

Abstract
Cerebral blood flow is highly sensitive to alterations in the partial pressures of O2 and CO2 (PO2 and PCO2, respectively) in the arterial blood. In humans, the extent to which nitric oxide (NO) is involved in this regulation is unclear. We hypothesized that the NO synthase (NOS) inhibitor NG-monomethyl-l-arginine (l-NMMA), attenuates the sensitivity of middle cerebral artery blood velocity (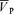 ) to isocapnic hypoxia (end-tidal PO2 = 50 Torr) and euoxic hypercapnia (end-tidal PCO2 =+9 Torr above resting values) in 10 volunteers (age, 28.7 ± 1.3 years; height, 179.2 ± 2.4 cm; weight, 78.0 ± 3.7 kg; mean ±s.e.m.). The techniques of transcranial Doppler ultrasound and dynamic end-tidal forcing were used to measure
) to isocapnic hypoxia (end-tidal PO2 = 50 Torr) and euoxic hypercapnia (end-tidal PCO2 =+9 Torr above resting values) in 10 volunteers (age, 28.7 ± 1.3 years; height, 179.2 ± 2.4 cm; weight, 78.0 ± 3.7 kg; mean ±s.e.m.). The techniques of transcranial Doppler ultrasound and dynamic end-tidal forcing were used to measure 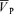 , and control end-tidal PO2 and end-tidal PCO2, respectively. At baseline (isocapnic euoxia), following intravenous administration of l-NMMA, mean arterial blood pressure (MAP) increased (76.3 ± 7.3 to 86.2 ± 9.4 mmHg) and heart rate (HR) decreased (59.5 ± 9.0 to 55.2 ± 9.5 beats min−1) but
, and control end-tidal PO2 and end-tidal PCO2, respectively. At baseline (isocapnic euoxia), following intravenous administration of l-NMMA, mean arterial blood pressure (MAP) increased (76.3 ± 7.3 to 86.2 ± 9.4 mmHg) and heart rate (HR) decreased (59.5 ± 9.0 to 55.2 ± 9.5 beats min−1) but 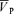 was unchanged. Hypoxia-induced increases in MAP, HR and
was unchanged. Hypoxia-induced increases in MAP, HR and 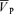 were similar with and without l-NMMA (5.0 ± 0.7 versus 7.1 ± 1.0 mmHg, 11.5 ± 1.4 versus 12.4 ± 1.5 beats min−1, 6.5 ± 0.8 versus 6.6 ± 0.8 cm s−1 for ΔMAP, ΔHR and Δ
were similar with and without l-NMMA (5.0 ± 0.7 versus 7.1 ± 1.0 mmHg, 11.5 ± 1.4 versus 12.4 ± 1.5 beats min−1, 6.5 ± 0.8 versus 6.6 ± 0.8 cm s−1 for ΔMAP, ΔHR and Δ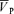 , respectively). Hypercapnia-induced increases in MAP, HR and
, respectively). Hypercapnia-induced increases in MAP, HR and 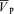 were similar with and without l-NMMA (7.4 ± 3.1 versus 8.1 ± 2.2 mmHg, 10.4 ± 4.6 versus 10.0 ± 4.2 beats min−1, 16.5 ± 1.5 versus 17.6 ± 1.5 cm s−1 for ΔMAP, ΔHR and Δ
were similar with and without l-NMMA (7.4 ± 3.1 versus 8.1 ± 2.2 mmHg, 10.4 ± 4.6 versus 10.0 ± 4.2 beats min−1, 16.5 ± 1.5 versus 17.6 ± 1.5 cm s−1 for ΔMAP, ΔHR and Δ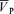 , respectively) but the sensitivity of the
, respectively) but the sensitivity of the  response at the removal of hypercapnia was attenuated with l-NMMA. In young healthy humans, pharmacological blockade of nitric oxide synthesis does not affect the increases in cerebral blood flow with hypoxia and hypercapnia, suggesting that nitric oxide is not required for the cerbrovascular responses to hypoxia and hypercapnia.
response at the removal of hypercapnia was attenuated with l-NMMA. In young healthy humans, pharmacological blockade of nitric oxide synthesis does not affect the increases in cerebral blood flow with hypoxia and hypercapnia, suggesting that nitric oxide is not required for the cerbrovascular responses to hypoxia and hypercapnia.
Cerebral blood flow (CBF) is highly sensitive to alterations in arterial blood gases (Kety & Schmidt, 1948; Heistad & Kontos, 1983; Poulin et al. 1996). A better understanding of the chemical regulation of CBF would help provide better therapeutic approaches in the prevention of stroke. In patients with cardiovascular disease risk factors, an attenuated CBF response to hypercapnia is restored by intravenous arginine (Zimmermann & Haberl, 2003), indicating that nitric oxide (NO) is involved in the chemical regulation of CBF. For instance, in animal studies it has been reported that blockade of NO synthesis using NO synthase (NOS) inhibitors attenuates the CBF responses to hypercapnia (Buchanan & Phillis, 1993; Iadecola & Xu, 1994; Iadecola & Zhang, 1994) and hypoxia (Audibert et al. 1995; Berger & von Kummer, 1998). In human studies of the NOS inhibitor NG-monomethyl-l-arginine (l-NMMA) on the CBF responses to hypoxia or hypercapnia, findings are inconsistent (Schmetterer et al. 1997; White et al. 1998; Van Mil et al. 2002) and the underlying reasons for these conflicting results are unclear. Possibilities include differences in study designs and methodologies, health status of cohorts, and poor control of blood gases. The latter consideration deserves particular attention since an increase in blood pressure associated with systemic administration of NOS inhibitor could attenuate ventilation (Cunningham et al. 1969; Heistad et al. 1972; Heistad et al. 1975), which may lead to an increase in the partial pressure of CO2 in the arterial blood (PaCO2). This is important because the sensitivity of CBF to alterations in PaCO2 is dependent on the level of PaCO2 itself (Ide et al. 2003), with higher sensitivity in the hypercapnic (Poulin et al. 1996; Ide et al. 2003) compared with the hypocapnic (Poulin et al. 1998; Ide et al. 2003) ranges.
This study investigated whether l-NMMA affects the CBF sensitivity to hypoxia and hypercapnia in humans. The technique of dynamic end-tidal forcing was used to control the end-tidal partial pressures of CO2(PET,CO2) and O2 (PET,O2) accurately and continuously. This allowed the examination of the CBF responses to dynamic and steady-state changes in PET,CO2 and PET,O2. Cerebral perfusion was evaluated by using transcranial Doppler ultrasound (TCD), a non-invasive tool for the evaluation of cerebral perfusion with a high temporal resolution. Together, these techniques enable the evaluation of both the steady-state and dynamic changes of the CBF responses to alterations in arterial blood gases.
Methods
Participants
Ten healthy male adults participated in this study. The average age, height and weight were 28.7 ± 4.5 (s.d.) years, 179.2 ± 7.7 cm and 78.0 ± 11.6 kg, respectively. None had a history of cardiovascular or respiratory disease, and all had normal systolic and diastolic blood pressure. Those eligible were enrolled following written informed consent. The study was approved by the Conjoint Ethics Committee (University of Calgary). The study was carried out according to Good Clinical Practice and the principles outlined in the Declaration of Helsinki.
Experiments were conducted at an elevation of 1103 m and a barometric pressure of 665 ± 5 Torr. Subjects visited the laboratory fasted on six occasions. On the first day, participants became familiarized with the apparatus and experimental procedures. During this initial visit, resting end-tidal gases and estimates of hypoxic and hypercapnic ventilatory responses were measured. The individual estimation of the ventilatory sensitivities were used to improve the calculated inspired gas concentrations that were required to produce the desired partial pressures during the experimental protocols (see details below). For the next five visits, subjects reported to the laboratory at the same time of day and conducted one of three experimental sessions (3 control days, 1 l-NMMA day, 1 phenylephrine day).
Air-breathing measurements
Each visit began with the measurement of the subject's normal PET,CO2 and PET,O2 while the subject was sitting quietly and comfortably for approximately 10 min. In brief, respired gases were sampled via a fine catheter held at the opening of one nostril by an adapted nasal O2 therapy kit. The gas was sampled continuously at a rate of 20 ml min−1 and analysed for PO2 and PCO2 by mass spectrometer (AMIS 2000, Innovision, Odense, Denmark). Values for PO2 and PCO2 were sampled by computer every 10 ms and PET,O2 and PET,CO2 were identified and recorded for each breath using a computer and dedicated software (Chamber v1.00, University Laboratory of Physiology, Oxford, UK), with pulse O2 saturation (SpO2) evaluated by oximetry (Ohmeda 3900 pulse oximeter, Ohmeda, Madison, WI, USA). On the two drug experimental days, these measurements were also done after placing an intravenous catheter. Measurements were started 10 min prior to drug administration and continued for another 15 min on the l-NMMA day and until MAP reached the desired level on the phenylephrine day.
