

Abstract
We recently generated a mouse model of facioscapulohumeral muscular dystrophy (FSHD) by selectively overexpressing FRG1, a candidate gene for FSHD, in skeletal muscle. The muscles of the FRG-1 mice did not show any plasmamembrane defect suggesting a novel pathogenetic mechanism for FSHD. Here, we study structure and function of muscle fibres from three lines of mice overexpressing FRG1 at different levels: FRG1-low, FRG1-med, FRG1-high. Cross-sectional area (CSA), specific force (Po/CSA) and maximum shortening velocity (Vo) of identified types of muscle fibres from FRG1-low and FRG1-med mice were analysed and found to be lower than in WT mice. Fast fibres and especially type 2B fibres (the fastest type) were preferentially involved in the dystrophic process showing a much larger force deficit than type 1 (slow) fibres. Consistent with the latter observation, the MHC isoform distribution of several muscles of the three FRG1 lines showed a shift towards slower MHC isoforms in comparison to WT muscle. Moreover, fast muscles showed a more evident histological deterioration, a larger atrophy and a higher percentage of centrally nucleated fibres than the soleus, the slowest muscle in mice. Interestingly, loss in CSA, Po/CSA and Vo of single muscle fibres and MHC isoform shift towards a slower phenotype can be considered early signs of muscular dystrophy (MD). They were, in fact, found also in FRG1-low mice which did not show any impairment of function in vivo and of muscle size in vitro and in soleus muscles, which had a completely preserved morphology. This study provides a detailed characterization of structure and function of muscle fibres in a novel murine model of one of the main human MDs and suggests that fundamental features of the dystrophic process, common to most MDs, such as the intrinsic loss of contractile strength of muscle fibres, the preferential involvement of fast fibres and the shift towards a slow muscle phenotype can occur independently from obvious alterations of the plasma membrane.
Muscular dystrophies (MDs) are a group of very heterogeneous genetic diseases characterized by skeletal muscle wasting and weakness. Most MDs have been linked to mutations in genes coding for proteins of the dystrophin-glycoprotein complex (DGC) (Durbeej & Campbell, 2002) or for enzymes likely to be involved in post-translational modification of such proteins (Muntoni et al. 2004). Whatever the mutation involved, the pathogenesis of most MDs can be traced back to a common path: a primary defect in the DGC would alter the link between cytoskeleton, plasmamembrane and extracellular matrix and would make muscle fibres more susceptible to contraction-induced damage. The latter phenomenon may depend either on a mechanical damage of the membrane and/or on altered ion homeostasis across the membrane ultimately bringing to calcium influx in muscle fibres and activation of proteolysis (Allen et al. 2005).
On the contrary, the genetic defect of facioscapulohumeral muscular dystrophy (FSHD), which is one of the three most common MDs, does not reside in any mutated genes. FSHD has been causally related to deletion of tandemly arrayed 3.3 kb repeat units (D4Z4) on chromosome 4q35. It has been shown that three genes located upstream of D4Z4, FRG2, FRG1 and ANT1, which have not been found to be mutated in FSHD subjects, are overexpressed in FSHD-affected human muscles (Gabellini et al. 2002). Very recently we demonstrated that transgenic overexpression of FRG1 in skeletal muscle was sufficient to cause MD in mice, whereas mice overexpressing the two other candidate genes, FRG2And ANT-1, did not show clear signs of MD (Gabellini et al. 2006). Moreover, the link between MD manifestations and FRG1 overexpression could not be ascribed to a plasma membrane defect, suggesting a novel pathogenetic mechanism for FSHD. However, similarly to other mouse models of MD, mice overexpressing FRG1 (FSHD mice) showed impaired mobility in vivo and dystrophic features, which appeared differentially distributed among skeletal muscles.
In this respect, the analysis of the structure and function of muscle fibres from the FSHD mice is relevant for at least two reasons. It will further the understanding of a novel mouse model of one of the main human MDs which will be used in the future to further our knowledge on the pathogenesis of the disease and to develop therapeutic strategies. Furthermore, the FSHD mouse model may help understanding of whether and to what extent fundamental features of the dystrophic process at muscle fibre level, such as the intrinsic loss of contractile strength, the preferential involvement of fast fibres, and the fast to slow shift in muscle phenotype are necessarily linked to alterations of the DCG complex or can be a common path of alternative pathogenetic mechanisms. The FSHD mouse represents in fact one model of MD displaying histological and functional dystrophic features similar to the most studied models of MDs (Gabellini et al. 2006) which cannot be linked to a plasma membrane defect and ultimately to altered calcium homeostasis across the membrane (Allen et al. 2005). The absence of a clear plasmamembrane defect, as demonstrated by no change in serum creatine kinase (Richard et al. 2000) and lack in Evans blue uptake, is found in other murine models of MDs, as calpain-3 deficiency (Richard et al. 2000; Fougerousse et al. 2003). Nevertheless, murine models in which mutations do not involve proteins of the DCG complex either ultimately show membrane alterations such as the collagen VI deficient mouse (Bonaldo et al. 1998; Irwin et al. 2003) or do not show any clear sign of skeletal muscle involvement, such as the Six5 deficient mouse, a model of myotonic MD (Klesert et al. 2000; Sarkar et al. 2000). Notwithstanding the large body of information accumulating on murine models of MD, key features of the disease are still far from being fully understood.
In particular, muscle weakness is generally attributed to replacement of degenerating and necrotic muscle fibres with non contractile material (connective tissue and fat), which ultimately causes a loss of contractile muscle mass (‘quantitative mechanism’) even in the presence of increased muscle weight (pseudo-hypertrophy) (Watchko et al. 2002). Whether a ‘qualitative mechanism’, namely a loss of the intrinsic capacity to develop force by a given amount of muscle mass, is also involved in generating muscle weakness is unclear. In fact, whereas some earlier studies suggested no loss in specific force of single muscle fibres of mdx mice (Takagi et al. 1992; Lynch et al. 2000), more recently we consistently showed a clear Po/CSA deficit in several mouse models of MD: mdx (Denti et al. 2006), scid-mdx (Torrente et al. 2004), α-sarcoglycan null mice (Sampaolesi et al. 2003). Interestingly, in humans affected by Duchenne MD, whereas Fink et al. (1990) found an impairment of specific force of muscle fibres, Horowits et al. (1990) failed to show such impairment.
The origin of force loss in muscular dystrophies appears to be far from understood. In particular an imbalance between protein synthesis and breakdown towards increased proteolysis (McKeran et al. 1977; Elia et al. 1981; Warnes et al. 1981) and the replacement of necrotic cells by weaker regenerating fibres which show different functional characteristics from uninjured fibres may contribute to reduced capacity to generate force (Gregorevic et al. 2004). Furthermore, in intact fibres, decreased evoked calcium release due to ongoing triadic disruption might also contribute to force loss at least in mdx mice (Allen et al. 2005). In any case, in all muscular dystrophies the loss in force and specific force appear as common endpoints of diverse primary defects.
In addition, it is generally believed that the dystrophic processes preferentially strike fast fibres, and among fast fibres the fastest, type 2B fibres (Webster et al. 1988). However, such a preferential involvement has not been definitely demonstrated by the comparative analysis of strength of slow and fast muscle fibres.
As regards the origin of fibre deterioration in MDs, an increased calcium influx across membrane tears and calcium permeable leak channels followed by activation of proteolysis is a candidate mechanism (Allen et al. 2005; Yeung et al. 2005). Nevertheless it is still unclear what is the link between the genetic defect and muscle fibre deterioration, which are the cellular pathways involved and whether alternative mechanisms can be identified in the process (Gillis, 1999).
