

Abstract
Human monoclonal antibodies derived from B cells of HCV infected individuals provide information on the immune response to native HCV envelope proteins as they are recognized during infection. Monoclonal antibodies have been useful in the determination of the function and structure of specific immunogenic domains of proteins and should also be useful for the structure/function characterization of HCV E1 and E2 envelope glycoproteins. The HCV E2 envelope glycoprotein has at least three immunodistinctive conformation domains, designated A, B, and C. Conformational epitopes within domain B and C are neutralizing antibody targets on HCV pseudoparticles as well as from infectious cell culture virus. In this study, a combination of differential surface modification and mass spectrometric limited proteolysis followed by alanine mutagenesis was used to provide insight into potential conformational changes within the E2 protein upon antibody binding. The arginine guanidine groups in the E2 protein were modified with CHD in both the affinity bound and free states followed by mass spectrometric analysis, and the regions showing protection upon antibody binding were identified. This protection can arise by direct contact between the residues and the monoclonal antibody, or by antibody-induced conformational changes. Based on the mass spectrometric data, site-directed mutagenesis experiments were performed which clearly identified additional amino acids residues on E2 distant from the site of antibody interaction, whose change to alanine inhibited antibody recognition by inducing conformational changes within the E2 protein.
Keywords: Hepatitis C E2 glycoprotein, human neutralizing and non-neutralizing antibodies, limited proteolysis, differential chemical modification, mass spectrometry, alanine scanning mutagenesis
1. Introduction
Hepatitis C virus (HCV) infects over 170 million people worldwide, and most of the infections develop into chronic hepatitis. This is one of the most prevalent causes of liver cirrhosis and represents the most frequent indication for liver transplantation. All cases of infection carry an increased risk of hepatocellular carcinoma, which may be further exacerbated by co-infection with hepatitis B [1]. HCV is a small, enveloped positive-strand RNA virus belonging to the Flaviviridae family [2]. The ~ 9.5 kb genome of HCV encodes a single polyprotein between 3010 and 3033 amino acids [1]. This polyprotein is processed co-and posttranslationally generating the structural proteins Core, E1, E2, and p7, as well as six nonstructural proteins. The two envelope proteins E1 and E2 are heavily N-glycosylated, with 6 and 11 sites of glycosylation respectively [3]. E1 and E2 are believed to be type 1 transmembrane proteins with N terminal ectodomains and C terminal hydrophobic anchors, and together they are expected to form the viral envelope [4].
Presently, the only available therapy for HCV infection is interferon α (IFN) in combination with ribavirin [5], but this treatment can have adverse side effects. Consequently, the development of a vaccine against hepatitis C remains a high priority goal. It has been reported that the presence of neutralizing antibodies against the E2 protein correlates with protection from HCV infection, suggesting that E2 is a good candidate for a vaccine against hepatitis C [6]. Thus, there has been a significant level of interest in characterizing the E2 protein and its antigenic regions. The majority of reported human monoclonal antibodies (HMAbs) and recombinant HMAbs against E2 have been characterized as recognizing conformational epitopes. These includes antibodies that are effective as well as ones that are ineffective in inhibiting binding of E2 to CD81, followed by subsequent entry into target cells of HCV pseudoparticles (HCVpp) or HCV cell culture infectious virus (HCVcc) [6-8].
Based on cross-competition binding studies of HMAbs against the E2 glycoprotein, at least three immunogenic conformational clusters of epitopes, designated as domains A, B and C, have been described that are accessible on the surface of the HCVpp [6, 7]. It has also been reported that epitopes within domains B and C are targets of HCVpp-and HCVcc-neutralizing antibodies [9]. Particularly, these two domains (B and C) contain epitopes that are conserved among diverse genotypes 1a, 1b, 2a and 2b [7]. Although domain A contains only non-neutralizing epitopes, it is rich in cysteines that are potentially involved in formation of a number of disulfide bonds believed to be important for the proper folding of the E2 protein. Yagnik et al. predicted that there are four disulfide bridges that are involved in maintaining the structure of the protein [10]. Interestingly, low pH-treated HCVpp lead to a greater exposure of domain A epitopes resulting in a 50% fold increase in antibody binding [6, 7]. Keck et al. investigated the functional relationship between the non-neutralizing domain A antibodies and the neutralizing domains B and C antibodies. They found that the epitopes recognized by domain A antibodies are in spatial proximity to domain C epitopes, as well as with a more sequentially distant epitope in domain B [6, 7]. The same studies indicated that, in a low pH environment the conformation of E2 changes and this might increase the exposure of certain amino acids that were previously buried.
Recently, a number of HMAbs to conformational epitopes on HCV proteins were found to be potential candidates with high virus neutralization potency [11, 12]. These antibodies recognize conserved epitopes across diverse HCV genotypes. Several studies indicated that increased viral diversity in the hypervariable region of the HCV E2 envelope gene is associated with lack of control of infection [13]. The outcome of the HCV infection may be dictated by escape mutations in the epitopes targeted by CD8+ cytotoxic T lymphocytes (CTL) [14, 15]. Moreover, amino acid substitutions can also alter CTL recognition of variant peptide major histocompatibility complexes (MHC) [16]. CTL escape mutants are found during HCV infection in humans. The loss of epitope phenotype can also occur when amino acid anchor residues required for MHC binding are mutated [14]. Amino acid substitutions within CTL epitopes can also alter proteosomal processing causing epitope destruction before transport to the endoplasmic reticulum (ER) for MHC binding [17]. These escape mutations can include not only mutations within the antibody binding region but also mutations outside of the recognition surface that induce conformational changes in the native structure leading to reduced access to the antibody recognition region. It has been shown by X-ray crystallography that binding of a monoclonal antibody (mAb) to a viral envelope protein, e.g., HIV gp120, can induce conformational changes [18].
Mass spectrometry (MS) is increasingly used to identify protein complexes and to elucidate protein structure and in particular for proteins that are not amenable to classical structural techniques. MS using MALDI and/or ESI [19-21] is becoming widely used in the determination of protein structures and of protein:protein interactions, especially when used in conjunction with limited proteolysis and chemical modification [22-25]. Differential surface modification experiments can identify differences in residue reactivity as a function of conformational changes of the protein upon interaction with the antibody, or by steric hindrance due to proximity of the antibody. Modification under conditions that retain the native conformation provide information on which residues are on the surface (accessible and reactive), and which are buried in the core of the protein (non-accessible and non-reactive). Only residues that are accessible or have higher reactivity will be modified chemically, however the extent of derivatization can also be affected by steric interactions [23, 26]. Additionally, free amino groups are highly sensitive to their chemical environments, and, therefore, their reactivity may be influenced by factors that alter their pKa or surface accessibility [26]. In addition to chemical modification, limited proteolysis experiments are particularly useful for studying antibody–antigen interactions, allowing for quick determination of the regions involved in antibody recognition [27]. In alanine scanning mutagenesis, the residues in a linear epitope are individually substituted with alanine, and binding to the antibody is probed. In addition, sites specific mutagenesis of the residues results in exchange either with a conservative substitution, or with a residue that changes the properties of the amino acid considerably. These approaches allow the characterization of the critical binding residues which are essential for interaction with the antibody [28].
In the present paper, the possibility of conformational changes on E2 upon antibody binding, as well as the elucidation of critical residues involved in antibody binding were investigated by a combination of differential surface modification and limited proteolysis with mass spectrometric characterization of the products, and by alanine scanning mutagenesis [29-31]. These results provide insight into conformational changes within the E2 protein as a result of antibody binding, as well as a more detailed look into the effect of mutational induced conformational changes on distant antibody binding.
2. Materials and methods
Materials
Sodium tetraborate-decahydrate, GnHCl, 1,2-cyclohexanedione (CHD), ammonium bicarbonate, dithiothreitol, GNA, p-nitrophenyl phosphate disodium hexa-hydrate (phosphatase substrate) and 96% formic acid were purchased from Sigma-Aldrich (St. Louis, MO). BS3 was purchased from Pierce, Rockford, IL. Alkaline phosphatase-conjugated goat anti-human IgG (H+L), alkaline phosphatase-conjugated goat anti-mouse IgG (H+L) and sequencing grade-modified trypsin were obtained from Promega (Madison, WI). Endoproteinase GluC was purchased from Roche Diagnostics Corporation (Indianapolis, IN). Acetonitrile was purchased from Caledon Laboratories, Ltd. (Georgetown, Ontario). Purified water (17.8 MΩ) was obtained from an in-house Hydro Picopure 2 system. All chemicals were used without further purification unless otherwise specified. The anti-E2 CBH-5, CBH-7, CBH-4D and -4G HMAbs have been previously described [7, 32]. The murine HMAb to the c-myc epitope was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). FITC-conjugated goat anti-human IgG, Fc γ fragment-specific and R-phycoerythrin-conjugated F(ab’)2 fragment goat anti-mouse IgG (H+L) were obtained from Jackson Immuno Research (West Grove, PA).
Protein
Hepatitis C virus E2 envelope glycoprotein (recombinant) was purchased from Austral Biologicals (San Ramon, CA). The protein was expressed in CHO cells, and it covers the HCV sequence from Ala384 to Lys715.
Cells and culture conditions
HEK 293T cells were obtained from the American Type Culture Collection (Manassas, Va.), and grown in Dulbecco’s minimal essential medium (Invitrogen, Carlsbad, CA) supplemented with 10% FCS (Gemini Bioproducts Inc. Calabasa, CA).
