

Abstract
The effect of voluntary hyperventilation-induced hypocapnic alkalosis (RALK) on pulmonary O2 uptake (V˙o2) kinetics and muscle deoxygenation was examined in young male adults (n = 8) during moderate-intensity exercise. Subjects performed five repetitions of a step-transition in work rate from 20 W cycling to a work rate corresponding to 90% of the estimated lactate threshold during control (CON; 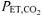 , ∼40 mmHg) and during hyperventilation (RALK;
, ∼40 mmHg) and during hyperventilation (RALK; 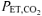 , ∼20 mmHg). V˙o2 was measured breath-by-breath and relative concentration changes in muscle deoxy- (ΔHHb), oxy- (ΔO2Hb) and total (ΔHbtot) haemoglobin were measured continuously using near-infrared (NIR) spectroscopy (Hamamatsu, NIRO 300). The time constant for the fundamental, phase 2, V˙o2 response (τV˙o2) was greater (P < 0.05) in RALK (48 ± 11 s) than CON (31 ± 9 s), while τHHb was similar between conditions (RALK, 12 ± 4 s; CON, 11 ± 4 s). The ΔHbtot was lower (P < 0.05) in RALK than CON, prior to (RALK, −3 ± 5 μmol l−1; CON, −1 ± 4 μmol l−1) and at the end (RALK, 1 ± 6 μmol l−1; CON, 5 ± 5 μmol l−1) of moderate-intensity exercise. Although slower adaptation of V˙o2 during RALK may be related to an attenuated activation of PDH (and other enzymes) and provision of oxidizable substrate to the mitochondria (i.e. metabolic inertia), the present findings also suggest a role for a reduction in local muscle perfusion and O2 delivery.
, ∼20 mmHg). V˙o2 was measured breath-by-breath and relative concentration changes in muscle deoxy- (ΔHHb), oxy- (ΔO2Hb) and total (ΔHbtot) haemoglobin were measured continuously using near-infrared (NIR) spectroscopy (Hamamatsu, NIRO 300). The time constant for the fundamental, phase 2, V˙o2 response (τV˙o2) was greater (P < 0.05) in RALK (48 ± 11 s) than CON (31 ± 9 s), while τHHb was similar between conditions (RALK, 12 ± 4 s; CON, 11 ± 4 s). The ΔHbtot was lower (P < 0.05) in RALK than CON, prior to (RALK, −3 ± 5 μmol l−1; CON, −1 ± 4 μmol l−1) and at the end (RALK, 1 ± 6 μmol l−1; CON, 5 ± 5 μmol l−1) of moderate-intensity exercise. Although slower adaptation of V˙o2 during RALK may be related to an attenuated activation of PDH (and other enzymes) and provision of oxidizable substrate to the mitochondria (i.e. metabolic inertia), the present findings also suggest a role for a reduction in local muscle perfusion and O2 delivery.
Oxidative phosphorylation and muscle O2 consumption adjust in an exponential manner during the transition to moderate-intensity exercise (Whipp & Mahler, 1980; Grassi et al. 1996). The provision of ADP to the muscle mitochondria is likely to exert control over the rate of this adaptation; however, additional mechanisms have been suggested to limit the rate of adjustment. It has been suggested that the convective and/or diffusive delivery of O2 to the muscle may be limiting at the onset of exercise (Hughson et al. 1996; Tschakovsky & Hughson, 1999). Alternatively, activation of specific enzymes limiting the provision of oxidative substrates to the mitochondrial tricarboxylic acid (TCA) cycle and electron transport chain (ETC) have been proposed to determine the initial rate of mitochondrial O2 utilization (Timmons et al. 1998; Howlett et al. 1999; Grassi, 2001; Hogan, 2001; Rossiter et al. 2003).
LeBlanc et al. (2002) reported that a 20 min period of voluntary hyperventilation-induced hypocapnic (i.e. respiratory) alkalosis (RALK) resulted in slowed activation of the mitochondrial pyruvate dehydrogenase (PDH) complex during the first minute of cycling exercise at 55% V˙o2,max. As PDH controls the entry of carbohydrate-derived acetyl CoA into the TCA cycle and thus the provision of reducing equivalents to the ETC (Spriet & Heigenhauser, 2002), slowed activation of PDH might be expected to result in a slower activation of mitochondrial oxidative phosphorylation and require a greater substrate-level phosphorylation to accommodate the instantaneous rise in ATP requirement early in the transition to exercise. This was demonstrated recently (Forbes et al. 2007), when we observed that RALK was accompanied by a slower time course and greater amplitude of muscle PCr breakdown during the transition to moderate-intensity plantar-flexion exercise; however, it remains to be determined whether RALK is also associated with a slowed activation of muscle O2 consumption.
Investigations of the effect of RALK on pulmonary O2 uptake (V˙o2) kinetics are limited to only two studies, with each reporting different findings. Ward et al. (1983) reported that pulmonary V˙o2 kinetics (as assessed by the half-time (t1/2) of the V˙o2 response) were similar when the exercise transition was preceded by normal breathing or by a period of hyperventilation. In this study (Ward et al. 1983), the hyperventilation manoeuvre lasted only 9 min and was stopped 15–20 s prior to the onset of exercise, with a normal breathing pattern adopted during the remaining rest–exercise transition. Alternatively, Hayashi et al. (1999) reported that pulmonary V˙o2 kinetics were slowed in RALK compared to either a control condition (with normal breathing pattern) or a normocapnic-hyperventilatory condition (with hyperventilation and CO2 breathing to prevent the hypocapnia). In this study (Hayashi et al. 1999), only a very brief period (i.e. 2 min) of hyperventilation preceded the start of exercise; however, the hyperventilation manoeuvre was maintained throughout the exercise transition. It is therefore unclear whether the brief hyperventilatory manoeuvre manifested a sufficient respiratory alkalosis to alter intramuscular pH and PDH activity (LeBlanc et al. 2002; Forbes et al. 2007). The slowing of V˙o2 kinetics in this study was attributed to impaired diffusion of O2 into muscle, consequent to a hypocapnia-induced leftward-shift of the oxy-haemoglobin dissociation curve and a greater O2 affinity for haemoglobin (Hayashi et al. 1999).
To resolve these issues and gain a better insight into the role of RALK in determining the rate of adaptation of pulmonary V˙o2 (and muscle O2 consumption), we used a more prolonged accommodation period of voluntary hyperventilation (similar to that used by LeBlanc et al. 2002) to lower body CO2 stores and to allow sufficient time for these stores to equilibrate to the new level, and maintained the hyperventilation manoeuvre throughout the exercise transition. Also, we used near-infrared spectroscopy (NIRS) to measure changes in concentration of muscle deoxy- (ΔHHb), oxy- (ΔO2Hb) and total (ΔHbtot) haemoglobin during the pre-exercise accommodation period and throughout the exercise transition (DeLorey et al. 2003; Grassi et al. 2003). The kinetics of the NIRS-derived ΔHHb signal reflect muscle O2 extraction and therefore depend on the balance between local muscle O2 delivery and O2 utilization. As phase 2 pulmonary V˙o2 reflects muscle O2 utilization (Grassi et al. 1996), it has been suggested that consideration of pulmonary V˙o2 and NIRS-derived oxy- and deoxygenation kinetics together would allow inferences to be drawn regarding the rate of adaptation of local muscle O2 utilization and microvascular perfusion (DeLorey et al. 2003; Ferreira et al. 2005) during the transition to exercise.
The purpose of the present investigation was to examine the effects of a hyperventilation-induced hypocapnic alkalosis (RALK) on pulmonary V˙o2 kinetics and muscle oxy- and deoxygenation (as determined by NIRS) during the transition from light- to moderate-intensity exercise. We hypothesized that during the transition to moderate-intensity exercise in RALK, pulmonary V˙o2 kinetics (reflecting muscle O2 consumption) would be slowed and thus attenuate the rate of deoxygenation within the working muscle.
Methods
Subjects
Eight healthy male volunteers (age, 24 ± 2 year; body mass, 82 ± 7 kg; V˙o2 peak, 3.77 ± 0.37 l min−1) participated in this study. Subjects were informed of the risks associated with the experimental protocol and provided written consent before participation in the study. This study was approved by The University of Western Ontario Review Board for Health Sciences Research Involving Human Subjects in accordance with the Declaration of Helsinki.
Exercise protocol
Subjects reported to the laboratory on 12 separate occasions. Subjects were asked to consume only a light meal and to refrain from caffeinated drinks and heavy-intensity exercise within 12 h prior to the testing sessions. During their initial visit, subjects familiarized themselves with the testing apparatus and completed an incremental ramp test (20 W min−1) to their limit of tolerance on an electromagnetically braked cycle ergometer (model H-300-R, Lode) to determine their estimated lactate threshold (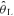 ) and peak O2 uptake (V˙o2,peak). V˙o2,peak was determined from the average V˙o2 obtained during the last 20 s of the incremental ramp test. The
) and peak O2 uptake (V˙o2,peak). V˙o2,peak was determined from the average V˙o2 obtained during the last 20 s of the incremental ramp test. The 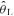 was determined by visual inspection and was defined as the V˙o2 at which CO2 output (V˙CO2) began to increase out of proportion relative to V˙o2 and the point at which the ventilatory equivalent for V˙o2 (V˙E/V˙o2) and end-tidal O2 (
was determined by visual inspection and was defined as the V˙o2 at which CO2 output (V˙CO2) began to increase out of proportion relative to V˙o2 and the point at which the ventilatory equivalent for V˙o2 (V˙E/V˙o2) and end-tidal O2 (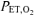 ) began to increase with no systematic increase in the ventilatory equivalent for V˙CO2 (V˙E/V˙CO2) or fall in end-tidal CO2 (
) began to increase with no systematic increase in the ventilatory equivalent for V˙CO2 (V˙E/V˙CO2) or fall in end-tidal CO2 (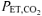 ). During an additional preliminary visit to the laboratory, subjects practiced the respiratory manoeuvres required to lower
). During an additional preliminary visit to the laboratory, subjects practiced the respiratory manoeuvres required to lower 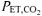 from a normal level of ∼35–40 mmHg to ∼18–20 mmHg (LeBlanc et al. 2002). Subjects maintained the required
from a normal level of ∼35–40 mmHg to ∼18–20 mmHg (LeBlanc et al. 2002). Subjects maintained the required 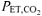 by monitoring their expired CO2 values, which were displayed continuously on a computer monitor (PowerLab Chart v4.2, ADInstruments Inc., Colorado Springs, CO, USA).
by monitoring their expired CO2 values, which were displayed continuously on a computer monitor (PowerLab Chart v4.2, ADInstruments Inc., Colorado Springs, CO, USA).