In this work, structure and function of identified types of single muscle fibres from the mouse model of FSHD were studied in comparison to corresponding types of WT fibres. The existence of several lines of FSHD, expressing FRG1 at different levels and showing a variable progression of MD, enabled us to relate structural and functional properties of muscle fibres to the progression of the disease. Results of this study (i) provide a detailed characterization of skeletal muscle fibres of a novel mouse model of one of the main human MDs and (ii) point toward the existence of alternative pathogenetic mechanisms, besides altered calcium homeostasis through the membrane and increased number of weaker regenerating fibres, which can give rise to fundamental features of the dystrophic process, common to other MDs, such as the intrinsic loss of contractile strength of muscle fibres, the preferential involvement of fast fibres and the shift towards a slow muscle phenotype.
Methods
Animals
Mice overexpressing FRG1 to variable extents under the control of the human skeletal α-actin (HSA) gene promoter were previously generated (Gabellini et al. 2006). Briefly, FRG1 open reading frames were PCR-amplified using the following primer: FRG1, HSA-FRG1-F 50-GA-TCTAGCGGCCGCCATGGCCGAGTACTCCTATGTGA-AGTCT-300. PCR products were sequenced and cloned into pBSX-HSAvpA, provided by J. Chamberlain. Transgene was excised from agarose gels and purified for microinjection into fertilized eggs recovered from C57BL/6 females crossed with C57BL/6 males. All procedures were performed at the University of Massachusetts Medical School Transgenic Animal Modelling Core Facility. In this work, we used the same three lines of mice we previously characterized (Gabellini et al. 2006) in which the transgene was overexpressed at levels within the range previously reported for FRG1 overexpression in FSHD patients (Gabellini et al. 2002): FRG1-low, FRG1-med, FRG1-high. In these lines RT-PCR and Southern blot analysis were used to quantify the FRG1 expression levels and the transgene copy number (3 for FRG1-low, 5 for FRG1-med; 9 for FRG1-high) which corresponded to 10- (FRG1-low), 30- (FRG1-med), and 50-fold (FRG1-high) over the endogenous level of the murine FRG1 gene (Gabellini et al. 2006).
This study was carried out on female FRG1 transgenic mice at 12 weeks of age; C57bL/6 mice of matched age served as controls. Animals were initially housed in the animal house facility of the Program in Gene Function and expression of the University of Massachusetts Medical School, under the care of the Department of Animal Medicine. At the age of 8 weeks mice were shipped (World Courier) to Italy avoiding extreme temperature during transportation. Once in Italy animals were individually housed in the animal house facility of the Department of Experimental Medicine (University of Pavia, Italy), maintained on a 12: 12 h light–dark cycle and given unlimited access to food (Dottori Piccioni Italy) and water for the duration of the study. On the day of the experiment, the animals were weighed and killed by cervical dislocation. Muscles (facials: masseter; respiratory: intercostals and diaphragm; anterior limbs: deltoids, biceps and triceps; posterior limbs: soleus, tibialis, vastus and gastrocnemius) were carefully dissected under a stereomicroscope, blotted on filter paper, weighed and stored for analysis. The experimental protocols were approved by the Animal Ethics Committee of the University of Pavia.
Morphological analysis
Muscle samples were included in OCT (Tissue-tek) embedding medium, frozen in liquid nitrogen and serial transverse sections (8 μm thick) were cut using a Leica CM 1850 cryostat. Sections were used for haematoxylin-eosin (H&E) staining, according to standard procedures (Gabellini et al. 2006). The percentage of centrally nucleated fibres was calculated in non-consecutive sections (at least five for each muscle) as (number of centrally nucleated fibres/total number of fibres considered) × 100 (no less than 600 for each group of mice, n = 5).
Analysis of force and velocity of single muscle fibres
Absolute (Po) and specific force (Po over the fibre cross-sectional area, Po/CSA) of single muscle fibres were measured in skinned preparations using an approach routinely used for years in our laboratory. All the procedures used have been previously described in detail (Bottinelli et al. 1991, 1996; D'Antona et al. 2006) except for the solutions which were modified. The skinning solution previously described (D'Antona et al. 2006) (150 mm potassium propionate, 5 mm KH2PO4, 5 mm magnesium acetate, 3 mm Na2ATP, 5 mm EGTA, pCa 9.0) was enriched with protease inhibitors (leupeptin 20 μm and E-64 10 μm). Pre-activating (PA) and activating (A) solutions had the following composition: PA: imidazole-propionate 10 mm, magnesium propionate 2.5 mm, potassium propionate 170 mm, K2-EGTA 5 mm; A: Imid-p 10 mm, magnesium propionate 2.2 mm, potassium phosphate 170 mm, K2-EGTA 0.11 mm, Ca-EGTA 4.8 mm. In both solutions, Na2ATP 5 mm and protease inhibitors leupeptin and E-64 were added. Ionic strength was 200 mm in all solutions. The pH of all solutions was set at 7.0.
Po was determined at pCa 4.5, 12°C and optimal sarcomeric length for force developing (2.5 μm) (Pellegrino et al. 2003). Po/CSA was expressed as kN m−2.
The set-up for single muscle fibre analysis was located on the stage of an inverted microscope which enabled us to view the specimen at ×320 magnification. CSA was determined, assuming a circular shape, from the mean of three diameters without correction for the swelling which is know to occur in demembranated specimens (Godt & Maughan, 1977). As discussed before (D'Antona et al. 2006), skeletal muscle fibres do not always have a circular cross-section and, due to the procedure used to mount the fibre in the set-up, only the largest of the two main diameters could be routinely measured. Therefore, an overestimation of CSA could occur and the degree of overestimation was bound to depend on the shape of the cross-section. To make sure that variation in the shape of the cross-section between WT and dystrophic fibres did not introduce a systematic error in the determination of CSA and affect the reliability of Po/CSA comparisons we used a procedure previously described (D'Antona et al. 2006). Briefly, immediately after dissection, muscles were frozen in liquid nitrogen for subsequent histological analysis. On such sections the larger and smaller diameter of a large population of muscle fibres from WT and FRG1-high mice were determined using freely available image analysis software (Scion Image, Scion Corp., USA). CSA was determined either using the largest diameter and assuming a circular shape or using both the largest and the smallest diameters and considering an elliptical shape for both WT and FRG1-med fibres. The ratio between the measurement based on a circular shape and the measurement based on an elliptical shape was not significantly different among WT (1.23 ± 0.05 n = 40 from 4 mice), FRG1-low (1.21 ± 0.07 n = 40) and FRG1-med (1.32 ± 0.06 n = 40 from 4 mice) fibres. As expected, the CSA determination assuming a circular shape somewhat underestimated Po/CSA, but could not introduce any systematic error in the comparison of Po/CSA (D'Antona et al. 2006).
To determine maximum shortening velocity (Vo), the slack test technique was employed (Bottinelli et al. 1996); Vo was expressed as fibre lengths per second (L s−1). In skinned fibres, disorder of the striation pattern can occur during maximal activation and this might affect determination of Vo. To ensure that specimens did not go through any significant sarcomere disorganization: (i) muscle fibres were viewed before, during and after each activation at ×320 and were discarded if non-uniformities were seen; (ii) fibres which went through a force loss of more than 10% between the first and the last (fifth–sixth) activation used to determine Vo were discarded even in the absence of clear non-uniformities. Indeed, as the fibre segments used for functional analysis were kept short (∼1.0 mm), were chosen on the basis of uniform CSA and were quickly and uniformly activated using a previously described procedure (Bottinelli et al. 1994), few fibres did not pass the quality controls and had to be discarded. The slack test manoeuvres enable us to determine the series compliance. As expected, series compliance ranged between 3.0% and 5.5% with no significant differences between corresponding fibre types of WT and FSHD mice.
At the end of the mechanical experiment, fibres were put in 20 μl of Leammli buffer (Laemmli, 1970) and stored at −20°C for MHC isoform content analysis.
