytosis within 30 ms of the onset of a depolarization (Zenisek et al. 2002).
Figure 4.
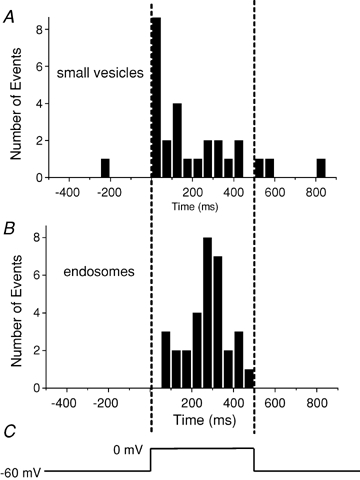
Times of fusion relative to a 0.5 s depolarization A, timing of exocytosis of presumptive synaptic vesicles loaded using protocol 1 followed by a 30–60 min wash, plotted as histogram. B, endosomes labelled using protocol 2. Vertical dashed lines indicate the beginning and end of the depolarization (C). Note that endosomes, but not synaptic vesicles, fuse after a delay.
Geometric size of endosomes
Spots seen after a short wait in strongly loaded terminals were clearly brighter than those after a long wait in weakly loaded terminals (Fig. 1A–C). Moreover, when they fused with the plasma membrane it became clear that they also contained more dye (Fig. 3C). In particular the latter finding suggests that the surface area of endosomes was larger than that of synaptic vesicles, provided that the lateral concentration of FM1-43 in the lipid bilayer does not change while endosomes are formed and processed (Murthy & Stevens, 1998). However, it is formally possible that endosomes rich in FM1-43 were no larger than synaptic vesicles, and instead resulted from a mechanism that concentrates FM1-43. This is not easily testable with a light microscope if the organelles are comparable in size or smaller than the point spread function of the microscope. At the signal/noise ratio achieved in our work, for example, the images of two vesicles 30 nm and 100 nm in diameter would be indistinguishable in size even though the latter has 10 times more surface area and may appear 10 times brighter.
In strong stimulus–short wash labelled terminals, some spots were nonetheless demonstrably larger than images of 20 nm beads labelled with FM1-43. An example is shown in Fig. 5, which plots the fluorescence as a function of distance from the centre, both for 20 nm beads (open circles) and for an endosome (filled circles). The beads were best fitted by Gaussian functions of average width (136 ± 22 nm, mean ±s.d.; n = 7 beads). The single organelle required a width of 231 nm, suggesting a diameter of 187 nm. The difference cannot generally be explained by noise. We expect that measurement uncertainty causes the width of bead images to appear > 180 nm, or 2 s.d.s larger than the mean, in 5% of the cases, and > 202 nm in 0.3% of the cases. By contrast, more than half of 16 strongly labelled organelles had a width larger than 180 nm, and nearly a third of them were wider than 202 nm. At least some organelles therefore are larger than 20 nm beads. They were also larger than synaptic vesicles. Images of synaptic vesicles stained by weak labelling required an average width (146 ± 5 nm, mean ±s.d., n = 14; see also Zenisek et al. 2002). In no case did a Gaussian fit indicate a width larger than 176 nm.
Figure 5.
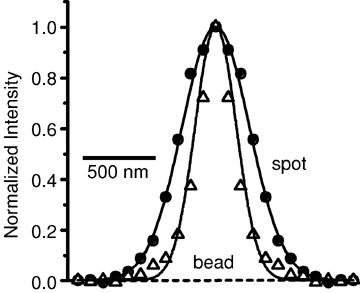
Some endosomes are larger than the diffraction limit Average fluorescence profiles (radial sweeps) of an endosome (•) and a 20 nm bead stained with FM1-43 (▵). Lines represent best-fit Gaussian curves through the data points. Radial sweep of endosome was taken from a single evanescent field image of a spot in a retinal bipolar cell terminal labelled using the strong labelling protocol, washed for 12 min and quenched using FM4-64. Bead profile was obtained from a single image of an FM1-43 stained bead. Note that bead chosen for this figure had a width (134 nm) similar to the mean for all beads we measured (136 ± 22 nm). The endosome was best fitted with a Gaussian with a width of 231 nm.