The protocol began with 5 min of normal breathing with gas exchange and muscle oxygenation being monitored continuously while the subject remained seated on the cycle ergometer (Pre-Accommodation). This period was followed by a 20 min accommodation period during which subjects either continued to breath normally (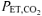 , ∼35–40 mmHg; CON) or they hyperventilated to achieve a target
, ∼35–40 mmHg; CON) or they hyperventilated to achieve a target 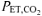 of ∼18–20 mmHg (RALK). During RALK, the lower
of ∼18–20 mmHg (RALK). During RALK, the lower 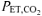 (∼18–20 mmHg) was maintained throughout the entire accommodation, exercise, and recovery periods.
(∼18–20 mmHg) was maintained throughout the entire accommodation, exercise, and recovery periods.
The CON and RALK conditions were presented in a randomized fashion. Subjects performed step-transitions in work rate from a baseline of 20 W cycling to an intensity corresponding to ∼90%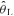 . Each step transition was 6 min in duration and was preceded and followed by 6 min of cycling at 20 W. Step changes in work rates were made instantaneously, without prior warning to the subject. The exercise protocol was repeated on five separate occasions for each of the CON and RALK conditions.
. Each step transition was 6 min in duration and was preceded and followed by 6 min of cycling at 20 W. Step changes in work rates were made instantaneously, without prior warning to the subject. The exercise protocol was repeated on five separate occasions for each of the CON and RALK conditions.
Materials
Inspired and expired airflows were measured using a low-resistance, low-dead-space (90 ml) bi-directional turbine and volume transducer (Alpha Technologies, Laguna Hills, CA, USA, VMM-110). The turbine and volume transducer were calibrated prior to each test with a syringe of known volume (3.0 l; Hans Rudolph, Kansas City, MO, USA). Respired gases were measured continuously at the mouth (1 ml s−1) and analysed for fractional concentrations of O2, CO2 and N2 by mass spectrometry (Innovision, AMIS 2000, Lindvedvej, Denmark). The mass spectrometer was calibrated against precision-analysed gases prior to each test. Analog signals from the mass spectrometer and turbine transducer were sampled at 50 Hz and stored on a computer for off-line breath-by-breath computations of V˙o2, V˙CO2, V˙E, and end-tidal 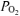 and
and 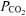 . Changes in gas concentrations were time-aligned with inspired and expired volumes by measuring the time delay for a square-wave bolus of gas to travel from the turbine transducer along a capillary of known length to the mass spectrometer system. Breath-by-breath alveolar gas exchange was calculated using the algorithms of Beaver et al. (1981).
. Changes in gas concentrations were time-aligned with inspired and expired volumes by measuring the time delay for a square-wave bolus of gas to travel from the turbine transducer along a capillary of known length to the mass spectrometer system. Breath-by-breath alveolar gas exchange was calculated using the algorithms of Beaver et al. (1981).
Heart rate was monitored by electrocardiogram (Life Pulse) using a three-lead arrangement. Data were recorded using PowerLab (Chart v4.2) on a separate computer.
Near-infrared (NIR) spectroscopy (Hamamatsu NIRO 300, Hamamatsu Photonics KK, Japan) was used to measure continuously the oxygenation status of the vastus lateralis muscle. The physical principles of tissue spectroscopy and the manner in which these are applied through the use of NIR spectroscopy instrumentation are described in detail by Elwell (1995). Briefly, four laser diodes produced NIR light at wavelengths of 775, 810, 850 and 910 nm. These wavelengths were pulsed in rapid succession and coupled to the tissue of interest by transmission through fibre optic bundles. Light was returned from the tissue through a separate fibre optic bundle to a highly sensitive photomultiplier tube and light intensity was measured. These values were used with known specific extinction coefficients and estimated optical path-lengths, to calculate changes in oxy- (ΔO2Hb), deoxy- (ΔHHb), and total (ΔHbtot) haemoglobin-myoglobin concentrations relative to resting, pre-accommodation baseline values. The tissue oxygenation index (TOI) was defined as (ΔO2Hb/ΔHbtot) × 100. Changes in light intensity were sampled and recorded every 500 ms (2 Hz). The raw attenuation signals were displayed and transferred to a computer for later analysis.
Prior to the start of the protocol, near-infrared (NIR) transmitting and receiving optodes were attached to the leg (see below) while subjects rested quietly on the cycle ergometer. NIR signals were allowed to stabilize for a period of at least 5 min, after which the NIR spectroscopy unit was initialized and the signals were set to ‘zero’.
Optodes were positioned on the vastus lateralis muscle of the right leg, midway between the lateral epicondyle and the greater trochanter of the femur (DeLorey et al. 2003). The optodes were housed in a black rubber holder designed to fix the optodes at a specific distance and ensure secure attachment to the skin surface. The holder was covered with a black pad and taped to the leg to reduce unwanted incoming visible light and to prevent the loss of NIR-transmitted light from the field of interrogation. The leg was wrapped with an elastic bandage to secure the position of the NIR probes on the leg while still allowing freedom of movement for cycling. The interoptode spacing was 5 cm. Changes in the concentration of O2Hb, HHb, and Hbtot from a ‘zero’ baseline determined at a time preceding the ‘pre-accommodation’ period, are reported as a delta (Δ) in units of μmol l−1.
During one trial in each condition, a percutaneous Teflon catheter (Angiocath, 21 gauge) was inserted into a dorsal hand vein for blood sampling. Arterialization of the venous blood was achieved by wrapping the forearm and hand in a heating pad with additional heat provided from a tungsten heating lamp. Blood samples were taken before and at the end of the 20 min accommodation period, during 20 W baseline cycling, and at specific times during the exercise on- and off-transitions. Arterialized-venous blood samples were drawn into 3 ml heparanized syringes, mixed and placed in an ice bath to be analysed shortly afterwards for plasma 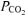 , pH, and [lactate−] (StatProfile 9 Plus blood gas-electrolyte analyser, Nova Biomedical Canada); the electrodes were calibrated before and during the analysis procedure. Plasma [H+] was calculated from the measured pH.
, pH, and [lactate−] (StatProfile 9 Plus blood gas-electrolyte analyser, Nova Biomedical Canada); the electrodes were calibrated before and during the analysis procedure. Plasma [H+] was calculated from the measured pH.
Data analysis
Gas exchange
V˙o2 data were filtered by removing aberrant data points that lay outside 4 standard deviations of the local mean. These points were justifiably removed because they did not conform to a Gaussian distribution as described by Lamarra et al. (1987) (see also, Whipp & Rossiter, 2005). The data for each transition then were linearly interpolated to 1 s intervals and time-aligned such that time ‘zero’ represented the onset of exercise. Data from each transition were ensemble-averaged to yield a single, averaged response for each subject. This transition was further time-averaged into 10 s bins to provide a single time-averaged response for each subject. The on-transient response for V˙o2 was modelled using a mono-exponential of the form:
| (1) |
where Y(t) represents V˙o2 at any time (t); YBsl is the baseline V˙o2 during 20 W cycling; Amp is the steady-state increase in V˙o2 above the baseline value; τ is the time constant defined as the duration of time for V˙o2 to increase to 63% of the steady-state increase, and TD is the time delay. The phase 1–phase 2 transition was determined as previously described (Rossiter et al. 1999; Gurd et al. 2005) and V˙o2 data were modelled from the beginning of phase 2 to 2 min (120 s) and 6 min (360 s) of the step transition. The model parameters were estimated by least-squares nonlinear regression (Origin, OriginLab Corp., Northampton, MA, USA) in which the best fit was defined by minimization of the residual sum of squares and minimal variation of residuals around the Y-axis (Y = 0). The 95% confidence interval (C95) for the estimated time constant was determined after a preliminary fit of the data with BSL, Amp, and TD constrained to the best-fit values and the τ allowed to vary.
Expired ventilation (V˙E) was time-aligned, ensemble-averaged and time-averaged into 10 s bins. The time course of V˙E was modelled using the exponential model described in eqn (1), and the data were fitted from the onset to the end of constant-load exercise.
Heart rate
Heart rate (HR) was determined from the R-R interval on a second-by-second basis and edited and modelled in the same manner as the V˙o2 data described above. The on-transient HR response was modelled from exercise onset to the end of exercise using the exponential model describe in eqn (1), but with the time delay (TD) constrained to ‘zero’ (i.e. TD = 0).
