

Abstract
Nicotinic acetylcholine receptors (nAChRs) are in the superfamily of Cys-loop ligand-gated ion channels, and are pentameric assemblies of five subunits, with each subunit arranged around the central ion-conducting pore. The binding of ACh to the extracellular interface between two subunits induces channel opening. With the recent 4 Å resolution of the Torpedo nAChR, and the crystal structure of the related molluscan ACh binding protein, much has been learned about the structure of the ligand binding domain and the channel pore, as well as major structural rearrangements that may confer channel opening. For example, the putative pathway coupling agonist binding to channel gating may include a major rearrangement of the C-loop within the ligand binding pocket, and the disruption of a salt bridge between an arginine residue at the end of the β10 strand and a glutamate residue in the β1–β2 linker. Here we will review and discuss the latest structural findings aiming to further refine the transduction pathway linking binding to gating for the nAChR channels, and discuss similarities and differences among the different members of this Cys-loop superfamily of receptors.
The structure of the nAChR ligand binding domain
Nicotinic acetylcholine receptors (nAChRs) are in the superfamily of Cys-loop ligand-gated ion channels that also include the serotonin 5-HT3, GABAA and GABAC, and glycine receptors. These channels are pentameric assemblies of five subunits that can be either homomeric (α subunits only) or heteromeric (both α and β for neuronal receptors and α, β, γ and δ for muscle or Torpedo receptors) (for review see Corringer et al. 2000; Giniatullin et al. 2005; Unwin, 2005; Sine & Engel, 2006). From decades of investigation, there is a good deal known about the structure and function of nAChRs using a variety of techniques including: electron microscopy (on 2D arrays of receptors from Torpedo marmorata), biochemistry, chemical labelling, site-directed mutagenesis and electrophysiology (for review see Lester et al. 2004; Unwin, 2005). However, two major advancements in the last few years have significantly increased our understanding of the structure/function characteristics of the nAChR ligand binding domain (LBD). First, the cloning and characterization of the molluscan ACh-binding protein (AChBP: Brejc et al. 2001; Smit et al. 2001) was a landmark event. The AChBP was found to be a pentamer analogous to the extracellular LBD of the Cys-loop family of receptors, and has the ability to bind nAChR ligands. The AChBP is a soluble protein and does not contain an ion channel pore or intracellular domains. However, when the AChBP is attached to the pore domain of the serotonin 5-HT3A receptor, ACh can activate the opening of this hybrid channel (Bouzat et al. 2004). The second major advancement has been the refined 4 Å resolution electron microscopy structure of the Torpedo nAChR (Unwin, 2005). This structure provided a complete picture of the nAChR in near-physiological conditions.
The major structural feature of the LBD is two sets of β strands sandwiched together with discrete loop regions between strands (Fig. 1). Two distinct structural regions appear to be responsible for ligand binding and subsequent transduction of receptor activation to the channel pore. First, the transmitter binding site, at the interface of two subunits, is composed of a pocket of aromatic and hydrophobic resides from both the principal and complimentary subunits, and is capped by the C-loop. Second, the transition zone is made up of several loops that come into close contact with the transmembrane region (transmembrane domains M1 to M4) including: the Cys-loop, the β1–β2 linker, the β8–β9 linker, the β10–M1 linker and the M2–M3 linker (from the transmembrane domain). These structural elements link the LBD to the pore region (M2) where gating of the channel is thought to occur (Fig. 1).
Figure 1.
