

Abstract
Background
Elevated phosphorylation of neurotophin-regulated transcription factors, such as cAMP-response Element Binding Protein (CREB), in the hippocampus has been proposed as a common mediator of antidepressant (ADT) efficacy in otherwise naïve rodents. The intracellular factors by which ADTs and glucocorticoids, causal factors in depression, regulate depression-like behavior remain unclear, but Extracellular signal-Regulated Kinase 1/2 (ERK1/2), upstream of CREB, is a likely candidate.
Methods
We explored the long-term consequences of glucocorticoid exposure and subsequent ADT treatment in a novel model of chronic depression. Motivated behaviors, immobility during tail suspension, and ERK1/2, known to be required for behavioral response to ADTs, were quantified.
Results
Chronic corticosterone (CORT) increased immobility, decreased responding in an operant conditioning task of motivation, and selectively reduced pERK1/2 in the dentate gyrus. Behavioral and biochemical measures were restored to baseline by amitriptyline (AMI) treatment. CORT regulated pERK1/2 on a timecourse that paralleled increases in heat-shock proteins associated with depression and decreased trkB receptor phosphorylation. Chronic AMI also produced regionally-dissociable effects on pERK1/2 in CA1/CA3, amygdala, and striatum, but not prefrontal cortex.
Conclusions
ADT efficacy in a motivational task and “behavioral despair” assay is associated with altered limbic pERK1/2, including restored pERK1/2 in the dentate gyrus after stress-related insult.
Introduction
Neurotrophic hypotheses of depression and ADT efficacy argue stress and ADTs chronically regulate mood via opposing action on intracellular and transcription factors associated with cellular growth, motility, and survival, primarily in the hippocampus (1). Consistent with this hypothesis, hippocampal CREB over-expression produces an ADT-like behavioral phenotype (2), while stress decreases BDNF expression (3,4) and suppresses local CREB phosphorylation (5). Enhancing cortical and hippocampal pCREB has been proposed as a common ADT mechanism (6), based largely on studies of ADT action in the naïve rodent (e.g.,7). ADT effects on transcription factor activation have largely not been verified in animal models of depression, however, nor have the intracellular mechanisms of action between receptor binding and transcription factor phosphorylation been determined, though a likely signaling candidate is ERK1/2.
The ERK MAP kinase pathway is an intracellular signaling cascade implicated in several forms of learning, memory, and neuroplasticity (8,9). ERK1/2 is activated by BDNF binding of the trkB receptor via the Ras-Raf-MEK-ERK cascade, inducing nuclear translocation and phosphorylation of target transcription factors. As such, ERK translates extracellular events, such as neurotransmitter and neurotrophin receptor binding, into gene transcription, synaptic remodeling, and behavioral events (10,11). For example, BDNF infusion into the dentate gyrus has ADT-like consequences that can be blocked by inhibiting ERK1/2 phosphorylation (12). Peripheral injection of MAP kinase kinase (MEK) inhibitors produces helplessness-like behaviors and eliminates response to ADTs in standard assays of “behavioral despair” (13), revealing a potential role for ERK1/2 in mood regulation and ADT therapeutic utility.
While ERK1/2 phosphorylation has been hypothesized as an intracellular signaling mechanism mediating ADT efficacy in depressed humans and animal models of depression, supporting evidence is largely limited to studies in otherwise naïve rodents or in vitro models (14,15, but see 16), despite evidence from post-mortem studies of depressive suicide subjects (17,18). It is not known if: 1) ERK1/2 is involved in reversal of the depression-like phenotype by ADT administration, and 2) if so, whether phosphorylation of both kinase isoforms is uniformly regulated throughout cortico-limbic circuits implicated in depression. We demonstrate AMI chronically regulates limbic ERK1/2 in a regionally- and isoform-specific manner consistent with enhancing neuroplastic processes, such as LTP. AMI treatment also restored progressive ratio (PR) responding in a motivation task in mice and increased mobility during tail suspension after corticosteroid exposure in a novel stress-related model of chronic depression.