Fibre typing and myosin isoform identification
The composition in myosin heavy chain (MHC) isoforms was determined in single fibres and in whole muscles by polyacrylamide-gel electrophoresis on 8% polyacrylamide slab gels after denaturation in sodium dodecyl sulphate (SDS-PAGE) using a procedure previously described (Bottinelli et al. 1994; Pellegrino et al. 2003). Four MHC isoforms (MHC-1, MHC-2A, MHC-2X and MHC-2B) could be separated on gels (Pellegrino et al. 2003). Single fibres were classified in pure fibre types (type 1, or slow, and type 2A, 2X and 2B or fast) and in hybrid fibre types (type 1–2A, 2AX and 2XB). Densitometric analysis of the MHC bands of whole muscle samples enabled us to assess their MHC isoform distribution (Harridge et al. 1996).
Treadmill
On the day of the experiment each mouse was placed on the belt of a six-lane motorized treadmill (Exer 3/6 Treadmill, Columbus Instruments, OH, USA) supplied with shocker plates which could be individually enabled or disabled for each lane. During each test the electrical stimulus was fixed at 200 ms duration, 0.34 mA amplitude and 1 Hz repetition rate. After a 10 min period of acclimation the treadmill was then started at 5 m min−1, 0 deg inclination for 5 min. After this period the speed was incrementally increased 1 m min−1 every minute until the mouse reached exhaustion. Exhaustion was defined as spending time on the shocker plate without attempting to re-engage the treadmill within 20 s. A video camera (Panasonic, NV-RX10EG) was positioned above the treadmill to record each test on a S-VHS video recorder (Toshiba V-729 W) at 25 frames s−1 and video recordings were subsequently used for analysis. The individual and the average time to exhaustion and velocity of running were measured.
Statistical analysis
Data were expressed as means ±s.e.m., unless otherwise stated. Statistical significance of the differences between means was assessed by ANOVA followed by the Student–Newman–Keuls test or by Student's unpaired t test. A probability of less than 5% was considered significant.
Results
Mice phenotype and function in vivo
As expected (Gabellini et al. 2006), at 12 weeks, i.e. the age of the mice used for structural and functional analysis, no difference was observed in body weight between FRG1-low mice (21.67 ± 0.28 g) and C57bL/6, WT mice (22.33 ± 0.57 g). The FRG1-med mice (17.71 ± 2.49 g) were smaller than WT mice, and the FRG1-high mice (13.66 ± 1.33 g) were significantly smaller than both WT and FRG1-med mice. Table 1 reports the weights of individual muscles of WT and FRG-1 mice. Overall, no signs of muscle atrophy were observed in FRG1-low mice, whereas muscle atrophy was clearly present in FRG1-med and FRG1-high, consistent with what has been previously observed (Gabellini et al. 2006).
Table 1.
Muscle weights (mg) from controls (WT) and FRG1 transgenic mice (FRG1-low expression; FRG1-med expression and FRG1-high expression) at 12 week of age (intercostals and masseter are not reported; see text)
| District | Muscle | WT | FRG1-low | FRG1-med | FRG1-high |
|---|---|---|---|---|---|
| Respiratory | Diaphragm | 95 ± 7 | 104 ± 14 | 74 ± 15* | 75 ± 5* |
| Anterior Limb | Biceps | 14 ± 2 | 14 ± 1 | 12 ± 3* | 9 ± 1† |
| Triceps | 66 ± 2 | 83 ± 8 | 46 ± 1* | 33 ± 2* | |
| Posterior Limb | Soleus | 7 ± 0.1 | 7 ± 0.1 | 7 ± 0.1 | 5 ± 0.1* |
| Tibialis | 42 ± 4 | 53 ± 12‡ | 32 ± 4 | 26 ± 1* | |
| Vastus | 111 ± 9 | 118 ± 9 | 61 ± 19* | 48 ± 11* | |
| Gastrocn. | 94 ± 19 | 108 ± 3 | 57 ± 4* | 46 ± 3* |
Significantly different from WT and FRG1-low
significantly different from WT, FRG1-low and FRG1-med
significantly different from WT and FRG1-med. Means ±s.d.P < 0.05, n = 5.
At the same age, tolerance to exercise, as evidenced in an incremental treadmill test to exhaustion (see Methods), was also in line with our previous observations (Gabellini et al. 2006), being significantly lower in FRG1-high (10.70 ± 1.57 min) and FRG1-med (13.50 ± 3.41 min) than in WT (20.80 ± 6.42 min). FRG1-low mice (22.30 ± 5.29 min) were not different from WT mice also in this respect.
As largely expected, the genotypic analysis and the above data confirm that the three lines of mice overexpressing FRG1 previously generated (Gabellini et al. 2006) and used in the present study maintained the same characteristics.
Contractile properties of single muscle fibres
A large population (n = 329) of single muscle fibres were dissected from the gastrocnemius and soleus muscles of WT, FRG1-low and FRG1-med mice at 12 weeks of age (n = 10). FRG1-high mice were not included in this study as the severity of the disease precluded dissection of large populations of well preserved individual muscle fibres. The mean values of cross-sectional area (CSA), specific force, and maximum shortening velocity and tension deficit of the single muscle fibres analysed were reported in Fig. 1. Interestingly, type 1 and type 2A single muscle fibres not only from FRG1-med, but also from FRG1-low mice, which did not show any clear phenotypic sign of MD and whose muscles were not smaller than WT, were smaller than corresponding fibre types of WT mice.
Figure 1.
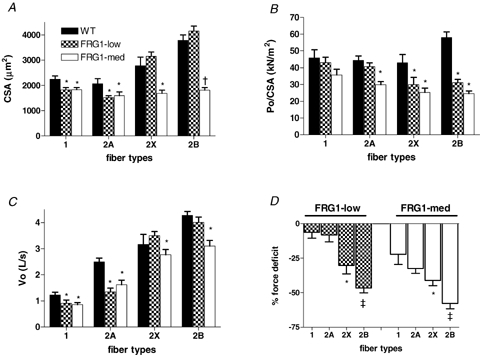
Cross-sectional area (CSA, A), specific force (Po/CSA, B), maximum shortening velocity (Vo, C) and tension deficit ((FRG1/force of WT) × 100, D) of single muscle fibres from WT, FRG1-low and FRG1-med mice All fibres (n = 329) were identified on the basis of MHC isoforms content determined by SDS-PAGE in pure type 1, 2A and 2B. CSA is expressed in μm, Po/CSA in kN m−1, Vo in length per second (L s−1) and force deficit as percentage force loss of corresponding fibres from WT mice. The height of each bar represents the mean (±s.e.m.). *Significantly different from corresponding fibres type of WT; †significantly different from corresponding fibres of WT and FRG1-low; ‡significantly different from type 1 and type 2A; #significantly different from type 1, 2A and 2X; P < 0.05 n = 10.
Figure 1B shows the mean values of Po/CSA of the same fibres reported in Fig. 1A. Types 2X and 2B fibres from both FRG1-low and FRG1-med transgenic mice were significantly weaker than those from WT animals, whereas type 2A fibres were significantly weaker only in FRG1-med transgenic mice. Although a trend towards a lower isometric force of type 1 fibres was reported in both FRG1-low and FRG1-med, it did not reach statistical significance. To more readily compare the impairment in strength of the different fibre types, the force deficit, i.e. force FRG1/force WT × 100 was calculated for each fibre type and reported in Fig. 1D. Force deficit increased in the order type 1 → 2A → 2X → 2B fibres. The latter results indicate that fast muscle fibres, and especially the fastest type 2B fibres, were preferentially involved in the dystrophic process.
Unloaded shortening velocity (Vo) was also impaired in dystrophic fibres. With the exception of type 2X and 2B fibres from FRG1-low, all fibre types of both FRG1 lines analysed (low and med) had lower Vo than corresponding fibre type of WT mice (Fig. 1C).
Alterations of single muscle fibre properties, being observed in FRG1-low mice that did not show clear manifestations of MD (body weight loss, muscle atrophy, decrease in exercise tolerance) can be considered early signs of MD.
