

Abstract
Calsequestrin (CS), the major Ca2+-binding protein in the sarcoplasmic reticulum (SR), is thought to play a dual role in excitation–contraction coupling: buffering free Ca2+ increasing SR capacity, and modulating the activity of the Ca2+ release channels (RyRs). In this study, we generated and characterized the first murine model lacking the skeletal CS isoform (CS1). CS1-null mice are viable and fertile, even though skeletal muscles appear slightly atrophic compared to the control mice. No compensatory increase of the cardiac isoform CS2 is detectable in any type of skeletal muscle. CS1-null muscle fibres are characterized by structural and functional changes, which are much more evident in fast-twitch muscles (EDL) in which most fibres express only CS1, than in slow-twitch muscles (soleus), where CS2 is expressed in about 50% of the fibres. In isolated EDL muscle, force development is preserved, but characterized by prolonged time-to-peak and half-relaxation time, probably related to impaired calcium release from and re-uptake by the SR. Ca2+-imaging studies show that the amount of Ca2+ released from the SR and the amplitude of the Ca2+ transient are significantly reduced. The lack of CS1 also causes significant ultrastructural changes, which include: (i) striking proliferation of SR junctional domains; (ii) increased density of Ca2+-release channels (confirmed also by 3H-ryanodine binding); (iii) decreased SR terminal cisternae volume; (iv) higher density of mitochondria. Taken together these results demonstrate that CS1 is essential for the normal development of the SR and its calcium release units and for the storage and release of appropriate amounts of SR Ca2+.
Calcium ions (Ca2+) are extremely versatile second messengers. Transient elevations of intracellular Ca2+ concentration ([Ca2+]i) play an important role in virtually all cell types and in many cellular functions: these processes include cell differentiation, gene transcription, generation of muscle force and metabolic regulation (Dolmetsch, 2003; Gerke et al. 2005). In striated muscles, the changes in [Ca2+]i that regulate myofibril function are caused by a large rapid Ca2+ release from internal stores, i.e. sarcoplasmic reticulum (SR), that follows depolarization of exterior membranes. The mechanism that links sarcolemmal depolarization to Ca2+ release is known as excitation–contraction (EC) coupling (Sandow, 1965; Schneider & Chandler, 1973; Rios et al. 1991), and is governed by a coordinated interaction among several proteins localized in highly specialized intracellular junctions named Ca2+ release units (CRUs).
In skeletal muscles, the CRU is a highly specialized system that finely controls the release and uptake of Ca2+ from the SR during muscle contraction and relaxation (Schneider, 1994; Protasi, 2002). In CRUs, two separate and well-organized membrane systems come in close contact with one another: the exterior membranes, i.e. sarcolemma and/or transverse-tubules (T-tubules), and the internal membranes, i.e. the SR. Several proteins are specifically localized in sites corresponding to these structures: the sarcolemmal slow voltage gated L-type Ca2+ channel (dihydropyridine receptor, DHPR), the SR Ca2+ release channel (ryanodine receptor, RyR1), and calsequestrin (CS) are three of the key elements in the EC coupling machinery (MacLennan & Wong, 1971; Lai et al. 1988; Jorgensen et al. 1989). DHPRs, organized in ordered arrays of tetrads in the T-tubule membrane, are thought to be physically coupled to RyR1s, which are clustered in ordered arrays corresponding with the SR terminal cisternae (Franzini-Armstrong, 1970; Saito et al. 1984; Block et al. 1988; Protasi et al. 1997; Protasi et al. 1998; Protasi et al. 2000). Calsequestrin, located in the SR lumen in close proximity to the junctional SR domains containing RyRs, is an acidic protein that binds Ca2+ with a moderate affinity, but with high capacity, concentrating it near the sites of Ca2+ release (Jorgensen et al. 1983; Franzini-Armstrong et al. 1987). Two isoforms of mammalian CS (Campbell et al. 1983; Damiani et al. 1990), which are products of two different genes, have been identified and characterized: a skeletal muscle and a cardiac muscle isoform, abbreviated CS1 and CS2, respectively. CS2 is the only isoform expressed in the heart at all developmental stages, whereas both cardiac and skeletal CS genes are differentially expressed in various skeletal muscles (Fliegel et al. 1987; Scott et al. 1988). In slow-twitch fibres, CS2, is the most abundant isoform in fetal and neonatal muscles, and is co-expressed with CS1 at a 1: 3 ratio in the adult (Damiani et al. 1990). In fast-twitch fibres, on the other hand, CS2 disappears completely after birth, and CS1 remains the only isoform in the adult (Sacchetto et al. 1993).
Active Ca2+ transport is limited by the intraluminal free Ca2+ concentration (Makinose & Hasselbach, 1965; Weber et al. 1966; Weber, 1971; Inesi & de Meis, 1989). CS functions as a buffer of Ca2+ in the SR lumen, keeping the free concentration relatively low and thus allowing more efficient inward transport by the sarco-endoplasmic reticulum calcium ATPase (SERCA) pumps. This is particularly important in fast-twitch fibres where the amount of Ca2+ released and taken up is much greater than in slow fibres (Fryer & Stephenson, 1996). The higher concentration of CS in fast fibres (Leberer & Pette, 1986; Leberer et al. 1988) reflects its important role. CS has also been implied in modulating the activity of the SR Ca2+ release channels (Ikemoto et al. 1989; Gilchrist et al. 1992; Beard et al. 2005; Dulhunty et al. 2006), but the details of this modulation and whether it is essential during EC coupling remain to be determined. Both activation (Kawasaki & Kasai, 1994; Ohkura et al. 1995; Szegedi et al. 1999; Herzog et al. 2000) and inhibition (Beard et al. 2002) of RyRs by CS have been reported. Given a possible dual role of CS as a Ca2+ buffer and RyR modulator, it is not surprising that up- or downregulation of CS expression levels results in alterations of Ca2+ release and Ca2+ reuptake as well store stability (Terentyev et al. 2003; Wang et al. 2006). Initial evidence from null CS mutations in C. elegans (Cho et al. 2000) and in cardiac muscle (Knollmann et al. 2006) would suggest that CS is not absolutely essential for muscle function. These studies, however, involved muscles in which Ca2+ entry from the extracellular space during activation is substantial. This leaves open the question about the importance of CS in the activity of fast-twitch skeletal muscle fibres that derive all the calcium needed for contraction from their internal stores. Can mammalian skeletal muscle fibres function in the absence of CS, as it occurs in C. elegans (Cho et al. 2000)? Will lack of CS affect fast- and slow-twitch fibres, which are known to handle different amounts of Ca2+ and at different rates during EC coupling, equally? How does the absence of CS1 change the structure and molecular composition of the CRU?
To address these specific questions and to elucidate the functional and structural roles of CS1 in skeletal muscle fibres, we developed a CS1 knockout model (CS1-null mouse). CS1-null mice are viable and fertile, and develop normally under standard housing conditions. Analysis of the skeletal muscles of CS1-null mice reveals structural alterations of the CRU and significant functional impairment in calcium handling, substantiating an important role of CS1 in calcium homeostasis, and revealing an important, probably indirect, structural regulation of the membranes involved in calcium release.
Methods
Creation of the CS1-null mouse
The SMCALSE1 gene trap allele (Fig. 1A) was generated, using random insertional mutagenesis with retroviral vector VICTR24, as part of the OmniBank gene trap database (Zambrowicz et al. 1998). This vector generates fusion proteins of neomycin with the 5′ end of the gene and a fusion with BTK at the 3′ end of the gene, introducing premature stop signals that prevent translation of the protein product. The OST82566 clone was identified as the one containing the mutated SMCALSE1 gene within the embryonic stem cell library of the 129/SvEvBrd mouse, in which the gene trap vector was randomly introduced (OmniBank Library, Lexicon Genetics). Inverse genomic polymerise chain reaction (PCR) (Silver & Keerikatte, 1989), was used to determine that the gene-trap vector had integrated in the intron between exons 3 and 4 (Fig. 1A). Listed below is a portion of the mouse genomic sequence (50 nucleotides of sequence on either side) surrounding the gene-trap insertion site, which is denoted with an asterisk (see Fig. 1A). 5′… GATGGGGGAAGGGTAGTTAGCAACAAGTCATCTGG-ACAGCAATAGCAAAG*AGTCAGCCACTAGATACTTC-AGAGTCTCTGGCAGGAATATTTGTCCCTGG… 3′ The SMCALSE1 null line was generated by microinjection of the OmniBank ES cell clone represented by OST82566 into host blastocysts, using standard methods. Chimeric mice resulting from the ES cell injections were bred to C57BL/6J albino mice for germline transmission of the SMCALSE1 mutation. Multiplex Quantitative real-time PCR was used to genotype knockout mice (Charles River Laboratories, Boston MA).
Figure 1. CS1-null mutation is not lethal and no compensatory increase of CS2 is detectable.

A, the SMCALSE1 gene trap allele was generated, using random insertional mutagenesis with retroviral vector VICTR24. The precise genomic insertion site of the gene-trap vector (asterisk) was determined by inverse genomic PCR. B, the CS1-null mice do not shown any significant behavioural alteration under standard housing conditions. C, Western blot analysis of total homogenates prepared from limb muscles shows that CS1 is missing in CS1-null muscle (right lane). D, representative Western blots of EDL and soleus homogenates from WT and CS1-null mice: lack of CS1 is confirmed and no sign of compensatory increase of CS2 expression is detectable.
CS1-null animals
All experiments involving animals were conducted according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and were approved by the animal welfare coordinator of our institution. C57BL/6J mice were obtained from the Charles River Laboratories Boston, MA, USA. Mice were maintained in an accredited animal care facility and examined daily. Mice were first killed by an overdose of the anaesthetic ethylic ether, and their muscles were rapidly dissected.