Protocols
Isocapnic euoxia
This served as a control protocol. PET,O2 was maintained at 88 Torr and PET,CO2 was held 1.5 Torr above the subject's normal value for 40 min.
Hypoxia and hypercapnia
The test protocols (isocapnic hypoxia and euoxic hypercapnia) started with a 10 min period during which PET,O2 was maintained at 88 Torr and PET,CO2 at 1.5 Torr above the subject's natural value as determined on that day. Then, PET,O2 or PET,CO2 was altered rapidly (over 2–3 breaths) to a new set of desired levels, according to the protocol described below, and maintained constant for 20 min. Finally, PET,O2 or PET,CO2 was returned (again within 2–3 breaths) to its initial euoxic and near-eucapnic value and maintained constant for a further 10 min. In a hypoxic protocol, PET,O2 was lowered to 50 Torr while PET,CO2 was maintained constant at 1.5 Torr above the subject's normal value. In a hypercapnic protocol, PET,O2 was held constant at 88 Torr while PET,CO2 was elevated by 7.5 Torr (i.e. 9.0 Torr above the subject's normal value).
The experiments were conducted contemporaneously with a fourth, hypocapnic protocol, as part of a larger study but those data are not presented here.
Control of inspired gases
The inspired and expired gases were sampled at a rate of 20 ml min−1 and analysed by mass spectrometer for fractional concentrations of O2 and CO2. Respiratory volumes and flow information were obtained by using a pneumotachograph and differential pressure transducer (RSS100-HR, Hans Rudolph, Kansas City, MO, USA). Respiratory flow direction and timing information were measured with a turbine volume transducer (VMM-400, Interface Associates). A computer sampled the experimental variables every 10 ms. Accurate control of the end-tidal gases was achieved by using the technique of dynamic end-tidal forcing (BreatheM version 2.07, University Laboratory of Physiology, Oxford, UK). A controlling computer generated the inspired partial pressure of O2 and CO2 predicted to give the desired end-tidal partial pressures by using a fast gas-mixing system. The controlling computer received feedback of the measured end-tidal partial pressures on a breath-by-breath basis as the experiment progressed. These measured end-tidal values were compared with the desired values, and the computer then adjusted the initial predicted inspired gas mixture by using an integral proportional feedback algorithm based on the deviations of the measured end-tidal values from the desired end-tidal values (Robbins et al. 1982a,b).
The protocols were randomly conducted with an order of hypoxic, control and hypercapnia protocols, or with an order of hypercapnia, control and hypoxic protocols. The order of the control days and l-NMMA day were randomly assigned. The phenylephrine day was always carried out after the l-NMMA day.
Drug administration
(a) NG-monomethyl-l-arginine(l-NMMA)
NOS inhibition was instituted by l-NMMA (Clinalfa AG, Läufelfingen, Switzerland) and administered intravenously, first by a bolus and then by infusion.
Reconstitution of l-NMMA (bolus, 5 mg kg−1)
The process of reconstituting l-NMMA was carried out immediately before each l-NMMA study day. A syringe and sterile needle was used to remove a small volume of sterile saline (0.9%) from a sterile 50 ml mini-infusion bag (Baxter Corporation). A different syringe and sterile needle was used to remove a further 1.0 ml of sterile 0.9% saline from the mini-infusion bag and this volume was added to the sterile vial of l-NMMA (239.4 or 263.5 mg in one vial), lyophilized powder (high purity l-NMMA; 99.6%, Clinalfa (AG)). The dissolved l-NMMA was removed from the sterile vial with a sterile needle and syringe and was injected into a sterile mini-infusion bag containing the 0.9% saline via one of two ports on the infusion bag. The second port was used for administering the product to the volunteer intravenously through a sterile cannula in a dorsal vein of the hand over 5 min via an infusion pump (IMED Gemini Infusion System, Alaris Medical Systems). This resulted in a concentration of 4.5 mg ml−1 (range, 183–270 mg (50 ml)−1), and given over 5 min, at an infusion rate of 10 ml min−1.
Reconsitution of l-NMMA (infusion, 50 μg kg−1 min−1)
The l-NMMA was mixed into a 250 ml sterile infusion bag (0.9% saline) according to the procedures outlined above, and was administered to the volunteer intravenously through a sterile cannula in a dorsal vein of the hand over 4 h via an infusion pump. This resulted in a concentration of 3.6 mg ml−1 (range, 732–1080 mg (250 ml)−1), and given over 240 min, is equivalent to 1.0 ml min−1.
After the bolus injection, l-NMMA was continuously infused at the rate stated above, until the end of the protocols. Before the bolus injection, air-breathing measurements were made for 10 min and continued for 15 min after the start of the bolus injection. These measurements included MAP, HR, PET,O2 and PET,CO2, SpO2, and the index of CBF (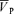 , see below for details). Approximately 20 min after starting the l-NMMA infusion, the first experimental protocol began.
, see below for details). Approximately 20 min after starting the l-NMMA infusion, the first experimental protocol began.
(b) Phenylephrine
The effect of an increase in blood pressure with l-NMMA was compared with that of phenylephrine, an α adrenoceptor agonist, on a different day at least 1 week after the l-NMMA day. After 10 min of basal measurements, the phenylephrine infusion began at a low rate of 0.3 μg kg−1 min−1 (0.1–0.6 μg kg−1 min−1) and adjusted to match the increases in blood pressure elicited with l-NMMA. This resulted in dose 0.6 ± 0.1 μg kg−1 min−1. Similar to the l-NMMA day, the first experimental protocol began approximately 20 min after the start of the phenylephrine infusion. Air-breathing measurements of blood pressure, HR, PET,O2 and PET,CO2, oxygen saturation of the arterial blood (SaO2), and 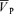 were made for 10 min before the bolus injection of phenylephrine and continued after the start of the injection of phenylephrine until MAP was titrated to the desired level (23 ± 7 min).
were made for 10 min before the bolus injection of phenylephrine and continued after the start of the injection of phenylephrine until MAP was titrated to the desired level (23 ± 7 min).
Haemodynamic measurements
Transcranial Doppler ultrasound
Backscattered Doppler signals from the right middle cerebral artery (MCA) were measured using a 2 MHz pulsed Doppler ultrasound system (PC Dop842, SciMed, Bristol, UK). The MCA was identified by an insonation pathway through the right temporal window just above the zygomatic arch by using search techniques previously described (Poulin et al. 1998; Ide et al. 2003). This procedure was repeated on each visit. The Doppler system was adapted by the manufacturer to make the Doppler signals available as analog signals sampled every 10 ms. Signals for maximum (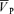 ) and intensity-weighted mean Doppler frequency shifts (
) and intensity-weighted mean Doppler frequency shifts (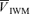 ) and power of Doppler signal (
) and power of Doppler signal (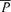 ) were available as analog signals and were updated each time a new spectrum was calculated every 10 ms. In this study, the maximum frequency of Doppler shift, namely peak blood velocity (
) were available as analog signals and were updated each time a new spectrum was calculated every 10 ms. In this study, the maximum frequency of Doppler shift, namely peak blood velocity (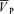 ), was taken as the primary index of CBF (Poulin & Robbins, 1996).
), was taken as the primary index of CBF (Poulin & Robbins, 1996).
Other haemodynamic parameters evaluated included mean arterial blood pressure (MAP) by photoplethysmography (Portapress, TPD Biomedical Instrumentation, the Netherlands) and heart rate (HR; Micromon 7142B monitor, Kontron Medical, Milton Keynes, UK) which were recorded by using a dedicated data acquisition system (Axoscope Digidata 1322A, Axon Instruments, CA, USA) and computer for off-line analyses. Measurements of MAP were also made every 3 min by using the auscultation method (Dynamap Compact S, Critikon, Germany).
Data analysis
Measurements during air-breathing
For data analysis during air breathing, PET,CO2, PET,O2, 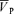 ,
, 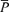 , SpO2 and MAP were averaged to one value for 10 min. To evaluate the effects of l-NMMA or phenylephrine administration on these variables, data were averaged over a 10 min period prior to administration of the drug and every 5 min after starting drug administration.
, SpO2 and MAP were averaged to one value for 10 min. To evaluate the effects of l-NMMA or phenylephrine administration on these variables, data were averaged over a 10 min period prior to administration of the drug and every 5 min after starting drug administration.
Effect of drug infusion when end-tidal gases are regulated near resting values
To average 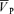 , HR and MAP when the end-tidal gases were regulated near resting values, these data were first averaged over each heart beat to give the beat-by-beat values for
, HR and MAP when the end-tidal gases were regulated near resting values, these data were first averaged over each heart beat to give the beat-by-beat values for 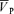 , HR and MAP. Likewise, the data for PET,O2 and PET,CO2 were determined for each breath to give breath-by-breath values. The beat-by-beat and breath-by-breath data were further averaged to give values for a 5 min baseline period before the onset of the hypoxic or hypercapnic stimulus.