Preparation of immobilized antibodies
CNBr activated sepharose beads were activated in accordance with the procedures described previously [33, 34]. Briefly, a 60 μl aliquot of washed beads was added to each of two CRCs (USB Corporation, Cleveland, OH). The CNBr-coupling product was prepared in a 0.8 ml micro-column which permits extensive washing without significant loss of material. Twenty micro-liters (48 μg) of the secondary antibody, anti-human Fc-specific IgG, was added to each column and incubated for 2 h in 80 μl of 100 mM NaHCO3, 150 mM NaCl, pH 8.2, with slow rotation. The columns were rinsed, and a 20 μl (50 μg) aliquot of primary antibody CBH-5 was added to one of the tubes while 50 μl of PBS (pH 7.2) was added to the other tube to serve as a control. The beads were incubated for 2 h at room temperature with slow rotation, drained, and washed three times with 0.5 ml of PBS. The CBH-5 HMAb antibody was affinity captured from solution and cross-linked to the Fc-specific antibody with BS3 as previously described [33]. A solution of 10 mM BS3 was prepared in PBS buffer (pH 7.2). A 10 μl aliquot was added to the beads and incubated in the dark with rotation for 45 min. The beads were washed twice with 100 μl of 100 mM Tris, pH 8.0, and then resuspended in 50 μl of PBS. The beads were washed three times with 0.4 ml of PBS. A 20 μl aliquot of the beads was set aside as a control, while the remainder was used to bind the E2 protein to form the immobilized antigen:antibody complex. A 100 μl aliquot of a protein solution containing 20 μg of E2 was added to the CRC. Both CRCs were rotated at room temperature for 3 h. The beads were then drained and rinsed with PBS.
Limited proteolysis
The immobilized antigen: antibody complex was digested for 2 h with 0.5 μg trypsin (Promega, Madison, WI) in 300 μl PBS buffer (pH 7.2) at room temperature and 1:200 enzyme: substrate ratio. Supernatant non-epitope peptides were drained by pressurizing the column with a syringe, and the matrix material was washed three times with 0.4 ml PBS buffer before MALDI-TOF MS analysis of the beads. Under the immobilization and proteolytic digestion conditions employed, IgG antibodies are usually highly stable towards degradation, as established in previous studies.
Site-directed mutagenesis
The cell surface expressed form of the E2 glycoprotein produced as an HA-1bE2384-661-cmyc fusion protein has been previously described [6, 35]. The plasmid pDisplay-38 carrying this fusion protein was used to construct the mutants. Residues located within regions identified by mass spectrometric analysis were subjected to alanine replacement mutagenesis by using the QuikChange II Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). The positions of the mutated residue are indicated followed by a number corresponding to the positions of the polyprotein reference strain H (GenBank access number AF009606) [36]. Two alanine residues at sites 490 and 499 were substituted with glycine. Mutagenic oligonucleotide primers were designed according to the manufacturer’s instruction and synthesized by Operon Biotechnologies (Huntsville, AL). The full-length E2 sequences of all the constructs were verified (Sequetech, Mountain View, CA). The resulting plasmids were transfected into HEK293T cells for transient protein expression using the calcium-phosphate method [37].
Chemical modification of arginine residues
Modification of arginine residues was done as previously described [23]. Briefly, in the control reaction 1 μM (7.5 μg) of E2 protein was modified with 30 mM CHD in 150 μl 100 mM sodium borate buffer (pH 9.0) at 37°C for 2 h, quenched with 1% trifluoroacetic acid and the mixture directly injected onto a C4 HPLC column. For the modification reaction of the immune complex, the antibody: antigen immune complex was first formed by mixing 1 μM E2 and 2 μM CBH-5 HMAb for 2h at room temperature. Afterwards, 30 mM CHD in 150 μl 100 mM sodium borate buffer (pH 9.0) was added to the immune complex solution and the reaction was performed at 37°C for 2 h and quenched with 1% trifluoroacetic acid. The modified E2 was separated from the antibody by liquid chromatography. Collected fractions were lyophilized, and then analyzed by MALDI-TOF MS. The fractions found to contain the E2 protein were combined and lyophilized to dryness. In both experiments, the E2 protein was denaturated by using a solution containing 4 M GnHCl and 10 mM dithiothreitol for 1.5 h at 65°C. The reaction mixture was cooled, and 200 μl of 100 mM Phosphate buffer (pH 7.5) were added and PNGaseF (2 units) (Sigma-Aldrich, St. Louis, MO) deglycosylation was performed for 24 h at 37°C. Further cleavage of the protein was accomplished with trypsin (using a 1:20 enzyme: substrate ratio) and endoproteinase GluC (1:20 enzyme: substrate ratio). nanoLC/MS/MS analyses were performed on the resulting peptides.
Binding of CBH-5 to E2 mutants by two-color flow cytometry analysis
E2 mutant-transfected HEK293T cells were incubated with anti-E2 CBH-5 HMAb at 5 μg/ml in 100 μl of staining solution (PBS plus 2% FCS) for 20 min at room temperature. After washing, the cells were incubated with FITC-conjugated Goat anti-human IgG and R-phycoerythrin conjugated goat anti-mouse IgG, diluted as recommended by the manufacturer, for 20 min at room temperature to detect E2 glycoprotein and c-myc expression respectively. After additional washing, cells were fixed in 1% paraformaldehyde and analyzed using a FACS Calibur apparatus (BD Biosciences, San Jose, CA). The ability of each E2 glycoprotein mutant to bind to CBH-5 was normalized by E2 glycoprotein expression as monitored by c-myc expression and by calculating the ratio of the mean FITC (CBH-5) intensity over the mean phycoerthyrin (PE) intensity. These values were then displayed as a percent of the values observed for wild type (wt) E2.
Binding of CBH-5 to E2 mutants by ELISA
The FACS data were corroborated by measuring the CBH-5 binding to the mutant E2 proteins by ELISA. Microtiter plates were coated with 500 ng per well of GNA, followed by blocking with BLOTTO, consisting of 2.5% nonfat dry milk and 2.5% normal goat serum in TBST (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% Tween 20) for 1 h. The expressed wt and mutant E2 glycoproteins present in transfected HEK293T cells were prepared by lysing cells in a buffer (10 mM Tris HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP 40, 0.5% sodium deoxycholate) on ice for 15 min. The lysates were then clarified by centrifuging at 18,000 g for 15 min. The lysis pellet was discarded and the supernatants were captured onto GNA coated plates. The bound E2 glycoproteins were detected by anti-E2 CBH-5 HMAb, followed by AP-conjugated goat anti-human IgG and further developed with p-nitrophenyl phosphate disodium hexa-hydrate. The absorbance was measured at 405 and 570 nm. Values for substrate development were derived by measuring absorbance at 405 nm and subtracting the background reading at 570 nm. These values for mutant E2 proteins were then displayed as a percent of the values observed for the wt E2.
Liquid chromatography
The separation and purification of modified proteins was achieved using a Hewlett Packard 1100 HPLC system (Wilmington, DE) equipped with a Vydak C4 column (4.6 mm i.d., 5 μm particles) (Grace Vydac, CA), a diode array detector, and a fraction collector were used. Gradient elution was carried out with two solvents: A: water, 0.1% trifluoroacetic acid and B: 90% acetonitrile, 10% water, 0.1% trifluoroacetic acid. A linear gradient at 1 ml/min from 5% B to 60% B over 50 min was used to purify the proteins. Fractions were collected, lyophilized, and redissolved in 50 μl 60% acetonitrile, 0.1% formic acid just prior to MALDI-TOF MS analysis.
Mass spectrometry
MALDI-TOF mass spectra were acquired on a Voyager DE-STR mass spectrometer from Applied Biosystems (Framingham, MA) equipped with a nitrogen laser (λ=337 nm). The measurements were performed in the reflector and linear positive ion modes. Samples (0.5 μl) were spotted onto a 100 sample stainless steel MALDI plate and mixed on target with 0.5 μl of a saturated solution of α-cyanohydroxycinnamic acid in 45:45:10 (v:v:v) water:ethanol:formic acid. Due to the small sample amounts and a relatively high salt content in the analyte solution, the salts were removed by washing the sample target with 50 mM ammonium acetate as follows. After complete drying, the target spot was covered with 5 μl of a 50 mM solution of ammonium acetate in water (pH 4.0), which was carefully absorbed after 10 s with a wiping tissue. The spot was then recrystallized by addition of 1 μl acetonitrile/0.1% formic acid (2:1); this resulted in a fine crystalline matrix. This sample preparation procedure significantly improved signal/noise ratios due to reduction of adduct signals, and generally resulted in highly reproducible spectra. The MS spectra were calibrated externally using a mixture of standard peptides with a mass accuracy of greater than 0.01%. Data were acquired using an accelerating voltage of 23000 V, a grid percentage of 65%, and a delay time of 200 nanoseconds for reflector ion mode and an accelerating voltage of 25000 V, a grid percentage of 85%, and a delay time of 400 nanoseconds for linear ion mode analyses.
NanoLC/MS/MS analyses were performed on a Waters Q-Tof Premier mass spectrometer equipped with a nanoAcquity UPLC system and a NanoLockspray source (Waters, Milford, MA). Separations were performed using a 3 μm nanoAquity Atlantis column dC18 100 μm × 100 mm (Waters) at a flow rate of 300 nl/min. A nanoAquity trapping column 5 μm C18 180 μm × 200 mm (Waters) was positioned in-line with the analytical column. Trapping of a 2 μl aliquot of the digested sample was performed for 3 min at 5 μl/min flow rate, and then separated on the analytical column. Peptides were eluted using a linear gradient from 98% A (water/0.1% formic acid (v/v)) and 2% B (acetonitrile/0.1% formic acid (v/v)) to 95% B over 60 minutes. Mass spectrometer settings for MS analyses were as follows: capillary voltage of 3.5 kV, cone voltage of 30 V, collision energy of 8.0 V, and scan range of 200-2000 Da. MS/MS data were acquired using a data dependent acquisition method selecting collision energies based on mass and charge state of the candidate ions. Peptides were identified by mass and charge measurement with in silico digestion using the GPMAW 6.0 General Protein/Mass Analysis for Windows (Lighthouse Data 2003, Odense, Denmark) [38], and the BioLynx Protein/ Peptide Editor, feature of MassLynx V4.0 (Micromass, UK).