Preparation of homogenate total membranes, electrophoresis, Western blot analysis, and 3H-ryanodine binding experiments
Preparation of total homogenates from soleus and EDL skeletal muscles
Extensor digitorum longus (EDL) and soleus muscles from control and CS1-null mice were homogenized in 3% SDS, 1 mM EGTA, boiled for 5 min and centrifuged at 900 g for 15 min. The protein concentration of the supernatants was quantified as described (Lowry et al. 1951).
Western blot analysis
For each sample, 20 μg of total protein were loaded on a 8% or 10% SDS-polyacrylamide gel, electrophoresed and transferred to nitrocellulose. Immunostaining of blots was performed using the following primary antibodies: monoclonal antibody for SERCA1 (Affinity Bioreagents, USA), rabbit polyclonal antibody reactive with both isoforms of CS (Affinity Bioreagents, USA); rabbit affinity-purified TRN6 antibody raised against residues 146–160 of mouse triadin (generous gift of L. A. Jones); secondary antibodies were anti–mouse or anti–rabbit AP-conjugated antibodies (SIGMA, Italia), respectively. Densitometric scans were analysed with Scion Image Software to quantify protein band intensities. Normalization was performed with anti-GAPDH antibody (Abcam, UK) or total protein concentration for CS and SERCA1, with consistent results. Quantification of the signal for the 95 kDa isoform of triadin and normalization to Ponceau Red staining was performed using QuantityOne Software from Bio-Rad Laboratories (Hercules, CA, USA).
Total membranes preparation
Total membranes (TMs) were prepared from a pool of 10–12 EDL muscles from either wild-type (WT) or CS1-null animals, as described (Damiani et al. 1991).
3H-ryanodine binding
Bound 3H-ryanodine was determined as described (Zorzato et al. 1989).
Cryostat sectioning and immunohistochemistry
EDL and soleus muscles were dissected from both CS1-null and WT mice (4–9 months of age) and wrapped in a small piece of bovine liver. The samples were then frozen in liquid nitrogen and cryoprotected with Tissue-Tek II OCT compound (Miles Inc. USA). Transverse sections 10–12 μm thick were cut in a Leica cryostat (CM 1850, Leica Microsystem, Austria) and fixed with 0.8% paraformaldehyde. Sections were then blocked with 1% BSA and 10% goat serum in PBS to avoid non-specific detection, and incubated with primary antibodies for 2 h, followed by secondary antibodies for 1 h, both at room temperature (CY3-conjugated goat anti–mouse and goat anti–rabbit, Jackson ImmunoResearch Laboratories, Lexington, KY, USA). The specimens were viewed on a fluorescence microscope (Leica DMLB) or a confocal microscope (LS510 META or LSM 5 Pascal, Zeiss, Germany). The following primary antibodies were used: pAB, reactive with both CS1 and CS2, diluted at 1: 800; 4B1, specific for CS1, 1: 400 (Jones et al. 1998); BA-F8 monoclonal antibody specific for myosin heavy chain (MHC) slow, supernatant diluted at 1: 10; SC-71 monoclonal antibody, specific for MHC IIA, supernatant diluted at 1: 40 (Schiaffino et al. 1989). pAB and 4B1 were a generous gift of L. A. Jones, BA-F8 and SC-71 were a generous gift of S. Schiaffino.
Force and contraction kinetics of isolated intact soleus and EDL
Soleus and EDL muscles were dissected from the hind limb of WT and CS1-null mice in warm oxygenated Krebs solution, and mounted between a force transducer (AME-801 SensorOne, Sausalito, CA, USA) and micromanipulator-controlled shaft in a small chamber where oxygenated Krebs solution was continuously circulated. The temperature was kept constant at 25°C. The stimulation conditions were optimized, and muscle length was increased until force development during tetanus was maximal. The responses to a single stimulus (twitch) or to a series of stimuli at various rates producing unfused or fused tetani were recorded. Time to peak tension, time to half relaxation, and peak tension were measured in single twitches. Tension was measured in completely fused maximal tetani and twitch/tetanus ratio was determined. The resistance to fatigue was tested by stimulating the muscles with a fatiguing protocol based on 0.5 s fused tetani with 1: 4 duty ratio (low-frequency fatigue).
Preparation of samples for electron microscopy (EM)
EDL and soleus muscles were carefully dissected from CS1-null and WT mice (4–9 months of age). Muscles were fixed at room temperature in 3.5% glutaraldehyde in 0.1 M Na-cacodylate buffer, pH 7.2 for 2 h. Small bundles of fixed fibres were then postfixed in 2% OsO4 in the same buffer for 2 h, and block-stained in aqueous saturated uranyl acetate. After dehydration, specimens were embedded in an epoxy resin (Epon 812). Ultrathin sections were cut in a Leica Ultracut R microtome (Leica Microsystem, Vienna, Austria) using a Diatome diamond knife (DiatomeLtd. CH-2501 Biel, Switzerland). After staining in 4% uranyl acetate and lead citrate, sections were examined with a Morgagni Series 268D electron microscope (FEI Company, Brno, Czech Republic), equipped with Megaview III digital camera.
Quantitative analysis of electron-micrographs
Micrographs, all at the same magnification (14 000× for counting CRUs and mitochondria; 28 000× for RyR frequency and SR terminal cisternae width) with no overlapping regions, were randomly collected from 5–10 different fibres for each analysed specimen, excluding peripheral areas, nuclei and Golgi regions. In both CS1-null and WT mice, three different time points for EDL and soleus muscles (n = 3) were quantitatively studied (ages: for CS1-null 5, 6.2 and 8.3-month-old-mice; for WT 5, 6 and 9.8-month-old-mice).
Average size of SR terminal cisternae lumen
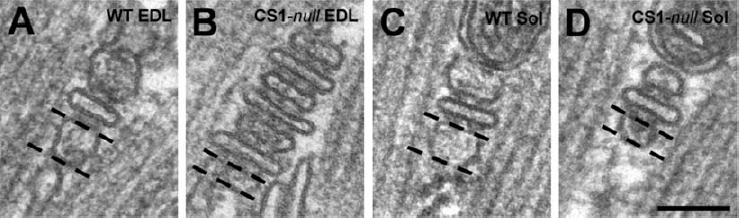
The width of the external SR vesicles was calculated using the Analysis Soft Imaging System (Germany), measuring the distance between the SR membrane facing the T-tubule and the opposite membrane in junctional SR vesicles at the outer borders of each CRU, as shown in panels A–D of Table 3, in which the two membranes are marked with dashed lines. The terminal cisternae to be measured were carefully selected based on clarity and definition of their outlines (Table 3, column I).
Table 3.
Ultrastructural morphometry of CRUs and mitochondria in EDL and soleus muscles
 | ||||
|---|---|---|---|---|
| Column I Junctional SR width (nm) | Column II No of RyRs (10 μm2 of sectional area)−1 | Column III Total SR volume (%) | Column IV No of mitochondria profiles (100 μm2 sectional area)−1 | |
| EDL | ||||
| Wild type (n = 3) | 62.4 ± 10.5 (n = 716) | 39.2 ± 16.7 (n = 143) | 5.66 ± 1.80 (n = 93) | 36.3 ± 14.2 (n = 98) |
| CS1-null (n = 3) | 25.0 ± 4.5† (n = 487) | 71.8 ± 26.3† (n = 111) | 5.25 ± 1.95 (n = 97) | 62.1 ± 21.7† (n = 125) |
| Soleus | ||||
| Wild type (n = 3) | 62.5 ± 11.7 (n = 308) | 36.0 ± 15.3 (n = 54) | – | 80.0 ± 18.8 (n = 85) |
| CS1-null (n = 3) | 30.0 ± 5.9† (n = 404) | 32.7 ± 13.9 (n = 74) | – | 81.4 ± 9.2 (n = 132) |
Column I, the profile of the SR terminal cisternae appears different and its width, measured as shown in the panels A–D, is much smaller in CS1-null than in WT fibres of both EDL and soleus. In soleus muscle, CRUs are still formed by three elements, whereas junctions in EDL fibres are often formed by multiple elements (panel B). Column II, this reorganization of CRUs results in a large increase of RyR content in EDL fibres. Column III, on the other hand, the total SR volume (in relation to the total fibre volume) in CS1-null fibres is still very similar to that of WT fibres. Column IV, CS1-null EDLs present also a large increase in the average density of mitochondria, that may be related to the decreased fatigability of these muscles (see Fig. 4). Values are mean ± s.d.
Significantly different from WT group at P < 0.0001. Scale bar: A–D, 0.1 μm.
Average number of RyRs in junctional arrays
In a set of micrographs at a higher magnification (28 000×), RyRs in the junctional gap were marked and counted, and their average number per sectional area was calculated (Table 3, column II).
Evaluation of the relative SR volume
Estimates of the ratio between total SR volume and fibre volume were obtained using the well-established stereology point-counting techniques (Loud et al. 1965; Mobley & Eisenberg, 1975). Data were obtained from the same micrographs used for the other quantitative analysis. The images were covered with an orthogonal array of dots at a spacing of 0.20 μm. The ratio of the numbers of dots falling within the SR profile to the total number of dots covering the image gave the ratio of the SR volume to the total volume. Data are presented in Table 3, column III as percentages of fibre volume occupied by the SR.
Density of mitochondria
The density of mitochondria was determined by counting the number of their sectioned profiles in EM images (14 000×), and referring them to the sectioned area. The same micrographs were used to calculate the percentage of fibres containing multiple junctions.