, HR and MAP. Likewise, the data for PET,O2 and PET,CO2 were determined for each breath to give breath-by-breath values. The beat-by-beat and breath-by-breath data were further averaged to give values for a 5 min baseline period before the onset of the hypoxic or hypercapnic stimulus.
Effect of drug infusion on responses to hypoxia and hypercapnia
The data were averaged according to the procedures in the previous section, including the entire 20 min period of the stimulus. These data included PET,O2 and PET,CO2, 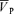 ,
, 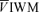 ,
, 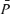 , MCA flow index (
, MCA flow index (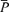 ·
·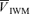 ), HR and MAP. In the normalization process for the flow index, beat-by-beat values were first averaged for 5 min, and deviations from this averaged value were calculated and then the 5 min baseline of normalized flow index was calculated.
), HR and MAP. In the normalization process for the flow index, beat-by-beat values were first averaged for 5 min, and deviations from this averaged value were calculated and then the 5 min baseline of normalized flow index was calculated.
The sensitivities of the MAP and HR responses to hypercapnia were calculated as the change in MAP over the change in PET,CO2 (ΔMAP/ΔPET,CO2) and the change in HR over the change in PET,CO2 (ΔHR/ΔPET,CO2). The sensitivities of the MAP and HR responses to hypoxia were calculated as ΔMAP/ΔSaO2 and ΔHR/ΔSaO2. For PET,CO2, the data over the 5 min baseline period were expressed as deviation from the baseline value and used to calculate the difference between the hypercapnic PET,CO2 and the baseline eucapnic PET,CO2 (ΔPET,CO2). Likewise, for SaO2, MAP and HR the same procedure was done to obtain ΔSaO2, ΔMAP and ΔHR.
Cerebrovascular resistance (CVR) was determined as MAP/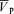 and calculated at baseline, the initial and last 5 min periods of hypercapnia or hypoxia and the initial 5 min period of recovery.
and calculated at baseline, the initial and last 5 min periods of hypercapnia or hypoxia and the initial 5 min period of recovery.
Modelling
Responses to isocapnic hypoxia
A dynamic single compartment (i.e. exponential) model was fitted to the 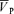 data to analyse the CBF response to hypoxia. A detailed description of this model has been provided elsewhere (Poulin et al. 1996).
data to analyse the CBF response to hypoxia. A detailed description of this model has been provided elsewhere (Poulin et al. 1996).
Responses to eucapnic hypercapnia
A simple proportional model of the response to CO2, with different time constants for the on- and off-responses was used to fit the 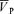 data to analyse the CBF response to hypercapnia. The description of the dynamic responses of
data to analyse the CBF response to hypercapnia. The description of the dynamic responses of 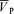 to step changes in PET,CO2 has been previously described (Poulin et al. 1996).
to step changes in PET,CO2 has been previously described (Poulin et al. 1996).
Statistical analyses
Data of the three control days were averaged to compare with those of the l-NMMA and phenylephrine days. The day-to-day variations in air breathing variables, the effects of l-NMMA infusion and phenylephrine infusion on baseline variables, model parameters, MAP and HR sensitivities, and CVR were analysed by using repeated measure ANOVA in conjunction with the Bonferoni post hoc test for multiple comparisons. The effects of l-NMMA infusion and phenylephrine infusion on CVR were evaluated by using a 2-way repeated measure ANOVA in conjunction with the Bonferoni post hoc test for multiple comparisons. A P value of < 0.05 was considered statistically significant. The statistical software package SPSS (SPSS Inc., version 13.0, IL, USA) was used for the repeated measure ANOVA and the 2-way repeated measure ANOVA.
Results
Air-breathing and effects of l-NMMA and phenylephrine administration
There were no differences in end-tidal gases (PET,CO2 and PET,O2), cerebrovascular (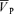 ) and cardiovascular (MAP and HR) variables during air-breathing between control, l-NMMA and phenylephrine days.
) and cardiovascular (MAP and HR) variables during air-breathing between control, l-NMMA and phenylephrine days.
Figure 1 illustrates the changes in end-tidal gases, 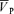 ,
, 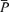 , MAP and HR before and after l-NMMA administration during air-breathing. Ten minutes (i.e. see 5–10 min, Fig. 1) after the start of the l-NMMA infusion, MAP increased from 80.0 ± 13.9 to 87.6 ± 15.3 mmHg (7.3%, P < 0.01) and HR decreased from 57.5 ± 10.9 to 51.6 ± 10.6 beats min−1 (10.3%, P < 0.01).
, MAP and HR before and after l-NMMA administration during air-breathing. Ten minutes (i.e. see 5–10 min, Fig. 1) after the start of the l-NMMA infusion, MAP increased from 80.0 ± 13.9 to 87.6 ± 15.3 mmHg (7.3%, P < 0.01) and HR decreased from 57.5 ± 10.9 to 51.6 ± 10.6 beats min−1 (10.3%, P < 0.01). 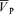 transiently increased 10 min (i.e. see 5–10 min, Fig. 1) after l-NMMA administration (54.7 ± 9.7 to 57.0 ± 10.0 cm s−1; 4.3%, P < 0.05) and then returned to values (56.2 ± 9.9 cm s−1) comparable to those prior to l-NMMA infusion (i.e. baseline values, Fig. 2).
transiently increased 10 min (i.e. see 5–10 min, Fig. 1) after l-NMMA administration (54.7 ± 9.7 to 57.0 ± 10.0 cm s−1; 4.3%, P < 0.05) and then returned to values (56.2 ± 9.9 cm s−1) comparable to those prior to l-NMMA infusion (i.e. baseline values, Fig. 2).
Figure 1. Changes in end-tidalPCO2 (PET,CO2) and PO2(PET,O2), middle cerebral artery blood velocity (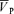 ), Doppler power signal (
), Doppler power signal (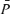 ), blood pressure (MAP) and heart rate (HR) before and after l-NMMA (red) and phenylephrine (blue) administration during air-breathing.
), blood pressure (MAP) and heart rate (HR) before and after l-NMMA (red) and phenylephrine (blue) administration during air-breathing.

Each symbol represents a group (n= 10 subjects), mean (n= 9) ±s.d.*Significant differences from baseline at P < 0.05 (red, l-NMMA; blue, phenylephrine).
Figure 2. Ensemble-averages of the time-related changes in end-tidalPCO2 (PET,CO2) and PO2 (PET,O2), and the responses of middle cerebral artery blood velocity (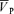 ), blood pressure (MAP) and heart rate (HR) to hypoxia and hypercapnia.
), blood pressure (MAP) and heart rate (HR) to hypoxia and hypercapnia.

Profiles represent group averages (n= 10 subjects) for control (black), l-NMMA (red), and phenylephrine (blue) days. Left panel (isocapnic euoxic), middle panel (isocapnic hypoxia), right panel (euoxic hypercapnia). Each symbol and error bar represents a 15 s mean ±s.d.
Figure 1 also illustrates the changes in end-tidal gases, 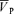 ,
, 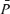 , MAP and HR before and after phenylephrine administration during air-breathing. Approximately 20 min (i.e. see 23 min, Fig. 1) after the start of the phenylephrine infusion, MAP increased from 76.4 ± 11.4 to 86.0 ± 8.2 mmHg (12.6%, P < 0.01) and HR decreased from 59.4 ± 10.6 to 54.4 ± 10.3 beats min−1 (8.4%, P < 0.05). End-tidal gases and
, MAP and HR before and after phenylephrine administration during air-breathing. Approximately 20 min (i.e. see 23 min, Fig. 1) after the start of the phenylephrine infusion, MAP increased from 76.4 ± 11.4 to 86.0 ± 8.2 mmHg (12.6%, P < 0.01) and HR decreased from 59.4 ± 10.6 to 54.4 ± 10.3 beats min−1 (8.4%, P < 0.05). End-tidal gases and 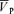 were unchanged.
were unchanged.