Myosin heavy chain isoform distribution of FRG1 mice muscles
The distribution of MHC isoforms, a widely used index of fibre type distribution, was determined in 10 muscles (facials: masseter; respiratory: intercostals and diaphragm; upper limbs: deltoids, biceps and triceps; lower limbs: soleus, tibialis, vastus and gastrocnemius) of WT and FRG1-med mice by densitometric analysis of MHC bands separated by SDS-PAGE (Table 2). Consistent with the observation that type 1, slow fibres were relatively spared by the dystrophic process whereas type 2B fibres were the most affected fibre type, FRG1-med mice showed a significant shift towards the expression of slower MHC isoforms in all muscles studied.
Table 2.
Myosin heavy chain (MHC) composition (%) of muscles from controls (WT) and FRG1-med transgenic mice at 12 week of age
| MHC-1 (%) | MHC-2A (%) | MHC-2X (%) | MHC-2B (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| District | Muscle | WT | FRG1-m | WT | FRG1-m | WT | FRG1-m | WT | FRG1-m |
| Facial | Masseter | 4.7 ± 5.5 | 9.3 ± 10.4 | 0 ± 0 | 16.1 ± 17.7* | 77.3 ± 8.0 | 68.9 ± 5.7* | 17.9 ± 12.6 | 5.7 ± 6.1* |
| Respiratory | Diaphragm | 11.6 ± 2.5 | 15.7 ± 0.8* | 39.5 ± 0.5 | 54.7 ± 2.0* | 37.7 ± 3.9 | 27.7 ± 2.3* | 11.1 ± 1.9 | 1.8 ± 0.7* |
| Intercostal | 6.1 ± 2.3 | 11.9 ± 1.8* | 17.3 ± 3.2 | 24.7 ± 3.9* | 19.9 ± 4.3 | 33 ± 1.5* | 56.7 ± 2.3 | 30.3 ± 3.7* | |
| Anterior limb | Deltoid | 11.6 ± 1.2 | 13.0 ± 2.2 | 8.0 ± 1.2 | 15.7 ± 1.4* | 14.0 ± 1.2 | 21.0 ± 1.5* | 66.4 ± 0.6 | 50.2 ± 4.7* |
| Biceps | 0 ± 0 | 0 ± 0 | 16.1 ± 2.9 | 27.2 ± 0.5* | 24.7 ± 1.3 | 27.2 ± 1.0* | 58.4 ± 4.8 | 45.8 ± 1.2* | |
| Triceps | 0 ± 0 | 28.7 ± 3.0* | 18.2 ± 1.9* | 0 ± 0 | 23.5 ± 2.9 | 27.6 ± 3.1* | 58.2 ± 3.7 | 45.7 ± 4.2* | |
| Posterior limb | Soleus | 37.1 ± 1.3 | 64.1 ± 3.5* | 51.5 ± 5.5 | 27.6 ± 3.6* | 11.4 ± 4.7 | 8.2 ± 2.1* | 0 ± 0 | 0 ± 0 |
| Tibialis | 0 ± 0 | 9.7 ± 10.4* | 5.4 ± 7.4 | 7.4.9 ± 8.0* | 21.9 ± 2.0 | 27.8 ± 4.8* | 72.7 ± 4.2 | 55.1 ± 22.* | |
| Vastus | 0 ± 0 | 2.3 ± 0.3* | 6.8 ± 1.9 | 17.0 ± 0.8* | 11.1 ± 2.0 | 28.7 ± 0.5* | 82.1 ± 1.3 | 52.0 ± 1.3* | |
| Gastrocn. | 0 ± 0 | 24.2 ± 2.2* | 7.3 ± 7.9 | 11.4 ± 12.3 | 16.1 ± 2.4 | 23.7 ± 2.7* | 76.6 ± 6.8 | 52.1 ± 3.5* | |
Significantly different from correspondent isoforms. Means ±s.d.P < 0.05, n = 10.
In masseter the shift was from MHC-2B and 2X towards 2A, in intercostals and deltoid from MHC-2B towards MHC-2X and 2A; in diaphragm the shift was from MHC-2B, 2X and 2A towards MHC-1. As regards anterior and posterior limb muscles, in biceps, triceps, vastus and gastrocnemius the shift was from MHC-2B towards
MHC-1; finally in soleus the shift was from MHC-2X and especially MHC-2A towards MHC-1 (Table 2).
To clarify whether a shift of muscle phenotype could be considered an early sign of FSHD, the MHC isoform distribution of soleus, as an example of a slow, mildly affected muscle, and of biceps and vastus (Fig. 2) as examples of fast significantly affected muscles was studied in all lines of FRG1 mice. Interestingly the overall shift towards a slower phenotype appeared evident in all muscles analysed and in all lines, even in soleus muscles (Fig. 2) of FRG1-low mice which did not show clear histological signs of MD (Fig. 3). Thus, change in MHC composition of the muscle can be considered an early sign of the disease.
Figure 2.
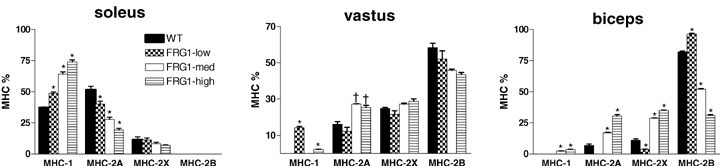
Myosin heavy chain (MHC) distribution of soleus, vastus and biceps in WT, FRG1-low, FRG1-med and FRG1-high mice MHC distribution determined from muscle samples (n = 5) of the four groups of mice by SDS-PAGE separation followed by densitometric analysis of MHC bands. The height of each bar represents the mean (±s.e.m.). *Significantly different from the other groups; †significantly different from WT and FRG1-low; P < 0.05, n = 5.
Figure 3.
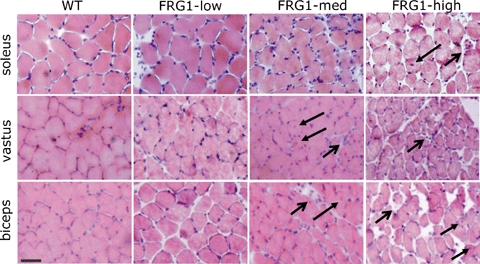
Heamatoxilyn–eosin stained sections from soleus, vastus and biceps muscle of WT, FRG1-low, FRG1-med and FRG1-high mice Distribution of muscle damage appeared to depend on the muscle considered and on the level of FRG1 expression. In all muscles no clear histological signs of muscular dystrophy were detectable in FRG1-low mice, whereas signs of fibre degeneration and necrosis (open arrows) as well as centrally nucleated fibres (close arrows) were detectable in biceps and vastus from FRG1-med and FRG1-high mice and appeared to be more evident in FRG1-high than FRG1-med mice. Bar represents 30 μm.
Muscle morphology of slow and fast muscles
The observation that the dystrophic process preferentially affected size and function of the fastest fibre type (2B) and relatively spared the slowest fibre type (1) should be reflected not only in a fast to slow transition in fibre type composition of the muscles, but also in a differential dystrophic deterioration of fast and slow muscle. To clarify whether this was actually the case, the morphology of the muscles, the ratio between muscle weight and body weight and the percentage of centrally nucleated fibres were compared through muscles and FRG-1 lines.
As expected (Gabellini et al. 2006), dystrophic histological features, namely fibre necrosis, number of centrally nucleated fibres, connective tissue, and increased fibre size variability were found to depend on FRG1 expression levels being almost absent in muscles of FRG1-low mice and increasingly more evident in FRG1-med and FRG1-high. More interestingly, soleus appeared much more spared by the disease process than vastus and biceps (Fig. 3).
To compare the degree of atrophy of different muscles, the ratio between muscle weight (MW) and body weight (BW) was determined and expressed relative to the MW/BW ratio of the same muscle of age-matched WT mice (Fig. 4). The MW/BW ratio of soleus (relatively spared by the disease process) was not smaller in FRG1-low and FRG1-high mice than in WT mice and larger in FRG1-med than in WT, suggesting compensatory hypertrophy. In FRG1-high apart from soleus and diaphragm all other muscles analysed showed significantly lower BW/MW ratio in comparison with WT mice. FRG1-med showed somewhat lower values and FRG1-low somewhat higher values than WT, but the difference did not reach statistical significance (Fig. 4). Some of the muscles (masseter, intercostals) were not included in the analysis of MW (Table 1) and MW/BW as their shape and architecture did not enable us to determine their weight consistently through experimental groups.