Intracellular Ca2+ measurements in single intact muscle fibres
Single FDB (flexor digitorum brevis) fibres were isolated with a modified collagenase/protease method as previously described (Defranchi et al. 2005) from CS1-null and WT mice. There was no difference in fibre yield between the two groups of mice. On the day of the experiment (generally 48 h after dissociation), isolated fibres were loaded with 5 μM Fura-2 acetoxymethyl ester (Molecular Probes, Invitrogen) in incubation buffer (mM: 125 NaCl, 5 KCl, 1 MgSO4, 1 KH2PO4, 5.5 glucose, 1 CaCl2, 20 Hepes and 1% bovine serum albumine, pH adjusted to 7.4 with NaOH) for 30 min at 37°C. After loading with Fura-2, fibres were washed twice for 10 min with incubation buffer without BSA at 37°C to retain the indicator in the cytosol. After a minimum of 30 min, calcium signals were recorded using a dual-beam excitation fluorescence photometry setup (IonOptix Corp.) at a temperature of 25°C. After 5–10 min of steady-state pacing at 0.5 Hz, 10 transients were recorded from each fibre, which was then removed with a micropipette and transferred to an Eppendorf test tube containing Laemmli solution for CS isoform identification. About five fibres were analysed from each Petri dish. Ca2+ transients were analysed using IonWizard software designed by IonOptixCorp (Milton, MA, USA). [Ca2+]i measurements are expressed as fluorescence ratio (F ratio) of the emission at 480 nm with reference to the excitation wavelengths of 360 and 380 nm, respectively.
Calcium release in single permeabilized muscle fibres
Single fibres were manually dissected free from the superficial layers of tibialis anterior muscle of CS1-null and WT mice, and segments of 1–1.5 mm length were mounted with small aluminium clips between a force transducer (AME-801 SensorOne, Sausalito, CA, USA) and an electromagnetic puller to control fibre length. The force transducer and the electromagnetic puller were part of a set-up composed of an aluminium plate equipped with seven small pedestals where drops containing different solutions were accommodated. The aluminium plate was placed on the stage of an inverted microscope (Axiovert 10, Zeiss, Germany). The fibre segment could be quickly moved from one small pool to the other, allowing a complete change of solution within 5 s. Dissection was carried out in a high-potassium solution containing EGTA, and the fibre was mounted in the same solution (drop n1) whereas the other six drops were, respectively, composed of skinning solution containing 5 mg ml−1 saponin (n2), relaxing solution (n3), loading solution (n4), washing solution (n5), releasing solution (n6), and maximal Ca2+ concentration activating solution (n7). The compositions of the solutions were identical to those described in a previous paper (Rossi et al. 2001). The floors of the pedestals were transparent, so that specimens could be viewed at 320× through the eyepieces of the microscope, and a video camera connected to a computer. Signals from the force and displacement transducer were displayed and recorded after A/D conversion (interface 1401 plus; CED) on a computer where the software Spike 2 (CED) was used for analysis.
For measuring Ca2+ release, fibres were transferred from the first drop (high-potassium solution) to the second drop to be permeabilized with saponin for 30 s. Fibres were then immersed in relaxing solution (n3), and then SR was loaded by immersing the fibres in a solution (n4) at pCa 6.45 in the presence of 5 mM ATP. After the fibres had been washed (n5) to remove excess EGTA, Ca2+ release was induced by transferring the fibre to solution n6 with low EGTA content (0.1 mM) containing caffeine in one of 5 variable concentrations from 0.1 to 20 mM (pCa 8). The fibre was finally transferred to activating solution (n7) to measure the ability to develop force during a maximal activation (pCa 4.7). The fibre was then brought back to relaxing solution (n3) to start a new cycle of loading and release. The release of Ca2+ was inferred by the transient tension development, and quantified by the tension–time area, according to a method, first developed by Endo (Endo & Iino, 1988) and widely used (Launikonis & Stephenson, 1997; Rossi et al. 2001). As discussed in a previous study (Rossi et al. 2001), experimental and model analyses point to the tension–time area as the best indicator of the amount of Ca2+ released from the SR and later removed by EGTA and diffusion. The tension–time area was normalized to the tension developed during maximal activation (pCa 4.7, n7) to account for the variability of the ability of the myofibrillar apparatus in each fibre to develop force. After normalization, the tension–time area was expressed in seconds. Dose–response curves were interpolated using the sigmoid curve:
where Y is the normalized area, X the caffeine concentration, T the maximal response amplitude and EC50 the concentration at which half-maximal response is achieved.
Statistical analysis
Data were expressed as mean ± standard deviation (s.d.), unless otherwise stated. Student's unpaired t test was used for comparisons between CS1-null and WT data, and statistical significance was set at P < 0.05. GraphPad Prism software (Site company and location) was used for curve fitting.
Results
CS1-null mouse
The CS1-null mutation is not lethal, mice are viable and fertile, and appear to develop and breed normally (Fig. 1B). Western blots of total homogenates prepared from limb muscles with an antibody reactive with both CS1 and CS2 show that in CS1-null tissue CS1 is missing, confirming the success of the knockout, whereas CS2 is still present (Fig. 1C, right lane). Although CS1-null mice do not show any significant behavioural alteration under standard housing conditions, they do display signs of muscle atrophy (Table 1). In fact, the average body weight of CS1-null mice is about 10% lower when compared to the WT group (Table 1, column I): 27.3 g versus 30.1 g (male animals, age 4–6 months, n, number of animals: CS1-null, n = 211; WT, n = 150; P < 0.0001). The average weight and muscle/body weight ratios of a predominantly fast-twitch muscle (EDL) in CS1-null mice is significantly lower than in WT mice of same sex and age (P < 0.0001) (Table 1, columns II and III). On the other hand, the soleus does not show a difference in mass between CS1-null and WT mice (Table 1, columns IV and V).
Table 1.
Average muscle of EDL and soleus muscle in WT and CS1-null mice
| Group | I Body weight (g) | II EDL weight (mg) | III Relative EDL muscle weight (%) | IV Soleus weight (mg) | V Relative soleus muscle weight (%) |
|---|---|---|---|---|---|
| Wild type | 30.1 ± 2.9 | 9.3 ± 1.4 | 0.036 ± 0.02 | 8.8 ± 0.8 | 0.034 ± 0.02 |
| CS1-null | 27.3 ± 2.0† | 7.5 ± 1.2† | 0.028 ± 0.03† | 8.6 ± 1.2 | 0.033 ± 0.03 |
Column I, CS1-null mice show a lower body weight than WT mice of same sex (male) and age (4–6 months, CS1-null, n = 211; WT, n = 150; n, number of animals). Columns II–V, the weights of EDL and soleus muscles are shown as absolute value (mg, columns II and IV) and relative to body weight (mg g−1, columns III and V). EDL muscles in CS1-null mice are on the average 20% smaller than in WT (columns II and III): this difference is highly significant (P < 0.0001). Soleus muscles on the other hand, do not show a significant difference between the two groups (columns IV and V). Data are means ± and s.d. of 35 muscles in CS1-null mice and 25 muscles in WT mice.
Significantly different from WT group at P < 0.0001.
Immunoblot and immunohistochemistry
Muscle-specific Western blots confirm the lack of CS1 in both EDL and soleus. As in WT muscle, CS2 is expressed in both muscle types, and the expression of CS2 is higher in soleus than in EDL (Fig. 1D). There was no detectable compensatory increase of CS2 in either of the two CS1-null muscles. Immunohistochemical staining of transverse cryosections with an antibody specific for CS1 confirms that CS1 expression is completely abolished in CS1-null mice (Fig. 2B and E). WT and CS1-null muscles were frozen next to each other: lack of fluorescence in CS1-null muscles is clearly evident in panels C and F (Fig. 2), which show the contact point between the two muscles. The fluorescence detectable in the interstitial spaces of CS1-null muscles is non-specific and is caused by our using a secondary antibody against murine immunoglobulin on mouse muscle sections. Immunostaining of sections with an antibody that recognizes both CS1 and CS2, indicates that CS2 – the only isoform expressed – is not present in all fibres, but is confined to a subpopulation (Fig. 3A and D) of fibres. Fibres expressing CS2 are rare in the EDL (5–20% depending on the section, n = 5, Fig. 3A), but are abundant in the soleus (about 40–50%, n = 4, Fig. 3D). Thus, most muscle fibres in the EDL of CS1-null muscle (80% or more) lack any CS, whereas in the soleus only about 50% of fibres do not express any CS.
Figure 2. Immunohistochemistry of CS1-null and WT muscles confirms the lack of CS1 in knockout muscles.

A and D, immunohistochemistry of transverse sections stained with the antibody specific for CS1 shows that all muscle fibres in WT EDL and soleus express CS1, even if the levels of expression are variable. B and E, muscle fibres of CS1-null EDL and soleus muscles do not express any CS1. C and F, CS1-null and WT muscles frozen next to one another and visible within the same sections.
Figure 3. CS2 is confined to a subpopulation of fibres, some of which are type IIA in EDL and mostly type I in soleus.

A and D, CS2 is not expressed in all fibres, but confined to a subpopulation of them, rare in EDL (5–20%), but abundant in soleus (40–50%). B, C, E and F, in soleus muscle, CS2 is mostly expressed in type I fibres, whereas in the EDL CS2 appears to be confined mostly to smaller fibres, some of which are type IIA. Corresponding fibres in the different sections are marked with the same numbers in paired panels.