General pattern of responses
Figure 2 shows ensemble-averages of the time-related changes in PET,O2 and PET,CO2 for each protocol, along with the responses of 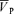 , MAP and HR to hypoxia and hypercapnia. Each profile represents group averages for the control, l-NMMA and phenylephrine days. There was no significant difference in the hypercapnic and hypoxic stimuli with or without drugs. During the hypercapnia protocol, PET,CO2 changed from 38.8 ± 1.9 to 46.2 ± 1.9 Torr and then back to 38.8 ± 1.9 Torr. During the hypercapnia protocol with l-NMMA, PET,CO2 changed from 38.4 ± 1.8 to 45.8 ± 1.7 Torr and then back to 38.5 ± 1.8 Torr. During the hypercapnia protocol with phenylephrine, PET,CO2 changed from 38.9 ± 1.7 to 46.3 ± 1.7 Torr and then back to 39.0 ± 1.8 Torr. During the hypoxia protocol, PET,O2 changed from 88.0 ± 0.2 to 50.3 ± 0.1 Torr. During the hypoxia protocol with l-NMMA, PET,O2 changed from 87.8 ± 0.3 to 50.3 ± 0.1 Torr. During the hypoxia protocol with phenylephrine, PET,O2 changed from 88.2 ± 1.1 to 50.4 ± 0.2 Torr. There were minor (non-significant) imperfections in the desired PET,O2 and PET,CO2 that were observed (i.e. lasting 1–2 breathes) particularly at the onset and relief of hypoxia or hypercapnia. However, the overall quality of input stimulation was very good and there were no difference in the PET,CO2 between the 10 min periods immediately preceding and following the hypoxic and hypercapnic periods. Further, the changes in blood pressure and heart rate elicited by l-NMMA and phenylephrine are clearly illustrated.
, MAP and HR to hypoxia and hypercapnia. Each profile represents group averages for the control, l-NMMA and phenylephrine days. There was no significant difference in the hypercapnic and hypoxic stimuli with or without drugs. During the hypercapnia protocol, PET,CO2 changed from 38.8 ± 1.9 to 46.2 ± 1.9 Torr and then back to 38.8 ± 1.9 Torr. During the hypercapnia protocol with l-NMMA, PET,CO2 changed from 38.4 ± 1.8 to 45.8 ± 1.7 Torr and then back to 38.5 ± 1.8 Torr. During the hypercapnia protocol with phenylephrine, PET,CO2 changed from 38.9 ± 1.7 to 46.3 ± 1.7 Torr and then back to 39.0 ± 1.8 Torr. During the hypoxia protocol, PET,O2 changed from 88.0 ± 0.2 to 50.3 ± 0.1 Torr. During the hypoxia protocol with l-NMMA, PET,O2 changed from 87.8 ± 0.3 to 50.3 ± 0.1 Torr. During the hypoxia protocol with phenylephrine, PET,O2 changed from 88.2 ± 1.1 to 50.4 ± 0.2 Torr. There were minor (non-significant) imperfections in the desired PET,O2 and PET,CO2 that were observed (i.e. lasting 1–2 breathes) particularly at the onset and relief of hypoxia or hypercapnia. However, the overall quality of input stimulation was very good and there were no difference in the PET,CO2 between the 10 min periods immediately preceding and following the hypoxic and hypercapnic periods. Further, the changes in blood pressure and heart rate elicited by l-NMMA and phenylephrine are clearly illustrated.
Effects of l-NMMA and phenylephrine administration on cerebrovascular and cardiovascular variables when end-tidal gases are regulated near resting values (i.e. isocapnia and euoxia)
Table 1 lists the cerebrovascular and cardiovascular variables during isocapnic euoxia. With drug infusions, MAP increased significantly for the l-NMMA day (P= 0.002) and phenyhephrine day (P= 0.02) compared with control days and HR decreased significantly for the l-NMMA day (P= 0.002) and phenyhephrine day (P= 0.005) compared with control days, while 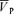 ,
, 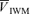 and
and 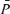 were unchanged.
were unchanged.
Table 1.
Cardiovascular and cerebrovascular variables during isocapnic euoxia
| Variables during lead-in (control, hypoxia, hypercapnia) | ||||||
|---|---|---|---|---|---|---|
| Day |
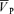 (cm s−1) (cm s−1) |
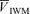 (cm s−1) (cm s−1) |
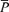 (V) (V) |
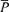 · ·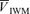 (%) (%) |
MAP (mmHg) | HR (beats min−1) |
| Control | 56.0 ± 9.5 | 38.1 ± 7.3 | 2.8 ± 0.5 | 100.0 ± 0.3 | 76.3 ± 7.5 | 59.5 ± 9.0 |
| l-NMMA | 57.6 ± 8.8 | 38.4 ± 5.4 | 2.7 ± 0.5 | 100.0 ± 0.2 | 86.2 ± 9.4† | 55.2 ± 9.5† |
| Phenylephrine | 58.7 ± 8.2 | 38.2 ± 5.9 | 2.7 ± 0.5 | 100.1 ± 0.2 | 85.4 ± 7.8† | 50.5 ± 9.3† |
Values are mean ±s.d. (n= 81 for control day, n= 27 for l-NMMA day, and n= 27 for phenylephrine day. Abbreviations: 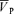 , middle cerebral artery blood velocity associated with the maximum frequency of the Doppler shift;
, middle cerebral artery blood velocity associated with the maximum frequency of the Doppler shift; 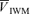 , middle cerebral artery blood velocity associated with the intensity-weighted mean frequency of the Doppler spectrum;
, middle cerebral artery blood velocity associated with the intensity-weighted mean frequency of the Doppler spectrum; 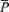 , Doppler power signal;
, Doppler power signal; 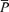 ·
·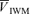 , product of
, product of 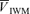 and
and 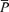 ; MAP, mean arterial blood pressure; HR, heart rate.
; MAP, mean arterial blood pressure; HR, heart rate.
denotes a significant difference from control.
Effects of drug infusions on cerebrovascular and cardiovascular responses
Cerebrovascular responses to hypoxia and hypercapnia
With hypoxia, 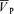 increased from 54.9 ± 8.5 to 61.5 ± 10.6 cm s−1 (Δ= 6.6 ± 2.4 cm s−1; 12.0%; P= 0.001). During hypoxia with l-NMMA,
increased from 54.9 ± 8.5 to 61.5 ± 10.6 cm s−1 (Δ= 6.6 ± 2.4 cm s−1; 12.0%; P= 0.001). During hypoxia with l-NMMA, 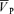 increased from 57.6 ± 8.9 to 64.1 ± 10.2 cm s−1 (Δ= 6.5 ± 2.4 cm s−1; 11.3%; P= 0.001), resulting in no effect of l-NMMA on changes in
increased from 57.6 ± 8.9 to 64.1 ± 10.2 cm s−1 (Δ= 6.5 ± 2.4 cm s−1; 11.3%; P= 0.001), resulting in no effect of l-NMMA on changes in 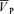 with hypoxia. During hypercapnia,
with hypoxia. During hypercapnia, 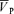 increased from 57.0 ± 9.6 to 74.7 ± 13.7 cm s−1 (Δ= 17.7 ± 4.7 cm s−1; 31.1%; P= 0.001). During hypercapnia with l-NMMA,
increased from 57.0 ± 9.6 to 74.7 ± 13.7 cm s−1 (Δ= 17.7 ± 4.7 cm s−1; 31.1%; P= 0.001). During hypercapnia with l-NMMA, 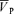 increased from 57.1 ± 9.6 to 73.6 ± 13.1 cm s−1 (Δ= 16.5 ± 4.6 cm s−1; 28.9%; P= 0.001) with no effect of l-NMMA on changes in
increased from 57.1 ± 9.6 to 73.6 ± 13.1 cm s−1 (Δ= 16.5 ± 4.6 cm s−1; 28.9%; P= 0.001) with no effect of l-NMMA on changes in 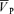 with hypercapnia. During hypoxia with phenylephrine,
with hypercapnia. During hypoxia with phenylephrine, 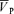 increased from 58.3 ± 9.4 to 64.7 ± 9.0 cm s−1 (Δ= 6.4 ± 2.2 cm s−1; 11.0%; P= 0.001) while during hypercapnia with phenylephrine,
increased from 58.3 ± 9.4 to 64.7 ± 9.0 cm s−1 (Δ= 6.4 ± 2.2 cm s−1; 11.0%; P= 0.001) while during hypercapnia with phenylephrine, 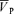 increased from 58.3 ± 3.0 to 76.0 ± 3.2 cm s−1 (Δ= 17.7 ± 4.5 cm s−1; 30.4%; P= 0.001) with no effect of phenylephrine on changes in
increased from 58.3 ± 3.0 to 76.0 ± 3.2 cm s−1 (Δ= 17.7 ± 4.5 cm s−1; 30.4%; P= 0.001) with no effect of phenylephrine on changes in 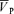 with either hypercapnia or hypoxia.
with either hypercapnia or hypoxia.