NIR spectroscopy
The NIRS-derived ΔHHb, ΔO2Hb, ΔHbtot and TOI data were analysed in a similar fashion to that of the gas exchange data. The ΔHHb profile has been described as consisting of, at the start of exercise, a time delay followed by an increase in the signal which is ‘exponential-like’ in its time course (DeLorey et al. 2003; Grassi et al. 2003). The time delay for the ΔHHb response (TD-HHb) was determined using second-by-second data, and corresponded to the time, after the onset of exercise, for the ΔHHb-signal to increase above one standard deviation of the mean response measured during 20 W baseline cycling (DeLorey et al. 2003). Determination of the TD-HHb was made on individual trials and averaged to yield a single value for each subject. The ΔHHb data were modelled from the TD-HHb to the end of exercise using an exponential model as described in eqn (1). The TD-HHb and τHHb described the time course for the increase in ΔHHb, and the mean response time (MRT = TD-HHb +τHHb) described the overall time course of the ΔHHb from the onset of exercise. The ‘exponential-like’ increase in the muscle ΔHHb signal and the residuals associated with that fit suggest that the mono-exponential model provides a reasonable representation of the time course of muscle deoxygenation during the transition to exercise. The profiles of the ΔO2Hb and ΔHbtot are not predicted to approximate to a known mathematical model and thus only the steady-state baseline and end-exercise values were determined for these signals.
Statistical analysis
All data are presented as means ±s.d. The kinetic parameter estimates for V˙o2, ΔHHb, HR and V˙E were analysed using a one-way ANOVA for repeated measures. A two-way ANOVA with repeated measures was used to establish differences between time and condition for the blood gas variables. Significant main effects were analysed by Tukey's pairwise multiple comparison procedure, and significance was accepted at P < 0.05.
Results
Respiratory and gas exchange measurements
The respiratory and gas exchange profiles for the entire pre-exercise accommodation and exercise periods during the CON and RALK conditions for a representative subject are shown in Fig. 1, with the group mean data for selected respiratory and gas exchange variables during the different periods presented in Table 1. The hyperventilation manoeuvre was maintained throughout the entire protocol and resulted in a reduced (P < 0.05) 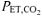 during the accommodation period (RALK, 19 ± 2 mmHg; CON, 34 ± 5 mmHg) and throughout exercise (RALK, 24 ± 1 mmHg; CON, 43 ± 1 mmHg). The reduction in
during the accommodation period (RALK, 19 ± 2 mmHg; CON, 34 ± 5 mmHg) and throughout exercise (RALK, 24 ± 1 mmHg; CON, 43 ± 1 mmHg). The reduction in 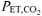 in RALK was achieved by an approximate doubling of V˙E (P < 0.05) (Table 1). The higher V˙E in RALK was achieved by a 75–100% higher breathing frequency (P < 0.05) with no significant change in tidal volume between conditions. The time course of the adaptation (τ) of V˙E was similar between RALK (56 ± 16 s) and CON (57 ± 30 s); however, the baseline and amplitude of V˙E was higher (P < 0.05) in RALK than in CON (Table 2).
in RALK was achieved by an approximate doubling of V˙E (P < 0.05) (Table 1). The higher V˙E in RALK was achieved by a 75–100% higher breathing frequency (P < 0.05) with no significant change in tidal volume between conditions. The time course of the adaptation (τ) of V˙E was similar between RALK (56 ± 16 s) and CON (57 ± 30 s); however, the baseline and amplitude of V˙E was higher (P < 0.05) in RALK than in CON (Table 2).
Figure 1.

The ventilatory and gas exchange profiles for a representative subject during the Control (CON; ○) and the hyperventilation-induced hypocapnic alkalosis (RALK; •) exercise protocols Dashed lines demarcate the transitions between the pre-accommodation period (Pre-Acc), accommodation period (Acc), baseline cycling (20 W), and constant-load moderate-intensity exercise (Mod).
Table 1.
Steady-state respiratory measures taken at rest before (Pre-Accommodation) and after 20 min normal or hyperventilation (Post-Accommodation), after 6 min baseline cycling at 20 W (20 W Baseline), and during the final minute of exercise at 90% 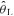 (End-Exercise) in Control (CON) and hyperventilation-induced hypocapnic alkalosis (RALK)
(End-Exercise) in Control (CON) and hyperventilation-induced hypocapnic alkalosis (RALK)
| Pre-Accommodation | Post-Accommodation | 20 W Baseline | End-Exercise | |||||
|---|---|---|---|---|---|---|---|---|
| CON | RALK | CON | RALK | CON | RALK | CON | RALK | |
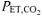 (mmHg) (mmHg) |
36 ± 3 | 35 ± 3 | 34 ± 5 | 19 ± 2*† | 39 ± 3‡ | 21 ± 1*† | 43 ± 1†‡# | 24 ± 1*†‡ |
| V˙E (l min−1) | 13.0 ± 2.5 | 13.4 ± 3.1 | 14.9 ± 5.0 | 27.7 ± 4.8*† | 24.7 ± 3.3†‡ | 52.0 ± 4.8*†‡ | 43.9 ± 5.5†‡# | 86.8 ± 5.3*†‡# |
| Vt (l/br) | 0.91 ± 0.3 | 0.88 ± 0.4 | 1.0 ± 0.7 | 1.1 ± 0.5 | 1.4 ± 0.5 | 1.6 ± 0.6† | 2.1 ± 0.4†‡# | 2.1 ± 0.7†‡ |
| Freq (br min−1) | 15 ± 5 | 15 ± 4 | 16 ± 5 | 28 ± 10*† | 19 ± 5 | 37 ± 13*† | 22 ± 5 | 46 ± 15*†‡ |
| V˙o2 (l min−1) | 0.44 ± 0.05 | 0.43 ± 0.05 | 0.46 ± 0.05 | 0.47 ± 0.06 | 0.96 ± 0.10†‡ | 0.98 ± 0.09†‡ | 1.87 ± 0.16†‡# | 1.94 ± 0.19*†‡# |
| V˙CO2 (l min−1) | 0.40 ± 0.04 | 0.40 ± 0.06 | 0.40 ± 0.04 | 0.48 ± 0.06* | 0.85 ± 0.07†‡ | 0.99 ± 0.09*†‡ | 1.76 ± 0.17†‡# | 1.86 ± 0.13*†‡# |
| RER | 0.93 ± 0.10 | 0.94 ± 0.14 | 0.87 ± 0.07 | 1.03 ± 0.08* | 0.89 ± 0.03 | 0.99*± 0.05 | 0.94 ± 0.04 | 0.96 ± 0.04 |
Values are means ±s.d. V˙o2, O2 uptake; V˙CO2, CO2 output; 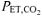 , end-tidal
, end-tidal 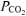 ; V˙o2, expired ventilation; Vt, tidal volume; Freq, breathing frequency; RER, respiratory exchange ratio.
; V˙o2, expired ventilation; Vt, tidal volume; Freq, breathing frequency; RER, respiratory exchange ratio.
Significant difference (P < 0.05) from Control
significant difference (P < 0.05) from Pre-Accommodation
significant difference (P < 0.05) from Post-Accommodation
significant difference (P < 0.05) from 20 W Baseline.
Table 2.
Kinetic parameter estimates for V˙E, V˙o2 and HR in Control (CON) and hyperventilation-induced hypocapnic alkalosis (RALK)
| Protocol | Bsl (l min−1) | Amp (l min−1) | τ (s) | C95 | Gain (ml min−1 W−1) |
|---|---|---|---|---|---|
| V˙E | |||||
| CON | 23.7 ± 2.2 | 22.1 ± 2.5 | 57 ± 30 | 6 ± 2 | — |
| RALK | 50.7 ± 4.8* | 39.4 ± 6.9* | 56 ± 16 | 6 ± 1 | — |
| V˙o2 (fit from ∼20 s to 360 s) | |||||
| CON | 0.96 ± 0.10 | 0.90 ± 0.18 | 31 ± 9 | 2 ± 1 | 10.3 ± 0.5 |
| RALK | 0.98 ± 0.09 | 0.96 ± 0.19* | 48 ± 11* | 3 ± 1 | 10.9 ± 0.6* |
| V˙o2 (fit from ∼20 s to 120 s) | |||||
| CON | 0.96 ± 0.10 | 0.89 ± 0.19 | 27 ± 8 | 2 ± 1 | 10.1 ± 0.4 |
| RALK | 0.98 ± 0.09 | 0.89 ± 0.19 | 34 ± 7* | 2 ± 1 | 10.1 ± 0.5 |
| Heart Rate | Bsl (b min−1) | Amp (b min−1) | τ (s) | C95 | |
| CON | 87 ± 6 | 30 ± 7 | 27 ± 11 | 3 ± 1 | |
| RALK | 85 ± 4 | 26 ± 6 | 28 ± 8 | 4 ± 1 | |
Values are means ±s.d. Baseline values are taken as the mean of the last 60 s of 20 W cycling. Bsl, baseline; Amp, amplitude; τ time constant; C95, 95% confidence interval; Gain, calculated as ΔV˙o2/ΔWR; TD, time delay. *Significant difference (P < 0.05) from Control.
Pulmonary 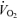 was similar between conditions during the accommodation period and at steady-state baseline cycling (Table 1), but was higher (P < 0.05) in RALK compared to CON during moderate-intensity exercise. Pulmonary V˙CO2 was higher (P < 0.05) in RALK than in CON throughout the period of hyperventilation (Table 1). The respiratory exchange ratio (RER) was higher (P < 0.05) in RALK (1.03 ± 0.08) than CON (0.87 ± 0.07) during accommodation and in baseline cycling, but was not different between conditions during steady-state, moderate-intensity exercise (Table 1).
was similar between conditions during the accommodation period and at steady-state baseline cycling (Table 1), but was higher (P < 0.05) in RALK compared to CON during moderate-intensity exercise. Pulmonary V˙CO2 was higher (P < 0.05) in RALK than in CON throughout the period of hyperventilation (Table 1). The respiratory exchange ratio (RER) was higher (P < 0.05) in RALK (1.03 ± 0.08) than CON (0.87 ± 0.07) during accommodation and in baseline cycling, but was not different between conditions during steady-state, moderate-intensity exercise (Table 1).