To determine which type of fibres express CS2, serial sections from both EDL and soleus muscles were labelled with antibodies specific for CS2 and with antibodies specific for either slow (type I) or fast IIA (type IIA) MHC. Corresponding fibres in the different sections are marked with the same numbers in paired panels (Fig. 3B and C, EDL; Fig. 3E and F, soleus). The results indicate that in soleus CS2 is almost exclusively expressed in type I fibres, whereas in the EDL, CS2 is predominantly expressed in a subset of type IIA fibres, i.e. oxidative fast-twitch. Labelling with the two anti-MHC antibodies does not suggest that there is any detectable fibre type switch towards type I fibres in CS1-null muscles when compared to WT muscles. This has also been confirmed by MHC isoform separation with gel electrophoresis (not shown). Thus, taking into account the known fibre type composition of murine EDL and soleus (Pellegrino et al. 2003), fibres lacking both CS1 and CS2 are mainly type IIX and IIB in EDL, whereas they are mostly type IIA and a few IIX in soleus.
Tension development and contraction kinetics of soleus and EDL
Maximum isometric tension in fused tetani of EDL and soleus muscles of CS1-null muscles is not reduced compared to WT (80 Hz in soleus and 100–120 Hz in EDL, Table 2, column II). However, twitch tension shows a trend to higher values in CS1-null than in WT muscles (Table 2, column I), and the twitch/tetanus ratio is significantly higher in EDL muscles of CS1-null compared to WT (Table 2, column III). This increase in the twitch/tetanus ratio is probably related to the altered kinetics of the contractile cycle in CS1-null muscle. These changes include a significant prolongation of both time to peak tension and time to half relaxation in EDL (Fig. 4A and B), but not in soleus muscles (Fig. 4D and E), suggesting a delayed Ca2+ release and delayed Ca2+ removal from the myofibrils. An interesting alteration of the contraction kinetics found in CS1-null EDL muscles is the highly significant increase in fatigue resistance (Fig. 4C): residual developed tension after 120 s of repetitive stimulation is 100% higher than in WT muscles. One possible explanation for this increased resistance to fatigue may be related to the increased mitochondria content in EDL muscles described below (see also Table 3). This does not occur in CS1-null soleus muscles, and they show no difference in fatigue resistance compared to WT.
Table 2.
Tension development of EDL and soleus muscles of WT and CS1-null mice
| I Twitch tension (mN mm−2) | II Tetanus tension (mN mm−2) | III Twitch/tetanus (ratio) | |
|---|---|---|---|
| EDL | |||
| Wild type (n = 12) | 43.5 ± 3.9 | 168.1 ± 14.3 | 0.263 ± 0.016 |
| CS1-null (n = 14) | 53.6 ± 6.1 | 153.3 ± 15.7 | 0.373 ± 0.020† |
| Soleus | |||
| Wild type (n = 13) | 25.1 ± 6.3 | 169.4 ± 28.7 | 0.147 ± 0.023 |
| CS1-null (n = 17) | 29.9 ± 4.5 | 190.6 ± 22.1 | 0.141 ± 0.009 |
Tension developed during isometric tetanus is not significantly different in the two muscles (column II), whereas twitch tension tends to be higher in CS1-null EDL than in WT EDL (column I) and twitch/tetanus ratio is significantly greater (column III). Means and s.e.m.
Significantly different at P < 0.05.
Figure 4. CS1-null EDL, but not soleus, muscles show slower contraction kinetics and higher resistance to fatigue.

Time to peak tension and half-relaxation time are prolonged in CS1-null EDL (A and B), but not in soleus (D and E) compared to WT. Force still developed after 120 s of stimulation with the fatigue protocol and is much greater in CS1-null EDL but not in soleus (C and F) compared to WT. Values are means ± s.e.m.*P < 0.05.
Ultrastructural features of the EC coupling apparatus
The overall architecture of the EC coupling apparatus in EDL and soleus is quite similar (compare Figs 5 and 6A and B). In WT skeletal muscle, the mature T-tubule network has a general transverse orientation, and is located at the edges of the A band, forming two transverse networks for each sarcomere. CRUs in mature skeletal muscle are usually in the form of triads, composed of two SR vesicles closely apposed to a T-tubule (Figs 5B and 6B). In these mature junctions, RyRs form two ordered rows along each side of the T-tubule (Fig. 5B, small arrows). Some general quantitative differences between CRUs of fast- and slow-twitch fibres involve a higher frequency of junctional SR–T-tubule apposition in the former, resulting in a higher overall density of ryanodine receptors (Appelt et al. 1989; Franzini-Armstrong et al. 1999).
Figure 5. CRUs in CS1-null EDL muscle are usually formed by multiple elements, which often contain multiple rows of RyRs.

A, CRUs in WT EDL are usually transversally oriented, evenly distributed within the fibres, and formed by three elements, i.e. triads (arrows). B, RyRs, the Ca2+-release channels of the SR, in WT muscle usually form two rows (small arrows). C, in CS1-null fibres the T-tubules (TT) often change direction forming CRUs that are more variably orientated (arrows). D, CRUs are often formed by multiple elements and couplons usually contain more than two rows of RyRs. The lumen of the SR terminal cisternae appears significantly narrower than in controls (compare panels B and D). Scale bars: A and C, 0.25 μm; B and D, 0.1 μm.
Figure 6. CRUs in CS1-null soleus muscle maintain the triadic structure, but the terminal cisternae appear much smaller.

In CS1-null fibres from soleus muscle (C and D), CRUs are mostly in the form of triads, as in WT soleus muscle (A and B), and maintain their normal location and orientation at the edges of the sarcomeric A band. The width of the SR terminal cisternae is, though, visibly smaller than in CS1-null fibres (B and D). Scale bars: A and C, 0.25 μm; B and D, 0.05 μm. TT, T-tubule.
In CS1-null EDL muscle the general shape of CRUs is strikingly altered
In CS1-null EDL the most noticeable difference is the presence of multiple stacks of alternating SR and T-tubule profiles, that occupy the place where triads are usually found. Junctions that are formed by five, seven or even nine elements (pentads, heptads and nonads, Fig. 5C and D) are seen in 71% of EDL fibres in CS1-null mice. In contrast, CRUs in soleus retain their usual triadic disposition and CRUs formed by more than three elements are quite rare. It must be noted that in order to deploy the stacked arrangement, the T-tubule in EDL muscles must bend repeatedly.
A second alteration seen in CS1-null muscles is in the width of SR terminal cisternae, which tend to be considerably narrower than in normal triads (Table 3, column I). In EDL, where multilayered junctions are frequent both the central elements of the stacks and those at the two borders are narrower than the junctional SR in WT triads. In soleus, the junctional SR cisternae are also narrower, although multiple stacks are not formed (Figs 5 and 6). The average width of the junctional SR cisternae is 25.0 ± 4.5 nm (n, number of measurements; n = 487) and 30.0 ± 5.9 nm (n = 404), respectively, in CS1-null EDL and soleus muscle, both of which are significantly lower than in their respective WT counterparts (EDL: 62.4 ± 10.5 nm; soleus: 62.5 ± 11.7 nm).
A third alteration, which is present only in CS1-null EDL is a change in the orientation of the main axis of the T-tubule within CRUs and thus a change in the axis of the whole CRU. CRUs often show transverse, oblique, and even longitudinal orientations with respect to the main axis of myofibrils (Fig. 5C), while still maintaining their usual position at the edges of the A band.
Due to the multiple stacking, many of the junctional SR cisternae in EDL are flanked by RyRs on two sides, both of which face T-tubules. This, together with the fact that the junctions are formed by multiple layers, and that RyRs form multiple rows, implies that CRUs of CS1-null EDL fibres contain a larger number of RyRs than triads in WT fibres. The density of RyRs related to the area of thin section, obtained by counting ‘feet’ directly in the images (see Methods) is proportional to the density of RyRs per fibre volume. RyR density is approximately double in CS1-null EDL when compared to WT fibres: 71.8 RyRs (10μm2)−1versus 39.2 RyRs (10μm2)−1 (Table 3, column II; n, number of micrographs: n = 111 and n = 143 for CS1-null and WT, respectively). In this analysis all EDL fibres are included, i.e. both fibres with multilayered CRUs and those that contain CRUs formed by three elements. By contrast, in soleus muscle the number of RyRs per unit area of section remains unchanged (Table 3, column II n = 54 and n = 74 for WT and CS1-null, respectively). The increase in RyR content found in EDL fibres was confirmed by an increase in Maximal Binding Capacity (Bmax) of 3H-ryanodine binding (see Fig. 7). Calculations based on morphometric analysis (Loud et al. 1965; Mobley & Eisenberg, 1975) show that the total SR volume relative to the total fibre volume is virtually unchanged in CS1-null EDL fibres compared to WT fibres (Table 3, column III): 5.25 ± 1.9% in CS1-null fibres versus 5.66 ± 1.8% in WT (n, number of micrographs; n = 97 and n = 93, respectively). The surprisingly unvaried total SR volume is the final result of two significant structural alterations: (a) shrinkage of the SR terminal cisternae (Table 3, column I), which alone would lead to a decrease in SR volume; and (b) proliferation of the junctional SR vesicles (Fig. 5C) that alone would lead to an increase in SR volume. The compounded effect of these two structural changes seems to result in an approximately unchanged total SR volume.
Figure 7. RyR expression, but not CS2, SERCA1, and triadin, is upregulated in EDL muscle.

A, binding of 3H-ryanodine shows that the Bmax is approximately doubled in CS1-null mice samples as compared to WT samples, whereas the Kd is virtually unchanged. Each data point represents the average of duplicate determinations. Bmax and Kd values are calculated by Scatchard plot analysis with Enzifitter version 1.03 Elsevier Biosoft program. B–D) quantitative analysis of total homogenates from WT and CS1-null EDL muscles (left panels) and graphical representation of the mean values of densitometric signals (right panels, arbitrary units). Data plotted in the bar graph are expressed as means ± s.d. for n = 4 WT EDL and n = 4 CS1-null EDL. Densitometric analysis shows a decrease of expression of CS2 in CS1-null EDLs, whereas SERCA1 and 95 kDa triadin contents are not significantly changed.