3. Results
Limited proteolysis
Limited proteolysis was carried out using E2 glycoprotein and immobilized CBH-5, a neutralizing HMAb recognizing an epitope on the E2 glycoprotein [6]. The sequence of the E2 glycoprotein that was used in this study is hepatitis C subtype 1a, and it covers from Ala383 to Lys715 of the HCV-1a polyprotein (GenBank access number AF009606). After binding the E2 glycoprotein to the immobilized CBH-5 the MALDI-TOF mass spectrum of the immobilized antibody beads showed the presence of the protein attached to the antibody beads (Figure 1). The 11 consensus glycosylation sites on E2 [3], are predominantly, [39] but not exclusively (paper submitted), occupied by high-mannose glycans. As shown in Figure 1, the MALDI-TOF mass spectrum of the intact protein contains broad peaks representing singly, doubly and triply charged ions. E2 has a theoretical molecular weight of 36.5 kDa, but the observed mass of the singly charged ion in the MALDI-TOF mass spectrum was approximately 50 kDa, indicating significant N-linked glycosylation with extensive heterogeneity. The protein was digested with trypsin; the supernatant was removed and retained, and the immobilized antibody beads were extensively washed. The beads were then analyzed by MALDI-TOF MS and only one large fragment, a singly charged ion of m/z 5532.40 was found to remain bound to the antibody beads (Figure 2). This observed molecular weight does not correspond to an unmodified tryptic peptide but does correspond to the tryptic fragment (462-500) containing a Man5GlcNAc2 (1216.6 Da) type glycan at position N476, determined with a mass accuracy better than 0.01% (Figure 2). Previous studies showed that the consensus glycosylation site N476 had a varying level of conservation of 97% among E2 genotype 1a, 18% for 1b and absent from the sequences of HCV E2 genotype 5 [3]. The tryptic peptide (462-500) did not undergo further cleavage with trypsin because arginines residues Arg483 and Arg492 are directly followed by a proline residue and trypsin will not cleave after arginine in this case. Further digestions with endoproteinase GluC (data not shown) lead to the loss of affinity, indicating that the peptides resulting from the two cleavages are insufficient for recognition by the antibody. This experiment was also performed with the deglycosylated E2 protein, but the protein did not bind to the antibody beads. This experiment indicated that some or all glycans are necessary but not sufficient for antibody binding, although they might not necessarily be a part of the CBH5 epitope. Figure 3 shows the amino acid sequence of the E2 protein along with the tryptic peptide (462-500) which was bound to CBH-5.
Fig. 1.
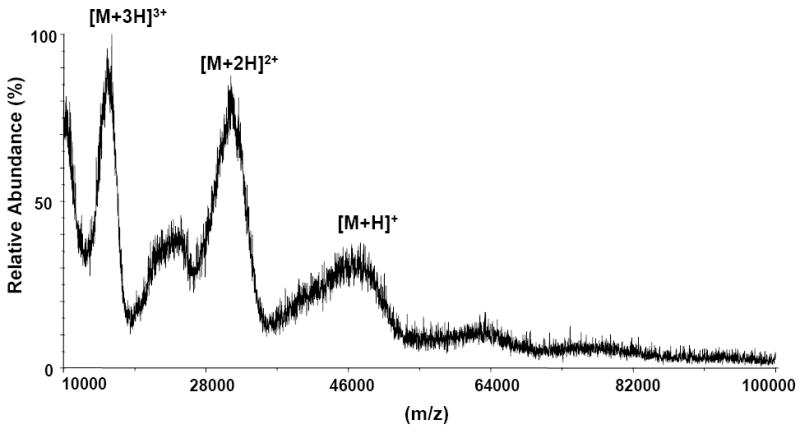
MALDI-TOF mass spectrum (mass range 10,000–100,000Da) of E2 glycoprotein affinity bound to CBH-5 HMAb on CNBr activated sepharose beads. The singly, doubly, and triply protonated molecule ions are marked. The broadness of the peaks is due to the extensive glycosylation of E2.
Fig. 2.
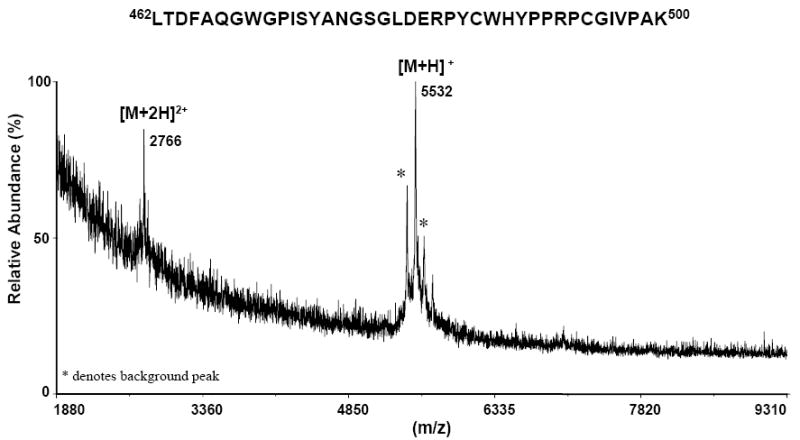
MALDI-TOF mass spectrum of the glycosylated peptide (462–500) that remained affinity bound to the beads after limited proteolysis of the E2/CBH-5 HMAb complex with endoproteinase trypsin. The spectrum shows the singly protonated peptide of m/z 5532 Da. The doubly doubly protonated molecule is also observed of m/z 2766. The mass difference of 1216 Da between the theoretical and experimental values is due to the presence of N-linked glycans at position N476, indicating the presence of a high-mannose type glycan (Man5GlcNAc2). The peaks marked with an asterisk * denote background ions.
Fig. 3.

Amino acid sequence of E2 protein (absolute numbering: amino acids 383–715) from strain HCV-1a. The positions of the residue are indicated corresponding to the positions of the polyprotein of reference strain H (GenBank access number AF009606) [36]. The peptide fragment that remained affinity bound to the CBH-5 HMAb in the limited proteolysis experiment (462-500) is indicated in bold and italics.
Modification of the arginine residues in free and affinity bound E2
To identify critical residues on E2, differential surface modification was applied; specifically, using arginine modification with CHD [23]. CHD-derivatized E2 was analyzed by nanoLC/MS/MS as described in the Materials and methods. Modification of arginines with CHD results in the formation of an N7,N8-(dihydroxy-1,2-cyclohexylidine)-derivative corresponding to a mass increase of 112 Da per modified arginine residue [23]. After HPLC purification, the derivatized protein was subjected to PNGaseF deglycosylation to remove the N-linked glycans, followed by trypsin and GluC digestion. These peptides were then separated and analyzed by nanoLC/MS/MS. Modified peptides were identified from the resulting chromatogram based on their mass shifts per modified guanidino group. The mass spectrometric analyses showed modification of nine of 19 arginines in free E2 antigen (control) (Table 1).
Table 1.
Arginine residues on E2 modified by 1,2-cyclohexanedione (CHD) identified by mass spectrometry
| Modified arginine residues | Shielded residues | |
|---|---|---|
| E2-control | CBH5-E2 complex | |
| 455, 460,521
587, 596, 630 639, 648, 651 |
455, 460, 521,
596, 639, 648 |
587
630, 651a |
The two-arginine residues Arg630 and Arg651 were CHD modified in both control and immune complex, but the peptides containing those residues showed decreased intensities in the immune complex sample, indicating that they are involved in the antibody binding.
In the immune complex sample, mass spectrometric peptide-mapping analyses showed selective modification of six arginines after CHD derivatization. Only three residues showed decreased accessibility in the immune complex in comparison with the control sample, indicating that those arginine residues are shielded upon antibody binding (Table 1). The GluC peptide (585-591) CFRKYPE containing Arg587, was only observed as CHD modified in the control sample (doubly charged ion of m/z 527.22) and only seen unmodified by CHD in the immune complex (doubly charged ion of m/z 471.22). This indicates that this Arg587 is part of the antibody recognition surface (which does not necessarily mean that the Arg is actively involved in binding to the antibody). The tandem mass spectrum shown in Figure 4 contains the fragment ions resulting from peptide (585-591) backbone cleavage [40]. The y5 fragment ion of m/z 804.42 and the b3 ion (m/z 500.23) resulting from the N-terminus fragmentation of the (585-591) peptide and corresponding to a mass increase of 112 Da compared to the unmodified protein also indicates that Arg587 has been modified.
Fig. 4.
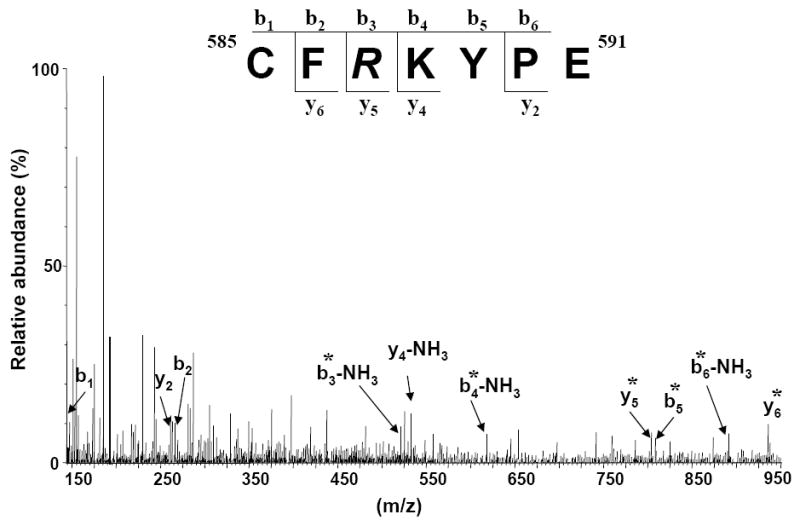
ESI-MS/MS of the precursor doubly protonated molecule of m/z 527.22 corresponding to the GluC peptide (585–591) with the sequence CFRKYPE. The Arg587 is modified with 1,2-cyclohexanedione. The modification took place only in the E2 control sample, in the immune complex Arg587 was not modified. Fragment ions b and y that are denoted by an asterisk * contain the modified arginine residue.