The analysis in Fig. 5 seeks to assay the geometric projection of organelles onto the image plane, in other words, their lateral size. Evanescent field illumination can provide information also about their vertical size, because the light emitted by a fluorescent molecule is steeply dependent on its distance from the coverslip on which the cell rests. After exocytosis, FM1-43 in the membrane of an exocytosing vesicle spreads into the plasma membrane (Fig. 3A–C and Zenisek et al. 2002), thereby approaching the coverslip, moving deeper into the evanescent field and experiencing stronger illumination. This is one of two reasons why fluorescence increases after fusion (Fig. 3C), the other being that FM1-43 molecules in the plasma membrane have a more uniform orientation than in a closed structure such as a vesicle (Zenisek et al. 2002).
To reach the plasma membrane, the average FM1-43 molecule must move a greater distance in larger organelles. On fusion, therefore, the fluorescence from dye in a large vesicle should increase more strongly than in a smaller vesicle. Indeed, the relative increase in fluorescence during exocytosis (9.24 ± 1.67-fold; 23 spots in 8 cells) was significantly larger during fusions in strongly labelled terminals than in weakly labelled terminals (4.0 ± 0.88-fold, 22 spots in 5 cells) (P < 0.01). The effect cannot be attributed solely to a change in molecular orientation, since this change is expected regardless of vesicle size. We conclude that endosomes are larger both in the vertical and lateral directions.
Large organelles reside near the plasma membrane in spontaneously active bipolar cells
Figure 6A shows an example of an electron micrograph from a synaptic terminal of a dissociated bipolar cell kept in our normal extracellular solution prior to fixation. As previously shown (Paillart et al. 2003; Holt et al. 2004), near-spherical membrane-bound organelles larger than synaptic vesicles were readily apparent near the plasma membrane. Figure 6B shows consecutive sections taken from the same cell.
Figure 6.
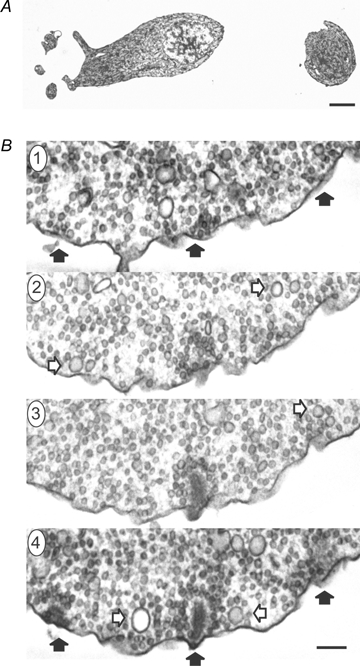
Large diameter vesicles are present in synaptic terminals of bipolar cells A shows an electron micrograph of an isolated bipolar cell whose dendrites and soma are positioned on the left, and the synaptic terminal is on the right (the axon is not captured in this section) (scale bar: 2 μm). B, four consecutive thin sections (thickness of 60 nm) through a synaptic terminal highlight large vesicles captured at their ‘widest’ diameter (open arrows). The filled black arrows in section 4 point to three synaptic ribbons, and the same regions are marked in section 1 to help orientate the viewer (the scale bar in B is 150 nm). Many of the large vesicles are near the plasma membrane and ribbons. For instance, note the two large vesicles surrounding the ribbon in the centre of section 4.
To measure the density and size of vesicles, the diameters of all vesicles within 250 nm of the plasma membrane were measured from EM sections. Figure 7 shows the distribution of vesicle sizes (outer diameter). A prominent peak is centred at ∼38 nm, which is similar to published estimates of synaptic vesicle diameter measured from goldfish dipolar cells (Lagnado et al. 1996). Additional larger sized vesicles skew the distribution to the right. Since the EM sections were judged to be ∼60 nm in thickness, vesicles wider than 60 nm could potentially span multiple sections and lead to an overestimate of the number of large vesicles and an underestimate of their true size; therefore, vesicles over 60 nm were analysed in serial sections to determine their widest diameter (see Methods). The average diameter of all vesicles over 60 nm was 90.50 ± 3.11 nm (±s.e.m.; n = 27 vesicles from 3 cells).