Methods
Subjects
Experimentally naïve C57bl/6J mice (n=159, 8–10 wks, housed 4–5/cage, Charles River Laboratories, Kingston, VA) were used in the following studies. Sixty-six were initially food-restricted (allowed 75-min. food access daily) to prevent weight gain during conditioning experiments and motivate operant responding for food reinforcers. Remaining animals had free access to food/water. During CORT exposure and ADT treatment, all mice had free access to food and water. Mice were maintained on a 12-hour light cycle (0700 on). Procedures were approved by the Yale University Animal Care and Use Committee.
Operant conditioning
Experimenters used standard aluminum operant chambers for mice (16x14x12.5 cm) with grid floors controlled by MedPC software (Med Associates Inc., George, VT). Each chamber was housed in a sound-attenuating outer chamber equipped with a white noise generator and fan. A house light illuminated the chamber, and a dispenser delivered grain-based food pellets (20 mg; Bio-Serv, Frenchtown, NJ) into the magazine. Head entries into the stimuli and magazine were detected by photocell.
Mice were initially allowed to consume reinforcer pellets in their home cages for 1 day. Mice were then habituated to the testing apparatus with 15 min unlimited food pellet access. Next, subjects were trained to nose poke, during which inserting the nose into one of three holes was reinforced; responding on the other holes had no consequences. The first 10 reinforcers were obtained on a fixed ratio 1 schedule, followed by a variable ratio 2 (VR2) schedule, in which 1, 2, or 3 responses were reinforced as randomly determined by the operating computer, for the duration of the 15-min session (see 19). Mice were required to retrieve each pellet before further reinforcement. The position of the active nose poke (left, right, or center) was counter-balanced between and within groups. Mice were required to display stable responding for 3 consecutive days to complete the acquisition phase. All mice acquired the task within 10–15 days; the last 10 are shown (fig. 1b).
Fig. 1.
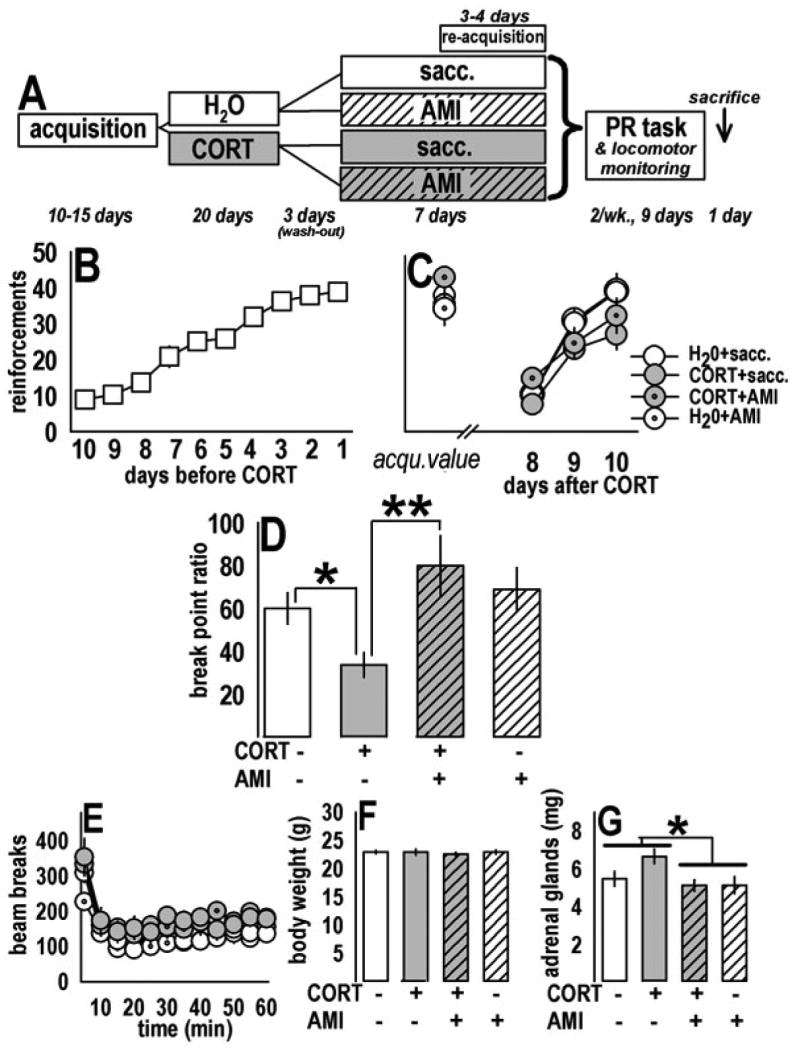
Prior CORT exposure suppressed motivated responding; subsequent chronic AMI treatment reversed the deficit. (a) Experimental timeline: Naïve mice were first trained to perform an operant responding task for food (“acquisition”). Animals were then exposed to CORT and subsequently treated with AMI; the contents of the drinking water after training and before progressive ratio (“PR”) testing are represented. “Sacc.” refers to saccharin, dissolved in the drinking water to increase the palatability of AMI. Mice were tested on 3–4 re-acquisition sessions and then 3 PR sessions over the course of 9 days, with locomotor monitoring on the last day. Mice were then sacrificed for post-mortem analyses (figs. 3,4). (b) The number of reinforcements food-restricted mice acquired during training is represented. After training, mice were