The final ultrastructural alteration in CS1-null muscles is that mitochondria are significantly more abundant in EDL fibres of CS1-null mice when compared to WT EDL fibres (62.1 versus 36.3 mitochondria (100μm2 of thin section area)−1. In contrast, no change is detected in soleus fibres (Table 3, column IV). On the whole, in agreement with the more pronounced lack of total CS, the reorganization of the SR, of the T-tubule/SR junctions, and of the mitochondrial apparatus, is much more evident in CS1-null EDL than in CS1-null soleus. Only the reduction in size of the SR terminal cisternae is similar in both muscles.
Expression levels of RyR, CS2, SERCA1, and triadin in EDL muscle
Since EDL muscles are more affected by the lack of CS1 both functionally and structurally, we limited this analysis to the EDL. A quantitative estimate of the RyR content is given by 3H-ryanodine binding of total membranes isolated from either WT or CS1-null EDL. As can be seen in Fig. 7Bmax(μg protein)−1 is approximately doubled in CS1-null samples as compared to WT samples, whereas Kd is virtually unchanged. The 3H-ryanodine-binding experiments confirm the ultrastructural results reported in Table 3, column II of an increase in the amount of RyR expression in EDL fibres. Densitometric analysis of total homogenates from WT and CS1-null EDL muscles was used to quantify the expression levels of CS2, SERCA1 and 95 kDa triadin. This quantitative analysis confirmed that there is no compensatory expression of CS2 in CS1-null muscle. Expression of CS2 in CS1-null EDLs seems actually slightly decreased, whereas SERCA1 and 95 kDa triadin contents are not significantly changed. Data plotted in the bar graphs of Fig. 7 are expressed as means ± s.d. for n = 4 WT EDL and n = 4 CS1-null EDL. Although the time to half relaxation is increased in CS1-null EDL, SERCA1 content is unchanged compared to WT (Fig. 7B and C), suggesting that the reduction in Ca2+ uptake must be due to the absence of CS1-buffering ability. The 95 kDa triadin isoform (Brandt et al. 1990; Kim et al. 1990; Caswell et al. 1991) is thought to be a CS1-binding protein. However, as can be seen from the average of four different experiments, there is no statistically significant difference in triadin-95 expression between CS1-null and WT EDL muscles (Fig. 7D).
Ca2+ kinetics in single muscle fibres
Intracellular free Ca2+ concentrations and Ca2+ transients in response to electrical stimulation were measured in single FDB fibres loaded with Fura-2 AM. Each fibre was later individually characterized by Western blot for its CS1 and CS2 content. Data obtained from WT fibres lacking CS2, and null fibres lacking both CS1 and CS2 are compared in Fig. 8A and D. Basal calcium levels at rest are not significantly different in CS1-null compared to WT fibres, but the amplitude of the transient induced by a single electrical stimulation is strongly and significantly reduced in the CS1-null group. Caffeine contractures of saponin skinned fast muscle fibres from the tibialis muscle, loaded with calcium, confirm that the amount of releasable calcium at maximal doses of caffeine is significantly reduced in null versus WT fibres (an example is given in Fig. 8B, caffeine 20 mM). The caffeine concentrations at which half of the maximal response is achieved (EC50) is not different in fibres lacking CS (Fig. 8E), so that when the dose–response curves are normalized to the maximal response they are virtually superimposable (Fig. 8F). These findings strongly support the view that, in spite of the ultrastructural reorganization of the CRUs and the increased density of RyR1s, the absence of calsequestrin reduces the amount of releasable Ca2+, and this in turn is responsible for the smaller amplitude of cytosolic Ca2+ transients.
Figure 8. Calcium transient amplitude and calcium release in response to caffeine in single muscle fibres.

A and D, CS1-null single muscle fibres display similar basal cytosolic free Ca2+ concentrations, but significantly smaller Ca2+ transients induced by electrical stimulation compared to WT fibres. Fura-2 AM is used as a cell-permeant free Ca2+ indicator (see Methods). B, the amount of Ca2+ released by caffeine (20 mM) from the SR of single fibres permeabilized with saponin is significantly smaller in CS1-null than in WT. The amount of Ca2+ released is evaluated by the tension–time area of the response to caffeine (see dose–response curve in E and F). E, the curves correspond to the equation Y =T/[1 + 10∧(logEC50−X)] where T = 7.06 ± 0.41 and logEC50= 0.24 ± 0.08 for CS1-null and T = 29.71 ± 1.19 and logEC50= 0.09 ± 0.06 for WT: the difference between the T values of CS1-null and WT is statistically significant. F, the dose–response curve for tension–time area normalized to the highest value reached with maximal caffeine concentration indicates no difference between CS1-null and WT. C, after all single-fibre experiments, the presence of CS isoforms has been verified in each fibre with Western blot. Examples of Western blot on single fibres (sf) are shown in comparison with total homogenate from WT FDB muscles. Only fibres lacking both CS1 and CS2 (as sf – a) have been used for comparison with WT fibres.
Discussion
The contribution of calsequestrin (CS) to the capacity of the SR to sequester and hold a large Ca2+ load and its specific location next to Ca2+ release channels are usually considered essential to support the massive Ca2+ release that occurs during normal activation of a muscle fibre (Somlyo et al. 1981; Hollingworth et al. 1996). In addition, it has been suggested that CS, which is held in proximity of the RyRs and presumably connected to them via triadin (Caswell et al. 1991; Liu & Pessah, 1994; Guo & Campbell, 1995), may have a direct regulatory role on the SR release channels, perhaps helping to shape the release event. However, available evidence is controversial, pointing either to an activation (Kawasaki & Kasai, 1994; Ohkura et al. 1995; Szegedi et al. 1999; Herzog et al. 2000) or an inhibition by CS of Ca2+ release through RyR1 (Beard et al. 2002). In addition, in SR vesicles that have been stripped of CS, the rate of ATP hydrolysis, indicative of active Ca2+ transport, falls off rapidly during the first seconds of uptake, due to back inhibition by accumulated Ca2+ (Weber, 1971; Inesi & de Meis, 1989), whereas SR Ca2+ capacity is increased by Ca2+-chelating agents, or Ca2+-buffering molecules such as CS (Makinose & Hasselbach, 1965).
Based on the above reasoning, the ablation of CS1 was expected to deeply impair contractile function. In this view, the small effect of the ablation of CS1 on the contractile response of skeletal muscles was totally unexpected, particularly in the case of the fast-twitch muscles in which the amount of Ca2+ cycled during a single twitch is about three times greater than in slow-twitch fibres (Baylor & Hollingworth, 2003). The knock-out of the CS1 gene in our mice results in muscles containing a large number of fibres virtually devoid of any CS (Figs 3 and 8). Nevertheless, their ability to generate a contractile response is maintained (Fig. 4). A careful investigation of the CRU's ultrastructure (and of the mitochondrial apparatus) in these CS-null muscles reveals significant adaptations that may to some extent compensate for the lack of CS1, particularly in fast-twitch muscles (i.e. EDL, FDB, tibialis anterior). These changes in CS1-null animals include multiple junctions presenting a wider profile and an almost doubled amount of Ca2+-release channels, compared to junctions in WT animals (Fig. 5 and Table 3). The proliferation of the SR junctional domains, i.e. the reorganization in multilayers of the Ca2+-release units, probably represent a compensation for the reduction in storage volume caused by the shrinkage of the terminal cisternae, since there is no variation in the relative SR volume (Table 3) and no variation in the relative amount of longitudinal SR (not shown). The increased number of release sites can also be ascribed to the compensatory changes, but it is important to underline that the striking proliferation of SR junctional domains and the increased abundance of RyR1 is not sufficient to completely compensate for the lack of CS1, as demonstrated by the lower amount of SR Ca2+ content and the reduced amplitude of the Ca2+ transients (Fig. 8). Nevertheless, the occurrence of compensatory changes of the SR/T-tubule junctional domains should be taken into consideration when comparing the contractile responses of WT and CS1-null muscles, since the structural rearrangement of the Ca2+-handling apparatus may reduce the functional impairment of the contractile response caused by the absence of CS1. The search for compensatory adaptations, however, has not been successful in all cases. Similarly, no apparent upregulation of Ca2+-binding proteins has been found in the hearts of CS2-null mice (Knollmann et al. 2006). Fibres completely lacking CS show a clear decrease in the amount of calcium release as shown by the reduced tension–time area of the response to maximal doses of caffeine and by the reduced amplitude of the Ca2+ transient measured with Fura-2 (Fig. 8). This means that, as mentioned above, the large increase in junctional domains (Fig. 5) and number of RyRs (Table 3 and Fig. 7) are not sufficient to completely compensate for the lack of CS1 in the SR lumen. It is important, however, to observe that in spite of the lower amount of Ca2+ released, tension developed by a twitch induced by electrical stimulation is not reduced (Fig. 4). The preservation of the peak tension reached during the twitch points to a second important effect of the lack of CS1, i.e. the prolongation of the twitch time course. The likely cause of the prolonged time-to-peak tension and half-relaxation time is the impairment of Ca2+ reuptake by the SR. In the absence of CS1, the increase of intraluminal Ca2+ concentration inhibits Ca2+ sequestration into SR (Weber, 1971; Inesi & de Meis, 1989). The delayed Ca2+ reuptake allows a prolonged activation of the myofibrils in CS1-null fibres and this, in turn, gives the necessary time to develop tension up to or even slightly above the peak value reached in WT fibres.
