

We read with great interest the study by Pereira et al. (2007) and the accompanying Perspectives by Bers (2007) which were recently published in the The Journal of Physiology. These reports discuss a novel role for Epac, a cAMP-activated guanine nucleotide exchange factor, in the regulation of Ca2+ release during cardiac excitation–contraction (EC) coupling. The study of Pereira et al. found that electrically evoked Ca2+ release in adult rat cardiomyocytes was reduced following Epac activation by the selective activator 8-CPT. Moreover, this decrease in Ca2+ release during EC coupling was due to a Ca2+–calmodulin kinase II (CaMKII)-dependent reduction in sarcoplasmic reticulum (SR) Ca2+ load that results from an increased frequency of Ca2+ sparks leading to SR Ca2+ leak and store depletion. While these results implicate an exciting and novel role for Epac activation in the regulation of cardiac EC coupling, they diverge with prior published results from our laboratories showing clear Epac-mediated potentiation of Ca2+ release in adult mouse ventricular myocytes (Oestreich et al. 2007). Given the divergent conclusions of the two studies, we feel that it is important to reconcile these differences by providing a fair and comprehensive comparison of the experiments conducted in both reports. Additionally, such an examination might lead to a more comprehensive understanding of the potential physiological role of the Epac signalling pathway in cardiac myocyte function.
Oestreich et al. (2007) demonstrated that acute activation of Epac by 8-CPT increased electrically evoked Ca2+ release in single mouse ventricular myocytes, while 8-CPT had no effect on Ca2+ release in myocytes isolated from mice lacking phosphatidylinositol-specific phospholipase Cɛ (PLCɛ). Moreover, adenoviral-mediated PLCɛ expression in myocytes from PLCɛ knockout mice restored 8-CPT enhancement of electrically evoked Ca2+ release. Thus, Epac-dependent regulation of electrically evoked Ca2+ release in the heart is entirely dependent on the presence of PLCɛ. This is logical because previous work by us and others demonstrated that PLCɛ enzymatic activity is directly regulated by Rap, making a direct connection between Epac and PLCɛ through Rap activation (Schmidt et al. 2001; Kelley et al. 2004). Importantly, inhibition of Rap activation with RapGAP significantly inhibited β-adrenergic receptor (AR)-dependent enhancement of Ca2+ release, placing this pathway downstream of βAR signalling. Consistent with a role for Epac and PLCɛ in acute βAR-dependent enhancement of Ca2+-induced Ca2+ release (CICR), β-adrenergic-dependent stimulation of CICR was significantly reduced in myocytes isolated from PLCɛ−/− mice. The relative role of the Epac and PKA pathways in βAR signalling was examined with specific inhibitors (H89, Rp-cAMPs) and activators (6-Bnz-cAMP) of PKA. Surprisingly, these experiments revealed that only 50–60% of βAR-dependent increase in evoked Ca2+ release could be attributed to PKA, with the remainder dependent on the novel Epac–Rap–PLCɛ pathway (Oestreich et al. 2007). This conflicts with some reports where H89 completely inhibits βAR-dependent increases in CICR (Curran et al. 2007). A potential explanation for this difference is that all of our experiments were performed during strong stimulation of the βAR pathway (1 μm isoproterenol (ISO; isoprenaline)). Thus, it is possible that the PKA-independent Epac pathway only operates during strong βAR signalling, consistent with the lower affinity of cAMP for Epac relative to PKA. A still unanswered question in these studies is the ultimate mechanism for how PLCɛ activity alters Ca2+ release during EC coupling.