V˙o2 kinetics
The moderate-intensity WR used in the present study elicited a steady-state V˙o2 in CON that represented 92% (± 4%) 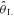 and 54% (± 4%) V˙o2 peak. The V˙o2 response profile with model fit and associated residuals for a representative subject during the transition to exercise is shown in Fig. 2. Kinetic parameter estimates of the V˙o2 response are presented in Table 2. Kinetic analysis showed that the V˙o2 measured during 20 W baseline cycling was not different between conditions (Table 2). When V˙o2 data were analysed for the entire exercise period (i.e. ∼20 s to 360 s), the V˙o2 amplitude was higher (P < 0.05) in RALK (0.96 ± 0.19 l min−1) than in CON (0.90 ± 0.18 l min−1), and resulted in a greater (P < 0.05) end-exercise V˙o2 (RALK, 1.95 ± 0.18 l min−1; CON, 1.87 ± 0.16 l min−1) and functional gain (ΔV˙o2/ΔWR) (RALK, 10.9 ± 0.6 ml min−1 W−1; CON, 10.3 ± 0.5 ml min−1 W−1) in this condition. The time constant for the phase 2 V˙o2 response (τV˙o2) was greater (P < 0.05) in RALK (48 ± 11 s) than CON (31 ± 9 s), with the difference between conditions being outside the 95% confidence (C95) interval for the estimation of the τV˙o2 (Table 2). A greater τV˙o2 in RALK than in CON was seen consistently in all eight subjects (Fig. 3). When this analysis was limited to the initial V˙o2 response (i.e. from the onset of phase 2 to 120 s), the magnitude of this kinetic slowing was reduced, but the τV˙o2 remained greater (P < 0.05) in RALK (34 ± 7 s) than in CON (27 ± 8 s) (Table 2).
and 54% (± 4%) V˙o2 peak. The V˙o2 response profile with model fit and associated residuals for a representative subject during the transition to exercise is shown in Fig. 2. Kinetic parameter estimates of the V˙o2 response are presented in Table 2. Kinetic analysis showed that the V˙o2 measured during 20 W baseline cycling was not different between conditions (Table 2). When V˙o2 data were analysed for the entire exercise period (i.e. ∼20 s to 360 s), the V˙o2 amplitude was higher (P < 0.05) in RALK (0.96 ± 0.19 l min−1) than in CON (0.90 ± 0.18 l min−1), and resulted in a greater (P < 0.05) end-exercise V˙o2 (RALK, 1.95 ± 0.18 l min−1; CON, 1.87 ± 0.16 l min−1) and functional gain (ΔV˙o2/ΔWR) (RALK, 10.9 ± 0.6 ml min−1 W−1; CON, 10.3 ± 0.5 ml min−1 W−1) in this condition. The time constant for the phase 2 V˙o2 response (τV˙o2) was greater (P < 0.05) in RALK (48 ± 11 s) than CON (31 ± 9 s), with the difference between conditions being outside the 95% confidence (C95) interval for the estimation of the τV˙o2 (Table 2). A greater τV˙o2 in RALK than in CON was seen consistently in all eight subjects (Fig. 3). When this analysis was limited to the initial V˙o2 response (i.e. from the onset of phase 2 to 120 s), the magnitude of this kinetic slowing was reduced, but the τV˙o2 remained greater (P < 0.05) in RALK (34 ± 7 s) than in CON (27 ± 8 s) (Table 2).
Figure 2.

The V˙o2 response profile with model line of best fit and associated residuals (at y = 0) for a representative subject in Control (CON; grey line) and hyperventilation-induced hypocapnic alkalosis (RALK; black line) Inset shows normalized V˙o2 profiles for the two conditions. Dashed line demarcates onset of moderate-intensity exercise.
Figure 3.

The time constant for the phase 2 V˙o2 response (τ V˙o2) for each subject in Control (CON) and hyperventilation-induced hypocapnic alkalosis (RALK) Continuous line represents group mean. *Significant difference (P < 0.05) from Control.
Heart rate kinetics
The parameter estimates for heart rate kinetics for the transition to moderate-intensity exercise are presented in Table 2. No differences (P > 0.05) were observed in the baseline, amplitude, or time constant for heart rate between RALK and CON.
Near-infrared spectroscopy measurements
The group mean responses for ΔHHb, ΔO2Hb, ΔHbtot and TOI for the entire RALK and CON protocols are shown in Fig. 4. Immediately following the start of the hyperventilation manoeuvre the ΔO2Hb and ΔHHb signals exhibited transient increases (∼440 s) and decreases (∼840 s), respectively, before returning to baseline levels. The ΔHbtot signal (which represents the sum of ΔO2Hb and ΔHHb) is dependent on the relative magnitude of the ΔO2Hb and ΔHHb signals, and thus, only small fluctuations were evident in this signal (Fig. 4). At the end of the accommodation period, ΔHHb, ΔO2Hb, ΔHbtot and TOI were not different between conditions (Table 3), but during baseline cycling and at end-exercise, the ΔO2Hb and ΔHbtot levels were lower (P < 0.05) in RALK than CON (Table 3).
Figure 4.

The group mean ΔHHb, ΔO2Hb, ΔHbtot and TOI responses for all subjects (n = 8) in Control (CON; ….) and hyperventilation-induced hypocapnic alkalosis (RALK; —) Dashed lines demarcate the pre-accommodation period (Pre-Acc), accommodation period (Acc), baseline cycling (20 W), and constant-load, moderate-intensity exercise (Mod).
Table 3.
Steady-state accommodation, baseline and end-exercise values for the NIRS-derived ΔHHb, ΔO2Hb, ΔHbtot and TOI signals in Control (CON) and hyperventilation-induced hypocapnic alkalosis (RALK)
| Pre-Accommodation | Post-Accommodation | 20 W Baseline | End-Exercise | |||||
|---|---|---|---|---|---|---|---|---|
| CON | RALK | CON | RALK | CON | RALK | CON | RALK | |
| ΔHHb (μmol l−1) | 0.3 ± 0.7 | 0.4 ± 0.6 | 0.7 ± 0.8 | −0.1 ± 1.1 | −3.2 ± 2.5†‡ | −3.4 ± 2.4†‡ | 3.2 ± 3.6# | 3.0 ± 4.4# |
| ΔO2Hb (μmol l−1) | 0.8 ± 0.3 | 0.7 ± 0.4 | 1.6 ± 0.6 | 1.9 ± 1.4 | 2.5 ± 3.0 | 0.6 ± 4.1* | 1.4 ± 4.3 | −2.1 ± 4.4*‡ |
| ΔHbtot (μmol l−1) | 1.1 ± 0.6 | 1.3 ± 0.5 | 2.2 ± 1.3 | 1.8 ± 1.6 | −0.7 ± 4.3 | −3.2 ± 5.0*†‡ | 4.7 ± 4.5# | 0.8 ± 5.8* |
| TOI | 61.5 ± 5.6 | 61.4 ± 3.7 | 61.3 ± 5.2 | 61.5 ± 2.0 | 65.1 ± 6.4†‡ | 64.2 ± 4.8†‡ | 61.4 ± 5.6# | 59.8 ± 5.0# |
ΔHHb, deoxy-haemoglobin; ΔO2Hb, oxy-haemoglobin; ΔHbtot, total haemoglobin; TOI, tissue oxygenation index. *Significant difference (P < 0.05) from Control. †significant difference (P < 0.05) from Pre-Accommodation; ‡significant difference (P < 0.05) from Post-Accommodation; #significant difference (P < 0.05) from 20 W Baseline.
During the transition to moderate-intensity exercise, there was no difference between CON and RALK for TD-HHb (RALK, 9 ± 5 s; CON, 10 ± 4 s), τHHb (RALK, 12 ± 4 s; CON, 11 ± 4 s) and the MRT for HHb (RALK, 21 ± 6 s; CON, 21 ± 7 s) (Table 4). Likewise, ΔHHb baseline and amplitude were not different between the conditions (Table 4).
Table 4.
Kinetic and steady-state parameters for the NIRS-derived deoxyhaemoglobin signal (ΔHHb) in Control (CON) and hyperventilation-induced hypocapnic alkalosis (RALK)
| CON | RALK | |
|---|---|---|
| Bsl (μmol l−1) | −3.0 ± 2.3 | −3.9 ± 2.4 |
| Amp (μmol l−1) | 6.1 ± 3.3 | 6.4 ± 3.7 |
| TD-HHb (s) | 10 ± 4 | 9 ± 5 |
| τ (s) | 11 ± 4 | 12 ± 4 |
| MRT (s) | 21 ± 6 | 21 ± 7 |
Values are means ±s.d. Bsl, Baseline (calculated during the final 60 s prior to the step-increase in work rate to 90%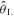 ); Amp, amplitude; TD, time delay; τ, time constant; MRT, mean response time (TD +τ).
); Amp, amplitude; TD, time delay; τ, time constant; MRT, mean response time (TD +τ).