By comparing the relative abundances of two peptides of interest by normalizing the data to an unrelated ion found in the mass spectra of both the control and immune complex samples, additional differences in the mass spectrometric data between the control and the immune complex were identified. Upon PNGaseF reaction, the glycosylated Asn623 (NYT) was converted to aspartic acid as observed in the mass spectrum by an increase in mass of 1 Da. The triply charged ion of m/z 968.19 was found to correspond to the GluC peptide (615-637) LWHYPCTINYTIFKVRMYVGGVE, with Arg630 CHD modified both in the control and immune complex samples. In the E2 control mass spectrum, the relative abundance of the peptide (615-637) is 25% when normalized to a non-arginine containing peptide, while the abundance decreases to 9% in the immune complex mass spectrum (Figure 5 A, B). These differences in peptide abundances suggest strong protection of Arg630 upon antibody binding. Considering that Arg630 was found modified in both experiments, it may be assumed that this particular arginine is not directly involved in antibody binding, but that it is either in close proximity to the CBH-5 binding site and protected by steric interactions between the antibody and antigen or that a conformational change is induced upon antibody binding which leads to reduced accessibility in the complex and correspondingly reduced reactivity. Moreover, the shorter tryptic peptide (615-628) (doubly charged ion of m/z 900.44), also showed a decreased abundance when the protein was bound to the antibody indicating reduced access to the cleavage site (data not shown). Furthermore, the triply charged ion of m/z 793.06 corresponding to the peptide (611-628) showed decreased abundance from the immune complex, in comparison with the control sample, indicating decreased access by the protease, even though Arg614 was not modified in either experiment (likely due to lack of a nearby proton acceptor). This result emphasizes the involvement of the same overlapping region LWHYPCTINYTIFK (615-628) in CBH-5 antibody binding. In the chemical modification experiment of the immune complex, residues Arg630 and 651 were CHD modified, but the peptides encompassing those residues showed some protection upon CBH-5 antibody binding. The partial modification of Arg630 and 651 was indicated only by the decreased abundance of the peptides (615-628), (615-637) and (649-655) in the immune complex sample. This result can be explained by conformational changes in either the protein structure or in the conformation of the glycans in the presence of the antibody. The CHD modification experiments were performed in triplicate, and the ions of interest were screened against three independent unrelated ions. The relative abundances of arginine 614, 630 and 651 in the free E2 and affinity bound antigen: antibody (E2/CBH-5) complex are presented in Table 2 and exhibit differences between the control and immune complex samples. The lower abundances of the peptides containing these residues in the immune complex sample suggests either that these amino acids are close to the affinity binding site of the neutralizing CBH-5 HMAb or that their accessibility is altered by a conformational change upon complex formation.
Fig. 5.
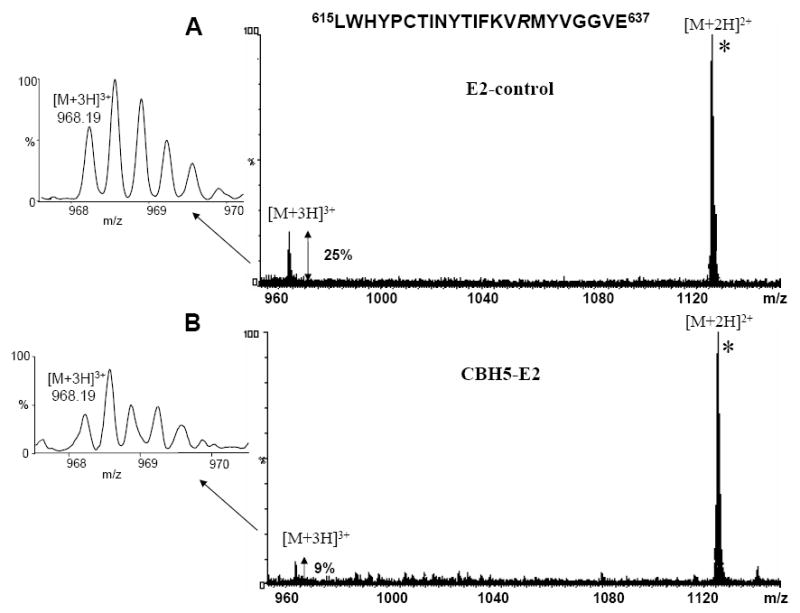
Mass spectrum representing the overlay of the peptide of interest (615-637) showing the triply protonated molecule of m/z 968.19, normalized to a non-arginine containing ion, a doubly charged ion of m/z 1135.18 (marked with an asterisk *) found in both experiments. Compared to the control sample (A), the mass spectrum shows a significant change in the relative abundance when the E2 protein was bound to the CBH-5 HMAb (B) indicating protection by the antibody.
Table 2.
The relative abundances ratios of the control versus the immune complex
| Peptide | Abundances % E2-controla | Abundances % CBH5-E2 complexa | Ratios b | |
|---|---|---|---|---|
| 615-628 | LWHYPCTINYTIFK | 60 | 14 | 0.23 |
| 611-628 | YPYRLWHYPCTINYTIFK | 28 | 8 | 0.28 |
| 615-637 | LWHYPCTINYTIFKVRMYVGGVE | 25 | 9 | 0.36 |
| 649-655 | GERCDLE | 23 | 7 | 0.30 |
Average of three independent mass spectrometric measurements of the ion of interest against an unrelated ion present in all measurements
Entries are the ratio of the relative abundance calculated between the control and the immune complex sample. The changes in abundances in the presence of the antibody were determined by dividing the values. Values significantly less than 1.0 indicate a decrease in reactivity, suggesting protection upon antibody binding.
Site-directed mutagenesis
In contrast to the MS experiments which identified residues whose reactivity is modified after antibody binding, alanine scanning mutagenesis was performed to define how mutations of specific residues on E2 may affect antibody binding and to identify specific residues that are critical to the interaction of E2 with CBH-5 either directly or via a conformational change. Based on the mass spectrometric data, a series of proteins mutated at sites between 481-500, 615-628 (which showed protection in the limited proteolysis and chemical modification) and four arginine residues, 587, 614, 630 and 651, (which showed protection by the antibody) were obtained by site-directed mutagenesis (Figure 6). For these mutants, the effect on CBH-5 binding was analyzed by transfecting cells with plasmids encoding E2-cmyc fusion protein. Mutant E2 proteins were assessed for their reactivity with CBH-5 by two-color flow cytometry, and confirmed by GNA capture ELISA (data not shown). Because c-myc is a C-terminal tag, the amount of c-myc detected by PE-anti-c-myc is a good indicator of full-length E2 expression. Thus, c-myc expression was used as an internal control to normalize the level of E2 expression. To evaluate the percentage of CBH-5 binding activity, E2/c-myc ratios were determined for each mutant and compared to the ratio obtained with wt protein.
Fig. 6.
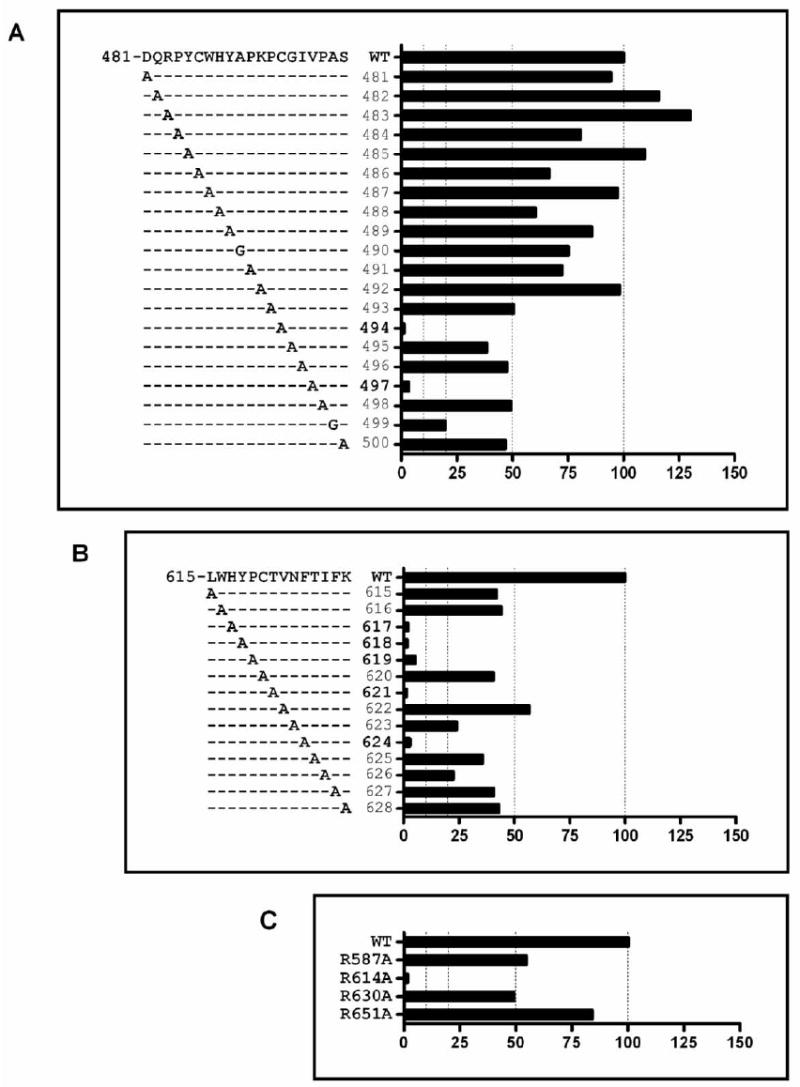
Alanine replacement based on MS data identifies a number of residues critical for CBH-5 binding. E2-c-myc fusion mutant proteins were expressed in HEK293T cells and cells were analyzed by FACS. The wt sequences that are illustrated correspond to the reference strain H (GenBank accession number AF009606). The y-axis of plots A, B and C show the position of the residues that are critical to the antibody binding. CBH-5 HMAb binding to each mutant is expressed as the percent of fluorescence intensity value normalized by the intensity of c-myc binding and divided by CBH-5 binding to the wild-type on the (x-axis). (A) and (B) are two discontinuous regions corresponding to two immune complex protection sites during tryptic digestion respectively at amino acids 481-500 and amino acids 615-628. (C) Four arginine sites, 587, 614, 630 and 651, that are protected from CHD modification or showed decreased intensities in the mass spectra upon antibody binding are illustrated. The experiments were done by two independent transfection and FACS followed by GNA capture ELISA.