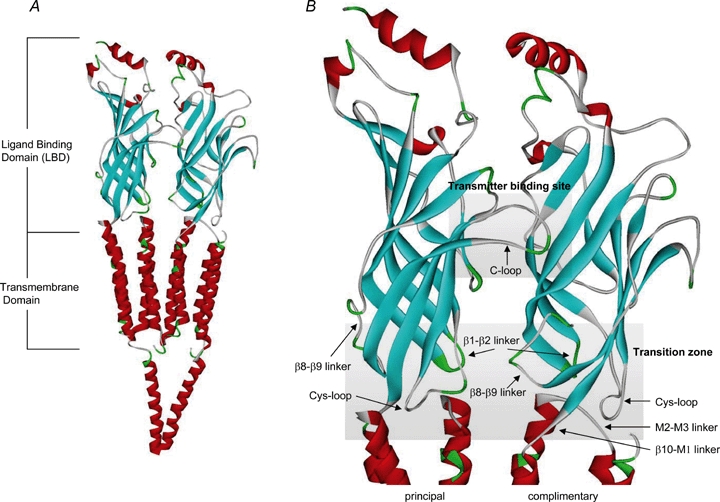
Ribbon diagrams of two subunits of the Torpedo nAChR (PDB:2BG9) A, the α helicies are shown in red and the β strands in blue. The extracellular ligand binding domain (LBD) and transmembrane domains are highlighted. B, two major structural elements of the LBD are shown: the transmitter binding site and the transition zone. The transmitter binding site is composed of a cluster of aromatic residues from both the principal and complimentary subunits and is capped by the C-loop. The transition domain consists of several loops including: Cys-loop, β1–β2 linker, β8–β9 linker, β10–M1 linker and the M2–M3 linker. These loops are involved in converting structural changes at the transmitter binding site down to the pore domain and inducing channel gating.
Structures of various states of the nAChR: closed, open and desensitized
To understand how the nAChR is functioning, it is important to know the structure of the receptor in its various states: the closed state (in the absence of agonist), the open state and the desensitized state (high-affinity ligand-bound but non-conducting state of the channel). The superposition of all crystal structures of the different species of AChBP to date, with a variety of ligands (both agonists and antagonists) and buffer molecules, fall into two groups (Dutertre & Lewis, 2006; Ulens et al. 2006). The first configuration seen with multiple toxin antagonists and some buffer molecules has the C-loop in an ‘open’ configuration, corresponding to the closed or resting state of the channel. The second configuration, seen when agonists are bound, has the C-loop in a ‘closed’ configuration, presumably the open or desensitized state of the receptor. In comparisons of agonist versus antagonist-bound crystal structures, the β1–β2 linker, the Cys-loop, and the β8–β9 linker all move, suggesting changes may be due to receptor activation. However, some variations may also result from differences between the species of AChBP used for crystallization (Unwin, 2005; Ulens et al. 2006). In addition, the inactive state of the Torpedo nAChR resembles antagonist-bound AChBP structures, with the C-loop in an open position, and similar relative locations of the β1–β2 linker and Cys-loop (Unwin, 2005).
Additionally, the currently accepted structure of the nAChR implies more global movements of the extracellular subunits. For the Torpedo nAChR, comparison of αversus non-α subunits indicated a clockwise rotation of ∼10 deg of the inner β strands. The two α subunits appear to be in a ‘distorted’ conformation, while the non-α subunits are in a more ‘relaxed’ conformation. Also, the non-α subunits appear to resemble the homomeric AChBP structures. Unwin suggests that upon agonist binding, the α subunits rotate to the non-α subunit conformation (creating a more uniform structure), and that this movement could lead to gating of the channel by displacing the β1–β2 linker, thereby affecting M2 and the channel pore (Unwin, 2005).
It has not yet been determined whether the AChBP structures with agonist bound are in either the open or desensitized receptor state, or even the agonist-bound closed state. It has been difficult to gain structural information for the nAChRs in the open versus desensitized state because after the addition of agonist, receptors open and then desensitize with very different rates (from milliseconds, to seconds and minutes) depending on the subtype of nAChRs (Giniatullin et al. 2005). However, the original Unwin structure (9 Å) is presumed to be in the open configuration because it was determined after only a 5 ms exposure to ACh (Unwin, 1995). In their comparison of multiple AChBP crystal structures, Dutertre & Lewis (2006) suggest that there are no noticeable structural differences between open and desensitized states. Ulens et al. (2006) attempted to distinguish the desensitized state of the AChBP by evaluating crystal structures containing α conotoxins that, according to functional data, were thought to favour the desensitized state of nAChRs, i.e. ImI and a mutant form of PnIA. However, crystals of these toxins with AChBP did not indicate significant differences from other antagonist-bound structures.