Heart rate responses to hypoxia and hypercapnia
During hypoxia HR increased from 60.4 ± 8.7 to 72.8 ± 9.6 beats min−1 (Δ= 12.4 ± 4.6 beats min−1; 20.5%; P= 0.001). During hypoxia with l-NMMA HR increased from 56.6 ± 10.6 to 68.1 ± 12.8 beats min−1 (Δ= 11.5 ± 4.4 beats min−1; 20.3%; P= 0.001), resulting in no effect of l-NMMA on changes in HR with hypoxia. During hypercapnia HR increased from 60.4 ± 8.9 to 70.4 ± 9.4 beats min−1 (Δ= 10.0 ± 4.2 beats min−1; 16.6%; P= 0.001). During hypercapnia with l-NMMA HR increased from 55.1 ± 9.8 to 65.5 ± 11.4 beats min−1 (Δ= 10.4 ± 4.6 beats min−1; 18.9%; P= 0.001) with no effect of l-NMMA on changes in HR with hypercapnia. During hypoxia with phenylephrine HR increased from 51.5 ± 11.1 to 64.4 ± 10.7 beats min−1 (Δ= 12.9 ± 4.3 beats min−1; 25.1%; P= 0.001) and during hypercapnia with phenylephrine HR increased from 50.1 ± 9.4 to 58.8 ± 13.3 beats min−1 (Δ= 8.7 ± 6.8 beats min−1; 17.4%; P= 0.003) with no effect of phenylephrine on changes in HR with either hypercapnia or hypoxia.
There were no significant differences in the sensitivities of the HR responses to hypercapnia (1.3 ± 0.6, 1.4 ± 0.6 and 1.2 ± 0.9 beats min−1 Torr−1 for the control, l-NMMA and phenylephrine days, respectively) or hypoxia (0.96 ± 0.35, 0.89 ± 0.35 and 1.00 ± 0.33 beats min−1 (% desaturation)−1 for the control, l-NMMA and phenylephrine days, respectively).
Mean arterial blood pressure responses to hypoxia and hypercapnia
During hypoxia MAP increased from 75.7 ± 6.9 to 83.0 ± 5.5 mmHg (Δ= 7.3 ± 3.1 mmHg; 9.6%; P= 0.001). During hypoxia with l-NMMA MAP increased from 83.8 ± 12.5 to 88.8 ± 12.5 mmHg (Δ= 4.9 ± 2.2 mmHg; 5.9%; P= 0.001), resulting in no effect of l-NMMA on changes in MAP with hypoxia. During hypercapnia MAP increased from 76.2 ± 8.7 to 84.2 ± 8.5 mmHg (Δ= 8.1 ± 2.2 mmHg; 10.6%; P= 0.001). During hypercapnia with l-NMMA MAP increased from 85.3 ± 15.5 to 92.7 ± 15.8 mmHg (Δ= 7.4 ± 3.1 mmHg; 8.7%; P= 0.001) with no effect of l-NMMA on changes in MAP with hypercapnia. During hypoxia with phenylephrine MAP increased from 85.1 ± 13.4 to 93.8 ± 12.4 mmHg (Δ= 8.7 ± 5.6 mmHg; 10.2%; P= 0.001) and during hypercapnia with phenylephrine MAP increased from 83.3 ± 6.9 to 92.9 ± 7.8 mmHg (Δ= 9.6 ± 3.2 mmHg; 11.5%; P= 0.001) with no effect of phenylehprine on changes in MAP with either hypercapnia or hypoxia.
There were no significant differences in the sensitivities of the MAP responses to hypercapnia (1.1 ± 0.3, 1.0 ± 0.4 and 1.3 ± 0.4 mmHg Torr−1 for the control, l-NMMA and phenylephrine days, respectively) or hypoxia (0.56 ± 0.24, 0.38 ± 0.17 and 0.68 ± 0.43 mmHg (% desaturation)−1 for the control, l-NMMA and phenylephrine days, respectively).
Cerebrovascular resistance responses to hypoxia and hypercapnia
Table 2 lists changes in CVR with hypoxia and hypercapnia. CVR was unchanged during hypoxia (control). With l-NMMA, CVR decreased (non-significant) at 5 and 20 min during hypoxia while CVR returned to baseline values during the recovery phase after hypoxia. With phenylephrine, CVR was unchanged during hypoxia.
Table 2.
Cerebrovascular resistance during hypoxia and hypercapnia with pharmacological interventions
| Hypoxia | Hypercapnia | |||||||
|---|---|---|---|---|---|---|---|---|
| Day | Lead-in | 5 min | 20 min | Recovery | Lead-in | 5 min | 20 min | Recovery |
| Control | 1.44 ± 0.34 | 1.40 ± 0.32 | 1.42 ± 0.32 | 1.52 ± 0.31 | 1.39 ± 0.32 | 1.14 ± 0.25* | 1.20 ± 0.26* | 1.63 ± 0.30* |
| l-NMMA | 1.51 ± 0.44 | 1.44 ± 0.43* | 1.42 ± 0.41* | 1.51 ± 0.46 | 1.56 ± 0.51 | 1.29 ± 0.41* | 1.32 ± 0.38* | 1.75 ± 0.50* |
| Phenylephrine | 1.50 ± 0.40 | 1.50 ± 0.37 | 1.48 ± 0.36 | 1.55 ± 0.35 | 1.47 ± 0.29 | 1.24 ± 0.22* | 1.26 ± 0.23* | 1.73 ± 0.25* |
Values (Torr (cm s−1)−1) are means ±s.d. Cerebrovascular resistance was calculated as MAP divided by 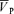 .
. 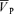 , middle cerebral artery blood velocity associated with the maximum frequency of the Doppler shift; MAP, mean arterial blood pressure.
, middle cerebral artery blood velocity associated with the maximum frequency of the Doppler shift; MAP, mean arterial blood pressure.
denotes a significant difference from lead-in at P < 0.05.
CVR decreased (non-significant) at 5 and 20 min during hypercapnia and increased during the recovery phase after hypercapnia. With l-NMMA, CVR decreased (non-significant) at 5 and 20 min during hypercapnia and increased during the recovery phase after hypercapnia. With phenylephrine, CVR decreased (non-significant) at 5 and 20 min during hypercapnia and increased during the recovery phase after hypercapnia.
Modelling
Responses to isocapnic hypoxia
There was no effect of l-NMMA on the model parameters of the 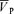 responses to hypoxia (Table 3). With l-NMMA 6/10 subjects showed a smaller gain term (G; i.e. sensitivity) (Fig. 3) while with phenylephrine 5/10 subjects showed a smaller G compared with that of control. With l-NMMA 7/10 subjects showed a slower time constant for the on-response (τon) and with phenylephrine 6/10 subjects showed faster τon. With l-NMMA and phenylephrine 7/10 subjects showed a slower τoff and 6/10 subjects showed a faster τoff. The time delay terms were unchanged.
responses to hypoxia (Table 3). With l-NMMA 6/10 subjects showed a smaller gain term (G; i.e. sensitivity) (Fig. 3) while with phenylephrine 5/10 subjects showed a smaller G compared with that of control. With l-NMMA 7/10 subjects showed a slower time constant for the on-response (τon) and with phenylephrine 6/10 subjects showed faster τon. With l-NMMA and phenylephrine 7/10 subjects showed a slower τoff and 6/10 subjects showed a faster τoff. The time delay terms were unchanged.
Table 3.
Estimated model parameters for cerebral blood flow response to hypoxia during control, l-NMMA and phenylephrine infusion
| Day | G | (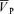 )B )B
|
τon | τoff | Td |
|---|---|---|---|---|---|
| Control | 0.58 ± 0.24 | 51.34 ± 7.74 | 41.95 ± 27.91 | 56.04 ± 45.12 | 5.74 ± 2.06 |
| l-NMMA | 0.53 ± 0.18 | 56.24 ± 9.01 | 45.89 ± 29.77 | 54.15 ± 52.86 | 4.41 ± 1.67 |
| Phenylephrine | 0.55 ± 0.18 | 54.2 ± 5.4 | 24.4 ± 15.4 | 73.6 ± 54.1 | 5.4 ± 3.7 |
Values are means ±s.d. Abbreviations. G, gain term for the response (cm s−1 (% desaturation)−1); (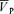 )B, baseline of
)B, baseline of 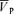 when no hypoxia is present (cm s−1); τon and τoff, time constants for the on and off responses (s); Td, time delay (s).
when no hypoxia is present (cm s−1); τon and τoff, time constants for the on and off responses (s); Td, time delay (s).
Figure 3. Gain terms of the middle cerebral artery blood velocity (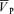 ) responses to isocapnic hypoxia for control days and l-NMMA day.
) responses to isocapnic hypoxia for control days and l-NMMA day.

Individual data are illustrated by numbers while the group mean (n= 9) is illustrated by filled circles.