Blood gas and acid–base status
Plasma [H+] was lower (P < 0.05) in RALK than in CON throughout the hyperventilation manoeuvre (Table 5). In both conditions, the plasma [H+] decreased (P < 0.05) during the 20 min accommodation period (RALK: Rest, 36 ± 0.1 nmol l−1; Post-Accommodation, 23 ± 0.2 nmol l−1; CON: Rest, 33 ± 0.3 nmol l−1; Post-Accommodation, 31 ± 0.3 nmol l−1); however, the decrease was greater (P < 0.05) in RALK (Table 5). The plasma [H+] increased (P < 0.05) during moderate-intensity exercise, with the plasma [H+] at end-exercise levels remaining below (P < 0.05) pre-accommodation levels in RALK, but being greater (P < 0.05) than pre-accommodation levels in CON (Table 5).
Table 5.
Blood gas parameters for Control (CON) and hyperventilation-induced hypocapnic alkalosis (RALK) during the pre- and post-accommodation period, at the end of 20 W cycling (20 W Baseline) and during the final minute of exercise at 90%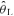
| Pre-Accommodation | Post-Accommodation | 20 W Baseline | End-Exercise | |||||
|---|---|---|---|---|---|---|---|---|
| CON | RALK | CON | RALK | CON | RALK | CON | RALK | |
| pH | 7.48 ± 0.04 | 7.44 ± 0.02 | 7.51 ± 0.05† | 7.63 ± 0.03*† | 7.47 ± 0.03‡ | 7.61 ± 0.01*† | 7.41 ± 0.02†‡# | 7.54 ± 0.03*†‡# |
| [H+] (nmol l−1) | 33 ± 0.3 | 36 ± 0.1 | 31 ± 0.3† | 23 ± 0.2*† | 34 ± 0.3‡ | 25 ± 0.1*† | 39 ± 0.2†‡# | 29 ± 0.2*†‡# |
|
(mmHg) |
36 ± 4 | 38 ± 2 | 34 ± 5 | 21 ± 4*† | 38 ± 4‡ | 21 ± 3*† | 40 ± 3†‡ | 23 ± 1*† |
| [La−] (mmol l−1) | 1.3 ± 0.6 | 1.4 ± 0.5 | 1.3 ± 0.7 | 1.9*± 0.7 | 1.2 ± 0.5 | 2.1 ± 0.7* | 3.1 ± 0.9†‡# | 5.2 ± 0.6*†‡# |
Values are means ±s.d. for plasma. [H+], hydrogen ion concentration; 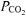 , arterialized-venous
, arterialized-venous 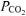 ; [La−], arterialized-venous lactate concentration.
; [La−], arterialized-venous lactate concentration.
Significant difference (P < 0.05) from Control
significant difference (P < 0.05) from Pre-Accommodation
significant difference (P < 0.05) from Post-Accommodation
significant difference (P < 0.05) from 20 W Baseline.
Plasma 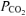 was lower (P < 0.05) in RALK than in CON throughout the hyperventilation manoeuvre; the end-exercise plasma
was lower (P < 0.05) in RALK than in CON throughout the hyperventilation manoeuvre; the end-exercise plasma 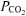 was 23 ± 1 mmHg (RALK) and 40 ± 3 mmHg (CON) (Table 5). Plasma [lactate−] was higher (P < 0.05) in RALK than CON throughout the period of hyperventilation (Fig. 5); end-exercise [lactate−] was 5.2 ± 0.6 mmol l−1 (RALK) and 3.1 ± 0.9 mmol l−1 (CON) (P < 0.05).
was 23 ± 1 mmHg (RALK) and 40 ± 3 mmHg (CON) (Table 5). Plasma [lactate−] was higher (P < 0.05) in RALK than CON throughout the period of hyperventilation (Fig. 5); end-exercise [lactate−] was 5.2 ± 0.6 mmol l−1 (RALK) and 3.1 ± 0.9 mmol l−1 (CON) (P < 0.05).
Figure 5.

Mean blood lactate concentration across time for Control (CON; ○) and hyperventilation-induced hypocapnic alkalosis (RALK; •) Dashed lines demarcate the pre-accommodation period (Pre-Acc), accommodation period (Acc), 20 W, and moderate-intensity (Mod) cycling exercise. *Significant difference (P < 0.05) from Control.
Discussion
The aim of the present investigation was to determine the effect of a voluntary hyperventilation-induced hypocapnic alkalosis (i.e. respiratory alkalosis, RALK) on the adaptation of pulmonary V˙o2 and muscle oxygenation during the transition to moderate-intensity exercise. The main findings of this study were that (i) the adaptation of pulmonary V˙o2 (as determined by τV˙o2), a reflection of the kinetics of muscle O2 consumption, was slowed during RALK; (ii) the adaptation of muscle deoxygenation (as determined by τΔHHb and MRT-ΔHHb), reflecting muscle O2 extraction and the balance between local muscle O2 delivery and local muscle O2 utilization, was similar in RALK and the control (CON, normal breathing) condition; and (iii) muscle oxygenated (ΔO2Hb) and total (ΔHbtot) haemoglobin concentrations during steady-state baseline and moderate-intensity exercise were lower during RALK than CON. Although slowed pulmonary V˙o2 kinetics (and muscle O2 consumption) are consistent with slowed activation of the mitochondrial PDH complex and metabolic inertia in RALK (see below; LeBlanc et al. 2002), the adaptation of muscle blood flow also may have been slowed and/or reduced by RALK, since slowed V˙o2 kinetics in association with similar kinetics of muscle O2 extraction suggests a mismatch between local muscle blood flow and muscle O2 consumption in RALK.
Respiratory alkalosis and pulmonary V˙o2 kinetics
In the present study, pulmonary V˙o2 kinetics were measured to provide a reliable estimate (i.e. within 10%) of the adaptation of muscle O2 consumption (Barstow et al. 1990; Grassi et al. 1996). During RALK, the adaptation of pulmonary V˙o2 (i.e. based on the τV˙o2), and thus the adaptation of muscle O2 consumption, was ∼17 s slower than in CON (Table 2; Fig. 3). This slowing of pulmonary V˙o2 was confirmed when the fitting ‘window’ for the data was confined to a time period restricted to the fundamental, phase 2 component of the V˙o2 response (i.e. ∼20–120 s) (Table 2). Regardless of the fitting strategy used (i.e. ∼20 s to either 120 s or 360 s), the τV˙o2 was greater in RALK than CON, with the differences between conditions exceeding the 95% confidence limits. Slower activation of muscle O2 consumption during hyperventilation-induced respiratory alkalosis was supported by recent findings showing that during the transition to moderate-intensity plantar-flexion exercise, the kinetics of muscle PCr breakdown (reflecting the adaptation of muscle O2 consumption) were slower and the amplitude of the response was greater compared to control (Forbes et al. 2007).
The slowing of the pulmonary V˙o2 on-transient response observed in RALK in the present study agrees with the findings of Hayashi et al. (1999), who reported an ∼16 s greater τV˙o2 during a hyperventilation-induced hypocapnia (τV˙o2, 37 s) than in control (τV˙o2, 21 s), but do not support the findings of Ward et al. (1983), who reported no difference in the half-time (t1/2) of the V˙o2 response between their hyperventilation (39 s) and control (31 s) conditions (the corresponding τV˙o2 for each condition was 56 s (hyperventilation) and 45 s (control)).
However, there were methodological differences in the hyperventilation protocols used in the present study and the studies of Hayashi et al. (1999) and Ward et al. (1983). In the study of Hayashi et al. (1999), hyperventilation was performed for only 2 min at rest before the start of exercise and throughout constant-load, moderate-intensity exercise, while in the study of Ward et al. (1983), the hyperventilation manoeuvre was performed at rest for only 9 min and was stopped 15–20 s before the signal to start exercise, with no hyperventilation performed during the exercise period, thereby allowing CO2 to rise during the transition to exercise. Removal and subsequent equilibration of CO2 stores following a sudden increase in ventilation occurs with time and is dependent on baseline metabolic conditions. Jones & Jurkowski (1979) reported that when exercising at 30% or 60% maximal V˙o2, ∼90% of the change in CO2 washout from body CO2 stores occurred by ∼4 min after the start of a voluntary hyperventilation protocol, but Brandi & Clode (1969) reported that, at rest, ∼15 min was required to remove 90% of the body CO2 stores. Therefore, in the present study a more prolonged period of hyperventilation was used than by Hayashi et al. (1999) and Ward et al. (1983), but similar to that used by LeBlanc et al. (2002) and Forbes et al. (2007). In the present study, the hyperventilation manoeuvre was maintained throughout the 20 min resting accommodation period, 6 min baseline exercise at 20 W and the 6 min constant-load, moderate-intensity exercise, thereby providing a longer period of time for body CO2 stores (and in particular, intramuscular CO2 stores) to equilibrate to the new, lower level (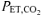 ∼20 mmHg) before the start of exercise and also maintaining these lower CO2 levels throughout the exercise transition. The effectiveness of this more prolonged period of voluntary hyperventilation is supported in our recent publication (Forbes et al. 2007) showing that intramuscular [H+] was reduced at rest as a consequence of a prolonged hyperventilation protocol similar to that used in this present study.
∼20 mmHg) before the start of exercise and also maintaining these lower CO2 levels throughout the exercise transition. The effectiveness of this more prolonged period of voluntary hyperventilation is supported in our recent publication (Forbes et al. 2007) showing that intramuscular [H+] was reduced at rest as a consequence of a prolonged hyperventilation protocol similar to that used in this present study.