Figure 4.
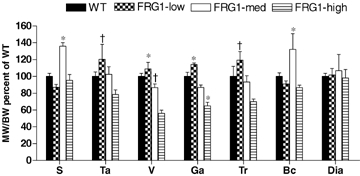
Ratio between muscle weight (MW) and body weight (BW) expressed relative to the MW/BW ratio of the same muscle of age-matched WT, FRG1-low, FRG1-med and FRG1-high mice S, soleus; Ta, tibialis anterior; Ga, gastrocnemius; Tr, triceps; Bc, biceps; Dia, diaphragm. The height of each bar represents the mean (±s.e.m.). *Significantly different from the other groups; †significantly different FRG1- high; P < 0.05, n = 10.
The degree of involvement of soleus, vastus and biceps was also quantified by determining the percentage of centrally nucleated fibres, considered an index of muscle fibre regeneration (Ferrari et al. 1998). Figure 5 shows that in soleus the percentage of centrally nucleated fibres was very similar in WT and in all FRG1 lines, whereas biceps and vastus had a highly significant increase of centrally nucleated fibres in all FRG1-med and FRG1-high in comparison with WT. In FRG1-low a significant increase of centrally nucleated fibres, but lower than in FRG1-med and high, was only observed in vastus.
Figure 5.
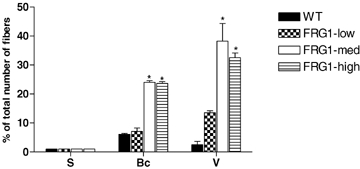
Percentage of centrally nucleated fibres in soleus (S), biceps (Bc) and vastus (V) of WT, FRG1-low, FRG1-med and FRG1-high mice At least 600 fibres were analysed from each group of mice (n = 5). The height of each bar represents the mean (±s.e.m.). *Significantly different from WT and FRG1- low, P < 0.05, n = 10.
Discussion
The present work studied structure and function of muscle fibres in a murine model of MD in which the pathogenesis of the disease cannot be initially traced back to a membrane defect. The main results indicate that: (i) muscle fibres are intrinsically weaker in the FSHD mice, and their loss of strength is an early sign of MD; (ii) fast muscle fibres are preferentially involved in the dystrophic process; (iii) the latter phenomenon can be a major determinant of the slower muscle phenotype of dystrophic muscles and can play a role in the differential distribution of the disease among muscles of the same animal.
Muscle weakness in MDs is not only due to a loss of contractile muscle mass
In the mouse model of FSHD, impaired mobility was ascribed to muscle atrophy (quantitative mechanism) which was observed at early stages of the disease in the absence of any, even transient, significant compensatory hypertrophy (Gabellini et al. 2006). The present findings, showing a loss of specific force (Po/CSA) of single muscle fibres, indicate the existence of a qualitative mechanism of muscle weakness based on a loss of intrinsic contractile strength. As type 2B fibres, which are the most represented fibre type in mice muscles (Pellegrino et al. 2003), lose approximately half of their intrinsic strength even in FRG1-low mice, the latter mechanism can play a major role, together with a loss of muscle mass, in determining muscle weakness in the FSHD mice in vivo. As muscle weakness in vivo, the loss of specific force of muscle fibres appeared related to the progression of the disease being more evident in FRG1-med than in FRG1-low mice.
The results obtained in this work confirm and extend previous findings. Earlier studies showed a loss of specific force in some population of skinned muscle fibres in mdx mice (Williams et al. 1993) and in an earlier murine model of MD, 129ReJ mice (Fink et al. 1986). Very recently, we consistently showed a loss of specific force in the fastest type 2B fibre type in several mouse models of MDs: mdx mice (Denti et al. 2006), α-sarcoglycan KO mice (Sampaolesi et al. 2003), scid-mdx mice (Torrente et al. 2004). It appears that the intrinsic loss of contractile strength is a major mechanism underlying muscle weakness in MD. Moreover, the latter phenomenon could be a fundamental feature of the dystrophic process as it is common to MDs having different pathogenetic mechanism and it is an early manifestation of the disease, at least in the only model, the FSHD mice, in which specific force deficit could be related to the severity of the disease.
The loss in specific force of muscle fibres observed in recent studies on mdx mice (Denti et al. 2006) is not in agreement with some earlier findings on the same mouse model (Takagi et al. 1992; Lynch et al. 2000). The dependence of muscle fibre alterations on fibre type and on the progression of the disease suggests possible causes for such discrepancy. In fact, in earlier works, muscle fibres were not precisely typed and results might have been affected by pooling and comparing muscle fibres of different types and therefore differentially affected by the disease.
Mechanisms underlying the loss of specific force are still unclear. Interestingly, the loss of Po/CSA in atrophic muscle fibres of elderly sedentary and immobilized humans has been shown to be linearly related to a decrease in myosin concentration within the fibres (Hook et al. 2001; D'Antona et al. 2003; D'Antona et al. 2006). As in MDs the balance between myofibrillar protein synthesis and degradation is shifted towards degradation (McKeran et al. 1977; Elia et al. 1981; Warnes et al. 1981), the latter phenomenon might be involved in MDs as well. Moreover a possible contribution to reduced specific force generation may also arise from increased number of weaker regenerating fibres which, in the presence of degeneration–regeneration cycles, initially replace necrotic fibres (Gregorevic et al. 2004). Nevertheless in FRG1 mice the observation of a limited amount of necrosis and regeneration, even in FRG1-high mice, suggests that this mechanism might play a minor role.
The observation that dystrophic fibres have not only lower specific force (Po/CSA), but also lower unloaded shortening velocity (Vo) than WT fibres is relevant as it can provide an additional explanation of muscle weakness and altered motility of FSHD mice. During movement and locomotion in vivo, muscles shorten against a load developing power (i.e. force times velocity). A loss of both Po/CSA and shortening velocity is bound to determine a much higher loss of mechanical power of the muscles and consequently a much more evident limitation of motility than a loss of force per se. It is unclear, whether a loss of Vo is a common features of all mouse models of MD as most studies so far focused on force. Recently it has been shown that diaphragm strips from mdx mice shorten at lower velocity than diaphragm strips from controls (Coirault et al. 1999). The origin of such impairment is unclear. In light of the modulator role played by myosin light chain (MLC) isoforms on Vo (Bottinelli et al. 1994), it might be due to a MD coupled shift in MLC composition of muscle fibres. However, it has been shown that Vo of muscle fibres and isolated myosin can change without a significant change in MLC content (Hook et al. 2001; D'Antona et al. 2003, 2006) possibly due post-translational modifications of the myosin molecule (Ramamurthy et al. 2001).
Fast muscle fibres are preferentially involved in the dystrophic process
An early morphological analysis of muscle fibres of patients affected by Duchenne MD (Webster et al. 1988) gave rise to the strong belief that the dystrophic process preferentially strikes fast fibres. The observation of a lower stretch-induced damage in slow (soleus) than in fast (EDL) muscles of mdx mice (Moens et al. 1993; Consolino & Brooks, 2004) supported such belief. However, no systematic comparative analysis of specific force of identified types of dystrophic fibres had been performed so far. The only data available suggested a significant force loss in slow muscle fibres from soleus, but not in fast muscle fibres from EDL muscles of mdx mice (Williams et al. 1993).
In this work, muscle fibres have been precisely identified on the basis of MHC isoform content, and contractile properties of three of the four types of muscle fibres expressed in skeletal muscle of the mice have been compared. The results indicate that in the FSHD mice the loss of specific force is significantly more evident in type 2B than in type 1, slow fibres (type 2A and 2X fibres being intermediate). Such findings support the general belief of a preferential involvement of fast fibres in the dystrophic process. Moreover, being observed in a murine model of MD with very peculiar pathogenetic features, the phenomenon appears a common feature of most MDs, regardless the underlying genetic defect.