While we are beginning to understand the structural variations between different states, we still lack definitive structures for the open and desensitized states of the nAChR (including the double ligand-bound closed and open states that most likely correspond to the receptor conformations assayed in functional studies) and other members of the Cys-loop ligand-gated ion channel family. Finally, based on a variety of functional data, multiple open and desensitized receptor states are expected to exist. Therefore, it is unlikely that crystal structures alone will be able to provide a complete understanding of nAChR motion upon ligand binding and channel gating, and thus caution must be taken when interpreting the data.
Recently, Dellisanti et al. (2007) reported a 1.94 Å resolution crystal structure of the mouse muscle α1 nAChR subunit bound to α-bungarotoxin. This structure is of a mutated single monomer of the extracellular domain, and provides useful comparisons with previous nAChR and AChBP structures, as well as interesting new results suggesting additional important structural features. When comparing this structure to that of the structures of AChBP and the Torpedo nAChR, they superimpose very well. Consistent with functional studies, the structure of the α1 extracellular domain indicates that K145 and D200 have a direct electrostatic interaction in this probably closed state of the receptor. Amino acid interactions in the transition zone are also similar, including a close proximity between R209 and E45. In addition, two new structural features are highlighted, a water molecule buried in the core of the subunit, and a well-ordered carbohydrate chain on the outside of the α subunit. Functional studies mutating the hydrophilic amino acids that interact with the water molecule suggest that this cavity may be important in channel gating. In addition, single channel experiments with deglycosylated receptors suggest that the carbohydrate chain may also regulate channel gating, as well as α-bungarotoxin binding. Both of these findings provide new avenues of investigation into the molecular mechanism controlling nAChR function.
Structural transitions during gating
Both the more recent structural data highlighted above, and a myriad of functional data over the last two decades, have provided significant insight into the transition state of nAChRs. As discussed above, in the AChBP, the binding of agonist to the ligand binding pocket results in a major rearrangement of the C-loop into the closed or capped position. Depending on whether an agonist or antagonist binds, the C-loop can swing by as much as 11 Å (Hansen et al. 2005). After ligand binding, a conserved tyrosine residue (Y185, for Lymnaea AChBP) in the C-loop is drawn closer to a conserved lysine residue (K139) in the β7 strand (Fig. 2B), breaking or weakening a previous interaction between this lysine and an aspartate residue (D194) in the β10 strand (K145 and D200 in the mouse muscle α1 subunit) (Sine & Engel, 2006). A variety of functional data over the years has suggested that movements such as these around the transmitter binding domain might propagate through the rigid β strands to cause rearrangements within the transition zone. These in turn may interact with the M2–M3 linker to cause channel opening (Lester et al. 2004). Recently, Lee & Sine (2005) proposed that agonist binding to the muscle nAChR can lead to the disruption of a salt bridge between an arginine residue at the end of the β10 strand (R209) and a glutamate residue (E45) in the β1–β2 linker (Fig. 2C). These residues are conserved among the various members of the Cys-loop ligand-gated ion channel family, and therefore suggest a common transduction mechanism between ligand binding and channel gating (Corringer et al. 2000; Absalom et al. 2003; Kash et al. 2003, 2004; Schofield et al. 2004; Xiu et al. 2005; Mercado & Czajkowski, 2006). In addition, for the 5-HT3 receptor, Lummis et al. (2005) have proposed that structural changes induced by ligand binding lead to the cis–trans isomerization of a conserved proline residue (P*) on the M2–M3 linker and subsequent channel opening. However, although such a proline exists for nAChRs, no similar proline exists in either the GABA or glycine members of this superfamily (Kash et al. 2003, 2004). Instead, electrostatic and hydrophobic interactions might be responsible for gating of these receptors (Lee & Sine, 2005; Sine & Engel, 2006). Thus, it has been proposed that upon agonist binding, the C-loop is pulled into to a ‘closed’ position leading to an interaction between K139 and Y185, a disruption of a salt bridge between the β10 strand and the β1–β2 linker, followed by the isomerization of a proline residue on the M2–M3 linker, leading to channel opening (Sine & Engel, 2006).
Figure 2.