Figure 7.
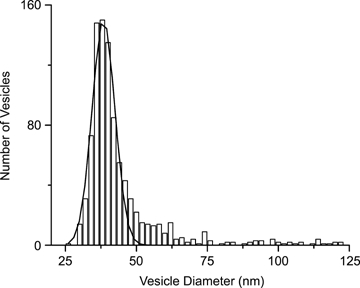
Electron microscopic images were used to measure the outer diameter of vesicles within 250 nm of the plasma membrane The distribution plotted here shows a prominent peak centred at ∼38 nm, and the peak is fitted with a single Gaussian to highlight the small vesicles. The plot also reveals larger diameter vesicles that significantly skew the distribution to the right. Three cells and 7 sections were analysed, yielding a total of 934 analysed vesicles.
To estimate the contribution of large vesicles to global measurements of exocytosis, we counted the number of membrane-bound organelles with a diameter greater than 60 nm that resided within 60 nm of the plasma membrane. We found that, on average, there were 3.52 large vesicles (> 60 nm diameter) per square micron of plasmalemma, meaning that a 10 μm diameter terminal would be expected to have 1107 spherical organelles with a diameter greater than 60 nm within 60 nm of the membrane. These organelles had a surface area (measured to middle of bilayer) of 0.024 μm2 on average, which would yield ∼27 μm2. If one assumes a specific capacitance of 10 fF μm−2, this would be equivalent to 270 fF of capacitance.
Discussion
Our results show that large membrane-bound organelles in the synaptic terminal of retinal bipolar neurons are capable of stimulated exocytosis. These organelles stain with FM1-43, were once surface exposed and therefore we designate as endosomes. Four lines of evidence indicate that the bright structures observed here represent organelles that are larger than synaptic vesicles. (1) The brighter fluorescence emerging from these organelles after exocytosis is most easily accounted for by a larger surface area. (2) After fusion, fluorescence from bright structures increases by a greater relative amount than from synaptic vesicles. (3) Dye is released more slowly from the brighter objects during exocytosis than from dimmer objects. (4) Gaussian fits to the organelles prior to exocytosis yielded spots, which in many cases were measurably larger than the image of a 20 nm bead.
The structures described here are probably identical to the cisternae and/or endosomes formed as a consequence of bulk endocytosis in some ribbon synapses following strong depolarizing stimuli (Lenzi et al. 2002; Holt et al. 2003, 2004; Paillart et al. 2003; but see also Rea et al. 2004). Using electron microscopy in bipolar neurons, Paillart et al. (2003) have demonstrated that most endocytosis following calcium action potentials or stimulation with elevated [K+] occurs via endosomes, which are thought to bud off directly from the plasma membrane. Indeed, under stimulation conditions similar to our strong stimulus protocol (50 mm KCl for 15–45 s), they found that 30 s after the stimulus most endocytosed membrane was taken into organelles, which were mostly larger than synaptic vesicles and often greater than 100 nm in diameter (see Fig. 6 in Paillart et al. 2003). Even in the absence of a depolarizing stimulus (although during spontaneous action potentials), the authors found internalization of a surface marker mostly in endosome-like structures. Similarly, Holt et al. (2003, 2004) found a major role for bulk endocytic mechanisms during stimulation with elevated potassium using light (Holt et al. 2003) and electron (Holt et al. 2004) microscopy techniques. The latter study estimated that 90% of all endocytosed membrane was taken up by bulk endocytosis following a 1 min depolarization with 50 mm KCl.