The slower V˙o2 on-kinetics observed in the present study with hyperventilation-induced hypocapnic alkalosis (RALK) differed from the findings of previous studies that examined the effects of a sodium bicarbonate-induced metabolic alkalosis (MALK) on pulmonary V˙o2 kinetics. Ingestion of sodium bicarbonate over several days (Oren et al. 1982) or within 1–2 h (Zoladz et al. 2005) prior to the transition to moderate-intensity cycling exercise resulted in no difference in the τV˙o2 when compared to a control condition (τV˙o2 MALK versus CON: 39 s versus 35 s: Oren et al. 1982; 26 s versus 24 s: Zoladz et al. 2005). Therefore, the V˙o2 response in moderate-intensity exercise may be dependent on whether the alkalosis was of a respiratory origin (i.e. by lowering plasma and intracellular CO2; Lindinger et al. 1990) or metabolic origin (i.e. by raising plasma strong cation concentration and the strong ion difference, [SID]; Lindinger et al. 1990). This is supported in recent studies using 31P-MRS to monitor intracellular acid–base status which reported that immediately prior to the start of exercise, the intracellular [H+] was lower during a hyperventilation-induced hypocapnic (respiratory) alkalosis (RALK) than in control (Forbes et al. 2007) but was not different from control during a sodium bicarbonate-induced metabolic alkalosis (MALK) (Forbes et al. 2005).
Respiratory alkalosis, muscle O2 delivery and blood flow
Hayashi et al. (1999) suggested that V˙o2 kinetics were slowed in hyperventilation as a result of a leftward-shift in the oxy-haemoglobin dissociation curve causing impaired O2 off-loading from haemoglobin, and thus impaired muscle O2 delivery, within the muscle microvascular space. The higher plasma pH and lower 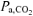 observed during RALK in the present study (Table 5) compared to CON would contribute to a leftward-shift in the oxy-haemoglobin dissociation curve. In the present study, an increase in ΔO2Hb (consistent with greater O2Hb affinity) was seen early in the hyperventilation accommodation period (i.e. ∼20 min before the start of exercise) without a significant change in ΔHbtot (Fig. 4), and this was accompanied by a decrease in ΔHHb. However, the increase in ΔO2Hb was only transient, lasting ∼440 s. In the study by Hayashi et al. (1999), the hyperventilation manoeuvre was initiated 120 s before the start of exercise (compared to 26 min before the start of moderate exercise in the present study, i.e. 20 min accommodation plus 6 min baseline exercise) and so the effect of hyperventilation on O2Hb affinity may have been more pronounced in that study. Because the ΔO2Hb signal in RALK returned to baseline levels (i.e. similar to that of CON) before the start of the exercise protocol in the present study, it appears unlikely that impaired unloading of O2 from haemoglobin can explain the slowed τV˙o2 observed in RALK at the onset of exercise in the present study. In fact, as discussed above, ΔHHb kinetics were similar between conditions in spite of slower muscle O2 consumption kinetics, suggesting that O2 extraction was faster relative to muscle O2 utilization.
observed during RALK in the present study (Table 5) compared to CON would contribute to a leftward-shift in the oxy-haemoglobin dissociation curve. In the present study, an increase in ΔO2Hb (consistent with greater O2Hb affinity) was seen early in the hyperventilation accommodation period (i.e. ∼20 min before the start of exercise) without a significant change in ΔHbtot (Fig. 4), and this was accompanied by a decrease in ΔHHb. However, the increase in ΔO2Hb was only transient, lasting ∼440 s. In the study by Hayashi et al. (1999), the hyperventilation manoeuvre was initiated 120 s before the start of exercise (compared to 26 min before the start of moderate exercise in the present study, i.e. 20 min accommodation plus 6 min baseline exercise) and so the effect of hyperventilation on O2Hb affinity may have been more pronounced in that study. Because the ΔO2Hb signal in RALK returned to baseline levels (i.e. similar to that of CON) before the start of the exercise protocol in the present study, it appears unlikely that impaired unloading of O2 from haemoglobin can explain the slowed τV˙o2 observed in RALK at the onset of exercise in the present study. In fact, as discussed above, ΔHHb kinetics were similar between conditions in spite of slower muscle O2 consumption kinetics, suggesting that O2 extraction was faster relative to muscle O2 utilization.
The slower pulmonary V˙o2 kinetics seen in the present study in RALK may be related to a lower blood flow response at the level of the working muscle (Kontos et al. 1972; Brice & Welch, 1985; Gustafsson et al. 1993; Karlsson et al. 1994). Kontos et al. (1972) observed a lower resting forearm blood flow in human subjects after 6 min voluntary hyperventilation compared to a control condition. While the effect of RALK on muscle blood flow in humans during exercise has yet to be determined, findings from animal studies suggest that both cardiac output (Karlsson et al. 1994) and skeletal muscle blood flow (Brice & Welch, 1985; Gustafsson et al. 1993) were reduced during exercise during hypocapnic alkalosis. Therefore, hyperventilation may act to reduce blood flow through a reduction in the plasma CO2 and [H+], potent vasodilators during exercise (Haddy & Scott, 1968), thereby maintaining a lower vascular conductance in exercise (Kontos et al. 1972). In the present study, data collected using near-infrared spectroscopy in combination with measures of pulmonary V˙o2 are consistent with the suggestion that muscle perfusion is reduced in RALK and thus may limit muscle O2 utilization in the transition to moderate-intensity exercise. Although pulmonary V˙o2 kinetics (and presumably muscle O2 consumption) were slowed in RALK (Table 2; Fig. 3), the adaptation of ΔHHb (reflecting muscle O2 extraction and the balance between muscle O2 delivery and muscle O2 utilization) was not different between conditions (Table 4), suggesting that O2 extraction was faster (relative to muscle O2 consumption kinetics), reflecting a slower adaptation of blood flow and O2 delivery relative to O2 consumption in RALK. Also, during steady-state baseline and constant-load, moderate-intensity exercise, NIRS-derived ΔHbtot and ΔO2Hb signals were lower in RALK than CON (Table 3), and the increase in ΔHHb/ΔHbtot ratio between steady-state baseline and constant-load, moderate-intensity exercise tended to be greater (P = 0.089) in RALK (1.6) than CON (1.2). These data suggest that the total volume of haemoglobin and oxygenation-state of haemoglobin were lower in RALK, and that the increase in O2 extraction relative to the increase in haemoglobin volume was greater in RALK, consistent with an RALK-induced hypo-perfusion and attenuated vascular conductance (or vasodilatation) within the region or impaired blood flow redistribution from less active muscle or splanchnic regions in RALK. In agreement, we recently reported that in spite of slower and greater muscle PCr breakdown kinetics during the transition to moderate-intensity plantar-flexion exercise, the kinetics of NIRS-derived ΔHHb increase was similar in RALK and control, but the ΔHHb amplitude and steady-state ΔHHb were greater, and the steady-state ΔO2Hb was lower in RALK (Forbes et al. 2007).
Respiratory alkalosis and mitochondrial PDH activation
Respiratory alkalosis also may contribute to slower activation of enzymes and provision of substrates (other than O2) required to support mitochondrial respiration (i.e. metabolic inertia). LeBlanc et al. (2002) demonstrated that the activation of the mitochondrial pyruvate dehydrogenase (PDH) enzyme complex was slowed during the first minute of cycling exercise at 55%V˙o2,max following a hyperventilation-induced hypocapnic alkalosis (RALK, 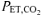 ∼20 mmHg). PDH is responsible for catalysing the non-reversible oxidative decarboxylation of pyruvate to acetyl CoA (Spriet & Heigenhauser, 2002) and is thought to play an important role in determining the rate of muscle O2 utilization. The hyperventilation protocol used in the present study was similar to that used by LeBlanc et al. (2002), which included a similar pre-exercise accommodation period (20 min) and similar exercise intensity (∼50%V˙o2,max) and resulted in a similar
∼20 mmHg). PDH is responsible for catalysing the non-reversible oxidative decarboxylation of pyruvate to acetyl CoA (Spriet & Heigenhauser, 2002) and is thought to play an important role in determining the rate of muscle O2 utilization. The hyperventilation protocol used in the present study was similar to that used by LeBlanc et al. (2002), which included a similar pre-exercise accommodation period (20 min) and similar exercise intensity (∼50%V˙o2,max) and resulted in a similar 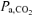 (∼20 mmHg) and arterial plasma pH (∼7.60). Importantly, Forbes et al. (2007) reported that intramuscular [H+], determined by 31P-MRS, was lower immediately before the onset of plantar-flexion exercise when preceded by a 20-min period of hyperventilation (
(∼20 mmHg) and arterial plasma pH (∼7.60). Importantly, Forbes et al. (2007) reported that intramuscular [H+], determined by 31P-MRS, was lower immediately before the onset of plantar-flexion exercise when preceded by a 20-min period of hyperventilation (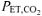 ∼17 mmHg). A shift to a more alkaline intramuscular pH could contribute to an activation of pyruvate dehydrogenase kinase (PDK) and inhibition of pyruvate dehydrogenase phosphatase (PDP). An increase in the ratio of active PDK-to-PDP would be expected to slow the rate of activation of PDH at the onset of exercise, and thus slow the provision of acetyl-CoA and reducing equivalents to the mitochondrial TCA cycle and ETC (see p.309 of LeBlanc et al. (2002) for discussion). Therefore, it is expected that the activation of PDH during the transition to exercise in RALK likely was slowed in the present study, which also could contribute to the slower activation of pulmonary V˙o2 (and muscle O2 consumption) in the RALK condition of the present study.
∼17 mmHg). A shift to a more alkaline intramuscular pH could contribute to an activation of pyruvate dehydrogenase kinase (PDK) and inhibition of pyruvate dehydrogenase phosphatase (PDP). An increase in the ratio of active PDK-to-PDP would be expected to slow the rate of activation of PDH at the onset of exercise, and thus slow the provision of acetyl-CoA and reducing equivalents to the mitochondrial TCA cycle and ETC (see p.309 of LeBlanc et al. (2002) for discussion). Therefore, it is expected that the activation of PDH during the transition to exercise in RALK likely was slowed in the present study, which also could contribute to the slower activation of pulmonary V˙o2 (and muscle O2 consumption) in the RALK condition of the present study.