Taken together the functional alterations demonstrate that two distinct effects follow the lack of CS1, on one hand the impaired Ca2+ release and on the other hand the impaired Ca2+ reuptake (see cartoon in Fig. 9). The impaired Ca2+ release can be attributed to the role played by CS in proximity to junctional membrane (Jorgensen et al. 1983; Franzini-Armstrong et al. 1987), to control either Ca2+ availability or the opening kinetics of RyRs (Ikemoto et al. 1989; Kawasaki & Kasai, 1994; Beard et al. 2002). The reason for the delayed Ca2+ reuptake can be found in the reduced Ca2+-buffering capacity of the SR. The reuptake rate is limited by intra-SR free [Ca2+] by back-inhibition on the Ca2+ pump as discussed above (Fryer & Stephenson, 1996). Fast-twitch fibres (superficial layers of the tibialis anterior and FDB) and predominantly fast muscles (EDL) are particularly sensitive to the lack of CS1, because the amount of Ca2+ released and taken up is greater than in slow fibres (Fryer & Stephenson, 1996), and because CS1 is in most cases the only isoform present. The only effective, although partial, compensation for the delayed removal of Ca2+ from the cytosol is probably given by the proliferation of the mitochondria in EDL fibres (see below).
Figure 9. Cartoon showing the structural and functional modifications occurring in fast-twitch muscle fibres following the removal of CS1.

Ultrastructural features of CRUs in CS1-null EDL fibres differ from those of WT muscle: junctions are often multilayered and longitudinal. RyR-feet and mitochondria are also increased in number (see also Fig. 5 and Table 3). Mitochondrial proliferation probably explains the surprising increase in fatigue resistance in CS1-null EDL (Fig. 4). Experiments in single fibres and in skinned fibres suggest that Ca2+ transient and total SR Ca2+ content (see ovals) are reduced in CS1-null muscle (see also Fig. 8). Abbreviations used: CS, calsequestrin; CRUs, calcium release units; SR, sarcoplasmic reticulum; RyRs, ryanodine receptors.
The structural reactions to lack of CS are quite interesting and may considered a combination of compensatory adaptations and developmental effects, in addition to some obvious geometrical alterations. A large decrease in size of the junctional SR cisternae is detected both in the EDL and soleus and is particularly prominent in the former, where very little CS2 is present. It is logical to assume that this volume decrease is directly due the lack of the luminal CS polymer that usually fills the junctional SR. In support of this hypothesis, other studies indicate that CS2 over-expression in murine myocardium induced the opposite effects, i.e. drastic swelling of SR terminal cisternae (Jones et al. 1998), whereas CS2 knockout in cardiomyocytes also caused significant alterations of the SR terminal cisternae, which were either slightly narrower or noticeably wider than in WT myocardium (Knollmann et al. 2006).
Interestingly, two changes that may seem to be direct compensatory responses to the reduced SR capacity for calcium are seen only in the EDL and are quite undistinguishable from the response of this muscle to other physiopathological stimuli (Franzini-Armstrong, 1991; Takekura et al. 1993; Takekura & Kasuga, 1999; Takekura et al. 2001; Boncompagni et al. 2006). The complex proliferation of the junctional SR and the convoluted (and often longitudinal) T-tubule path has the effect of increasing the density of Ca2+-release channels while maintaining the total SR volume constant (despite the fact that the cisternae are smaller in size, see above). The same abundance and, interestingly, also the same change in orientation of the junctional SR is seen in the EDL, but not in soleus, as a swift response to short-term denervation that rapidly returns to normal when the muscle is innervated again (Takekura & Kasuga, 1999). Thus, the formation of complex junctions and the increased density of RyRs may be in response to complex stimuli rather than simply as a compensation for the lowered calcium content of the SR.
It is interesting that CS1-null EDL shows a doubling of mitochondria content, with the resulting increase in fatigue resistance. In contrast, mitochondria content is unchanged in CS1-null soleus or CS2-null cardiomyocytes (Knollmann et al. 2006). Skeletal muscle mitochondria take up calcium during a single twitch (Rudolf et al. 2004) and can affect the time course of relaxation if sufficiently abundant, e.g. in mitochondria-rich slow-twitch fibres (Gillis, 1997). Mitochondrial biogenesis in fast-twitch fibres has previously been shown to be stimulated by the absence of the cyoplasmic calcium buffering protein parvalbumin (Racay et al. 2006) and in junctate-overexpressing mice (Divet et al. 2007). Conceivably, prolonged presence of Ca2+ in the cytosol, even if only slight, induces mitochondrial proliferation as suggested by Rohas et al. (2007). However, an increased mitochondrial volume, while perhaps helpful in accelerating relaxation, does not fully solve the problem due to reduced SR capacity for calcium, since the ions need to be sequestered in the SR, not the mitochondria, to be available for subsequent release.
The preserved ability to develop tension even in fibres completely devoid of CS is, in our view, one of the most remarkable findings stemming from the analysis of CS1-null mice. The reduced Ca2+ release and the decreased cytosolic Ca2+ transient support the view that the Ca2+-storage capacity of the SR is impaired, whereas the prolongation of the contractile response is consistent with a defective Ca2+ reuptake. Such functional alterations are present in spite of the reorganization of the CRUs, the large increase in RyR content and the increased abundance of mitochondria. Thus, Ca2+ buffering in the SR is unequivocally one of the essential functions of CS1 in skeletal muscles, in agreement with the very recent study by Pape et al. (2007). The evidence pointing to an impaired Ca2+ release is not sufficient to make conclusions about the modulatory function of CS1 on RyR kinetics. The dissection of the Ca2+ release and uptake of single muscle fibres or myotubes in culture and a detailed analysis of compensatory mechanisms at transcriptomic and proteomic level will be the goal of future studies on the CS1-null model.
Acknowledgments
We thank the Charles River Laboratories (Boston, MA), Ronit Hirsch and Santwana Mukherjee (P. D. Allen laboratory) for technical support in managing the mice colony and the genotyping. We also thank L. R. Jones (Indiana University) and S. Schiaffino (University of Padova) for the generous gift of antibodies used to mark CS, triadin, and MHC in our experiments; Luana Toniolo for help with single-fibre experiments; Giorgio Fanò for the lab space and financial support that have allowed us to start working at the University of Chieti; and Dante Tatone for his technical help in setting up our new laboratory in the CeSI building. This study was supported by: (a) Research Grant # GGP030289 from the Italian Telethon Foundation and Research Funds from the University G. d'Annunzio of Chieti to Feliciano Protasi; (b) FIRB to Pompeo Volpe; and (c) NIH P01AR AR17605-05 to Paul D. Allen.
References
- Appelt D, Buenviaje B, Champ C, Franzini-Armstrong C. Quantitation of ‘junctional feet’ content in two types of muscle fiber from hind limb muscles of the rat. Tissue Cell. 1989;21:783–794. doi: 10.1016/0040-8166(89)90087-6. [DOI] [PubMed] [Google Scholar]
- Baylor SM, Hollingworth S. Sarcoplasmic reticulum calcium release compared in slow-twitch and fast-twitch fibres of mouse muscle. J Physiol. 2003;551:125–138. doi: 10.1113/jphysiol.2003.041608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beard NA, Casarotto MG, Wei L, Varsanyi M, Laver DR, Dulhunty AF. Regulation of ryanodine receptors by calsequestrin: effect of high luminal Ca2+ and phosphorylation. Biophys J. 2005;88:3444–3454. doi: 10.1529/biophysj.104.051441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beard NA, Sakowska MM, Dulhunty AF, Laver DR. Calsequestrin is an inhibitor of skeletal muscle ryanodine receptor calcium release channels. Biophys J. 2002;82:310–320. doi: 10.1016/S0006-3495(02)75396-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block BA, Imagawa T, Campbell KP, Franzini-Armstrong C. Structural evidence for direct interaction between the molecular components of the transverse tubule/sarcoplasmic reticulum junction in skeletal muscle. J Cell Biol. 1988;107:2587–2600. doi: 10.1083/jcb.107.6.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boncompagni S, d'Amelio L, Fulle S, Fano G, Protasi F. Progressive disorganization of the excitation–contraction coupling apparatus in aging human skeletal muscle as revealed by electron microscopy: a possible role in the decline of muscle performance. J Gerontol A Biol Sci Med Sci. 2006;61:995–1008. doi: 10.1093/gerona/61.10.995. [DOI] [PubMed] [Google Scholar]