Since cisternae form soon after endocytosis, and in some cells even appear connected with the external space (Takei et al. 1996; Richards et al. 2000; Teng & Wilkinson, 2000), it is likely that they form directly from the plasma membrane without the benefit of the sorting provided by clathrin-mediated endocytosis (Lenzi et al. 2002; Holt et al. 2003, 2004; Paillart et al. 2003). Hence, it has been proposed that cisternae are a mixture of plasma membrane and vesicle membrane and probably contain proteins belonging to both compartments (Miller & Heuser, 1984; Takei et al. 1996). When synaptic vesicles bud off cisternae and presumably take with them their full complement of membrane proteins, they leave behind structures containing mostly surface membrane components (Miller & Heuser, 1984). Although diminished in size, the cisternae are still larger than synaptic vesicles. The exocytosis observed here returns them to the plasma membrane. Since endosomes are likely to at least transiently contain vesicular neurotransmitter transporters, exocytosis of endosomes may be expected to give rise to a postsynaptic response. Consistent with this idea, after tetanic stimulation or treatment with vinblastine, giant miniature endplate potentials (mEPPs) have been observed at the frog neuromuscular junction; these giant mEPPs have been suggested to arise from exocytosis of endosomes (Heuser, 1974; Pecot-Dechavassine, 1976). It is noteworthy that large mEPSCs have also been reported postsynaptic to retinal bipolar neurons (Gao & Wu, 1999).
Exocytosis of endosomes at physiological membrane potentials
In this study, bipolar neurons were specifically selected for giant synaptic terminals (8–12 μm in diameter). Morphological studies indicate that these neurons most probably belong to the Mb class of mixed-input on-type bipolar neurons (Ishida et al. 1980; Sherry & Yazulla, 1993). Typically, on-type bipolar cells operate over membrane potentials between −45 mV and −15 mV (Werblin & Dowling, 1969; Ashmore & Falk, 1980; Lasansky, 1992; Protti et al. 2000), thus depolarizations to 0 mV, while typical for studies of exocytosis in these neurons, are outside of the normal operating range of retinal bipolar neurons. Bipolar cells depolarized to −25 mV or held at −60 mV and in the absence of a patch pipette also exhibited endosomal fusion events indicating that these events also occur within the normal voltage range of retinal bipolar neurons, albeit less frequently than at 0 mV. It is important to note that the events described here were limited to those organelles labelled at one point in time and endocytic structures larger than synaptic vesicles are commonly observed at the electron microscope level, even in spontaneously firing cells (Paillart et al. 2003; Holt et al. 2004). Since bulk loading seems to be elicited specifically by strong stimuli (Holt et al. 2003), the presence of large organelles in unstimulated cells probably results in response to spontaneous calcium action potentials (Burrone & Lagnado, 1997; Zenisek & Matthews, 1998), which can give rise to large amounts of membrane turnover (Burrone & Lagnado, 1997).
Consequences for studying membrane recycling
Our results also demonstrate that synaptic vesicles are not the only FM1-43 stained organelles undergoing exocytosis. The stimulated release of FM1-43 is often equated with the exocytosis of synaptic vesicles in studies using either epifluorescence and/or confocal microscopy. Our results indicate that this assumption may not be correct under all conditions. Similarly, membrane capacitance measurements are largely assumed to arise solely from synaptic vesicle exocytosis. Given that endosomes have been observed near the membrane in ribbon-type presynaptic terminals not exposed to external depolarizing stimuli (Lenzi et al. 2002; Paillart et al. 2003; Holt et al. 2004), our results also suggest that this assumption may not always hold true.
The degree to which endosome fusion will contaminate measurements of exocytosis will be dependent upon several factors including the activity of the cell prior to establishing whole-cell recordings, the synapse being studied and the stimulus conditions used to evoke exocytosis. Our results indicate that endosome fusion events in goldfish bipolar cells are most prevalent between 200 and 400 ms after the initiation of a step depolarization to 0 mV and indicate that briefer depolarizations or depolarizations to lower membrane potentials are unlikely to be significantly contaminated by endosomal fusion events. We can estimate the potential impact of such events on capacitance measurements if we assume that all of the large (> 60 nm diameter) membrane-bound structures we observe at the electron microscopy level are capable of exocytosis and fuse at the same rate we observe for endosomes labelled with FM1-43. We find that in each square micron, 3.52 of these organelles reside within 60 nm of the plasma membrane, roughly equivalent to the depth of the evanescent field used in these experiments. Summed together, these organelles contain 27 μm2 of surface area. Since we find that 9% of visible endosomes fuse during a 0.5 s depolarization to 0 mV, our results suggest that endosomes could give rise to a capacitance increase of 24 fF during such stimuli. By comparison, a 500 ms depolarization to 0 mV evokes a capacitance jump of approximately 150 fF (von Gersdorff & Matthews, 1994). It is important to note that our estimate is probably an upper limit on the potential contribution, since some large membrane-bound organelles observed near the membrane by electron microscopy may not be fusion competent.