A significant reduction in CBH-5 HMAb binding was observed when Cys494, Val497 (Figure 6 A), His617, Tyr618, Pro619, Thr621, Phe624 (Figure 6 B), and Arg614 (Figure 6 C) were replaced by an alanine. In fact, CBH-5 binding to these mutants was reduced to less than 10% of that bound to the wt protein. Because the Cys494 mutant showed a global effect on maintaining E2 structure in our previous studies (unpublished data), the role that it plays in CBH-5 antibody binding is unclear at this point, although it’s involvement in disulfide bonds would indicate a critical role in maintaining the protein’s native conformation. In addition, mutations of the residues located at positions Ala499 (Figure 6 A), Asn623 and Ile626 (Figure 6 B) led to approximately 80% reduction in binding of CBH-5 to E2 compared to wt. Interestingly, substitutions in a larger region covering residues at positions Pro493, Gly495, Ile496, Pro498, Ser500 (Figure 6 A), Leu615, Trp616, Cys620, Val622, Thr625, Phe627, Lys628 (Figure 6 B), Arg587 and Arg630 (Figure 6 C), could weaken CBH-5 interaction with E2 by approximately 50%, as compared to the wt protein, indicating that these residues can affect CBH-5 HMAb binding to different HCV genotypes and/or isolates. Collectively, these findings suggest that eight critical amino acids (Cys494, Val497, Arg614, His617, Tyr618, Pro619, Thr621 and Phe624) significantly affect binding of CBH-5 HMAb.
In addition to the alanine scanning results described in Figure 6, we assessed whether the amino acids mutated above specifically affect only the interaction of E2 with CBH-5 or also have an affect on the interaction of E2 with other HMAbs, which might indicate a potential conformational change of the E2 protein affecting antibody binding. To examine this hypothesis, the alanine scanning mutagenesis was expanded just beyond the region identified by mass spectrometry, starting with amino acid Lys500 to Thr510, as well as the region encompassing Tyr611 to Arg614 and Met631 to Glu637. For this study three other HMAbs against E2 were tested, CBH-4D and CBH-4G, which are non-neutralizing and directed against domain A, as well as the neutralizing CBH-7 HMAb directed against a different epitope within domain C [7, 9]. The GNA analysis results of the binding between domain A, B and C HMAbs to the alanine mutants are presented in Figure 7. These results indicated that the eight knock down mutations identified and described above have similar effects on recognition by antibodies to all three domains, showing a decreased or complete loss of affinity towards all four antibodies that were tested. To differentiate whether a global conformation change occurred with each highlighted mutation (in Figure 7); HCVpp were produced with each mutant. Wild-type HCVpp is a robust means to analyze functional envelope proteins involved in virus attachment and entry [41]. These HCVpp contain fully functional envelope glycoproteins that preferentially infect human hepatocytes and hepatocellular cell lines. The envelope proteins are at least in part noncovalent E1E2 heterodimers and their recognition by a panel of human monoclonal antibodies (HMAbs) to conformational epitopes on E2 confirms the expression of native antigenic structures [35]. None of these HCVpp mutants were infectious compared to wt HCVpp consistent with a global change at these sites (Figure 8).
Fig. 7.
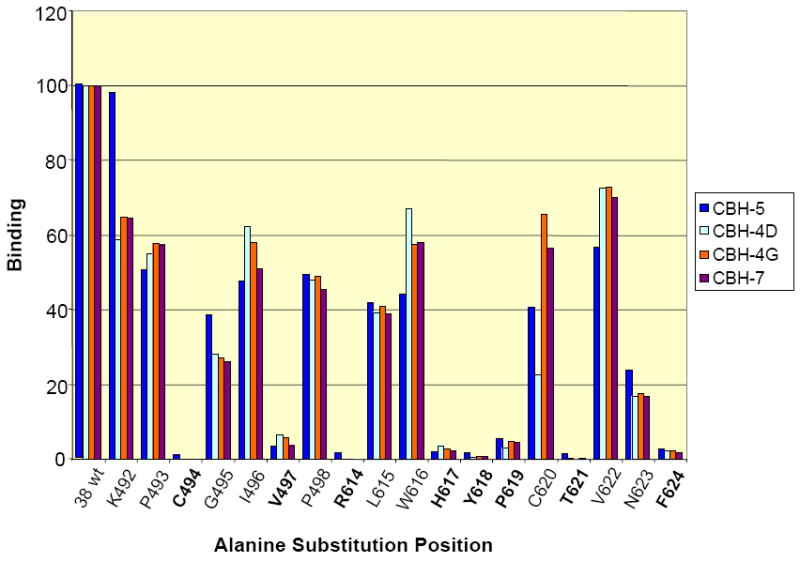
GNA analysis of binding of domain A, B and C antibodies to the alanine mutants 492-498 and 614-624. The eight amino acids that are critical for the antibody binding are presented in bold. The numbers in the beginning of the peptides correspond to the position in the polyprotein of reference strain H (GenBank accession number AF009606). The expressed wt and mutated E2 glycoproteins present in HEK293T cell lysates were captured onto GNA coated plates. The bound E2 glycoproteins were detected by anti-E HMAbs, followed by AP-conjugated goat anti-human IgG and further developed with p-nitrophenyl phosphate disodium hexa-hydrate. E2 concentration was normalized by quantification of a c-terminal myc tag on the recombinant proteins. The y-axis depicts binding values for each E2 alanine mutant as a percentage of the binding observed with wt E2.
Fig. 8.
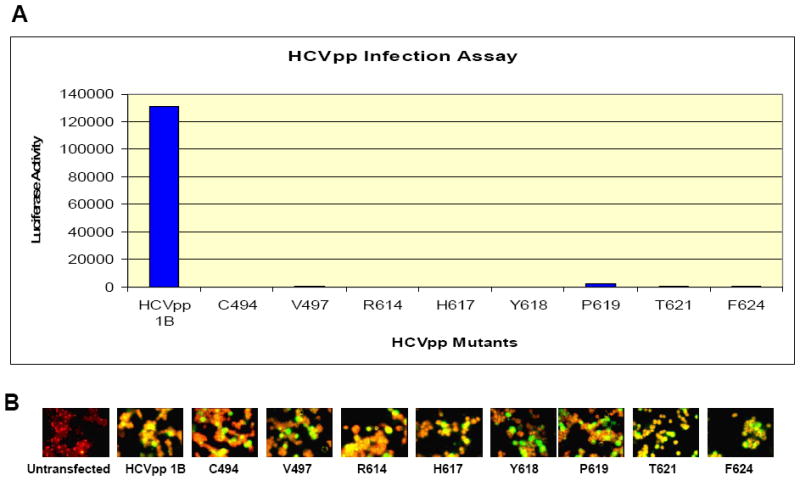
HCVpp mutants do not infect Huh-7.5 cells: (A) the luciferase activity of the HCVpp mutants in which the eight amino acids found to be critical for mAb CBH-5 binding have been mutated to Ala was measured against the genotype HCVpp 1B. The activity assay is measured on the y-axis, and it shows that the mutants had no effect on the Huh-7.5 cells. (B) Immuno-Fluorescence staining indicating the HCV E2 expression against the mAb AP33: the staining of the control HCVpp 1B and the mutants shows that HCVpp mutants have no effect on the E2 protein expression which is unchanged for all eight mutants analyzed.
Moreover, an extended alanine scanning mutagenesis experiment starting with Lys500 through Thr510 was performed indicating specific amino acids in this extended region that, upon alanine mutation, results in a partial decrease or complete loss of binding affinity to the CBH-5 HMAb (Figure 9). Substitutions at Val502 and from Gly504 to Phe509 led to reduced or no binding with both CBH-5 and -4G. In the Cys503A mutant, binding to CBH-5 was eliminated although binding to CBH-4G was retained. These results suggest that Cys503 could be involved in a disulfide bridge leading to a more localized structural change affecting only CBH-5. Interestingly, C508A mutation destroys binding to CBH-4G as well as it does with CBH-5 HMAb (Figure 9). Again, these results would be consistent with either a significant conformational change or possibly a change in binding due to a change in hydrophobicity/ hydrophilicity or polarity of the residues. Further experiments similar to ones described above were performed on the extended alanine mutation in the regions Tyr611 to Arg614, and Lys628 to Glu637. Major differences in the loss of affinity were seen with mutation at amino acids Tyr611 and Arg614. As previously discussed Arg614 was found to be partially shielded by the antibody in the mass spectrometric differential modification experiments.
Fig. 9.