The preferential involvement of the fastest fibre types in the dystrophic process is consistent with several concomitant findings. The morphological signs of MDs were less evident in soleus, the slowest muscle of the mice (45% MHC-1) than in fast muscles such as the biceps (27% MHC-2X and 46% MHC-2B) and the vastus (11% MHC-2X and 82% MHC-2B). MHC isoform distribution of a large population of skeletal muscles was shifted from faster to slower isoforms. The percentage of centrally nucleated fibres was normal in soleus and diaphragm (15% MHC-1 and 40% MHC-2A), but was much higher in fast muscle such as the vastus and the biceps.
In FRG1 mice a possible contribution to the observed selective involvement of fast fibres and fast muscles may take place from higher susceptibility of such fibres and muscles to eccentric contraction-induced damage as previously observed in mdx mice (Moens et al. 1993; Raymackers et al. 2003; Yeung et al. 2003). Indeed, the preferential deterioration of the fastest fibre types could be a one of the major determinants of the latter features common to most MDs.
Lack of plasmamembrane defect and muscle fibre deterioration
This study suggests that fundamental features of the dystrophic process are common to the FSHD mice and to the most studied models of MD such as the mdx mice and the α-SG-KO mice. The loss of intrinsic strength of muscle fibres (Sampaolesi et al. 2003; Torrente et al. 2004; Denti et al. 2006), the preferential involvement of fast fibres (Webster et al. 1988), the fast to slow shift in muscle phenotype (Petrof et al. 1993; Muller et al. 2001), and the differential involvement of skeletal muscles with soleus being less or later involved in MD (Nguyen et al. 2002; Briguet et al. 2004) were, in fact, previously observed in mdx and, to a certain extent, in α-SG-KO mice. Among the dystrophic features observed, only muscle atrophy is rather distinctive of the FSHD mice (Gabellini et al. 2006) and the mouse model of myotonic dystrophy (Mankodi et al. 2000; Seznec et al. 2001). Pseudo-hypertrophy is mostly observed, in fact, in the other mouse models (Watchko et al. 2002).
In the FSHD mice we previously demonstrated sarcolemmal integrity based on lack of Evans blue uptake by muscle fibres, normal creatine kinase serum levels and integrity of the DCG complex (Gabellini et al. 2006). This feature is common to other murine models of MDs, as calpain-3 deficiency, which also fails to show a clear increase in serum creatine kinase (Richard et al. 2000) and Evans blue uptake (Fougerousse et al. 2003). On the contrary a consistent increase of creatine kinase serum levels and the appearance of Evans blue positive cells are considered distinctive features in animal models of MDs due to alterations of the DCG complex (Sampaolesi et al. 2003; Denti et al. 2006).
In FRG1 mice, lack of obvious sarcolemmal impairment appeared in contrast with the observation of unspecific signs of fibre degeneration–regeneration. The extent of unspecific histopathological signs of MD correlated with FRG1 overexpression but appeared somehow moderate relative to mdx or α-SG-KO mice (Sampaolesi et al. 2003; Denti et al. 2006). In particular quantification of the number of centrally nucleated fibres showed, in all muscles analysed, a moderate level of central nuclei in FRG1 transgenic mice relative to mdx and α-SG-KO even in the presence of FRG1-high expression (Gabellini et al. 2006). Furthermore no regenerating myosins (neonatal or developmental MyHCs) have been detected in all FRG1 lines thus suggesting a low level of muscle degeneration–regeneration.
Nevertheless coexistence of unspecific dystrophic changes and sarcolemmal integrity does not represent a unique feature of the FRG1 mouse and FSHD as it has been already found in the mouse model of myotonic dystrophy (Mankodi et al. 2000; Seznec et al. 2001) and myotonic dystrophy (Casanova & Jerusalem, 1979).
Overall sarcolemmal integrity in FRG1-mice suggests that the dystrophic features observed so far cannot be ascribed to an obvious change of calcium levels followed by activation of proteolysis as in MDs due to alteration of DGC (Allen et al. 2005; Deconinck & Dan, 2007). Nevertheless no data on extracellular and intracellular calcium levels are currently available for FRG1-mice and the possible contribution of altered calcium homeostasis in determining the observed histological changes should be investigated.
Furthermore the present findings strongly suggest that fundamental features of the dystrophic process very similar to those observed in presence of DCG alterations can be the common outcome of alternative pathogenetic mechanisms. Such fundamental manifestations (loss of specific force, preferential involvement of fast muscle fibres, fast to slow shift in muscle phenotype, differential involvement of muscles) occur at early stages of the disease at least in the FSHD mice whereas histopathological signs of MD increase with age (Gabellini et al. 2006). Therefore, it can be ruled out that they are common in different MDs just because they necessarily appear in any heavily compromised muscle whatever the underlying myopathic process.
The physiological role of FRG1 and the mechanism by which FRG1 overexpression leads to the myopathic response are largely obscure. FRG1 is a nuclear protein identified as a component of purified spliceosomes (Rappsilber et al. 2002). We found that FRG1 overexpression is responsible for abnormal alternative splicing of specific pre-mRNAs and this can play a major role in the pathogenesis of FSHD (Gabellini et al. 2006). In particular it is likely that both in FRG1 mice and FSHD patients the increased levels of aberrant proteins may contribute to the histopathological features arising.
Interestingly abnormal mRNA processing plays a pathogenetic role in other neuromuscular disorders such as oculopharyngeal muscular dystrophy (Calado et al. 2000) and myotonic MD (Ranum & Day, 2004) and a recent study on muscle fibres of patients affected by myotonic MD (Krivickas et al. 2000) showed a loss of specific force, although limited to slow fibres. The latter observation strengthens the idea that abnormal mRNA processing can determine the structural and functional alterations at the fibre level. However the causal link between aberrant pre-mRNA processing and loss in force of dystrophic fibres is still unknown.
In conclusion, the analysis of the FSHD mice provided a detailed characterization of a novel mouse model of a major human MD which is bound to be used to further study both the pathogenesis and the therapy of such important disease. Moreover, the observation that fundamental features of the dystrophic process are common to the FSHD mice and to the most studied murine models of MD strongly suggest they can be on a common path of different pathogenetic mechanisms.
Acknowledgments
This work has been supported by grants from the Italian Ministry of University and Research (PRIN 2005) and from Cariplo Foundation, Italy. The authors wish to thank Dr Raffaella Adami for help in single fibre experiments.