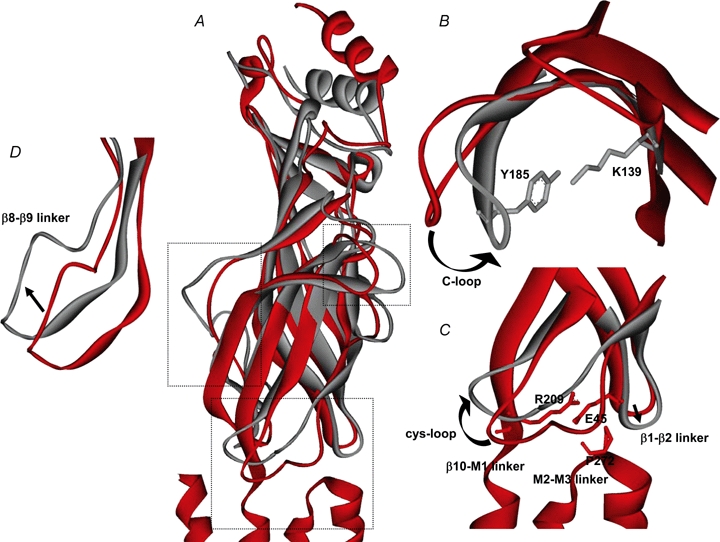
Possible movements within the ligand binding domain upon agonist binding A, the closed receptor state (Torpedo nAChR α subunit; PDB:2BG9) is in red and a possible active or desensitized receptor state (Lymnaea AChBP bound to carbamylcholine; PDB:1UV6) for the LBD is in grey. Boxes indicate areas that are expanded in B–D. B, upon agonist binding, the C-loop moves in toward the channel, bringing Y185 and K139 (AChBP) into close contact, breaking a previous interaction between K139 and D194 (not shown). C, movement of the β1–β2 linker and the β10–M1 linker leads to the disruption of a salt-bridge between E45 and R209 (Torpedo nAChR), allowing for the possible isomerization of P272 (Torpedo nAChR) in some Cys-loop receptors. In addition, the Cys-loop may shift position in the presence of agonist. D, the β8–β9 linker also is thought to shift in a clockwise direction. These cartoons are derived from overlays of current crystal structures and while these are suggested movements based on agonist binding to the nAChR, some of these changes could be species related.
Other regions of the receptor have also been proposed to be involved in the transition between ligand binding and channel gating (Lee & Sine, 2005; Mukhtasimova et al. 2005). For the chick α7 nAChR, various regions in the outer β strands were investigated (Lyford et al. 2003; McLaughlin et al. 2006). A glutamate residue in the β8–β9 linker (E172), a known site for modulation by divalent cations (Galzi et al. 1996), was found to undergo agonist-dependent movements during receptor activation (Lyford et al. 2003; Sine & Engel, 2006). Similarly, using fluorescence anisotropy decay to study the segmental motion of side chains in AChBP, Hibbs et al. (2006) demonstrated that agonists (but not antagonists) induced changes in conformational dynamics in the β8–β9 linker.
Above are highlights of recent insights into the structural mechanisms of receptor transitions during gating from an array of functional studies on nAChRs or other members in the Cys-loop ligand-gated ion channel family. While not covered in detail in this review, there have been a large number of studies combining a number of techniques (including receptor point mutations, ligand binding, pharmacological modifications and single channel analysis) that have provided information about possible transition states of nAChRs. It is important to understand that these functional studies give us a framework within which the structural data of various receptor states can be understood. For example, the extensive work of Auerbach and colleagues has provided an overall picture of the sequential nature of receptor gating for muscle nAChRs. By making point mutations in both the LBD and the pore domain, followed by measurements of rate-equilibrium free energy relationships (Zhou et al. 2005), they have been able to suggest blocks of coordinated motions starting with the β4–β5 linker, the β7–β8 linker, and the C-loop (which increases affinity for agonists), through the transition zone (the Cys-loop and the β1–β2 linker), to the pore region (M2) and gating of the channel (Grosman et al. 2000; Chakrapani et al. 2004; Purohit et al. 2007). This conformational wave propagates throughout the nAChR via Brownian motion in ∼1 μs (Grosman et al. 2000; Chakrapani & Auerbach, 2005), and provides a more complete view of nAChR gating than that which exists from crystal structures alone. In addition, functional studies have suggested movements that occur during the transition from the closed to open state of the nAChR that have yet to be identified through comparison of crystal structures (e.g. the β4–β5 linker, which contains the A-loop (Purohit et al. 2007)).