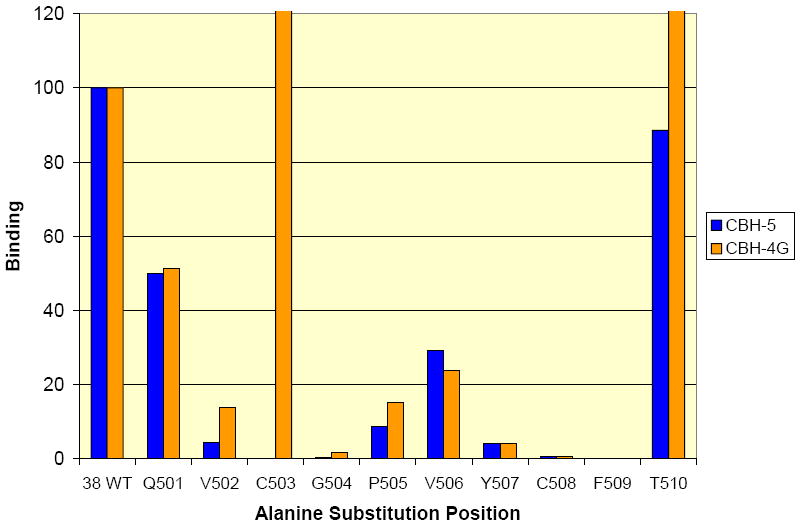
GNA analysis of binding of domain A and B antibodies to the alanine mutants 501-510. The numbers in the beginning of the peptides correspond to the position in the polyprotein of reference strain H (GenBank accession number AF009606). The expressed wt and mutated E2 glycoproteins present in HEK293T cell lysates were captured onto GNA coated plates. The bound E2 glycoproteins were detected by anti-E2 HMAbs, followed by AP-conjugated goat anti-human IgG and further developed with p-nitrophenyl phosphate disodium hexa-hydrate. E2 concentration was normalized by quantification of a c-terminal Myc tag on the recombinant proteins. The y-axis depicts binding values for each E2 alanine mutant as a percentage of the binding observed with wt E2.
4. Discussion
Known conformational changes in Flaviviridae
To date there is no crystal structure available for the HCV E2 protein, a protein belonging to the Flaviviridae family [2, 42]. It was reported that a significant amount of structural data relating to potential sites of interaction between the antibodies and HCV E2 could be obtained by using neutralization escape mutants [14, 17]. These data were used to model the structure of the Flavivirus E protein [43]. Interestingly, similar to the HCV E2 protein, each of the three domains contributing to the exterior face of the E protein can induce and bind neutralizing antibodies. Furthermore, antibodies against Flavivirus E protein primarily recognize a site involving residues of the highly conserved fusion peptide loop, which is a cryptic epitope and largely inaccessible on the surface of native infectious virions [43]. Moreover, similar viruses, such as the Tick Borne Encephalitis Virus (TBEV) have been previously described [44, 45]. These studies showed that Flavivirus envelope glycoprotein E from TBEV shows functional similarity to HCV E2, and the application of fold recognition structures highlighted the similarity between TBEV and HCV E2 [10, 44]. Furthermore, both viruses undergo structural rearrangements at low pH environments that hypothetically lead to the exposure of initially buried hydrophobic residues. This pH-induced process is believed to be part of an endocytosis entry pathway [46]. HCV E2 envelope glycoprotein has been shown to undergo conformational changes as a result of a change in pH [10]. Keck et al. have also shown that a neutralizing HMAb, CBH-5, cross-competes to a moderate degree with a related group of non-neutralizing HMAbs, designated as domain A [7]. One hypothesis to explain these results is that when a domain A HMAb binds, it induces a conformational change in the E2 protein. To explore this possibility and to obtain more structural information about the E2 glycoprotein, a series of experiments designed to probe potential conformational changes in E2 were undertaken.
Experimental approach
In the present study, critical residues in the E2 protein that affect and/or are affected by binding of HMAbs were identified. Limited proteolysis and differential chemical modification combined with mass spectrometric product elucidation was used to identify residues either directly involved in the interaction between the antibody CBH-5 and E2 and/or whose reactivity is affected by the presence of the antibody, either by steric hindrance or by a conformational change. In the differential modification approach a specific reagent is used to modify solvent-accessible residues which can provide structural information by determining which residues are more reactive and, thus, on the surface of the molecule [22-25]. Therefore, these experiments are a valuable method for determining critical amino acids involved in antibody binding and/or to modify residues whose surface accessibility (and, thus, reactivity) is affected by interaction with the mAb.
A complementary set of alanine-scanning mutagenesis experiments was performed on the critical regions identified by mass spectrometry. This mutagenesis approach identified residues for which a mutation to alanine affects antibody binding either by affecting E2 residues involved in HMAb recognition or by causing a change in E2 conformation that inhibits subsequent antibody interaction. That is, the alanine scanning experiments uncover effects occurring prior to and affecting antibody binding, while the MS experiments uncover effects subsequent to antibody binding.
Changes in conformation due to antibody binding
The mass spectrometric analyses indicated that peptides (462-500) and (615-628) interact strongly with the CBH-5 HMAb. Interestingly, these peptides overlap the predicted E1/E2 dimerization domains [47]. The CHD arginine modification experiments furthermore revealed three arginine residues that showed decreased abundances when the protein was bound to the antibody: Arg587, 630 and 651 located near domain A which is required for recognition by all mAbs. Moreover, our results indicate that a significant conformational change occurs upon CBH-5 binding, strongly affecting the region 579-644 whose presence is required for recognition by all of the antibodies as previously determined [9]. The alanine scanning mutagenesis data indicate that the following residues strongly affect E2/CBH-5 binding: Cys494, Val497, His617, Tyr618, Pro619, Thr621 and Phe624. In the E2 homology model described by Yagnik et al, the locations of these amino acids that are critical for maintaining the native structure of the E2 protein (as determined by alanine scanning mutagenesis) are found to encompass two different regions of the molecule: one in the N-terminal part of the molecule, near the hyper variable region 2 (HVR2) and a second fragment located in the middle of the E2 glycoprotein sequence, in the previously defined domain B [9, 10]. Furthermore, previous epitope mapping studies on HCV showed that the mAb D32.10 produced by immunizing mice with an HCV-enriched pellet binds the HCV particles derived from serum of different type HCV 1a- and HCV 1b-infected patients [48]. Moreover, this mAb has been shown to recognize both HCV envelope proteins E1 and E2. The epitope recognized by D32.10 on E2 determined using a phage display peptide library showed that the following regions in E2 are involved in binding the antibody: (480-494) and (613-621). The mass spectrometric results obtained using the CBH-5 HMAb also indicate interaction with similar regions of the protein, (462-500) and (615-628). Overall, these results show the critical amino acids which were recognized by antibodies against all three domains of the HCV E2 protein. In addition, our data indicate two distinct regions that are important in maintaining the native structure of the E2 protein, which are also similar to the regions involved in/or affected by CBH-5 HMAb binding. Furthermore, these results suggest that binding of CBH-5 HMAb to native E2 induces a conformational change in the protein structure.
Changes in conformation induced by alanine scanning mutagenesis
In the alanine scanning mutagenesis experiments, the structure of the protein is altered prior to the binding of the antibody, so the possibility of a conformational change of the antigen that may affect binding of similar antibodies directed against different E2 domains cannot be excluded. These experiments indicated that structural alterations of E2 protein result in partially competitive binding of antibodies directed to different E2 domains. Present data indicate that the interaction of the antibody with the antigen triggers conformational changes in the viral E2 envelope protein. Several studies have indicated that alanine mutations may interfere with antibody recognition and binding. Zimbwa et al., have shown that polymorphism within known antigenic sites results in loss of immune recognition in an HIV type 1 antigen processing mutant [49]. Furthermore, amino acid substitution can enable immune escape by interfering with the processing of the optimal peptide antigen recognized by a specific antibody. Cao et al., indicated that mutation of some conserved amino acids in HIV gp41 leads to changes in structure and function of the protein [50]. A single amino acid mutation in the viral envelope glycoprotein was reported to abolish virus infectivity in CD4+ T lymphocytes, as well as to significantly decrease the stability of the mutant protein compared with the wt virus [51]. Our data suggest that, upon alanine mutation, loss in binding is due to a conformational change which affects antibody recognition as well as correct folding of the protein. However, these data indicate that the loss in binding is due to both removal of contact residues as well as to a major conformational change of the protein upon alanine mutation.
Glycosylation and conformational changes
In the early secretory pathway, the glycans play a role in protein folding and certain sorting events. Previous studies showed that viral envelope proteins usually contain N-linked glycans that may play a major role in their folding, entry functions or in modulating the immune response [46, 52, 53]. In a recent paper, Helle et al. describes the neutralizing activity of anti-HCV antibodies, and they found that this neutralization is modulated by specific glycans on the E2 protein [54]. In addition, they observed that HCVpp containing non-glycosylation mutants at positions N476 and N540 were more sensitive to neutralization by all the mAb tested, among them the CBH-5 HMAb. A decreased level of inhibition of the wt N476 glycosylation site compared with the non-glycosylated alanine mutants might be explained by a local conformational change induced by the alanine mutations. This observation seems in contradiction with the mass spectrometric results which indicated that the N476 glycosylation site might be involved in the antibody binding. However, the mass spectrometric analyses were performed with intact, glycosylated wt E2 protein, whereas Helle et al. used single glycosylation mutants to test their neutralizing effect against different antibodies [54]. Thus, alanine mutation of a single glycosylation site has no apparent effect on the ability of the antibody to bind E2 protein, in comparison with the intact wt glycosylated E2 protein which most probably has a conformation that is required for antibody-antigen recognition. Furthermore, the mutation-binding data suggests that there is a conformational change induced by the single site mutation, which reduces but does not eliminate the affinity of neutralizing antibodies for the E2 glycoprotein. Moreover, initial removal of the glycans of E2 glycoprotein before binding the protein to the antibody results in a significant change in conformation and abolishes CBH-5 HMAb binding.