Responses to euoxic hypercapnia
There was no effect of l-NMMA on all the model parameters of the 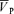 responses to hypercapnia except for the gain of the off-response (Table 4). With l-NMMA, 7/10 subjects showed a smaller gain term for the on-response (Gon) (Fig. 4) while with phenylephrine, 5/10 subjects showed smaller Gon compared with that of control. With l-NMMA and phenylephrine 7/10 subjects showed a faster τon. The gain terms of the off-responses to hypercapnia (Goff) were 2.99 ± 0.92 cm s−1 Torr−1 (control days), 2.56 ± 0.82 cm s−1 Torr−1 (P < 0.05, l-NMMA day), and 3.03 ± 0.89 cm s−1 Torr−1 (phenylephrine day). With l-NMMA and phenylephrine 9/10 subjects and 8/10 subjects showed a larger Goff. With l-NMMA and phenylephrine 5/10 subjects and 6/10 subjects showed a slower τoff.
responses to hypercapnia except for the gain of the off-response (Table 4). With l-NMMA, 7/10 subjects showed a smaller gain term for the on-response (Gon) (Fig. 4) while with phenylephrine, 5/10 subjects showed smaller Gon compared with that of control. With l-NMMA and phenylephrine 7/10 subjects showed a faster τon. The gain terms of the off-responses to hypercapnia (Goff) were 2.99 ± 0.92 cm s−1 Torr−1 (control days), 2.56 ± 0.82 cm s−1 Torr−1 (P < 0.05, l-NMMA day), and 3.03 ± 0.89 cm s−1 Torr−1 (phenylephrine day). With l-NMMA and phenylephrine 9/10 subjects and 8/10 subjects showed a larger Goff. With l-NMMA and phenylephrine 5/10 subjects and 6/10 subjects showed a slower τoff.
Table 4.
Estimated model parameters for cerebral blood flow response to hypercapnia during control, l-NMMA, and phenylephrine infusion
| Day | Gon | F1 | τon | Td | Goff | F2 | τoff |
|---|---|---|---|---|---|---|---|
| Control | 2.55 ± 0.71 | 56.87 ± 9.52 | 42.05 ± 22.41 | 5.94 ± 0.67 | 2.99 ± 0.92 | 52.30 ± 7.06 | 6.68 ± 1.57 |
| l-NMMA | 2.37 ± 0.64 | 57.24 ± 9.06 | 34.29 ± 21.34 | 6.07 ± 2.67 | 2.56 ± 0.82† | 55.16 ± 8.10 | 6.58 ± 2.77 |
| Phenylephrine | 2.50 ± 0.56 | 58.71 ± 9.12 | 15.06 ± 16.77 | 6.26 ± 1.06 | 3.03 ± 0.89 | 53.95 ± 4.38 | 6.49 ± 2.58 |
Values are means ±s.d. Abbreviations: Gon and Goff, gain terms for the on- and off-responses (cm s−1 Torr−1); F1 and F2, 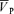 before and after hypercapnia (cm s−1); τon and τoff, time constant for the on- and off-responses (s); Td, time delay.
before and after hypercapnia (cm s−1); τon and τoff, time constant for the on- and off-responses (s); Td, time delay.
denotes a significant difference from control at P < 0.05.
Figure 4. Gain terms of the middle cerebral artery blood velocity (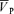 ) responses to euoxic hypercapnia for the on-response (Gain-on, left panel) and off-response (Gain-off, right panel) for control and l-NMMA days.
) responses to euoxic hypercapnia for the on-response (Gain-on, left panel) and off-response (Gain-off, right panel) for control and l-NMMA days.

Individual data are illustrated by numbers while the group means (n= 9) are illustrated by filled circles.
Discussion
Major findings
This study reports two new findings. First, the non-selective NOS inhibitor l-NMMA does not affect the response of 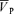 to step changes of isocapnic hypoxia, when PET,CO2 is controlled near resting levels. Second, the effect of l-NMMA on changes in
to step changes of isocapnic hypoxia, when PET,CO2 is controlled near resting levels. Second, the effect of l-NMMA on changes in 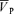 associated with step changes in PET,CO2 becomes apparent only during the off-transient, when the hypercapnic stimulus is withdrawn.
associated with step changes in PET,CO2 becomes apparent only during the off-transient, when the hypercapnic stimulus is withdrawn.
Effects of NOS inhibitors on the CBF response to hypoxia
Studies on the effects of l-NMMA or l-NAME on the CBF response to hypoxia are controversial. Our findings of no effect of l-NMMA on the hypoxic responses are in agreement with those of others (Kozniewska et al. 1992; Buchanan & Phillis, 1993) who reported no effects of l-NAME and l-NMMA on the CBF response to hypoxic stimuli in rats. Those studies are in contrast to others (Audibert et al. 1995; Reid et al. 1995; Berger & von Kummer, 1998) who reported substantial effects of l-NMMA on the CBF response to hypoxia in rats. The reason(s) for the lack of agreement in the animal studies is unclear. In humans, Van Mil et al. (2002) found that the CBF response to hypoxia, determined by phase-contrast magnetic resonance imaging, was attenuated by l-NMMA. A major difference between the study by Van Mil et al. (2002) and ours lies in the control of PET,CO2, where we controlled the PET,CO2 near resting baseline, while Van Mil et al. (2002) used a poikilocapnic protocol (i.e. no attempt to control the CO2 to prevent it from falling). A recent study from our group (Steinback & Poulin, 2007) clearly illustrates a substantial decline (∼15–20%) in PET,CO2 during hypoxia under similar conditions to those reported by Van Mil et al. (2002). However, to our knowledge a comparison of the effects of l-NMMA on the cerebral blood flow responses to poikilocapnic and isocapnic hypoxia has not been completed and it remains unclear whether this might explain some of the differences between the Van Mil study and the current study.
Alternative explanation for the lack of effect of l-NMMA on the CBF responses to hypercapnia and hypoxia
Although numbers of studies using an animal model has suggested that NO would be involved in the regulation of CBF during hypoxia and hypercapnia, we did not demonstrate a significant role of NO in regulating the CBF responses to these stimuli. We speculate that there may be compensatory pathways that may be activated with NOS inhibition. First, it has been reported that the agonist (ATP)-induced increase in intracellular Ca2+ ([Ca2+]i) in endothelial cells was enhanced in the presence of NOS inhibitors (Shin et al. 1992). Second, phospholipase activity is highly dependent on [Ca2+]i of endothelial cells. Therefore, epoxyeicosatrienoic acid, one of the putative endothelial-derived hyperpolarizing factors (EDHFs), may take over the control of vessel tone in the presence of NOS inhibitors (Bauersachs et al. 1996). Moreover, it has been reported that in the human umbilical vein, flow-induced prostacyclin production was enhanced in the presence of a NOS inhibitor (Osanai et al. 2000). Furthermore, histamine-induced NO production was enhanced in the presence of indomethacin (Bolz & Pohl, 1997). Finally, in rats with hypercholesterolaemia, NO production by endothelial cells of the carotid artery is attenuated, whereas EDHF and calcium-dependent potassium channels maintain normal vasodilatory responses to acetylcholine (Najibi et al. 1994).
In a study by Kamper et al. (2004), NO blockade by using l-NMMA decreased CBF during supine rest in elderly individuals, whereas there was no such effect on healthy young adult. In studies by Zimmerman & Haberl (2003) and Lavi et al. (2006), the CBF response to hypercapnia was restored by arginine infusion in patients with cardiovascular risk factors. It may be that the cerebral circulation relies more on NO production in the elderly and patients with cardiovascular disease, when redundant multiple mechanisms are either impaired or absent. We speculate that this might be at least one reason why elderly individuals are more susceptible to cerebrovascular events.
Effects of l-NMMA on the off-response of cerebral blood flow after hypercapnia
Compared with that of control, the Goff for 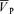 was attenuated by l-NMMA. There is no clear indication why l-NMMA attenuated the off-response of (CBF). However, we propose two possible explanations. First, we speculate that this attenuation might be associated with a blunted vasoconstriction of the arteriolar bed by l-NMMA. Second, it is possible that large cerebral arteries such as the middle cerebral artery (MCA) might dilate via NO synthesis when small resistance vessels constrict and CBF decreases below baseline following the decrease in PET,CO2 from hypercapnia. Such differences in the behaviour of cerebral vessels with hypercapnia are dependent on vessel size, with smaller cerebral vessels responding more than larger vessels (Wei et al. 1980). Further, it appears that relatively large cerebral vessels dilate more in response to small decreases in blood pressure without any change in the diameter of the smaller cerebral arteries (Kontos et al. 1978).
was attenuated by l-NMMA. There is no clear indication why l-NMMA attenuated the off-response of (CBF). However, we propose two possible explanations. First, we speculate that this attenuation might be associated with a blunted vasoconstriction of the arteriolar bed by l-NMMA. Second, it is possible that large cerebral arteries such as the middle cerebral artery (MCA) might dilate via NO synthesis when small resistance vessels constrict and CBF decreases below baseline following the decrease in PET,CO2 from hypercapnia. Such differences in the behaviour of cerebral vessels with hypercapnia are dependent on vessel size, with smaller cerebral vessels responding more than larger vessels (Wei et al. 1980). Further, it appears that relatively large cerebral vessels dilate more in response to small decreases in blood pressure without any change in the diameter of the smaller cerebral arteries (Kontos et al. 1978).