Structural transitions during desensitization
This review is focused primarily on movements within the extracellular domain of nAChRs during gating. However, there is a significant body of work that has focused on identifying key residues within the pore domain of nAChRs involved in desensitization, the high-affinity, ligand-bound but non-conducting state of the channel reached during sustained agonist application (Giniatullin et al. 2005). First of all, there are probably multiple functional desensitized states, the properties of which can depend on various factors including subunit make-up and modulation by local factors (including signal transduction cascades; Giniatullin et al. 2005). In addition, it is important to keep in mind that if receptor mutants display altered desensitization kinetics, this does not necessarily indicate that such sites are the structural motifs responsible for desensitization since macroscopic desensitization kinetics can be affected by not only the microscopic desensitization rate constants, but also by agonist binding and channel gating (Giniatullin et al. 2005).
Auerbach & Akk (1998) first suggested that there are two separate gates in the nAChR, a resting gate and a desensitization gate. Wilson & Karlin (2001), who were studying structural changes in mouse muscle nAChR in resting, open and desensitized states utilizing electrophysiology and site-directed mutagenesis, suggested a resting state gate near the mouth of the channel, and a desensitization gate further up into the M2 region near to the 9′ location. The 9′ location (residue L247 in the chick α7 nAChR), within the pore domain (M2), was previously shown to control fast desensitization (Revah et al. 1991; Giniatullin et al. 2005). The 9′ mutation dramatically decreased desensitization onset and increased agonist affinity, which was suggested to be due to the stabilization of a conducting, desensitized state of the receptor (Revah et al. 1991).
Besides the pore domain, regions in the extracellular domain of nAChRs might also be involved in receptor desensitization. Recently we found that mutating W55 of the rat α7 nAChR subunit to alanine dramatically slowed the rate of onset of desensitization; kinetic modelling indicated that the rate of transition of the receptor from the open to the desensitized state decreased by > 30-fold, and the rate of recovery from desensitization increased by ∼2-fold (Gay EA, Giniatullin R, Skorinkin A & Yakel J, unpublished data). Interestingly, this W residue is within the β2 strand and is considered one of the possible aromatic residues that make up the ligand binding pocket. Up until now, identification of individual residues involved in desensitization of the α7 nAChR has been limited to the pore domain. However, the use of chimeric receptors has demonstrated that regions in the extracellular domain of non-α7 nAChRs are involved in defining receptor desensitization kinetics (Bohler et al. 2001; Giniatullin et al. 2005). For the 5-HT3 receptor channel, Reeves et al. (2005) proposed that recovery from desensitization may require reformation of an interaction between the β1–β2 linker and the M2–M3 linker. Therefore, for the nAChRs, any structural change that interferes with the reformation of this β1–β2 linker/M2–M3 linker interaction might have an affect on the kinetics of desensitization.
Interestingly ImI and a mutant form of PnIA, which are antagonists at wildtype α7 receptors, can activate the non-desensitizing α7-L247T nAChR mutant, signifying that they may stabilize a ‘desensitized’ state of this receptor (Ulens et al. 2006). In addition, when exposed to these peptides, this 9′ mutant activates and then desensitizes in the continued presence of ligand. These data suggest there are multiple desensitized states of the α7 nAChR, one of which involves the 9′ location in the M2 pore domain, while others may involve different amino acids in the LBD such as W55.