- Brandt NR, Caswell AH, Wen SR, Talvenheimo JA. Molecular interactions of the junctional foot protein and dihydropyridine receptor in skeletal muscle triads. J Membr Biol. 1990;113:237–251. doi: 10.1007/BF01870075. [DOI] [PubMed] [Google Scholar]
- Campbell KP, MacLennan DH, Jorgensen AO, Mintzer MC. Purification and characterization of calsequestrin from canine cardiac sarcoplasmic reticulum and identification of the 53 000 dalton glycoprotein. J Biol Chem. 1983;258:1197–1204. [PubMed] [Google Scholar]
- Caswell AH, Brandt NR, Brunschwig JP, Purkerson S. Localization and partial characterization of the oligomeric disulfide-linked molecular weight 95 000 protein (triadin) which binds the ryanodine and dihydropyridine receptors in skeletal muscle triadic vesicles. Biochemistry. 1991;30:7507–7513. doi: 10.1021/bi00244a020. [DOI] [PubMed] [Google Scholar]
- Cho JH, Oh YS, Park KWYuJ, Choi KY, Shin JY, Kim DH, Park WJ, Hamada T, Kagawa H, Maryon EB, Bandyopadhyay J, Ahnn J. Calsequestrin, a calcium sequestering protein localized at the sarcoplasmic reticulum, is not essential for body-wall muscle function in Caenorhabditis elegans. J Cell Sci. 2000;113:3947–3958. doi: 10.1242/jcs.113.22.3947. [DOI] [PubMed] [Google Scholar]
- Damiani E, Tobaldin G, Volpe P, Margreth A. Quantitation of ryanodine receptor of rabbit skeletal muscle, heart and brain. Biochem Biophys Res Commun. 1991;175:858–865. doi: 10.1016/0006-291x(91)91644-r. [DOI] [PubMed] [Google Scholar]
- Damiani E, Volpe P, Margreth A. Coexpression of two isoforms of calsequestrin in rabbit slow-twitch muscle. J Muscle Res Cell Motil. 1990;11:522–530. doi: 10.1007/BF01745219. [DOI] [PubMed] [Google Scholar]
- Defranchi E, Bonaccurso E, Tedesco M, Canato M, Pavan E, Raiteri R, Reggiani C. Imaging and elasticity measurements of the sarcolemma of fully differentiated skeletal muscle fibres. Microsc Res Tech. 2005;67:27–35. doi: 10.1002/jemt.20177. [DOI] [PubMed] [Google Scholar]
- Divet A, Paesante S, Grasso C, Cavagna D, Tiveron C, Protasi F, Huchet-Cadiou C, Treves S, Zorzato F. Increased Ca2+ storage capacity of the skeletal muscle sarcoplasmaic reticulum of transgenic mice over-expressing membrane bound calcium binding protein junctate. J Cell Physiol. 2007;May 21 doi: 10.1002/jcp.21121. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Dolmetsch R. Excitation-transcription coupling: signaling by ion channels to the nucleus. Sci STKE. 2003;2003:PE4. doi: 10.1126/stke.2003.166.pe4. [DOI] [PubMed] [Google Scholar]
- Dulhunty AF, Beard NA, Pouliquin P, Kimura T. Novel regulators of RyR Ca2+ release channels: insight into molecular changes in genetically-linked myopathies. J Muscle Res Cell Motil. 2006;27:351–365. doi: 10.1007/s10974-006-9086-1. [DOI] [PubMed] [Google Scholar]
- Endo M, Iino M. Measurement of Ca2+ release in skinned fibers from skeletal muscle. Meth Enzymol. 1988;157:12–26. doi: 10.1016/0076-6879(88)57064-7. [DOI] [PubMed] [Google Scholar]
- Fliegel L, Ohnishi M, Carpenter MR, Khanna VK, Reithmeier RA, MacLennan DH. Amino acid sequence of rabbit fast-twitch skeletal muscle calsequestrin deduced from cDNA and peptide sequencing. Proc Natl Acad Sci U S A. 1987;84:1167–1171. doi: 10.1073/pnas.84.5.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzini-Armstrong C. Studies of the triad. J Cell Biol. 1970;47:488–499. doi: 10.1083/jcb.47.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzini-Armstrong C. Simultaneous maturation of transverse tubules and sarcoplasmic reticulum during muscle differentiation in the mouse. Dev Biol. 1991;146:353–363. doi: 10.1016/0012-1606(91)90237-w. [DOI] [PubMed] [Google Scholar]
- Franzini-Armstrong C, Kenney LJ, Varriano-Marston E. The structure of calsequestrin in triads of vertebrate skeletal muscle: a deep-etch study. J Cell Biol. 1987;105:49–56. doi: 10.1083/jcb.105.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzini-Armstrong C, Protasi F, Ramesh V. Shape, size, and distribution of Ca2+ release units and couplons in skeletal and cardiac muscles. Biophys J. 1999;77:1528–1539. doi: 10.1016/S0006-3495(99)77000-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer MW, Stephenson DG. Total and sarcoplasmic reticulum calcium contents of skinned fibres from rat skeletal muscle. J Physiol. 1996;493:357–370. doi: 10.1113/jphysiol.1996.sp021388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerke V, Creutz CE, Moss SE. Annexins: linking Ca2+ signalling to membrane dynamics. Nat Rev Mol Cell Biol. 2005;6:449–461. doi: 10.1038/nrm1661. [DOI] [PubMed] [Google Scholar]
- Gilchrist JS, Belcastro AN, Katz S. Intraluminal Ca2+ dependence of Ca2+ and ryanodine-mediated regulation of skeletal muscle sarcoplasmic reticulum Ca2+ release. J Biol Chem. 1992;267:20850–20856. [PubMed] [Google Scholar]
- Gillis JM. Inhibition of mitochondrial calcium uptake slows down relaxation in mitochondria-rich skeletal muscles. J Muscle Res Cell Motil. 1997;18:473–483. doi: 10.1023/a:1018603032590. [DOI] [PubMed] [Google Scholar]
- Guo W, Campbell KP. Association of triadin with the ryanodine receptor and calsequestrin in the lumen of the sarcoplasmic reticulum. J Biol Chem. 1995;270:9027–9030. doi: 10.1074/jbc.270.16.9027. [DOI] [PubMed] [Google Scholar]
- Herzog A, Szegedi C, Jona I, Herberg FW, Varsanyi M. Surface plasmon resonance studies prove the interaction of skeletal muscle sarcoplasmic reticular Ca2+ release channel/ryanodine receptor with calsequestrin. FEBS Lett. 2000;472:73–77. doi: 10.1016/s0014-5793(00)01431-9. [DOI] [PubMed] [Google Scholar]
- Hollingworth S, Zhao M, Baylor SM. The amplitude and time course of the myoplasmic free [Ca2+] transient in fast-twitch fibers of mouse muscle. J Gen Physiol. 1996;108:455–469. doi: 10.1085/jgp.108.5.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikemoto N, Ronjat M, Meszaros LG, Koshita M. Postulated role of calsequestrin in the regulation of calcium release from sarcoplasmic reticulum. Biochemistry. 1989;28:6764–6771. doi: 10.1021/bi00442a033. [DOI] [PubMed] [Google Scholar]
- Inesi G, de Meis L. Regulation of steady state filling in sarcoplasmic reticulum. Roles of back-inhibition, leakage, and slippage of the calcium pump. J Biol Chem. 1989;264:5929–5936. [PubMed] [Google Scholar]
- Jones LR, Suzuki YJ, Wang W, Kobayashi YM, Ramesh V, Franzini-Armstrong C, Cleemann L, Morad M. Regulation of Ca2+ signaling in transgenic mouse cardiac myocytes overexpressing calsequestrin. J Clin Invest. 1998;101:1385–1393. doi: 10.1172/JCI1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen AO, Shen AC, Arnold W, Leung AT, Campbell KP. Subcellular distribution of the 1,4-dihydropyridine receptor in rabbit skeletal muscle in situ: an immunofluorescence and immunocolloidal gold-labeling study. J Cell Biol. 1989;109:135–147. doi: 10.1083/jcb.109.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen AO, Shen AC, Campbell KP, MacLennan DH. Ultrastructural localization of calsequestrin in rat skeletal muscle by immunoferritin labeling of ultrathin frozen sections. J Cell Biol. 1983;97:1573–1581. doi: 10.1083/jcb.97.5.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki T, Kasai M. Regulation of calcium channel in sarcoplasmic reticulum by calsequestrin. Biochem Biophys Res Commun. 1994;199:1120–1127. doi: 10.1006/bbrc.1994.1347. [DOI] [PubMed] [Google Scholar]
- Kim KC, Caswell AH, Talvenheimo JA, Brandt NR. Isolation of a terminal cisterna protein which may link the dihydropyridine receptor to the junctional foot protein in skeletal muscle. Biochemistry. 1990;29:9281–9289. doi: 10.1021/bi00491a025. [DOI] [PubMed] [Google Scholar]
- Knollmann BC, Chopra N, Hlaing T, Akin B, Yang T, Ettensohn K, Knollmann BE, Horton KD, Weissman NJ, Holinstat I, Zhang W, Roden DM, Jones LR, Franzini-Armstrong C, Pfeifer K. Casq2 deletion causes sarcoplasmic reticulum Volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest. 2006;116:2510–2520. doi: 10.1172/JCI29128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai FA, Erickson HP, Rousseau E, Liu QY, Meissner G. Purification and reconstitution of the calcium release channel from skeletal muscle. Nature. 1988;331:315–319. doi: 10.1038/331315a0. [DOI] [PubMed] [Google Scholar]
- Launikonis BS, Stephenson DG. Effect of saponin treatment on the sarcoplasmic reticulum of rat, cane toad and crustacean (yabby) skeletal muscle. J Physiol. 1997;504:425–437. doi: 10.1111/j.1469-7793.1997.425be.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leberer E, Hartner KT, Pette D. Postnatal development of Ca2+-sequestration by the sarcoplasmic reticulum of fast and slow muscles in normal and dystrophic mice. Eur J Biochem. 1988;174:247–253. doi: 10.1111/j.1432-1033.1988.tb14090.x. [DOI] [PubMed] [Google Scholar]
- Leberer E, Pette D. Immunochemical quantification of sarcoplasmic reticulum Ca-ATPase, of calsequestrin and of parvalbumin in rabbit skeletal muscles of defined fiber composition. Eur J Biochem. 1986;156:489–496. doi: 10.1111/j.1432-1033.1986.tb09607.x. [DOI] [PubMed] [Google Scholar]