The recent work by Pereira et al. potentially sheds new insight on this question. This work shows that activation of Epac by 8-CPT leads to CaMKII phosphorylation at the auto-phosphorylation site and subsequent CaMKII-dependent phosphorylation of the ryanodine receptor (RyR2). Epac activation also increased Ca2+ spark frequency in a CaMKII-dependent manner. Since an increase in spark frequency reflects a higher sensitivity of RyR2 to activation by basal Ca2+ levels, this effect could account for the enhanced electrically evoked Ca2+ release observed in our prior study (Oestreich et al. 2007). In fact, CaMKII activation has been shown to contribute to ISO-dependent increases in CICR in the heart (Wang et al. 2004; Maier et al. 2007; Curran et al. 2007). How Epac activates CaMKII is not clear from the Pereira et al. study. Consideration of our previous work demonstrating PLCɛ dependence of the acute effects of Epac activation on cardiac Ca2+ handling suggests that the missing link between Epac activation and CaMKII activation may involve products of PLCɛ activity. PLCɛ hydrolyses PIP2 to produce IP3 and DAG leading to subsequent IP3 receptor-dependent Ca2+ release or PKC activation, respectively. Interestingly, O-Uchi et al. (2005) found that α1-adrenoceptor stimulation in the heart leads to CaMKII activation in a PKC-dependent manner. However, further work will be required to determine if the Epac activation of CaMKII observed in the experiments of Pereira et al. results from an Epac–PLCɛ–PKC pathway. Additionally, PLCɛ is a unique PLC isoform in that in addition to its traditional catalytic function it also contains a guanine nucleotide exchange factor domain that functions as an activator of Rap. Clearly, delineating the relative role of the different PLCɛ-dependent signalling elements in CaMKII activation during βAR stimulation will require further investigation.
A caveat of this discussion is that the observed effect of Epac activation on Ca2+ release during cardiac EC coupling are diametrically opposed between the studies of Oestreich et al. (2007) and Pereira et al. (2007) (see Fig. 1). Specifically, we found that acute treatment with 8-CPT increased electrically evoked Ca2+ release whereas the work by Pereira et al. showed a reduction. The reason for these disparate results are not completely clear, but one possible explanation is that the experiments conducted by Oestreich et al. involved acute exposure to 8-CPT for only 20 s prior to 60 s of 1 Hz electrical stimulation. Significant SR Ca2+ depletion may not occur during this brief exposure period, and in fact, we have observed that caffeine-induced responses were not different between control and 8-CPT-treated myocytes (authors' unpublished results). On the other hand, Pereira et al. found that significant SR Ca2+ depletion was observed using a longer (5 min) treatment protocol. However, it is important to consider that βAR stimulation of the Epac pathway occurs in parallel with PKA stimulation, which would limit SR Ca2+ depletion through PKA effects on phospholamban phosphorylation and SERCA-mediated SR Ca2+ reuptake. Alternatively, differences between the two studies might also reflect inherent species differences between rat and mouse myocytes.
Figure 1.
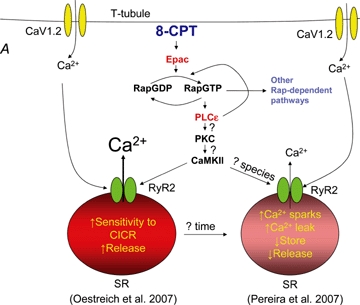
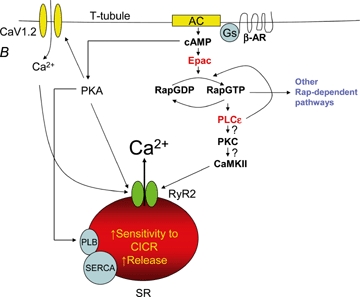
Roles for the Epac–PLCɛ pathway in cardiac calcium release A, treatment of isolated cardiac myocytes with 10 μm 8-CPT activates Epac and PLCɛ, ultimately leading to CaMKII-dependent phosphorylation of RyR2 and either enhancement (Oestreich et al. 2007) or inhibition (Pereira et al. 2007) of Ca2+-induced Ca2+ release (CICR). PKC activation of CaMKII is proposed to provide a link between the observed PLCɛ- and CaMKII-dependent effects. PLCɛ Rap guanine nucleotide exchange factors (GEF) activity is also shown to potentiate Rap activation and trigger additional downstream signalling pathways that may impact susceptibility to hypertrophy. B, Oestreich et al. provide evidence that activation of the Epac–Rap–PLCɛ pathway occurs in parallel with PKA activation under conditions of strong β-AR stimulation. Thus, during concerted PKA and Epac activation, RyR2 activity is shown to be influenced by both PKA and CaMKII phosphorylation and SR depletion prevented by PKA-dependent SERCA activation.