Based on Yagnik’s homology model, it is hypothesized that the glycans on E2 define the extracellular surface of the molecule [10]. Consequently, the critical residues that were determined herein should be located on the same side of the protein as the glycan molecules. Furthermore, changes that were observed in surface reactivity of these residues not located on the same side of the E2 molecule in the homology model might be due to large conformational changes of E2 protein rather than protection by the HMAb. Moreover, based on the homology model, the critical residues were found in the same region where CD81 binding occurs [55], as well as in the region of the E1/E2 association sites, regions which are physically located in close proximity in the model. It was shown that these regions are exposed in the E2 homology model and they contain a high number of charged polar residues [10]. Both these regions contain the “WHY” motif which has been shown to be involved in the heteromeric association between HCV-E1 and E2 proteins [47]. Mutations of residues in these regions, especially cysteine residues, most likely trigger a significant conformational change. One possible explanation for our results is that the E2 protein has two (or more) conformationally stable states with a relatively low energy barrier between them. Disruption of key structural residues (alanine scanning mutagenesis), increased steric interactions (antibody binding experiments) or changes in electrostatic interactions (by pH changes) can then lead to collapse of one conformation into an alternate low energy conformation.
Summary
The data presented in this manuscript suggest that, upon antibody binding, there is a significant conformational change in E2. Moreover, by coupling mass spectrometry-based approaches with mutagenesis techniques, critical residues on E2 protein that are recognized by antibodies against all three domains were identified, and two distinct regions that are important to maintain a native structure of the protein have also been determined. More detailed conformational studies on the E2 glycoprotein and the epitope recognized by the CBH-5 HMAb are currently underway in our laboratories. In conclusion, the information presented in this paper provides new insights to the unsolved structure of the E2 glycoprotein, and these results should lead to a better understanding of the hepatitis C virus E2 envelope glycoprotein function and antibody-antigen recognition structure.
Acknowledgments
this research was supported in part by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences, and by National Institutes of Health grants HL079381 and AI47355 to SKHF. The authors want to thank Dr. Leesa Deterding for informative discussions throughout the project, Dr. Arvind Patel for supplying the AP33 antibody and Dr. Ta-Kai Li for technical assistance. The authors also want to thank Dr. James G. Smedley, III and Dr. Allison Schorzman for critical review of this manuscript.
Abbreviations
- GNA
galanthus nivalis antigen
- AP
alkaline phospathase
- FACS
fluorescent-activated cell sorting
- FITC
fluorescein isothiocyante
- FCS
fetal calf serum
- BS3
bis(sulfosuccinimidyl)suberate
- GnHCl
guanidine hydrochloride
- PBS
phosphate buffered saline
- CRC
compact reaction columns
- MALDI
matrix assisted laser desorption/ ionization
- ESI
electrospray ionization
- TOF
time of flight
- ESI-MS/MS
electrospray-tandem mass spectrometry
- LC/MS/MS
liquid chromatography-tandem mass spectrometry
- HPLC
high performance liquid chromatography
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rehermann B, Nascimbeni M. Immunology of hepatitis B virus and hepatitis C virus infection. Nat Rev Immunol. 2005;5:215–229. doi: 10.1038/nri1573. [DOI] [PubMed] [Google Scholar]
- 2.Simmonds P. Genetic diversity and evolution of hepatitis C virus--15 years on. The Journal of general virology. 2004;85:3173–3188. doi: 10.1099/vir.0.80401-0. [DOI] [PubMed] [Google Scholar]
- 3.Goffard A, Dubuisson J. Glycosylation of hepatitis C virus envelope proteins. Biochimie. 2003;85:295–301. doi: 10.1016/s0300-9084(03)00004-x. [DOI] [PubMed] [Google Scholar]
- 4.Keck ZY, Sung VM, Perkins S, Rowe J, Paul S, Liang TJ, Lai MM, Foung SK. Human monoclonal antibody to hepatitis C virus E1 glycoprotein that blocks virus attachment and viral infectivity. Journal of virology. 2004;78:7257–7263. doi: 10.1128/JVI.78.13.7257-7263.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gish RG, Qian KP, Quan S, Xu YL, Pike I, Polito A, DiNello R, Lau JY. Concordance between hepatitis C virus serotyping assays. J Viral Hepat. 1997;4:421–422. doi: 10.1046/j.1365-2893.1997.00069.x. [DOI] [PubMed] [Google Scholar]
- 6.Keck ZY, Op De Beeck A, Hadlock KG, Xia J, Li TK, Dubuisson J, Foung SK. Hepatitis C virus E2 has three immunogenic domains containing conformational epitopes with distinct properties and biological functions. Journal of virology. 2004;78:9224–9232. doi: 10.1128/JVI.78.17.9224-9232.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keck ZY, Li TK, Xia J, Bartosch B, Cosset FL, Dubuisson J, Foung SK. Analysis of a highly flexible conformational immunogenic domain a in hepatitis C virus E2. Journal of virology. 2005;79:13199–13208. doi: 10.1128/JVI.79.21.13199-13208.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clayton RF, Owsianka A, Aitken J, Graham S, Bhella D, Patel AH. Analysis of antigenicity and topology of E2 glycoprotein present on recombinant hepatitis C virus-like particles. Journal of virology. 2002;76:7672–7682. doi: 10.1128/JVI.76.15.7672-7682.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keck ZY, Xia J, Cai Z, Li TK, Owsianka AM, Patel AH, Luo G, Foung SK. Immunogenic and functional organization of hepatitis C virus (HCV) glycoprotein E2 on infectious HCV virions. Journal of virology. 2007;81:1043–1047. doi: 10.1128/JVI.01710-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yagnik AT, Lahm A, Meola A, Roccasecca RM, Ercole BB, Nicosia A, Tramontano A. A model for the hepatitis C virus envelope glycoprotein E2. Proteins. 2000;40:355–366. doi: 10.1002/1097-0134(20000815)40:3<355::aid-prot20>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 11.Allander T, Drakenberg K, Beyene A, Rosa D, Abrignani S, Houghton M, Widell A, Grillner L, Persson MA. Recombinant human monoclonal antibodies against different conformational epitopes of the E2 envelope glycoprotein of hepatitis C virus that inhibit its interaction with CD81. The Journal of general virology. 2000;81:2451–2459. doi: 10.1099/0022-1317-81-10-2451. [DOI] [PubMed] [Google Scholar]
- 12.Habersetzer F, Fournillier A, Dubuisson J, Rosa D, Abrignani S, Wychowski C, Nakano I, Trepo C, Desgranges C, Inchauspe G. Characterization of human monoclonal antibodies specific to the hepatitis C virus glycoprotein E2 with in vitro binding neutralization properties. Virology. 1998;249:32–41. doi: 10.1006/viro.1998.9202. [DOI] [PubMed] [Google Scholar]
- 13.Farci P, Shimoda A, Coiana A, Diaz G, Peddis G, Melpolder JC, Strazzera A, Chien DY, Munoz SJ, Balestrieri A, Purcell RH, Alter HJ. Science. Vol. 288. New York, N.Y: 2000. The outcome of acute hepatitis C predicted by the evolution of the viral quasispecies; pp. 339–344. [DOI] [PubMed] [Google Scholar]
- 14.Erickson AL, Kimura Y, Igarashi S, Eichelberger J, Houghton M, Sidney J, McKinney D, Sette A, Hughes AL, Walker CM. The outcome of hepatitis C virus infection is predicted by escape mutations in epitopes targeted by cytotoxic T lymphocytes. Immunity. 2001;15:883–895. doi: 10.1016/s1074-7613(01)00245-x. [DOI] [PubMed] [Google Scholar]
- 15.Shoukry NH, Cawthon AG, Walker CM. Cell-mediated immunity and the outcome of hepatitis C virus infection. Annual review of microbiology. 2004;58:391–424. doi: 10.1146/annurev.micro.58.030603.123836. [DOI] [PubMed] [Google Scholar]
- 16.Bowen DG, Walker CM. Adaptive immune responses in acute and chronic hepatitis C virus infection. Nature. 2005;436:946–952. doi: 10.1038/nature04079. [DOI] [PubMed] [Google Scholar]
- 17.Kimura Y, Gushima T, Rawale S, Kaumaya P, Walker CM. Escape mutations alter proteasome processing of major histocompatibility complex class I-restricted epitopes in persistent hepatitis C virus infection. Journal of virology. 2005;79:4870–4876. doi: 10.1128/JVI.79.8.4870-4876.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wyatt R, Kwong PD, Desjardins E, Sweet RW, Robinson J, Hendrickson WA, Sodroski JG. The antigenic structure of the HIV gp120 envelope glycoprotein. Nature. 1998;393:705–711. doi: 10.1038/31514. [DOI] [PubMed] [Google Scholar]
- 19.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Science. Vol. 246. New York, N.Y: 1989. Electrospray ionization for mass spectrometry of large biomolecules; pp. 64–71. [DOI] [PubMed] [Google Scholar]
- 20.Smith RD, Loo JA, Edmonds CG, Barinaga CJ, Udseth HR. New developments in biochemical mass spectrometry: electrospray ionization. Anal Chem. 1990;62:882–899. doi: 10.1021/ac00208a002. [DOI] [PubMed] [Google Scholar]
- 21.Hillenkamp F, Karas M, Beavis RC, Chait BT. Matrix-assisted laser desorption/ionization mass spectrometry of biopolymers. Anal Chem. 1991;63:1193A–1203A. doi: 10.1021/ac00024a002. [DOI] [PubMed] [Google Scholar]
- 22.Steiner RF, Albaugh S, Fenselau C, Murphy C, Vestling M. A mass spectrometry method for mapping the interface topography of interacting proteins, illustrated by the melittin-calmodulin system. Anal Biochem. 1991;196:120–125. doi: 10.1016/0003-2697(91)90127-f. [DOI] [PubMed] [Google Scholar]