- Liu G, Pessah IN. Molecular interaction between ryanodine receptor and glycoprotein triadin involves redox cycling of functionally important hyperreactive sulfhydryls. J Biol Chem. 1994;269:33028–33034. [PubMed] [Google Scholar]
- Loud AV, Barany WC, Pack BA. Quantitative evaluation of cytoplasmic structures in electron micrographs. Laboratory Invest. 1965;14:996–1008. [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- MacLennan DH, Wong PT. Isolation of a calcium-sequestering protein from sarcoplasmic reticulum. Proc Natl Acad Sci U S A. 1971;68:1231–1235. doi: 10.1073/pnas.68.6.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makinose M, Hasselbach W. [The influence of oxalate on calcium transport of isolated sarcoplasmic reticular vesicles. Biochem Z. 1965;343:360–382. [PubMed] [Google Scholar]
- Mobley BA, Eisenberg BR. Sizes of components in frog skeletal muscle measured by methods of stereology. J Gen Physiol. 1975;66:31–45. doi: 10.1085/jgp.66.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkura M, Ide T, Furukawa K, Kawasaki T, Kasai M, Ohizumi Y. Calsequestrin is essential for the Ca2+ release induced by myotoxin alpha in skeletal muscle sarcoplasmic reticulum. Can J Physiol Pharmacol. 1995;73:1181–1185. doi: 10.1139/y95-167. [DOI] [PubMed] [Google Scholar]
- Pape PC, Fenelon K, Lamboley CR, Stachura D. Role of calsequestrin evaluated from changes in free and total calcium concentrations in the sarcoplasmic reticulum of frog cut skeletal muscle fibres. J Physiol. 2007;581:319–367. doi: 10.1113/jphysiol.2006.126474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrino MA, Canepari M, Rossi R, D'Antona G, Reggiani C, Bottinelli R. Orthologous myosin isoforms and scaling of shortening velocity with body size in mouse, rat, rabbit and human muscles. J Physiol. 2003;546:677–689. doi: 10.1113/jphysiol.2002.027375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protasi F. Structural interaction between RYRs and DHPRs in calcium release units of cardiac and skeletal muscle cells. Front Biosci. 2002;7:d650–658. doi: 10.2741/A801. [DOI] [PubMed] [Google Scholar]
- Protasi F, Franzini-Armstrong C, Allen PD. Role of ryanodine receptors in the assembly of calcium release units in skeletal muscle. J Cell Biol. 1998;140:831–842. doi: 10.1083/jcb.140.4.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protasi F, Franzini-Armstrong C, Flucher BE. Coordinated incorporation of skeletal muscle dihydropyridine receptors and ryanodine receptors in peripheral couplings of BC3H1 cells. J Cell Biol. 1997;137:859–870. doi: 10.1083/jcb.137.4.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protasi F, Takekura H, Wang Y, Chen SR, Meissner G, Allen PD, Franzini-Armstrong C. RYR1 and RYR3 have different roles in the assembly of calcium release units of skeletal muscle. Biophys J. 2000;79:2494–2508. doi: 10.1016/S0006-3495(00)76491-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racay P, Gregory P, Schwaller B. Parvalbumin deficiency in fast-twitch muscles leads to increased ‘slow-twitch type’ mitochondria, but does not affect the expression of fiber specific proteins. FEBS J. 2006;273:96–108. doi: 10.1111/j.1742-4658.2005.05046.x. [DOI] [PubMed] [Google Scholar]
- Rios E, Ma JJ, Gonzalez A. The mechanical hypothesis of excitation-contraction (EC) coupling in skeletal muscle. J Muscle Res Cell Motil. 1991;12:127–135. doi: 10.1007/BF01774031. [DOI] [PubMed] [Google Scholar]
- Rohas LM, St-Pierre J, Uldry M, Jager S, Handschin C, Spiegelman BM. A fundamental system of cellular energy homeostasis regulated by PGC-1α. Proc Natl Acad Sci U S A. 2007;104:7933–7938. doi: 10.1073/pnas.0702683104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi R, Bottinelli R, Sorrentino V, Reggiani C. Response to caffeine and ryanodine receptor isoforms in mouse skeletal muscles. Am J Physiol Cell Physiol. 2001;281:C585–C594. doi: 10.1152/ajpcell.2001.281.2.C585. [DOI] [PubMed] [Google Scholar]
- Rudolf R, Mongillo M, Magalhaes PJ, Pozzan T. In vivo monitoring of Ca2+ uptake into mitochondria of mouse skeletal muscle during contraction. J Cell Biol. 2004;166:527–536. doi: 10.1083/jcb.200403102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacchetto R, Volpe P, Damiani E, Margreth A. Postnatal development of rabbit fast-twitch skeletal muscle: accumulation, isoform transition and fibre distribution of calsequestrin. J Muscle Res Cell Motil. 1993;14:646–653. doi: 10.1007/BF00141561. [DOI] [PubMed] [Google Scholar]
- Saito A, Seiler S, Chu A, Fleischer S. Preparation and morphology of sarcoplasmic reticulum terminal cisternae from rabbit skeletal muscle. J Cell Biol. 1984;99:875–885. doi: 10.1083/jcb.99.3.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandow A. Excitation-contraction coupling in skeletal muscle. Pharmacol Rev. 1965;17:265–320. [PubMed] [Google Scholar]
- Schiaffino S, Gorza L, Sartore S, Saggin L, Ausoni S, Vianello M, Gundersen K, Lomo T. Three myosin heavy chain isoforms in type 2 skeletal muscle fibres. J Muscle Res Cell Motil. 1989;10:197–205. doi: 10.1007/BF01739810. [DOI] [PubMed] [Google Scholar]
- Schneider MF. Control of calcium release in functioning skeletal muscle fibers. Annu Rev Physiol. 1994;56:463–484. doi: 10.1146/annurev.ph.56.030194.002335. [DOI] [PubMed] [Google Scholar]
- Schneider MF, Chandler WK. Voltage dependent charge movement of skeletal muscle: a possible step in excitation–contraction coupling. Nature. 1973;242:244–246. doi: 10.1038/242244a0. [DOI] [PubMed] [Google Scholar]
- Scott BT, Simmerman HK, Collins JH, Nadal-Ginard B, Jones LR. Complete amino acid sequence of canine cardiac calsequestrin deduced by cDNA cloning. J Biol Chem. 1988;263:8958–8964. [PubMed] [Google Scholar]
- Silver J, Keerikatte V. Novel use of polymerase chain reaction to amplify cellular DNA adjacent to an integrated provirus. J Virol. 1989;63:1924–1928. doi: 10.1128/jvi.63.5.1924-1928.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somlyo AV, Gonzalez-Serratos HG, Shuman H, McClellan G, Somlyo AP. Calcium release and ionic changes in the sarcoplasmic reticulum of tetanized muscle: an electron-probe study. J Cell Biol. 1981;90:577–594. doi: 10.1083/jcb.90.3.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szegedi C, Sarkozi S, Herzog A, Jona I, Varsanyi M. Calsequestrin: more than ‘only’ a luminal Ca2+ buffer inside the sarcoplasmic reticulum. Biochem J. 1999;337:19–22. [PMC free article] [PubMed] [Google Scholar]
- Takekura H, Fujinami N, Nishizawa T, Ogasawara H, Kasuga N. Eccentric exercise-induced morphological changes in the membrane systems involved in excitation–contraction coupling in rat skeletal muscle. J Physiol. 2001;533:571–583. doi: 10.1111/j.1469-7793.2001.0571a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takekura H, Kasuga N. Differential response of the membrane systems involved in excitation-contraction coupling to early and later postnatal denervation in rat skeletal muscle. J Muscle Res Cell Motil. 1999;20:279–289. doi: 10.1023/a:1005447317302. [DOI] [PubMed] [Google Scholar]
- Takekura H, Shuman H, Franzini-Armstrong C. Differentiation of membrane systems during development of slow and fast skeletal muscle fibres in chicken. J Muscle Res Cell Motil. 1993;14:633–645. doi: 10.1007/BF00141560. [DOI] [PubMed] [Google Scholar]
- Terentyev D, Viatchenko-Karpinski S, Gyorke I, Volpe P, Williams SC, Gyorke S. Calsequestrin determines the functional size and stability of cardiac intracellular calcium stores: Mechanism for hereditary arrhythmia. Proc Natl Acad Sci U S A. 2003;100:11759–11764. doi: 10.1073/pnas.1932318100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Xu L, Duan H, Pasek DA, Eu JP, Meissner G. Knocking down type 2 but not type 1 calsequestrin reduces calcium sequestration and release in C2C12 skeletal muscle myotubes. J Biol Chem. 2006;281:15572–15581. doi: 10.1074/jbc.M600090200. [DOI] [PubMed] [Google Scholar]
- Weber A. Regulatory mechanisms of the calcium transport system of fragmented rabbit sarcoplasmic rticulum. I. The effect of accumulated calcium on transport and adenosine triphosphate hydrolysis. J Gen Physiol. 1971;57:50–63. doi: 10.1085/jgp.57.1.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber A, Herz P, Reiss I. Study on the kinetics of calcium transport by isolated sarcoplasmic reticulum. Biochem Z. 1966;345:329–369. [Google Scholar]
- Zambrowicz BP, Friedrich GA, Buxton EC, Lilleberg SL, Person C, Sands AT. Disruption and sequence identification of 2000 genes in mouse embryonic stem cells. Nature. 1998;392:608–611. doi: 10.1038/33423. [DOI] [PubMed] [Google Scholar]
- Zorzato F, Volpe P, Damiani E, Quaglino D, Jr, Margreth A. Terminal cisternae of denervated rabbit skeletal muscle: alterations of functional properties of Ca2+ release channels. Am J Physiol Cell Physiol. 1989;257:C504–C511. doi: 10.1152/ajpcell.1989.257.3.C504. [DOI] [PubMed] [Google Scholar]