While in our study we did not find evidence of changes in the cross-sectional area of the MCA, it has previously been reported that the MCA may dilate during early recovery from combined hypercapnia and hypoxia (Poulin et al. 1996). Therefore, it is possible that NO might become an important regulator to counteract vasoconstriction when CBF decreases rapidly, in instances such as following hypotension or rapid decreases in end-tidal (i.e. arterial) PCO2 (Kontos et al. 1978; Wei et al. 1980; Poulin et al. 1996).
Effects of NOS inhibitors on the cerebral blood flow response to hypercapnia
We found no effect of l-NMMA on the on-response of CBF to hypercapnia, a finding consistent with that of White et al. (1998). On the other hand, a study by Schmetterer et al. (1997) reported that the CBF response to hypercapnia was attenuated by l-NMMA. The exact mechanism(s) underlying these discrepancies is difficult to delineate given the limitations of studies in humans regarding the temporal nature and extent of NOS inhibition in the cerebral circulation.
Dynamic changes in the CBF responses to hypoxia and hypercapnia
In addition to the steady state CBF responses to hypoxia and hypercapnia, we also evaluated the dynamic changes in the CBF responses to changes in PET,O2 and PET,CO2. Results of these dynamic components of the responses including time delays and time constants suggest that NO does not affect the transient components of the CBF responses to hypoxia and hypercapnia.
Cerebrovascular resistance
The decreases observed in CVR in response to hypercapnia were statistically significant. Further, the changes in CVR were comparable regardless of whether l-NMMA was administered. This finding is consistent with our observations of a non-significant effect of l-NMMA on the sensitivities of CBF to hypercapnia. CVR did not decrease significantly in response to hypoxia during control and phenylephrine days. However, CVR did decrease significantly in response to hypoxia when l-NMMA was administered. It is unclear whether this is due to the higher (non-significant) CVR values at baseline with l-NMMA compared with control.
Methodological considerations and limitations
We employed the technique of TCD to evaluate changes in CBF. The changes in blood flow velocity reflect changes in underlying CBF only if the diameter of the insonated vessel remains constant. The validity of this measurement under a wide range of PET,CO2 and PET,O2 values is well established by us (Poulin et al. 1996; Poulin & Robbins, 1996) and others (Aaslid et al. 1982; Djurberg et al. 1998; Serrador et al. 2000). Some studies have suggested the possibility that blood velocity determined by this technique does not reflect changes in CBF when production of NO is affected by NO donors and NOS inhibitors. In our study, there was a transient increase in 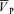 after the commencement of l-NMMA but this was not associated with a change in Doppler power (Fig. 1), suggesting that the cross-sectional area of the MCA remained unchanged (Poulin et al. 1996; Poulin & Robbins, 1996). This might suggest a transient impairment of cerebral autoregulation after l-NMMA administration, whereby a transient passive increase in cerebral blood flow follows the increase in blood pressure (Fig. 1). Finally, it is unlikely that l-NMMA would constrict the MCA in a condition when a vasodilator stimulus such as hypoxia and hypercapnia is administered.
after the commencement of l-NMMA but this was not associated with a change in Doppler power (Fig. 1), suggesting that the cross-sectional area of the MCA remained unchanged (Poulin et al. 1996; Poulin & Robbins, 1996). This might suggest a transient impairment of cerebral autoregulation after l-NMMA administration, whereby a transient passive increase in cerebral blood flow follows the increase in blood pressure (Fig. 1). Finally, it is unlikely that l-NMMA would constrict the MCA in a condition when a vasodilator stimulus such as hypoxia and hypercapnia is administered.
The power of our study in detecting a specified clinically relevant difference of 25% (moderate effect) is sufficiently high (i.e. > 0.95) for Gon and Goff. However, since significance was not achieved for Gon, our results can be interpreted as probably indicating a negligible relevant effect of l-NMMA, at least in young healthy humans, on the cerebral blood flow sensitivity to hypercapnia. With respect to the sensitivity (i.e. G) of the cerebral blood flow response to hypoxia, power was below 0.80 in detecting a moderate (25%) effect of l-NMMA. This result can be interpreted as probably indicating inadequate power, possibly related to our small sample size (n= 10). In our study, sample size was determined as a matter of concession between the available resources (i.e. safety, substantial cost of l-NMMA, etc.) and the study's specific objectives. Thus, future studies on the effects of l-NMMA on the cerebral blood flow responses to hypoxia in humans must give careful consideration to sample size.
Summary and relevance
We found significant individual variability in the effects of l-NMMA on the 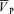 responses to hypoxia and hypercapnia. However, overall, there was no effect of l-NMMA on the increases in CBF or dynamic changes in CBF in response to either hypoxia or hypercapnia, suggesting that the involvement of NO in this regulation is small in healthy humans. In contrast, there was an effect of l-NMMA on the decrease in CBF with a step decrease in PET,CO2. This might indicate that in the cerebral circulation, NO might play an important role in counteracting vessel constrictors in an effort to normalize CBF.
responses to hypoxia and hypercapnia. However, overall, there was no effect of l-NMMA on the increases in CBF or dynamic changes in CBF in response to either hypoxia or hypercapnia, suggesting that the involvement of NO in this regulation is small in healthy humans. In contrast, there was an effect of l-NMMA on the decrease in CBF with a step decrease in PET,CO2. This might indicate that in the cerebral circulation, NO might play an important role in counteracting vessel constrictors in an effort to normalize CBF.
Acknowledgments
We thank Gisela Engels for assistance with statistical analysis. This project was supported by the Alberta Heritage Foundation for Medical Research (AHFMR), Heart and Stroke Foundation of Alberta, NWT and Nunavut, the Canadian Institutes of Health Research (CIHR) and the Canada Foundation for Innovation. K.I. was supported by an AHFMR PDF. T.J.A. and M.J.P. are AHFMR Senior Medical Scholars. M.W. was supported by a Royal Australian College of Physician travelling fellowship.
Authors' current addresses
K. Ide: Laboratory of Exercise Physiology, Department of Sports and Health Sciences, Fukuoka University, 8-19-1, Nanakuma, Jyonan-ku, Fukuoka-shi, Fukuoka-ken, Japan. Email: ide_kojiro@hotmail.com
M. I. Worthley: Cardiovascular Investigation Unit, Royal Adelaide Hospital, North Terrace, Adelaide, South Australia, Australia. Email: matthew.worthley@adelaide.edu.au
References
- Aaslid R, Markwalder T-M, Nornes H. Noninvasive transcranial Doppler ultrasound recording of flow velocity in basal cerebral arteries. J Neurosurg. 1982;57:769–774. doi: 10.3171/jns.1982.57.6.0769. [DOI] [PubMed] [Google Scholar]
- Audibert G, Saunier CG, Siat J, Hartemann D, Lambert J. Effect of the inhibitor of nitric oxide synthase, NG-nitro-L-arginine methyl ester, on cerebral and myocardial blood flows during hypoxia in the awake dog. Anesth Analg. 1995;81:945–951. doi: 10.1097/00000539-199511000-00009. [DOI] [PubMed] [Google Scholar]
- Bauersachs J, Popp R, Hecker M, Sauer E, Fleming I, Busse R. Nitric oxide attenuates the release of endothelium-derived hyperpolarizing factor. Circulation. 1996;94:3341–3347. doi: 10.1161/01.cir.94.12.3341. [DOI] [PubMed] [Google Scholar]