allowed free access to food and were exposed to CORT and subsequent AMI in the drinking water. (c) One week after CORT, mice successfully re-acquired the task to previous (“acqu.”) levels. (d) In the PR task, however, break point ratios were decreased after CORT; this deficit was reversed by subsequent AMI treatment. AMI alone had no effect on PR responding. (e) Ambulatory activity was modestly increased in CORT-exposed mice, with no effect of AMI, suggesting decreased PR responding in the CORT+sacc. group cannot be attributed to decreased locomotor activity. (f) Body weights did not differ at the time of test. (g) In weighing adrenal glands, a main effect of AMI was detected, suggesting AMI decreased HPA activity as has been described previously (ref. 25). Notably, no effect of CORT was observed, suggesting the prior CORT exposure did not grossly alter HPA axis activity after wash-out, despite long-terms behavioral and biochemical consequences. Bars represent means (±SEM) per treatment group (*p≤0.05, **p≤0.001).
CORT (25 μg/ml free base; 4-pregnen-11β 21-DIOL-3 20-DIONE 21-hemisuccinate; Steraloids, Inc., Newport, RI) was then dissolved in tap water and neutralized to a pH of 7.0–7.4 with HCl. Group-housed mice were presented with CORT in place of normal drinking water for 14 days, resulting in a dose of approximately 6.9 mg/kg/day, p.o. Animals were weaned with 3 days of 12.5 μg/ml, then 3 days with 6.25 μg/ml, to allow for gradual recovery of endogenous CORT secretion. Three days after CORT, mice were administered AMI hydrochloride (200 μg/ml; Sigma Aldrich, St. Louis, MO) dissolved in 2% w/v saccharin (SACC; Sigma) to increase palatability for 1 wk. This dose has been shown to produce blood plasma concentrations of AMI in C57bl/6J mice comparable to those in AMI-treated humans (20). Control mice received SACC in the drinking water. Bottles were weighed daily. Solutions were no more than 3 days old and stored at 4°C.
Responding was restored to training levels with 3–4 conditioning sessions identical to acquisition trials; the last 3 sessions are shown (fig. 1c). Starting 11 days after CORT (=1 day after last AMI administration), PR responding was assessed every 3 days. Operant responding on a PR schedule is thought to quantify the reinforcing properties of the reinforcement and an animal’s motivation to acquire that reinforcement (21). In this test, the response requirement progressively increased on a linear scale (i.e., 1, 5, 9, x+4 responses:reinforcement) such that mice had to respond progressively more for subsequent reinforcement. The tests ended when no active responses were emitted for 5 min, or the mouse had not “timed out” within 4 hrs (n=3 across all sessions). The highest ratio of responses:reinforcements earned is termed the “break point ratio” and serves as the dependent variable. Data were analyzed by two-factor (CORTxADT) ANOVA with repeated measures when appropriate and Student’s post-hoc comparisons.