Molecular dynamic simulations
A series of molecular dynamic simulations of the α7 nAChR have provided corroborating insight into the structural rearrangements that may occur during agonist binding and subsequent channel gating (Henchman et al. 2005; Law et al. 2005; Taly et al. 2005; Cheng et al. 2006). For example, simulations of a model α7 nAChR predict a closer interaction between K145 and Y188 (equivalent to K139 and Y185 in the AChBP; Fig. 2B) after C-loop closure. In addition when the C-loop moves into the ligand-bound conformation, this induced an up- and outward movement of the lower part of the β10 strand, which initially broke the salt-bridge between R206 and E45, but which then reformed a more stable hydrogen bond between these amino acids (Cheng et al. 2006). In this simulation, after about 4 ns, there was a ∼10 deg rotation of M2–M3 linker, resulting in an increase in pore size from ∼1.9 to ∼3.0 Å. Additionally, Henchman et al. (2005) observe global outward movements of the bottom half of the LBD with agonists. Overall, these simulations suggest that only small movements are necessary to produce significant changes in channel gating as the process is energy -efficient and easily modulated by agonist binding and unbinding.
In conclusion, the exact nature of the structure of the Cys-loop ligand-gated ion channel subunits, and the movements/rearrangements observed during and after ligand binding, gating and desensitization are still unknown. Nevertheless, a general hypothesis has emerged that indicates agonist binding induces closure of the C-loop, which is conveyed to the M2 pore region, resulting in channel opening. Thus, the transduction pathway involves many regions of the channel. With the continued use of a variety of experimental, structural and modelling techniques, including the recent crystallization of the extracellular domain of the mouse muscle α1 nAChR subunit (Dellisanti et al. 2007), major advances are expected in the near future.
Acknowledgments
We would like to thank C. Erxleben and S. Dudek for advice in preparing the manuscript. Research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences.
References
- Absalom NL, Lewis TM, Kaplan W, Pierce KD, Schofield PR. Role of charged residues in coupling ligand binding and channel activation in the extracellular domain of the glycine receptor. J Biol Chem. 2003;278:50151–50157. doi: 10.1074/jbc.M305357200. [DOI] [PubMed] [Google Scholar]
- Auerbach A, Akk G. Desensitization of mouse nicotinic acetylcholine receptor channels. A two-gate mechanism. J Gen Physiol. 1998;112:181–197. doi: 10.1085/jgp.112.2.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohler S, Gay S, Bertrand S, Corringer PJ, Edelstein SJ, Changeux JP, Bertrand D. Desensitization of neuronal nicotinic acetylcholine receptors conferred by N-terminal segments of the β2 subunit. Biochemistry. 2001;40:2066–2074. doi: 10.1021/bi0020022. [DOI] [PubMed] [Google Scholar]
- Bouzat C, Gumilar F, Spitzmaul G, Wang HL, Rayes D, Hansen SB, Taylor P, Sine SM. Coupling of agonist binding to channel gating in an ACh-binding protein linked to an ion channel. Nature. 2004;430:896–900. doi: 10.1038/nature02753. [DOI] [PubMed] [Google Scholar]
- Brejc K, Van Dijk WJ, Klaassen RV, Schuurmans M, Van Der Oost J, Smit AB, Sixma TK. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature. 2001;411:269–276. doi: 10.1038/35077011. [DOI] [PubMed] [Google Scholar]
- Chakrapani S, Auerbach A. A speed limit for conformational change of an allosteric membrane protein. Proc Natl Acad Sci U S A. 2005;102:87–92. doi: 10.1073/pnas.0406777102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrapani S, Bailey TD, Auerbach A. Gating dynamics of the acetylcholine receptor extracellular domain. J Gen Physiol. 2004;123:341–356. doi: 10.1085/jgp.200309004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Lu B, Grant B, Law RJ, McCammon JA. Channel opening motion of α7 nicotinic acetylcholine receptor as suggested by normal mode analysis. J Mol Biol. 2006;355:310–324. doi: 10.1016/j.jmb.2005.10.039. [DOI] [PubMed] [Google Scholar]