Acknowledgments
This work was supported by National Institutes of Health grants EY014990 (D.Z.) and MH60600 (W.A.) and grants from the McKnight and Kinship Foundations (D.Z.).
Supplementary material
Online supplemental material for this paper can be accessed at:
http://jp.physoc.org/cgi/content/full/jphysiol.2007.140848/DC1
and
http://www.blackwell-synergy.com/doi/suppl/10.1113/jphysiol.2007.140848
References
- An S, Zenisek D. Regulation of exocytosis in neurons and neuroendocrine cells. Curr Opin Neurobiol. 2004;14:522–530. doi: 10.1016/j.conb.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Andrews NW. Lysosomes and the plasma membrane: trypanosomes reveal a secret relationship. J Cell Biol. 2002;158:389–394. doi: 10.1083/jcb.200205110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashmore JF, Falk G. Responses of rod bipolar cells in the dark-adapted retina of the dogfish, Scyliorhinus canicula. J Physiol. 1980;300:115–150. doi: 10.1113/jphysiol.1980.sp013155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelrod D. Total internal reflection fluorescence microscopy in cell biology. Traffic. 2001;2:764–774. doi: 10.1034/j.1600-0854.2001.21104.x. [DOI] [PubMed] [Google Scholar]
- Betz WJ, Bewick GS. Optical analysis of synaptic vesicle recycling at the frog neuromuscular junction. Science. 1992;255:200–203. doi: 10.1126/science.1553547. [DOI] [PubMed] [Google Scholar]
- Burrone J, Lagnado L. Electrical resonance and Ca2+ influx in the synaptic terminal of depolarizing bipolar cells from the goldfish retina. J Physiol. 1997;505:571–584. doi: 10.1111/j.1469-7793.1997.571ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange RP, de Roos AD, Borst JG. Two modes of vesicle recycling in the rat calyx of Held. J Neurosci. 2003;23:10164–10173. doi: 10.1523/JNEUROSCI.23-31-10164.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried RC, Blaustein MP. Retrieval and recycling of synaptic vesicle membrane in pinched-off nerve terminals (synaptosomes) J Cell Biol. 1978;78:685–700. doi: 10.1083/jcb.78.3.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gad H, Low P, Zotova E, Brodin L, Shupliakov O. Dissociation between Ca2+-triggered synaptic vesicle exocytosis and clathrin-mediated endocytosis at a central synapse. Neuron. 1998;21:607–616. doi: 10.1016/s0896-6273(00)80570-x. [DOI] [PubMed] [Google Scholar]
- Gao F, Wu SM. Multiple types of spontaneous excitatory synaptic currents in salamander retinal ganglion cells. Brain Res. 1999;821:487–502. doi: 10.1016/s0006-8993(99)01067-7. [DOI] [PubMed] [Google Scholar]
- Grabner CP, Price SD, Lysakowski A, Fox AP. Mouse chromaffin cells have two populations of dense core vesicles. J Neurophysiol. 2005;94:2093–2104. doi: 10.1152/jn.00316.2005. [DOI] [PubMed] [Google Scholar]
- Heidelberger R, Matthews G. Calcium influx and calcium current in single synaptic terminals of goldfish retinal bipolar neurons. J Physiol. 1992;447:235–256. doi: 10.1113/jphysiol.1992.sp019000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser JE. A possible origin of the ‘giant’ spontaneous potentials that occur after prolonged transmitter release at the frog neuromuscular junctions. J Physiol. 1974;293:106–108P. doi: 10.1113/jphysiol.1974.sp010593. [DOI] [PubMed] [Google Scholar]