- 23.Suckau D, Mak M, Przybylski M. Protein surface topology-probing by selective chemical modification and mass spectrometric peptide mapping. Proc Natl Acad Sci U S A. 1992;89:5630–5634. doi: 10.1073/pnas.89.12.5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Young MM, Tang N, Hempel JC, Oshiro CM, Taylor EW, Kuntz ID, Gibson BW, Dollinger G. High throughput protein fold identification by using experimental constraints derived from intramolecular cross-links and mass spectrometry. Proc Natl Acad Sci U S A. 2000;97:5802–5806. doi: 10.1073/pnas.090099097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sinz A. Chemical cross-linking and mass spectrometry to map three-dimensional protein structures and protein-protein interactions. Mass Spectrom Rev. 2006;25:663–682. doi: 10.1002/mas.20082. [DOI] [PubMed] [Google Scholar]
- 26.Glocker MO, Borchers C, Fiedler W, Suckau D, Przybylski M. Molecular characterization of surface topology in protein tertiary structures by amino-acylation and mass spectrometric peptide mapping. Bioconjug Chem. 1994;5:583–590. doi: 10.1021/bc00030a014. [DOI] [PubMed] [Google Scholar]
- 27.Suckau D, Kohl J, Karwath G, Schneider K, Casaretto M, Bitter-Suermann D, Przybylski M. Molecular epitope identification by limited proteolysis of an immobilized antigen-antibody complex and mass spectrometric peptide mapping. Proc Natl Acad Sci U S A. 1990;87:9848–9852. doi: 10.1073/pnas.87.24.9848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zwick MB, Jensen R, Church S, Wang M, Stiegler G, Kunert R, Katinger H, Burton DR. Anti-human immunodeficiency virus type 1 (HIV-1) antibodies 2F5 and 4E10 require surprisingly few crucial residues in the membrane-proximal external region of glycoprotein gp41 to neutralize HIV-1. Journal of virology. 2005;79:1252–1261. doi: 10.1128/JVI.79.2.1252-1261.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Williams JG, Tomer KB, Hioe CE, Zolla-Pazner S, Norris PJ. The antigenic determinants on HIV p24 for CD4+ T cell inhibiting antibodies as determined by limited proteolysis, chemical modification, and mass spectrometry. J Am Soc Mass Spectrom. 2006;17:1560–1569. doi: 10.1016/j.jasms.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 30.Parker CE, Papac DI, Trojak SK, Tomer KB. Epitope mapping by mass spectrometry: determination of an epitope on HIV-1 IIIB p26 recognized by a monoclonal antibody. J Immunol. 1996;157:198–206. [PubMed] [Google Scholar]
- 31.Parker CE, Tomer KB. MALDI/MS-based epitope mapping of antigens bound to immobilized antibodies. Mol Biotechnol. 2002;20:49–62. doi: 10.1385/MB:20:1:049. [DOI] [PubMed] [Google Scholar]
- 32.Hadlock KG, Lanford RE, Perkins S, Rowe J, Yang Q, Levy S, Pileri P, Abrignani S, Foung SK. Human monoclonal antibodies that inhibit binding of hepatitis C virus E2 protein to CD81 and recognize conserved conformational epitopes. Journal of virology. 2000;74:10407–10416. doi: 10.1128/jvi.74.22.10407-10416.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peter JF, Tomer KB. A general strategy for epitope mapping by direct MALDI-TOF mass spectrometry using secondary antibodies and cross-linking. Anal Chem. 2001;73:4012–4019. doi: 10.1021/ac010258n. [DOI] [PubMed] [Google Scholar]
- 34.Hochleitner EO, Gorny MK, Zolla-Pazner S, Tomer KB. Mass spectrometric characterization of a discontinuous epitope of the HIV envelope protein HIV-gp120 recognized by the human monoclonal antibody 1331A. J Immunol. 2000;164:4156–4161. doi: 10.4049/jimmunol.164.8.4156. [DOI] [PubMed] [Google Scholar]
- 35.Op De Beeck A, Voisset C, Bartosch B, Ciczora Y, Cocquerel L, Keck Z, Foung S, Cosset FL, Dubuisson J. Characterization of functional hepatitis C virus envelope glycoproteins. Journal of virology. 2004;78:2994–3002. doi: 10.1128/JVI.78.6.2994-3002.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuiken C, Combet C, Bukh J, Shin IT, Deleage G, Mizokami M, Richardson R, Sablon E, Yusim K, Pawlotsky JM, Simmonds P. A comprehensive system for consistent numbering of HCV sequences, proteins and epitopes. Hepatology. 2006;44:1355–1361. doi: 10.1002/hep.21377. [DOI] [PubMed] [Google Scholar]
- 37.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning. 2. Cold Spring Harbor Laboratory Press; Cold Spring Harbor NY: 1989. [Google Scholar]
- 38.Peri S, Steen H, Pandey A. GPMAW--a software tool for analyzing proteins and peptides. Trends Biochem Sci. 2001;26:687–689. doi: 10.1016/s0968-0004(01)01954-5. [DOI] [PubMed] [Google Scholar]
- 39.Duvet S, Cocquerel L, Pillez A, Cacan R, Verbert A, Moradpour D, Wychowski C, Dubuisson J. Hepatitis C virus glycoprotein complex localization in the endoplasmic reticulum involves a determinant for retention and not retrieval. J Biol Chem. 1998;273:32088–32095. doi: 10.1074/jbc.273.48.32088. [DOI] [PubMed] [Google Scholar]
- 40.Roepstorff P, Fohlman J. Proposal for a common nomenclature for sequence ions in mass spectra of peptides. Biomed Mass Spectrom. 1984;11:601. doi: 10.1002/bms.1200111109. [DOI] [PubMed] [Google Scholar]
- 41.Bartosch B, Dubuisson J, Cosset FL. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. The Journal of experimental medicine. 2003;197:633–642. doi: 10.1084/jem.20021756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blok J, McWilliam SM, Butler HC, Gibbs AJ, Weiller G, Herring BL, Hemsley AC, Aaskov JG, Yoksan S, Bhamarapravati N. Comparison of a dengue-2 virus and its candidate vaccine derivative: sequence relationships with the flaviviruses and other viruses. Virology. 1992;187:573–590. doi: 10.1016/0042-6822(92)90460-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stiasny K, Kiermayr S, Heinz FX. Entry functions and antigenic structure of flavivirus envelope proteins. Novartis Foundation symposium. 2006;277:57–65. discussion 65-73, 251-253. [PubMed] [Google Scholar]
- 44.Rey FA, Heinz FX, Mandl C, Kunz C, Harrison SC. The envelope glycoprotein from tick-borne encephalitis virus at 2 A resolution. Nature. 1995;375:291–298. doi: 10.1038/375291a0. [DOI] [PubMed] [Google Scholar]
- 45.Garry RF, Dash S. Proteomics computational analyses suggest that hepatitis C virus E1 and pestivirus E2 envelope glycoproteins are truncated class II fusion proteins. Virology. 2003;307:255–265. doi: 10.1016/s0042-6822(02)00065-x. [DOI] [PubMed] [Google Scholar]
- 46.Hebert DN, Zhang JX, Chen W, Foellmer B, Helenius A. The number and location of glycans on influenza hemagglutinin determine folding and association with calnexin and calreticulin. J Cell Biol. 1997;139:613–623. doi: 10.1083/jcb.139.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yi M, Nakamoto Y, Kaneko S, Yamashita T, Murakami S. Delineation of regions important for heteromeric association of hepatitis C virus E1 and E2. Virology. 1997;231:119–129. doi: 10.1006/viro.1997.8516. [DOI] [PubMed] [Google Scholar]
- 48.Petit MA, Jolivet-Reynaud C, Peronnet E, Michal Y, Trepo C. Mapping of a conformational epitope shared between E1 and E2 on the serum-derived human hepatitis C virus envelope. J Biol Chem. 2003;278:44385–44392. doi: 10.1074/jbc.M304047200. [DOI] [PubMed] [Google Scholar]
- 49.Zimbwa P, Milicic A, Frater J, Scriba TJ, Willis A, Goulder PJ, Pillay T, Gunthard H, Weber JN, Zhang HT, Phillips RE. Precise identification of a human immunodeficiency virus type 1 antigen processing mutant. Journal of virology. 2007;81:2031–2038. doi: 10.1128/JVI.00968-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cao J, Bergeron L, Helseth E, Thali M, Repke H, Sodroski J. Effects of amino acid changes in the extracellular domain of the human immunodeficiency virus type 1 gp41 envelope glycoprotein. Journal of virology. 1993;67:2747–2755. doi: 10.1128/jvi.67.5.2747-2755.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee YM, Tang XB, Cimakasky LM, Hildreth JE, Yu XF. Mutations in the matrix protein of human immunodeficiency virus type 1 inhibit surface expression and virion incorporation of viral envelope glycoproteins in CD4+ T lymphocytes. Journal of virology. 1997;71:1443–1452. doi: 10.1128/jvi.71.2.1443-1452.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wei X, Decker JM, Wang S, Hui H, Kappes JC, Wu X, Salazar-Gonzalez JF, Salazar MG, Kilby JM, Saag MS, Komarova NL, Nowak MA, Hahn BH, Kwong PD, Shaw GM. Antibody neutralization and escape by HIV-1. Nature. 2003;422:307–312. doi: 10.1038/nature01470. [DOI] [PubMed] [Google Scholar]
- 53.Ohuchi M, Ohuchi R, Feldmann A, Klenk HD. Regulation of receptor binding affinity of influenza virus hemagglutinin by its carbohydrate moiety. Journal of virology. 1997;71:8377–8384. doi: 10.1128/jvi.71.11.8377-8384.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Helle F, Goffard A, Morel V, Duverlie G, McKeating J, Keck ZY, Foung S, Penin F, Dubuisson J, Voisset C. The neutralizing activity of anti-HCV antibodies is modulated by specific glycans on the E2 envelope protein. Journal of virology. 2007 doi: 10.1128/JVI.00127-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Owsianka AM, Timms JM, Tarr AW, Brown RJ, Hickling TP, Szwejk A, Bienkowska-Szewczyk K, Thomson BJ, Patel AH, Ball JK. Identification of conserved residues in the E2 envelope glycoprotein of the hepatitis C virus that are critical for CD81 binding. Journal of virology. 2006;80:8695–8704. doi: 10.1128/JVI.00271-06. [DOI] [PMC free article] [PubMed] [Google Scholar]