- Corringer PJ, Le Novere N, Changeux JP. Nicotinic receptors at the amino acid level. Annu Rev Pharmacol Toxicol. 2000;40:431–458. doi: 10.1146/annurev.pharmtox.40.1.431. [DOI] [PubMed] [Google Scholar]
- Dellisanti CD, Yao Y, Stroud JC, Wang ZZ, Chen L. Crystal structure of the extracellular domain of nAChR α1 bound to α-bungarotoxin at 1.94 A resolution. Nat Neurosci. 2007;10:953–962. doi: 10.1038/nn1942. [DOI] [PubMed] [Google Scholar]
- Dutertre S, Lewis RJ. Toxin insights into nicotinic acetylcholine receptors. Biochem Pharmacol. 2006;72:661–670. doi: 10.1016/j.bcp.2006.03.027. [DOI] [PubMed] [Google Scholar]
- Galzi JL, Bertrand S, Corringer PJ, Changeux JP, Bertrand D. Identification of calcium binding sites that regulate potentiation of a neuronal nicotinic acetylcholine receptor. EMBO J. 1996;15:5824–5832. [PMC free article] [PubMed] [Google Scholar]
- Giniatullin R, Nistri A, Yakel JL. Desensitization of nicotinic ACh receptors: shaping cholinergic signaling. Trends Neurosci. 2005;28:371–378. doi: 10.1016/j.tins.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Grosman C, Zhou M, Auerbach A. Mapping the conformational wave of acetylcholine receptor channel gating. Nature. 2000;403:773–776. doi: 10.1038/35001586. [DOI] [PubMed] [Google Scholar]
- Hansen SB, Sulzenbacher G, Huxford T, Marchot P, Taylor P, Bourne Y. Structures of Aplysia AChBP complexes with nicotinic agonists and antagonists reveal distinctive binding interfaces and conformations. EMBO J. 2005;24:3635–3646. doi: 10.1038/sj.emboj.7600828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henchman RH, Wang HL, Sine SM, Taylor P, McCammon JA. Ligand-induced conformational change in the α7 nicotinic receptor ligand binding domain. Biophys J. 2005;88:2564–2576. doi: 10.1529/biophysj.104.053934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbs RE, Johnson DA, Shi J, Taylor P. Structural dynamics of the acetylcholine binding protein: hydrodynamic and fluorescence anisotropy decay analyses. J Mol Neurosci. 2006;30:73–74. doi: 10.1385/JMN:30:1:73. [DOI] [PubMed] [Google Scholar]
- Kash TL, Jenkins A, Kelley JC, Trudell JR, Harrison NL. Coupling of agonist binding to channel gating in the GABAA receptor. Nature. 2003;421:272–275. doi: 10.1038/nature01280. [DOI] [PubMed] [Google Scholar]
- Kash TL, Kim T, Trudell JR, Harrison NL. Evaluation of a proposed mechanism of ligand-gated ion channel activation in the GABAA and glycine receptors. Neurosci Lett. 2004;371:230–234. doi: 10.1016/j.neulet.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Law RJ, Henchman RH, McCammon JA. A gating mechanism proposed from a simulation of a human α7 nicotinic acetylcholine receptor. Proc Natl Acad Sci U S A. 2005;102:6813–6818. doi: 10.1073/pnas.0407739102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WY, Sine SM. Principal pathway coupling agonist binding to channel gating in nicotinic receptors. Nature. 2005;438:243–247. doi: 10.1038/nature04156. [DOI] [PubMed] [Google Scholar]
- Lester HA, Dibas MI, Dahan DS, Leite JF, Dougherty DA. Cys-loop receptors: new twists and turns. Trends Neurosci. 2004;27:329–336. doi: 10.1016/j.tins.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Lummis SC, Beene DL, Lee LW, Lester HA, Broadhurst RW, Dougherty DA. Cis-trans isomerization at a proline opens the pore of a neurotransmitter-gated ion channel. Nature. 2005;438:248–252. doi: 10.1038/nature04130. [DOI] [PubMed] [Google Scholar]
- Lyford LK, Sproul AD, Eddins D, McLaughlin JT, Rosenberg RL. Agonist-induced conformational changes in the extracellular domain of a α7 nicotinic acetylcholine receptors. Mol Pharmacol. 2003;64:650–658. doi: 10.1124/mol.64.3.650. [DOI] [PubMed] [Google Scholar]