References
- Allen DG, Whitehead NP, Yeung EW. Mechanisms of stretch-induced muscle damage in normal and dystrophic muscle: role of ionic changes. J Physiol. 2005;567:723–735. doi: 10.1113/jphysiol.2005.091694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaldo P, Braghetta P, Zanetti M, Piccolo S, Volpin D, Bressan GM. Collagen VI deficiency induces early onset myopathy in the mouse: an animal model for Bethlem myopathy. Hum Mol Genet. 1998;7:2135–2140. doi: 10.1093/hmg/7.13.2135. [DOI] [PubMed] [Google Scholar]
- Bottinelli R, Betto R, Schiaffino S, Reggiani C. Unloaded shortening velocity and myosin heavy chain and alkali light chain isoform composition in rat skeletal muscle fibres. J Physiol. 1994;478:341–349. doi: 10.1113/jphysiol.1994.sp020254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottinelli R, Canepari M, Pellegrino MA, Reggiani C. Force-velocity properties of human skeletal muscle fibres: myosin heavy chain isoform and temperature dependence. J Physiol. 1996;495:573–586. doi: 10.1113/jphysiol.1996.sp021617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottinelli R, Schiaffino S, Reggiani C. Force-velocity relations and myosin heavy chain isoform compositions of skinned fibres from rat skeletal muscle. J Physiol. 1991;437:655–672. doi: 10.1113/jphysiol.1991.sp018617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briguet A, Courdier-Fruh I, Foster M, Meier T, Magyar JP. Histological parameters for the quantitative assessment of muscular dystrophy in the mdx-mouse. Neuromuscul Disord. 2004;14:675–682. doi: 10.1016/j.nmd.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Calado A, Tome FM, Brais B, Rouleau GA, Kuhn U, Wahle E, Carmo-Fonseca M. Nuclear inclusions in oculopharyngeal muscular dystrophy consist of poly(A) binding protein 2 aggregates which sequester poly(A) RNA. Hum Mol Genet. 2000;9:2321–2328. doi: 10.1093/oxfordjournals.hmg.a018924. [DOI] [PubMed] [Google Scholar]
- Casanova G, Jerusalem F. Myopathology of myotonic dystrophy. A morphometric study. Acta Neuropathol (Berl) 1979;45:231–240. doi: 10.1007/BF00702675. [DOI] [PubMed] [Google Scholar]
- Coirault C, Lambert F, Marchand-Adam S, Attal P, Chemla D, Lecarpentier Y. Myosin molecular motor dysfunction in dystrophic mouse diaphragm. Am J Physiol Cell Physiol. 1999;277:C1170–C1176. doi: 10.1152/ajpcell.1999.277.6.C1170. [DOI] [PubMed] [Google Scholar]
- Consolino CM, Brooks SV. Susceptibility to sarcomere injury induced by single stretches of maximally activated muscles of mdx mice. J Appl Physiol. 2004;96:633–638. doi: 10.1152/japplphysiol.00587.2003. [DOI] [PubMed] [Google Scholar]
- D'Antona G, Lanfranconi F, Pellegrino MA, Brocca L, Adami R, Rossi R, Moro G, Miotti D, Canepari M, Bottinelli R. Skeletal muscle hypertrophy and structure and function of skeletal muscle fibres in male body builders. J Physiol. 2006;570:611–627. doi: 10.1113/jphysiol.2005.101642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Antona G, Pellegrino MA, Adami R, Rossi R, Carlizzi CN, Canepari M, Saltin B, Bottinelli R. The effect of ageing and immobilization on structure and function of human skeletal muscle fibres. J Physiol. 2003;552:499–511. doi: 10.1113/jphysiol.2003.046276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deconinck N, Dan B. Pathophysiology of duchenne muscular dystrophy: current hypotheses. Pediatric Neurol. 2007;36:1–7. doi: 10.1016/j.pediatrneurol.2006.09.016. [DOI] [PubMed] [Google Scholar]
- Denti MA, Rosa A, D'Antona G, Sthandier O, De Angelis FG, Nicoletti C, Allocca M, Pansarasa O, Parente V, Musaro A, Auricchio A, Bottinelli R, Bozzoni I. Body-wide gene therapy of Duchenne muscular dystrophy in the mdx mouse model. Proc Natl Acad Sci U S A. 2006;103:3758–3763. doi: 10.1073/pnas.0508917103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbeej M, Campbell KP. Muscular dystrophies involving the dystrophin-glycoprotein complex: an overview of current mouse models. Curr Opin Genet Dev. 2002;12:349–361. doi: 10.1016/s0959-437x(02)00309-x. [DOI] [PubMed] [Google Scholar]
- Elia M, Carter A, Bacon S, Winearls CG, Smith R. Clinical usefulness of urinary 3-methylhistidine excretion in indicating muscle protein breakdown. Br Med J (Clin Res Ed) 1981;282:351–354. doi: 10.1136/bmj.282.6261.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari G, Cusella-De Angelis G, Coletta M, Paolucci E, Stornaiuolo A, Cossu G, Mavilio F. Muscle regeneration by bone marrow-derived myogenic progenitors. Science. 1998;279:1528–1530. doi: 10.1126/science.279.5356.1528. [DOI] [PubMed] [Google Scholar]
- Fink RH, Stephenson DG, Williams DA. Calcium and strontium activation of single skinned muscle fibres of normal and dystrophic mice. J Physiol. 1986;373:513–525. doi: 10.1113/jphysiol.1986.sp016060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink RH, Stephenson DG, Williams DA. Physiological properties of skinned fibres from normal and dystrophic (Duchenne) human muscle activated by Ca2+ and Sr2+ J Physiol. 1990;420:337–353. doi: 10.1113/jphysiol.1990.sp017916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fougerousse F, Gonin P, Durand M, Richard I, Raymackers JM. Force impairment in calpain 3-deficient mice is not correlated with mechanical disruption. Muscle Nerve. 2003;27:616–623. doi: 10.1002/mus.10368. [DOI] [PubMed] [Google Scholar]
- Gabellini D, D'Antona G, Moggio M, Prelle A, Zecca C, Adami R, Angeletti B, Ciscato P, Pellegrino MA, Bottinelli R, Green MR, Tupler R. Facioscapulohumeral muscular dystrophy in mice overexpressing FRG1. Nature. 2006;439:973–977. doi: 10.1038/nature04422. [DOI] [PubMed] [Google Scholar]
- Gabellini D, Green MR, Tupler R. Inappropriate gene activation in FSHD: a repressor complex binds a chromosomal repeat deleted in dystrophic muscle. Cell. 2002;110:339–348. doi: 10.1016/s0092-8674(02)00826-7. [DOI] [PubMed] [Google Scholar]
- Gillis JM. Understanding dystrophinopathies: an inventory of the structural and functional consequences of the absence of dystrophin in muscles of the mdx mouse. J Muscle Res Cell Motil. 1999;20:605–625. doi: 10.1023/a:1005545325254. [DOI] [PubMed] [Google Scholar]
- Godt RE, Maughan DW. Swelling of skinned muscle fibers of the frog. Experimental observations. Biophys J. 1977;19:103–116. doi: 10.1016/S0006-3495(77)85573-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorevic P, Plant DR, Stupka N, Lynch GS. Changes in contractile activation characteristics of rat fast and slow skeletal muscle fibres during regeneration. J Physiol. 2004;558:549–560. doi: 10.1113/jphysiol.2004.066217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harridge SD, Bottinelli R, Canepari M, Pellegrino MA, Reggiani C, Esbjornsson M, Saltin B. Whole-muscle and single-fibre contractile properties and myosin heavy chain isoforms in humans. Pflugers Arch. 1996;432:913–920. doi: 10.1007/s004240050215. [DOI] [PubMed] [Google Scholar]
- Hook P, Sriramoju V, Larsson L. Effects of aging on actin sliding speed on myosin from single skeletal muscle cells of mice, rats, and humans. Am J Physiol Cell Physiol. 2001;280:C782–C788. doi: 10.1152/ajpcell.2001.280.4.C782. [DOI] [PubMed] [Google Scholar]
- Horowits R, Dalakas MC, Podolsky RJ. Single skinned muscle fibers in Duchenne muscular dystrophy generate normal force. Ann Neurol. 1990;27:636–641. doi: 10.1002/ana.410270609. [DOI] [PubMed] [Google Scholar]