Complete resolution of the physiological impact of Epac activation on Ca2+ dynamics during EC coupling requires further analyses. Additional insight into the physiological role of this novel pathway could be gleaned from the cardiac phenotype of PLCɛ−/− mice (Wang et al. 2005). Since the Epac-dependent Ca2+ release pathway is completely dependent on PLCɛ(Oestreich et al. 2007), these mice lack Epac signalling in the heart. Isoproterenol-dependent enhancement of left ventricular developed pressure is significantly reduced in PLCɛ−/− mice, consistent with the Epac–Rap–PLCɛ pathway being regulated by βAR-dependent activation of Epac through cAMP (Wang et al. 2005; Oestreich et al. 2007). Surprisingly, PLCɛ−/− mice are more sensitive to the development of hypertrophy. This result is inconsistent with the putative prohypertrophic role of CaMKII (Maier & Bers, 2007; Anderson, 2007) and the previously reported prohypertrophic effects of Epac activation (Morel et al. 2005), but is consistent with a protective role for Epac activation described by Dodge-Kafka et al. (2005). One possibility is that Epac activation of PLCɛ may activate additional cardioprotective pathways that balance the prohypertrophic effects of CaMKII activation. Overall, the recent studies of Epac regulation of cardiac Ca2+ regulation (Wang et al. 2005; Oestreich et al. 2007; Pereira et al. (2007) provide exciting evidence for a novel signalling mechanism for the control of cardiac Ca2+ dynamics during EC coupling in health and disease.
References
- Anderson ME. Cardiovasc Res. 2007;73:657–666. doi: 10.1016/j.cardiores.2006.12.009. [DOI] [PubMed] [Google Scholar]
- Bers DM. J Physiol. 2007;583:415–416. doi: 10.1113/jphysiol.2007.140764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran J, Hinton MJ, Rios E, Bers DM, Shannon TR. Circ Res. 2007;100:391–398. doi: 10.1161/01.RES.0000258172.74570.e6. [DOI] [PubMed] [Google Scholar]
- Dodge-Kafka KL, Soughayer J, Pare GC, Carlisle Michel JJ, Langeberg LK, Kapiloff MS, Scott JD. Nature. 2005;437:574–578. doi: 10.1038/nature03966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley GG, Reks SE, Smrcka AV. Biochem J. 2004;378:129–139. doi: 10.1042/BJ20031370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier LS, Bers DM. Cardiovasc Res. 2007;73:631–640. doi: 10.1016/j.cardiores.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Morel E, Marcantoni A, Gastineau M, Birkedal R, Rochais F, Garnier A, Lompre AM, Vandecasteele G, Lezoualc'h F. Circ Res. 2005;97:1296–1304. doi: 10.1161/01.RES.0000194325.31359.86. [DOI] [PubMed] [Google Scholar]
- Oestreich EA, Wang H, Malik S, Kaproth-Joslin KA, Blaxall BC, Kelley GG, Dirksen RT, Smrcka AV. J Biol Chem. 2007;282:5488–5495. doi: 10.1074/jbc.M608495200. [DOI] [PubMed] [Google Scholar]
- O-Uchi J, Komukai K, Kusakari Y, Obata T, Hongo K, Sasaki H, Kurihara S. Proc Natl Acad Sci U S A. 2005;102:9400–9405. doi: 10.1073/pnas.0503569102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira L, Métrich M, Fernández-Velasco M, Lucas A, Leroy J, Perrier R, Morel E, Fischmeister R, Richard S, Benitah JP, Lezoualc'h F, Gómez AM. J Physiol. 2007;583:685–694. doi: 10.1113/jphysiol.2007.133066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M, Evellin S, Weernink PA, von Dorp F, Rehmann H, Lomasney JW, Jakobs KH. Nat Cell Biol. 2001;3:1020–1024. doi: 10.1038/ncb1101-1020. [DOI] [PubMed] [Google Scholar]
- Wang H, Oestreich EA, Maekawa N, Bullard TA, Vikstrom KL, Dirksen RT, Kelley GG, Blaxall BC, Smrcka AV. Circ Res. 2005;97:1305–1313. doi: 10.1161/01.RES.0000196578.15385.bb. [DOI] [PubMed] [Google Scholar]
- Wang W, Zhu W, Wang S, Yang D, Crow MT, Xiao RP, Cheng H. Circ Res. 2004;95:798–806. doi: 10.1161/01.RES.0000145361.50017.aa. [DOI] [PubMed] [Google Scholar]