Respiratory alkalosis and plasma lactate accumulation
Lactate accumulation is a consequence of an imbalance between the rate of pyruvate production via glycolysis and the oxidative removal of pyruvate, catalysed by the mitochondrial PDH reaction (Howlett et al. 1999). The plasma lactate− concentration of ∼3 mmol l−1 during CON is in agreement with previous reports examining plasma lactate− concentrations during steady-state, moderate-intensity cycling exercise (Scheuermann et al. 1998; LeBlanc et al. 2002). Also, the higher plasma lactate− concentrations observed in the present study in RALK compared to CON (Fig. 4) are similar to those reported by LeBlanc et al. (2002) and are likely to reflect a delayed activation of PDH with a higher rate of glycogenolysis and pyruvate production, lower rate of pyruvate oxidation, greater muscle lactate accumulation (LeBlanc et al. 2002), along with greater lactate− efflux from muscle (Spriet et al. 1986) and lower lactate− clearance from plasma. Lactate− efflux from the muscle occurs via a membrane-bound monocarboxylate transporter with transport kinetics determined by the [H+] gradient across the sacrolemma (Spriet et al. 1986; Lindinger et al. 1990; Roth & Brooks, 1990; Juel, 1997; Juel & Halestrap, 1999). Lactate− efflux from the muscle would therefore be favoured in a hyperventilation-mediated reduction in blood [H+]. A greater lactate− accumulation in muscle (LeBlanc et al. 2002), and lower plasma [H+] (present study) but higher end-exercise intracellular [H+] (Forbes et al. 2007) in RALK would contribute to a higher muscle-to-plasma lactate− concentration gradient and muscle-to-plasma [H+] gradient, both of which would promote lactate− efflux from muscle in RALK, and thus contribute to the higher plasma lactate− concentration which was observed in RALK. Also, the higher plasma [lactate−] in RALK may be a consequence of a lower tissue blood flow (see above) and lactate− delivery to inactive muscle (and other tissues), thereby reducing tissue lactate− uptake and removal (Nielsen et al. 2002).
Hyperventilation and V˙o2
A higher end-exercise pulmonary V˙o2 was observed in RALK (1.95 l min−1) compared to CON (1.87 l min−1) in spite of the same work rate and, presumably, leg ATP requirement in both conditions. Pulmonary V˙o2 reflects whole-body O2 requirements, and in RALK, this includes the extra O2 cost of respiratory muscle work. Thus, the increased O2 cost of breathing in RALK could contribute to the higher end-exercise V˙o2. Aaron et al. (1992) reported an O2 cost of breathing of 1.8 ml O2 min−1 l−1V˙E for exercise intensities requiring a V˙E∼75 l min−1 (∼70%V˙o2max). Using this estimate (Aaron et al. 1992), the ∼43 l min−1 greater end-exercise V˙E in RALK (87 l min−1) than in CON (44 l min−1) would require an additional ∼77 ml O2 min−1; essentially identical to the 80 ml min−1 greater V˙o2 observed in the steady-state of RALK compared to CON. Therefore, the increased work of breathing and higher V˙o2 associated with RALK in the present study is in agreement with the linear, inverse relationship that exists between 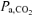 and V˙o2 (Karetzky & Cain, 1970; Cain, 1970; Harken, 1976). Alternatively, Cain (1970) and Harken (1976) observed a higher V˙o2 in association with reduced
and V˙o2 (Karetzky & Cain, 1970; Cain, 1970; Harken, 1976). Alternatively, Cain (1970) and Harken (1976) observed a higher V˙o2 in association with reduced 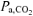 in their dog preparations suggesting that the factors intrinsic to muscle may be affected by a lower
in their dog preparations suggesting that the factors intrinsic to muscle may be affected by a lower 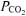 and contribute to the higher V˙o2 in RALK.
and contribute to the higher V˙o2 in RALK.
During the transition to exercise, the contribution of the extra O2 cost of breathing to total pulmonary V˙o2 will depend on the difference in absolute V˙E and on the rate of increase of V˙E (i.e. τV˙E) during the transition to exercise. In the present study, although the V˙E was controlled during the hyperventilation in order to lower 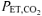 to a required level of ∼20 mmHg, the τV˙E was not different in RALK (56 s) and CON (57 s) suggesting that differences in the adaptation of V˙E likely do not contribute to the slow pulmonary V˙o2 response seen in RALK.
to a required level of ∼20 mmHg, the τV˙E was not different in RALK (56 s) and CON (57 s) suggesting that differences in the adaptation of V˙E likely do not contribute to the slow pulmonary V˙o2 response seen in RALK.
Conclusion
The present findings demonstrate that the adaptation of pulmonary V˙o2 and presumably muscle O2 consumption at the onset of moderate-intensity exercise were significantly slowed when exercise was accompanied by a period of voluntary hyperventilation-induced hypocapnic alkalosis (RALK). While the mechanism responsible for the slower V˙o2 kinetics cannot be definitively elucidated in this study, a greater metabolic inertia in RALK consequent to a slower rate of provision of oxidative substrate to the mitochondrial TCA cycle and ETC (because of an attenuated activation of PDH) is expected. However, our pulmonary V˙o2 data used in combination with NIRS measures of muscle deoxygenation also suggest that the adaptation of blood flow is slower and possibly lower in RALK, which may also contribute to slower pulmonary V˙o2 kinetics and activation of muscle O2 consumption in RALK.
Acknowledgments
This study was supported by operating grants from the Natural Sciences and Engineering Research Council (NSERC) of Canada. Additional support was provided by a UWO Academic Development Fund Grant and infrastructure support of the Canadian Foundation for Innovation (CFI) and Ontario Innovation Trust (OIT). We wish to acknowledge the technical support provided by Brad Hansen and the subjects for their participation.
References
- Aaron EA, Johnson BD, Seow CK, Dempsey JA. Oxygen cost of exercise hyperpnea: measurement. J Appl Physiol. 1992;72:1810–1817. doi: 10.1152/jappl.1992.72.5.1810. [DOI] [PubMed] [Google Scholar]
- Barstow TJ, Lamarra N, Whipp BJ. Modulation of muscle and pulmonary O2 uptakes by circulatory dynamics during exercise. J Appl Physiol. 1990;68:979–989. doi: 10.1152/jappl.1990.68.3.979. [DOI] [PubMed] [Google Scholar]
- Beaver WL, Lamarra N, Wasserman K. Breath-by-breath measurement of true alveolar gas exchange. J Appl Physiol. 1981;51:1662–1675. doi: 10.1152/jappl.1981.51.6.1662. [DOI] [PubMed] [Google Scholar]
- Brandi G, Clode M. CO2 washout during hyperventilation in man. Respir Physiol. 1969;7:163–172. doi: 10.1016/0034-5687(69)90003-6. [DOI] [PubMed] [Google Scholar]
- Brice AG, Welch HG. Effect of respiratory alkalosis on skeletal muscle metabolism in the dog. J Appl Physiol. 1985;58:658–664. doi: 10.1152/jappl.1985.58.2.658. [DOI] [PubMed] [Google Scholar]
- Cain SM. Increased oxygen uptake with passive hyperventilation of dogs. J Appl Physiol. 1970;28:4–7. doi: 10.1152/jappl.1970.28.1.4. [DOI] [PubMed] [Google Scholar]
- DeLorey DS, Kowalchuk JM, Paterson DH. Relationship between pulmonary O2 uptake kinetics and muscle deoxygenation during moderate-intensity exercise. J Appl Physiol. 2003;95:113–120. doi: 10.1152/japplphysiol.00956.2002. [DOI] [PubMed] [Google Scholar]
- Elwell CE. A Practical User's Guide to Near Infrared Spectroscopy. London: Hamamatsu Photonics KK; 1995. [Google Scholar]
- Ferreira LF, Townsend DK, Lutjemeier BJ, Barstow TJ. Muscle capillary blood flow kinetics estimated from pulmonary O2 uptake and near-infrared spectroscopy. J Appl Physiol. 2005;98:1820–1828. doi: 10.1152/japplphysiol.00907.2004. [DOI] [PubMed] [Google Scholar]
- Forbes SC, Kowalchuk JM, Thompson RT, Marsh GD. Effects of hyperventilation on phosphocreatine kinetics and muscle deoxygenation during moderate-intensity plantar flexion exercise. J Appl Physiol. 2007;102:1565–1573. doi: 10.1152/japplphysiol.00895.2006. [DOI] [PubMed] [Google Scholar]
- Forbes SC, Raymer GH, Kowalchuk JM, Marsh GD. NaHCO3-induced alkalosis reduces the phosphocreatine slow component during heavy-intensity forearm exercise. J Appl Physiol. 2005;99:1668–1675. doi: 10.1152/japplphysiol.01200.2004. [DOI] [PubMed] [Google Scholar]