- Heuser JE, Reese TS. Evidence for recycling of synaptic vesicle membrane during transmitter release at the frog neuromuscular junction. J Cell Biol. 1973;57:315–344. doi: 10.1083/jcb.57.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt M, Cooke A, Neef A, Lagnado L. High mobility of vesicles supports continuous exocytosis at a ribbon synapse. Curr Biol. 2004;14:173–183. doi: 10.1016/j.cub.2003.12.053. [DOI] [PubMed] [Google Scholar]
- Holt M, Cooke A, Wu MM, Lagnado L. Bulk membrane retrieval in the synaptic terminal of retinal bipolar cells. J Neurosci. 2003;23:1329–1339. doi: 10.1523/JNEUROSCI.23-04-01329.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida AT, Stell WK, Lightfoot DO. Rod and cone inputs to bipolar cells in goldfish retina. J Comp Neurol. 1980;191:315–335. doi: 10.1002/cne.901910302. [DOI] [PubMed] [Google Scholar]
- Kristensson K. Retrograde axonal transport of horseradish peroxidase. Uptake at mouse neuromuscular junctions following systemic injection. Acta Neuropathol (Berl) 1977;38:143–147. doi: 10.1007/BF00688561. [DOI] [PubMed] [Google Scholar]
- Lagnado L, Gomis A, Job C. Continuous vesicle cycling in the synaptic terminal of retinal bipolar cells. Neuron. 1996;17:957–967. doi: 10.1016/s0896-6273(00)80226-3. [DOI] [PubMed] [Google Scholar]
- Lasansky A. Properties of depolarizing bipolar cell responses to central illumination in salamander retinal slices. Brain Res. 1992;576:181–196. doi: 10.1016/0006-8993(92)90679-4. [DOI] [PubMed] [Google Scholar]
- LaVail JH, LaVail MM. The retrograde intraaxonal transport of horseradish peroxidase in the chick visual system: a light and electron microscopic study. J Comp Neurol. 1974;157:303–357. doi: 10.1002/cne.901570304. [DOI] [PubMed] [Google Scholar]
- Lenzi D, Crum J, Ellisman MH, Roberts WM. Depolarization redistributes synaptic membrane and creates a gradient of vesicles on the synaptic body at a ribbon synapse. Neuron. 2002;36:649–659. doi: 10.1016/s0896-6273(02)01025-5. [DOI] [PubMed] [Google Scholar]
- McNeil PL, Miyake K, Vogel SS. The endomembrane requirement for cell surface repair. Proc Natl Acad Sci U S A. 2003;100:4592–4597. doi: 10.1073/pnas.0736739100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marxen M, Volknandt W, Zimmermann H. Endocytic vacuoles formed following a short pulse of K+-stimulation contain a plethora of presynaptic membrane proteins. Neuroscience. 1999;94:985–996. doi: 10.1016/s0306-4522(99)00351-6. [DOI] [PubMed] [Google Scholar]
- Miller TM, Heuser JE. Endocytosis of synaptic vesicle membrane at the frog neuromuscular junction. J Cell Biol. 1984;98:685–698. doi: 10.1083/jcb.98.2.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy VN, Stevens CF. Synaptic vesicles retain their identity through the endocytic cycle. Nature. 1998;392:497–501. doi: 10.1038/33152. [DOI] [PubMed] [Google Scholar]
- Paillart C, Li J, Matthews G, Sterling P. Endocytosis and vesicle recycling at a ribbon synapse. J Neurosci. 2003;23:4092–4099. doi: 10.1523/JNEUROSCI.23-10-04092.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecot-Dechavassine M. Action of vinblastine on the spontaneous release of acetylcholine at the frog neuromuscular junction. J Physiol. 1976;261:31–48. doi: 10.1113/jphysiol.1976.sp011547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protti DA, Flores-Herr N, von Gersdorff H. Light evokes Ca2+ spikes in the axon terminal of a retinal bipolar cell. Neuron. 2000;25:215–227. doi: 10.1016/s0896-6273(00)80884-3. [DOI] [PubMed] [Google Scholar]