Twenty-four hours after the last PR session, locomotor activity was monitored to verify differences in responding were not due to locomotor differences between groups. With mice still food-restricted, ambulatory activity in a clean cage was quantified using the automated Omnitech Digiscan Micromonitor system equipped with 16 photocells (Columbus, OH). Data are represented as photobeams broken across 1 hr.
An additional group of mice was trained to perform the operant response, then subjected to 3 PR sessions before CORT exposure and 3 sessions after to test whether CORT selectively decreases responding when the PR task is novel, i.e., when the response contingency is first learned. Also, because the PR task has been suggested to test responding in extinction, and not primary motivation, non-reinforced responding was analyzed in another group of mice on days 14–19 after CORT exposure. In this test, the food hopper and magazine were disconnected; all other conditions were identical to those during training.
Tail Suspension Test (TST)
The TST is a standard assay of ADT efficacy, and was conducted to complement our PR studies, as PR responding is not conventionally associated with ADT efficacy or animal models of depression, even though decreased motivation is a hallmark symptom of depression. Mice were first exposed to CORT as described, expected to increase immobility. After exposure, AMI (200 μg/ml) or fluoxetine (FLX; 160 μg/ml) for comparison to a more popularly prescribed ADT was dissolved in the drinking water with 2% w/v SACC for 2 wks. ADTs were removed the day before testing. This protocol would not necessarily be expected to decrease baseline immobility (22), but was hypothesized to restore any CORT-induced increase in immobility to control levels. Mice were individually suspended by securely taping the tail 1/2 cm from the tip to flexible plastic tubing secured to the edge of a table in a quiet room. Immobility time was scored by a blinded observer over the 6 min test.
Tissue harvest and preparation
Still food-restricted, mice in the initial PR experiment (fig. 1) were sacrificed 20 days after CORT and at the same time of day at which PR testing would have occurred. Immediately after decapitation, brains were frozen on dry ice. Adrenal glands were excised from surrounding tissue and weighed as an approximate reflection of recent hypothalamic-pituitary-adrenal (HPA) axis activity to verify that the CORT weaning protocol described above was effective.
An additional group of behaviorally naïve mice was sacrificed 1 or 14 days after CORT exposure for a timecourse analysis of ERK1/2 and trkB receptor phosphorylation in the dentate gyrus. Glucose-regulated protein 78 (GRP78) and calreticulin (calr.), heat-shock proteins associated with cellular stress and depression in humans (23,24) but not characterized in the depressive-like rodent, were also assayed. Trunk blood was collected, and serum CORT was analyzed by ELISA (Assay Designs, Ann Arbor, MI). After initial analysis, an additional group of mice was exposed to water+SACC, CORT+SACC, or CORT+AMI (2 wks, 200 μg/ml) to evaluate whether GRP78 and calr. were sensitive to ADT. Control groups did not differ and are combined for representative/statistical purposes.
Frozen brains were cut into 1 mm slices using a chilled brain matrix (Plastics One, Roanoke, VA). Bilateral punches (1 mm diameter, Fine Science Tools, Foster City, CA) were aimed at the dentate gyrus (−1.46mm AP, ±0.5mm ML, −1.9mm DV), CA1/CA3 (−2.46mm AP, ±2.9mm ML, −2.9mm DV), amygdala (−1.34mm AP, ±3.0mm ML, −4.75mm DV), and dorsal striatum (+2.75mm AP, ±1.4mm ML, −2.9mm DV) and rapidly refrozen on dry ice. Samples from the medial prefrontal cortex (mPFC) were collected with a midline punch (+1.8mm AP, −2.3mm DV). Samples were sonicated in lysis buffer (200 μl: 137 mM NaCl, 20 mM tris-Hcl [pH=8], 1% igepal, 10% glycerol, 1:100 Protease and Phosphatase Inhibitor Cocktails 1 and 2 [Sigma]) and stored at −80°C. Protein concentrations were determined using a Bradford colorimetric assay kit (Pierce, Rockland, IL).