- McLaughlin JT, Fu J, Sproul AD, Rosenberg RL. Role of the outer β-sheet in divalent cation modulation of α7 nicotinic receptors. Mol Pharmacol. 2006;70:16–22. doi: 10.1124/mol.106.023259. [DOI] [PubMed] [Google Scholar]
- Mercado J, Czajkowski C. Charged residues in the α1 and β2 pre-M1 regions involved in GABAA receptor activation. J Neurosci. 2006;26:2031–2040. doi: 10.1523/JNEUROSCI.4555-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhtasimova N, Free C, Sine SM. Initial coupling of binding to gating mediated by conserved residues in the muscle nicotinic receptor. J Gen Physiol. 2005;126:23–39. doi: 10.1085/jgp.200509283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit P, Mitra A, Auerbach A. A stepwise mechanism for acetylcholine receptor channel gating. Nature. 2007;446:930–933. doi: 10.1038/nature05721. [DOI] [PubMed] [Google Scholar]
- Reeves DC, Jansen M, Bali M, Lemster T, Akabas MH. A role for the β1-β2 loop in the gating of 5-HT3 receptors. J Neurosci. 2005;25:9358–9366. doi: 10.1523/JNEUROSCI.1045-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revah F, Bertrand D, Galzi JL, Devillers-Thiery A, Mulle C, Hussy N, Bertrand S, Ballivet M, Changeux JP. Mutations in the channel domain alter desensitization of a neuronal nicotinic receptor. Nature. 1991;353:846–849. doi: 10.1038/353846a0. [DOI] [PubMed] [Google Scholar]
- Schofield CM, Trudell JR, Harrison NL. Alanine-scanning mutagenesis in the signature disulfide loop of the glycine receptor α1 subunit: critical residues for activation and modulation. Biochemistry. 2004;43:10058–10063. doi: 10.1021/bi036159g. [DOI] [PubMed] [Google Scholar]
- Sine SM, Engel AG. Recent advances in Cys-loop receptor structure and function. Nature. 2006;440:448–455. doi: 10.1038/nature04708. [DOI] [PubMed] [Google Scholar]
- Smit AB, Syed NI, Schaap D, Van Minnen J, Klumperman J, Kits KS, Lodder H, Van Der Schors RC, Van Elk R, Sorgedrager B, Brejc K, Sixma TK, Geraerts WP. A glia-derived acetylcholine-binding protein that modulates synaptic transmission. Nature. 2001;411:261–268. doi: 10.1038/35077000. [DOI] [PubMed] [Google Scholar]
- Taly A, Delarue M, Grutter T, Nilges M, Le Novere N, Corringer PJ, Changeux JP. Normal mode analysis suggests a quaternary twist model for the nicotinic receptor gating mechanism. Biophys J. 2005;88:3954–3965. doi: 10.1529/biophysj.104.050229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulens C, Hogg RC, Celie PH, Bertrand D, Tsetlin V, Smit AB, Sixma TK. Structural determinants of selective α-conotoxin binding to a nicotinic acetylcholine receptor homolog AChBP. Proc Natl Acad Sci U S A. 2006;103:3615–3620. doi: 10.1073/pnas.0507889103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unwin N. Acetylcholine receptor channel imaged in the open state. Nature. 1995;373:37–43. doi: 10.1038/373037a0. [DOI] [PubMed] [Google Scholar]
- Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J Mol Biol. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- Wilson G, Karlin A. Acetylcholine receptor channel structure in the resting, open, and desensitized states probed with the substituted-cysteine-accessibility method. Proc Natl Acad Sci U S A. 2001;98:1241–1248. doi: 10.1073/pnas.031567798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiu X, Hanek AP, Wang J, Lester HA, Dougherty DA. A unified view of the role of electrostatic interactions in modulating the gating of Cys loop receptors. J Biol Chem. 2005;280:41655–41666. doi: 10.1074/jbc.M508635200. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Pearson JE, Auerbach A. Φ-Value analysis of a linear, sequential reaction mechanism: theory and application to ion channel gating. Biophys J. 2005;89:3680–3685. doi: 10.1529/biophysj.105.067215. [DOI] [PMC free article] [PubMed] [Google Scholar]