- Berger C, von Kummer R. Does NO regulate the cerebral blood flow response in hypoxia? Acta Neurol Scand. 1998;97:118–125. doi: 10.1111/j.1600-0404.1998.tb00620.x. [DOI] [PubMed] [Google Scholar]
- Bolz SS, Pohl U. Indomethacin enhances endothelial NO release – evidence for a role of PGI2 in the autocrine control of calcium-dependent autacoid production. Cardiovasc Res. 1997;36:437–444. doi: 10.1016/s0008-6363(97)00197-1. [DOI] [PubMed] [Google Scholar]
- Buchanan JE, Phillis JW. The role of nitric oxide in the regulation of cerebral blood flow. Brain Res. 1993;610:248–255. doi: 10.1016/0006-8993(93)91408-k. [DOI] [PubMed] [Google Scholar]
- Cunningham DJ, Howson MG, Pickering TG, Sleight P, Petersen ES. The effect of raising arterial blood pressure on ventilation in man. J.Physiol. 1969;204:89P. [PubMed] [Google Scholar]
- Djurberg HG, Seed RF, Evans DA, Brohi FA, Pyper DL, Tjan GT, al Moutaery KR. Lack of effect of CO2 on cerebral arterial diameter in man. J Clin Anesth. 1998;10:646–651. doi: 10.1016/s0952-8180(98)00107-x. [DOI] [PubMed] [Google Scholar]
- Heistad D, Abboud FM, Mark AL, Schmid PG. Effect of baroreceptor activity on ventilatory response to chemoreceptor stimulation. J Appl Physiol. 1975;39:411–416. doi: 10.1152/jappl.1975.39.3.411. [DOI] [PubMed] [Google Scholar]
- Heistad DD, Kontos HA. Cerebral circulation. In: Shepherd JT, Abbond FM, Geiger JR, editors. Peripheral Circulation and Organ Blood Flow. Bethesda, MD: American Physiological Society; 1983. pp. 137–182. [Google Scholar]
- Heistad DD, Wheeler RC, Mark AL, Schmid PG, Abboud FM. Effects of adrenergic stimulation on ventilation in man. J Clin Invest. 1972;51:1469–1475. doi: 10.1172/JCI106943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Xu X. Nitro-L-arginine attenuates hypercapnic cerebrovasodilation without affecting cerebral metabolism. Am J Physiol Regul Integr Comp Physiol. 1994;266:R518–R525. doi: 10.1152/ajpregu.1994.266.2.R518. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Zhang F. Nitric oxide-dependent and independent components of cerebrovasodilation elicited by hypercapnia. Am J Physiol Regul Integr Comp Physiol. 1994;266:R546–R552. doi: 10.1152/ajpregu.1994.266.2.R546. [DOI] [PubMed] [Google Scholar]
- Ide K, Eliasziw M, Poulin MJ. The relationship between middle cerebral artery blood velocity and end-tidal PCO2 in the hypocapnic-hypercapnic range in humans. J Appl Physiol. 2003;95:129–137. doi: 10.1152/japplphysiol.01186.2002. [DOI] [PubMed] [Google Scholar]
- Kamper AM, Spilt A, de Craen AJ, Van Buchem MA, Westendorp RG, Blauw GJ. Basal cerebral blood flow is dependent on the nitric oxide pathway in elderly but not in young healthy men. Exp Gerontol. 2004;39:1245–1248. doi: 10.1016/j.exger.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Kety SS, Schmidt CF. The effects of altered arterial tensions of carbon dioxide and oxygen on cerebral blood flow and cerebral oxygen consumption of normal young men. J Clin Invest. 1948;27:484–492. doi: 10.1172/JCI101995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontos HA, Wei EP, Navari RM, Levasseur JE, Rosenblum WI, Patterson JL. Responses of cerebral arteries and arterioles to acute hypotension and hypertension. Am J Physiol Heart Circ Physiol. 1978;234:H371–H383. doi: 10.1152/ajpheart.1978.234.4.H371. [DOI] [PubMed] [Google Scholar]
- Kozniewska E, Oseka M, Stys T. Effects of endothelium-derived nitric oxide on cerebral circulation during normoxia and hypoxia in the rat. J Cereb Blood Flow Metab. 1992;12:311–317. doi: 10.1038/jcbfm.1992.43. [DOI] [PubMed] [Google Scholar]
- Lavi S, Gaitini D, Milloul V, Jacob G. Impaired cerebral CO2 vasoreactivity: association with endothelial dysfunction. Am J Physiol Heart Circ Physiol. 2006;291:H1856–H1861. doi: 10.1152/ajpheart.00014.2006. [DOI] [PubMed] [Google Scholar]
- Najibi S, Cowan CL, Palacino JJ, Cohen RA. Enhanced role of potassium channels in relaxations to acetylcholine in hypercholesterolemic rabbit carotid-Artery. Am J Physiol Heart Circ Physiol. 1994;266:H2061–H2067. doi: 10.1152/ajpheart.1994.266.5.H2061. [DOI] [PubMed] [Google Scholar]
- Osanai T, Fujita N, Fujiwara N, Nakano T, Takahashi K, Guan W, Okumura K. Cross talk of shear-induced production of prostacyclin and nitric oxide in endothelial cells. Am J Physiol Heart Circ Physiol. 2000;278:H233–H238. doi: 10.1152/ajpheart.2000.278.1.H233. [DOI] [PubMed] [Google Scholar]
- Poulin MJ, Liang P-J, Robbins PA. Dynamics of the cerebral blood flow response to step changes in end-tidal PCO2 and PO2 in humans. J Appl Physiol. 1996;81:1084–1095. doi: 10.1152/jappl.1996.81.3.1084. [DOI] [PubMed] [Google Scholar]
- Poulin MJ, Liang P-J, Robbins PA. Fast and slow components of the cerebral blood flow response to step decreases in end-tidal PCO2 in humans. J Appl Physiol. 1998;85:388–397. doi: 10.1152/jappl.1998.85.2.388. [DOI] [PubMed] [Google Scholar]
- Poulin MJ, Robbins PA. Indexes of flow and cross-sectional area of the middle cerebral artery using Doppler ultrasound during hypoxia and hypercapnia in humans. Stroke. 1996;27:2244–2250. doi: 10.1161/01.str.27.12.2244. [DOI] [PubMed] [Google Scholar]
- Reid JM, Davies AG, Ashcroft FM, Paterson DJ. Effect of L-NMMA, cromakalim, and glibenclamide on cerebral blood flow in hypercapnia and hypoxia. Am J Physiol Heart Circ Physiol. 1995;269:H916–H922. doi: 10.1152/ajpheart.1995.269.3.H916. [DOI] [PubMed] [Google Scholar]
- Robbins PA, Swanson GD, Howson MG. A prediction correction scheme for forcing alveolar gases along certain time courses. J Appl Physiol. 1982a;52:1353–1357. doi: 10.1152/jappl.1982.52.5.1353. [DOI] [PubMed] [Google Scholar]
- Robbins PA, Swanson GD, Micco AJ, Schubert WP. A fast gas-mixing system for breath-to-breath respiratory control studies. J Appl Physiol. 1982b;52:1358–1362. doi: 10.1152/jappl.1982.52.5.1358. [DOI] [PubMed] [Google Scholar]
- Schmetterer L, Findl O, Strenn K, Graselli U, Kastner J, Eichler H-G, Wolzt M. Role of NO in the O2 and CO2 responsiveness of cerebral and ocular circulation in humans. Am J Physiol Regul Integr Comp Physiol. 1997;273:R2005–R2012. doi: 10.1152/ajpregu.1997.273.6.R2005. [DOI] [PubMed] [Google Scholar]
- Serrador JM, Picot PA, Rutt BK, Shoemaker JK, Bondar RL. MRI measures of middle cerebral artery diameter in conscious humans during simulated orthostasis. Stroke. 2000;31:1672–1678. doi: 10.1161/01.str.31.7.1672. [DOI] [PubMed] [Google Scholar]
- Severinghaus JW. Simple, accurate equations for human blood O2 dissociation computations. J Appl Physiol. 1979;46:599–602. doi: 10.1152/jappl.1979.46.3.599. [DOI] [PubMed] [Google Scholar]
- Shin WS, Sasaki T, Kato M, Hara K, Seko A, Yang WD, Shimamoto N, Sugimoto T, Toyo-oka T. Autocrine and paracrine effects of endothelium-derived relaxing factor on intracellular Ca2+ of endothelial cells and vascular smooth muscle cells. Identification by two-dimensional image analysis in coculture. J Biol Chem. 1992;267:20377–20382. [PubMed] [Google Scholar]
- Steinback CD, Poulin MJ. Ventilatory responses to isocapnic and poikilocapnic hypoxia in humans. Respir Physiol Neurobiol. 2007;155:104–113. doi: 10.1016/j.resp.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Van Mil AH, Spilt A, Van Buchem MA, Bollen EL, Teppema L, Westendorp RG, Blauw GJ. Nitric oxide mediates hypoxia-induced cerebral vasodilation in humans. J Appl Physiol. 2002;92:962–966. doi: 10.1152/japplphysiol.00616.2001. [DOI] [PubMed] [Google Scholar]
- Wei EP, Kontos HA, Patterson JL., Jr Dependence of pial arteriolar response to hypercapnia on vessel size. Am J Physiol. 1980;238:697–703. doi: 10.1152/ajpheart.1980.238.5.H697. [DOI] [PubMed] [Google Scholar]
- White RP, Deane C, Vallance P, Markus HS. Nitric oxide synthase inhibition in humans reduces cerebral blood flow but not the hyperemic response to hypercapnia. Stroke. 1998;29:467–472. doi: 10.1161/01.str.29.2.467. [DOI] [PubMed] [Google Scholar]
- Zimmermann C, Haberl RL. L-arginine improves diminished cerebral CO2 reactivity in patients. Stroke. 2003;34:643–647. doi: 10.1161/01.STR.0000056526.35630.47. [DOI] [PubMed] [Google Scholar]