- Irwin WA, Bergamin N, Sabatelli P, Reggiani C, Megighian A, Merlini L, Braghetta P, Columbaro M, Volpin D, Bressan GM, Bernardi P, Bonaldo P. Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nat Genet. 2003;35:367–371. doi: 10.1038/ng1270. [DOI] [PubMed] [Google Scholar]
- Klesert TR, Cho DH, Clark JI, Maylie J, Adelman J, Snider L, Yuen EC, Soriano P, Tapscott SJ. Mice deficient in Six5 develop cataracts: implications for myotonic dystrophy. Nat Genet. 2000;25:105–109. doi: 10.1038/75490. [DOI] [PubMed] [Google Scholar]
- Krivickas LS, Ansved T, Suh D, Frontera WR. Contractile properties of single muscle fibers in myotonic dystrophy. Muscle Nerve. 2000;23:529–537. doi: 10.1002/(sici)1097-4598(200004)23:4<529::aid-mus11>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lynch GS, Rafael JA, Chamberlain JS, Faulkner JA. Contraction-induced injury to single permeabilized muscle fibers from mdx, transgenic mdx, and control mice. Am J Physiol Cell Physiol. 2000;279:C1290–C1294. doi: 10.1152/ajpcell.2000.279.4.C1290. [DOI] [PubMed] [Google Scholar]
- Mankodi A, Logigian E, Callahan L, McClain C, White R, Henderson D, Krym M, Thornton CA. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science. 2000;289:1769–1773. doi: 10.1126/science.289.5485.1769. [DOI] [PubMed] [Google Scholar]
- McKeran RO, Halliday D, Purkiss P. Increased myofibrillar protein catabolism in Duchenne muscular dystrophy measured by 3-methylhistidine excretion in the urine. J Neurol Neurosurg Psychiatry. 1977;40:979–981. doi: 10.1136/jnnp.40.10.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moens P, Baatsen PH, Marechal G. Increased susceptibility of EDL muscles from mdx mice to damage induced by contractions with stretch. J Muscle Res Cell Motil. 1993;14:446–451. doi: 10.1007/BF00121296. [DOI] [PubMed] [Google Scholar]
- Muller J, Vayssiere N, Royuela M, Leger ME, Muller A, Bacou F, Pons F, Hugon G, Mornet D. Comparative evolution of muscular dystrophy in diaphragm, gastrocnemius and masseter muscles from old male mdx mice. J Muscle Res Cell Motil. 2001;22:133–139. doi: 10.1023/a:1010305801236. [DOI] [PubMed] [Google Scholar]
- Muntoni F, Brockington M, Torelli S, Brown SC. Defective glycosylation in congenital muscular dystrophies. Curr Opin Neurol. 2004;17:205–209. doi: 10.1097/00019052-200404000-00020. [DOI] [PubMed] [Google Scholar]
- Nguyen HH, Jayasinha V, Xia B, Hoyte K, Martin PT. Overexpression of the cytotoxic T cell GalNAc transferase in skeletal muscle inhibits muscular dystrophy in mdx mice. Proc Natl Acad Sci U S A. 2002;99:5616–5621. doi: 10.1073/pnas.082613599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrino MA, Canepari M, Rossi R, D'Antona G, Reggiani C, Bottinelli R. Orthologous myosin isoforms and scaling of shortening velocity with body size in mouse, rat, rabbit and human muscles. J Physiol. 2003;546:677–689. doi: 10.1113/jphysiol.2002.027375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrof BJ, Stedman HH, Shrager JB, Eby J, Sweeney HL, Kelly AM. Adaptations in myosin heavy chain expression and contractile function in dystrophic mouse diaphragm. Am J Physiol Cell Physiol. 1993;265:C834–C841. doi: 10.1152/ajpcell.1993.265.3.C834. [DOI] [PubMed] [Google Scholar]
- Ramamurthy B, Hook P, Jones AD, Larsson L. Changes in myosin structure and function in response to glycation. FASEB J. 2001;15:2415–2422. doi: 10.1096/fj.01-0183com. [DOI] [PubMed] [Google Scholar]
- Ranum LP, Day JW. Myotonic dystrophy. RNA pathogenesis comes into focus. Am J Human Genet. 2004;74:793–804. doi: 10.1086/383590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Ryder U, Lamond AI, Mann M. Large-scale proteomic analysis of the human spliceosome. Genome Res. 2002;12:1231–1245. doi: 10.1101/gr.473902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymackers JM, Debaix H, Colson-Van Schoor M, De Backer F, Tajeddine N, Schwaller B, Gailly P, Gillis JM. Consequence of parvalbumin deficiency in the mdx mouse: histological, biochemical and mechanical phenotype of a new double mutant. Neuromuscul Disord. 2003;13:376–387. doi: 10.1016/s0960-8966(03)00031-2. [DOI] [PubMed] [Google Scholar]
- Richard I, Roudaut C, Marchand S, Baghdiguian S, Herasse M, Stockholm D, Ono Y, Suel L, Bourg N, Sorimachi H, Lefranc G, Fardeau M, Sebille A, Beckmann JS. Loss of calpain 3 proteolytic activity leads to muscular dystrophy and to apoptosis-associated IκBα/nuclear factor κB pathway perturbation in mice. J Cell Biol. 2000;151:1583–1590. doi: 10.1083/jcb.151.7.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampaolesi M, Torrente Y, Innocenzi A, Tonlorenzi R, D'Antona G, Pellegrino MA, Barresi R, Bresolin N, De Angelis MG, Campbell KP, Bottinelli R, Cossu G. Cell therapy of α-sarcoglycan null dystrophic mice through intra-arterial delivery of mesoangioblasts. Science. 2003;301:487–492. doi: 10.1126/science.1082254. [DOI] [PubMed] [Google Scholar]
- Sarkar PS, Appukuttan B, Han J, Ito Y, Ai C, Tsai W, Chai Y, Stout JT, Reddy S. Heterozygous loss of Six5 in mice is sufficient to cause ocular cataracts. Nat Genet. 2000;25:110–114. doi: 10.1038/75500. [DOI] [PubMed] [Google Scholar]
- Seznec H, Agbulut O, Sergeant N, Savouret C, Ghestem A, Tabti N, Willer JC, Ourth L, Duros C, Brisson E, Fouquet C, Butler-Browne G, Delacourte A, Junien C, Gourdon G. Mice transgenic for the human myotonic dystrophy region with expanded CTG repeats display muscular and brain abnormalities. Hum Mol Genet. 2001;10:2717–2726. doi: 10.1093/hmg/10.23.2717. [DOI] [PubMed] [Google Scholar]
- Takagi A, Kojima S, Ida M, Araki M. Increased leakage of calcium ion from the sarcoplasmic reticulum of the mdx mouse. J Neurol Sci. 1992;110:160–164. doi: 10.1016/0022-510x(92)90023-e. [DOI] [PubMed] [Google Scholar]
- Torrente Y, Belicchi M, Sampaolesi M, Pisati F, Meregalli M, D'Antona G, Tonlorenzi R, Porretti L, Gavina M, Mamchaoui K, Pellegrino MA, Furling D, Mouly V, Butler-Browne GS, Bottinelli R, Cossu G, Bresolin N. Human circulating AC133+ stem cells restore dystrophin expression and ameliorate function in dystrophic skeletal muscle. J Clin Invest. 2004;114:182–195. doi: 10.1172/JCI20325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnes DM, Tomas FM, Ballard FJ. Increased rates of myofibrillar protein breakdown in muscle-wasting diseases. Muscle Nerve. 1981;4:62–66. doi: 10.1002/mus.880040111. [DOI] [PubMed] [Google Scholar]
- Watchko JF, O'Day TL, Hoffman EP. Functional characteristics of dystrophic skeletal muscle: insights from animal models. J Appl Physiol. 2002;93:407–417. doi: 10.1152/japplphysiol.01242.2001. [DOI] [PubMed] [Google Scholar]
- Webster C, Silberstein L, Hays AP, Blau HM. Fast muscle fibers are preferentially affected in Duchenne muscular dystrophy. Cell. 1988;52:503–513. doi: 10.1016/0092-8674(88)90463-1. [DOI] [PubMed] [Google Scholar]
- Williams DA, Head SI, Lynch GS, Stephenson DG. Contractile properties of skinned muscle fibres from young and adult normal and dystrophic (mdx) mice. J Physiol. 1993;460:51–67. doi: 10.1113/jphysiol.1993.sp019458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung EW, Head SI, Allen DG. Gadolinium reduces short-term stretch-induced muscle damage in isolated mdx mouse muscle fibres. J Physiol. 2003;552:449–458. doi: 10.1113/jphysiol.2003.047373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung EW, Whitehead NP, Suchyna TM, Gottlieb PA, Sachs F, Allen DG. Effects of stretch-activated channel blockers on [Ca2+]i and muscle damage in the mdx mouse. J Physiol. 2005;562:367–380. doi: 10.1113/jphysiol.2004.075275. [DOI] [PMC free article] [PubMed] [Google Scholar]