- Grassi B. Regulation of oxygen consumption at exercise onset: is it really controversial? Exerc Sport Sci Rev. 2001;29:134–138. doi: 10.1097/00003677-200107000-00009. [DOI] [PubMed] [Google Scholar]
- Grassi B, Pogliaghi S, Rampichini S, Quaresima V, Ferrari M, Marconi C, Cerretelli P. Muscle oxygenation and pulmonary gas exchange kinetics during cycling exercise on-transitions in humans. J Appl Physiol. 2003;95:149–158. doi: 10.1152/japplphysiol.00695.2002. [DOI] [PubMed] [Google Scholar]
- Grassi B, Poole DC, Richardson RS, Knight DR, Erickson BK, Wagner PD. Muscle O2 uptake kinetics in humans: implications for metabolic control. J Appl Physiol. 1996;80:988–998. doi: 10.1152/jappl.1996.80.3.988. [DOI] [PubMed] [Google Scholar]
- Gurd BJ, Scheuermann BW, Paterson DH, Kowalchuk JM. Prior heavy-intensity exercise speeds VO2 kinetics during moderate-intensity exercise in young adults. J Appl Physiol. 2005;98:1371–1378. doi: 10.1152/japplphysiol.01028.2004. [DOI] [PubMed] [Google Scholar]
- Gustafsson U, Sjoberg F, Lewis DH, Thorborg P. The effect of hypocapnia on skeletal muscle microcirculatory blood flow, oxygenation and pH. Int J Microcirc Clin Exp. 1993;12:131–141. [PubMed] [Google Scholar]
- Haddy FJ, Scott JB. Metabolically linked vasoactive chemicals in local regulation of blood flow. Physiol Rev. 1968;48:688–707. doi: 10.1152/physrev.1968.48.4.688. [DOI] [PubMed] [Google Scholar]
- Harken AH. Hydrogen ion concentration and oxygen uptake in an isolated canine hindlimb. J Appl Physiol. 1976;40:1–5. doi: 10.1152/jappl.1976.40.1.1. [DOI] [PubMed] [Google Scholar]
- Hayashi N, Ishihara M, Tanaka A, Yoshida T. Impeding O2 unloading in muscle delays oxygen uptake response to exercise onset in humans. Am J Physiol Regul Integr Comp Physiol. 1999;277:R1274–R1281. doi: 10.1152/ajpregu.1999.277.5.R1274. [DOI] [PubMed] [Google Scholar]
- Hogan MC. Fall in intracellular PO2 at the onset of contractions in Xenopus single skeletal muscle fibers. J Appl Physiol. 2001;90:1871–1876. doi: 10.1152/jappl.2001.90.5.1871. [DOI] [PubMed] [Google Scholar]
- Howlett RA, Heigenhauser GJ, Hultman E, Hollidge-Horvat MG, Spriet LL. Effects of dichloroacetate infusion on human skeletal muscle metabolism at the onset of exercise. Am J Physiol Endocrinol Metab. 1999;277:E18–E25. doi: 10.1152/ajpendo.1999.277.1.E18. [DOI] [PubMed] [Google Scholar]
- Hughson RL, Shoemaker JK, Tschakovsky ME, Kowalchuk JM. Dependence of muscle VO2 on blood flow dynamics at onset of forearm exercise. J Appl Physiol. 1996;81:1619–1626. doi: 10.1152/jappl.1996.81.4.1619. [DOI] [PubMed] [Google Scholar]
- Jones NL, Jurkowski JE. Body carbon dioxide storage capacity in exercise. J Appl Physiol. 1979;46:811–815. doi: 10.1152/jappl.1979.46.4.811. [DOI] [PubMed] [Google Scholar]
- Juel C. Lactate-proton cotransport in skeletal muscle. Physiol Rev. 1997;77:321–358. doi: 10.1152/physrev.1997.77.2.321. [DOI] [PubMed] [Google Scholar]
- Juel C, Halestrap AP. Lactate transport in skeletal muscle – role and regulation of the monocarboxylate transporter. J Physiol. 1999;517:633–642. doi: 10.1111/j.1469-7793.1999.0633s.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karetzky MS, Cain SM. Effect of carbon dioxide on oxygen uptake during hyperventilation in normal man. J Appl Physiol. 1970;28:8–12. doi: 10.1152/jappl.1970.28.1.8. [DOI] [PubMed] [Google Scholar]
- Karlsson T, Stjernstrom EL, Stjernstrom H, Norlen K, Wiklund L. Central and regional blood flow during hyperventilation. An experimental study in the pig. Acta Anaesthesiol Scand. 1994;38:180–186. doi: 10.1111/j.1399-6576.1994.tb03863.x. [DOI] [PubMed] [Google Scholar]
- Kontos HA, Richardson DW, Raper AJ, Zubair-ul-Hassan, Patterson JL., Jr Mechanisms of action of hypocapnic alkalosis on limb blood vessels in man and dog. Am J Physiol. 1972;223:1296–1307. doi: 10.1152/ajplegacy.1972.223.6.1296. [DOI] [PubMed] [Google Scholar]
- Lamarra N, Whipp BJ, Ward SA, Wasserman K. Effect of interbreath fluctuations on characterizing exercise gas exchange kinetics. J Appl Physiol. 1987;62:2003–2012. doi: 10.1152/jappl.1987.62.5.2003. [DOI] [PubMed] [Google Scholar]
- LeBlanc PJ, Parolin ML, Jones NL, Heigenhauser GJ. Effects of respiratory alkalosis on human skeletal muscle metabolism at the onset of submaximal exercise. J Physiol. 2002;544:303–313. doi: 10.1113/jphysiol.2002.022764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindinger MI, Heigenhauser GJ, Spriet LL. Effects of alkalosis on muscle ions at rest and with intense exercise. Can J Physiol Pharmacol. 1990;68:820–829. doi: 10.1139/y90-125. [DOI] [PubMed] [Google Scholar]
- Nielsen HB, Clemmesen JO, Skak C, Ott P, Secher NH. Attenuated hepatosplanchnic uptake of lactate during intense exercise in humans. J Appl Physiol. 2002;92:1677–1683. doi: 10.1152/japplphysiol.00028.2001. [DOI] [PubMed] [Google Scholar]
- Oren A, Whipp BJ, Wasserman K. Effect of acid-base status on the kinetics of the ventilatory response to moderate exercise. J Appl Physiol. 1982;52:1013–1017. doi: 10.1152/jappl.1982.52.4.1013. [DOI] [PubMed] [Google Scholar]
- Rossiter HB, Ward SA, Doyle VL, Howe FA, Griffiths JR, Whipp BJ. Inferences from pulmonary O2 uptake with respect to intramuscular [phosphocreatine] kinetics during moderate exercise in humans. J Physiol. 1999;518:921–932. doi: 10.1111/j.1469-7793.1999.0921p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossiter HB, Ward SA, Howe FA, Wood DM, Kowalchuk JM, Griffiths JR, Whipp BJ. Effects of dichloroacetate on VO2 and intramuscular 31P metabolite kinetics during high-intensity exercise in humans. J Appl Physiol. 2003;95:1105–1115. doi: 10.1152/japplphysiol.00964.2002. [DOI] [PubMed] [Google Scholar]
- Roth DA, Brooks GA. Lactate and pyruvate transport is dominated by a pH gradient-sensitive carrier in rat skeletal muscle sarcolemmal vesicles. Arch Biochem Biophys. 1990;279:386–394. doi: 10.1016/0003-9861(90)90506-t. [DOI] [PubMed] [Google Scholar]
- Scheuermann BW, Kowalchuk JM, Paterson DH, Cunningham DA. O2 uptake kinetics after acetazolamide administration during moderate- and heavy-intensity exercise. J Appl Physiol. 1998;85:1384–1393. doi: 10.1152/jappl.1998.85.4.1384. [DOI] [PubMed] [Google Scholar]
- Spriet LL, Heigenhauser GJ. Regulation of pyruvate dehydrogenase (PDH) activity in human skeletal muscle during exercise. Exerc Sport Sci Rev. 2002;30:91–95. doi: 10.1097/00003677-200204000-00009. [DOI] [PubMed] [Google Scholar]
- Spriet LL, Lindinger MI, Heigenhauser GJ, Jones NL. Effects of alkalosis on skeletal muscle metabolism and performance during exercise. Am J Physiol Regul Integr Comp Physiol. 1986;251:R833–R839. doi: 10.1152/ajpregu.1986.251.5.R833. [DOI] [PubMed] [Google Scholar]
- Timmons JA, Gustafsson T, Sundberg CJ, Jansson E, Greenhaff PL. Muscle acetyl group availability is a major determinant of oxygen deficit in humans during submaximal exercise. Am J Physiol Endocrinol Metab. 1998;274:E377–E380. doi: 10.1152/ajpendo.1998.274.2.E377. [DOI] [PubMed] [Google Scholar]
- Tschakovsky ME, Hughson RL. Interaction of factors determining oxygen uptake at the onset of exercise. J Appl Physiol. 1999;86:1101–1113. doi: 10.1152/jappl.1999.86.4.1101. [DOI] [PubMed] [Google Scholar]
- Ward SA, Whipp BJ, Koyal S, Wasserman K. Influence of body CO2 stores on ventilatory dynamics during exercise. J Appl Physiol. 1983;55:742–749. doi: 10.1152/jappl.1983.55.3.742. [DOI] [PubMed] [Google Scholar]
- Whipp BJ, Mahler M. Dynamics of pulmonary gas exchange during exercise. In: West JB, editor. Pulmonary Gas Exchange. New York: Academic Press; 1980. pp. 33–96. [Google Scholar]
- Whipp BJ, Rossiter HB. The kinetics of oxygen uptake: physiological inferences from the parameters. In: Jones AM, Poole DC, editors. Oxygen Uptake Kinetics in Sport, Exercise and Medicine. New York: Taylor & Francis Inc.; 2005. pp. 62–94. [Google Scholar]
- Zoladz JA, Szkutnik Z, Duda K, Majerczak J, Korzeniewski B. Preexercise metabolic alkalosis induced via bicarbonate ingestion accelerates VO2 kinetics at the onset of a high-power-output exercise in humans. J Appl Physiol. 2005;98:895–904. doi: 10.1152/japplphysiol.01194.2003. [DOI] [PubMed] [Google Scholar]