- Rea R, Li J, Dharia A, Levitan ES, Sterling P, Kramer RH. Streamlined synaptic vesicle cycle in cone photoreceptor terminals. Neuron. 2004;41:755–766. doi: 10.1016/s0896-6273(04)00088-1. [DOI] [PubMed] [Google Scholar]
- Richards DA, Guatimosim C, Betz WJ. Two endocytic recycling routes selectively fill two vesicle pools in frog motor nerve terminals. Neuron. 2000;27:551–559. doi: 10.1016/s0896-6273(00)00065-9. [DOI] [PubMed] [Google Scholar]
- Rouze NC, Schwartz EA. Continuous and transient vesicle cycling at a ribbon synapse. J Neurosci. 1998;18:8614–8624. doi: 10.1523/JNEUROSCI.18-21-08614.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai T. Relation between thickness and interference colors of biological ultrathin section. J Electron Microsc (Tokyo) 1980;29:369–375. [PubMed] [Google Scholar]
- Sankaranarayanan S, Ryan TA. Real-time measurements of vesicle-SNARE recycling in synapses of the central nervous system. Nat Cell Biol. 2000;2:197–204. doi: 10.1038/35008615. [DOI] [PubMed] [Google Scholar]
- Sherry DM, Yazulla S. Goldfish bipolar cells and axon terminal patterns: a Golgi study. J Comp Neurol. 1993;329:188–200. doi: 10.1002/cne.903290204. [DOI] [PubMed] [Google Scholar]
- Tachibana M, Okada T, Arimura T, Kobayashi K, Piccolino M. Dihydropyridine-sensitive calcium current mediates neurotransmitter release from bipolar cells of the goldfish retina. J Neurosci. 1993;13:2898–2909. doi: 10.1523/JNEUROSCI.13-07-02898.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei K, Mundigl O, Daniell L, De Camilli P. The synaptic vesicle cycle: a single vesicle budding step involving clathrin and dynamin. J Cell Biol. 1996;133:1237–1250. doi: 10.1083/jcb.133.6.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teichberg S, Holtzman E, Crain SM, Peterson ER. Circulation and turnover of synaptic vesicle membrane in cultured fetal mammalian spinal cord neurons. J Cell Biol. 1975;67:215–230. doi: 10.1083/jcb.67.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng H, Cole JC, Roberts RL, Wilkinson RS. Endocytic active zones: hot spots for endocytosis in vertebrate neuromuscular terminals. J Neurosci. 1999;19:4855–4866. doi: 10.1523/JNEUROSCI.19-12-04855.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng H, Wilkinson RS. Clathrin-mediated endocytosis near active zones in snake motor boutons. J Neurosci. 2000;20:7986–7993. doi: 10.1523/JNEUROSCI.20-21-07986.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Gersdorff H, Matthews G. Dynamics of synaptic vesicle fusion and membrane retrieval in synaptic terminals. Nature. 1994;367:735–739. doi: 10.1038/367735a0. [DOI] [PubMed] [Google Scholar]
- Werblin FS, Dowling JE. Organization of the retina of the mudpuppy, Necturus maculosus. II. Intracellular recording. J Neurophysiol. 1969;32:339–355. doi: 10.1152/jn.1969.32.3.339. [DOI] [PubMed] [Google Scholar]
- Zenisek D, Matthews G. Calcium action potentials in retinal bipolar neurons. Vis Neurosci. 1998;15:69–75. doi: 10.1017/s0952523898151064. [DOI] [PubMed] [Google Scholar]
- Zenisek D, Steyer JA, Almers W. Transport, capture and exocytosis of single synaptic vesicles at active zones. Nature. 2000;406:849–854. doi: 10.1038/35022500. [DOI] [PubMed] [Google Scholar]
- Zenisek D, Steyer JA, Feldman ME, Almers W. A membrane marker leaves synaptic vesicles in milliseconds after exocytosis in retinal bipolar cells. Neuron. 2002;35:1085–1097. doi: 10.1016/s0896-6273(02)00896-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.