Western blotting
Twenty μg of issue was diluted 4:1 with 5X laemmili buffer (20% glycerol, 2% SDS, Bromphenol blue), and loaded onto 10% or 8–16% gradient tris-glycine gels (Invitrogen, Carlsbad, CA) for SDS-polyacrylamide gel electrophoresis separation. After transfer, nitrocellulose membranes (0.2 μM, Bio-Rad, Hercules, CA) were immunoblotted with anti-phospho-ERK1/2 (Ms; 1:1,000; Cell Signaling, Beverly, MA). ERK was determined with anti-ERK1/2 (Rb; 1:2,000; Cell Signaling), and GRP78, calreticulin, ptrkB, and trkB with anti-GRP78 (Rb; 1:3,000, Stressgen Bioreagents, Ann Arbor, MI), anti-calreticulin (Ms; 1:5,000, Stressgen), anti-phospho-trkB(Tyr490) (1:500; Cell Signaling) and anti-trkB (Ms; 1:1,000; BD Biosciences, San Jose, CA), respectively. GAPDH was the loading control (Ms; 1:80,000; Advanced Immunochemical Inc., Long Beach, CA). For antibody detection by the Odyssey infrared imaging system (LI-COR, Lincoln, NE), membranes were incubated with IRDye 700 Dx Anti-Rb IgG and IRDye 800 Dx Anti-Ms IgG (1:5,000; Rockland Immunochemicals, Gilbertsville, PA). Fluorescence was detected by the Odyssey Imager; visible bands were quantified using Odyssey software. To control for variance between gels, data were converted to percent of control samples on the same gel. pERK signals were quantified as percent total ERK, which did not change in any analysis. ptrkB, trkB, GRP78, and calreticulin were calculated as percent of GAPDH loading control values. Data were analyzed by one- or two-factor ANOVA, with Student’s post-hoc comparisons. Statistical outliers were identified using standard criteria (falling >2 standard deviations outside the mean) and excluded (≤2/analysis). Amygdalar and CA1/CA3 data were square-root transformed to preserve required homogeneity of variance.
Results
Operant conditioning, PR responding, and ambulatory activity
Mice successfully acquired the task, as measured by reinforcements earned over the last 10 days of training [F(9,288)=60.1, p<0.001] (fig. 1b) with no differences between mice designated to the four treatment groups [F(3,3)<1]. Following CORT, 2-factor RM-ANOVA revealed successful task reacquisition, as indicated by an increasing number of reinforcements earned each day [F(2,64)=61.0, p<0.001, post-hoc ps<0.05] with no main effects of CORT or AMI, and no CORTxAMI interaction (ps≥0.18) (fig. 1c).
To analyze PR responding, each animal’s 3 break point ratios were averaged and analyzed, revealing a significant CORTxAMI interaction (F(1,1)=4.42, p=0.04). Post-hoc analyses revealed: 1) CORT exposure decreased break point ratios (p=0.03), and 2) the deficit was restored to control levels by subsequent AMI treatment (p=0.001) with no effects of AMI alone on PR responding (p=0.5 compared to water controls) (fig. 1d). Ambulatory activity in a novel environment was decreased across time in all subjects [F(11,375)=25.95, p<0.001] with a main effect of CORT [F(1,1)=9.2, p=0.005] and no interaction effect of CORT and AMI [F(1,375)<1] (fig. 1e). Nonetheless, the PR responding deficit after CORT is unlikely to be due to gross differences in locomotor activity, as these data indicate CORT exposure increases, not decreases, activity. At the time of test, body weights did not differ between groups [interaction F(1,1)<1; main effect Fs(1,1)<1] (fig. 1f). Post-mortem analysis of adrenal gland weights revealed a main effect of AMI [F(1,1)=6.0, p=0.02] but only a trend for an interaction effect [F(1,1)=2.5, p=0.1], suggesting AMI non-specifically decreased adrenal CORT secretion (fig. 1g), as previously reported (25), but prior CORT had no effects.
Another cohort of mice was tested in the PR task 3 days before and after CORT exposure. Two-factor RM-ANOVA revealed no main effect of CORT on break point ratios under these conditions [F(1,1)<1] and no interaction of drug and time relative to exposure [F(5,5)=1.2, p=0.35] (fig. 2a). Similarly, CORT did not affect responding in extinction [F(1,1)<1], and no drug x day interaction was observed [F(4,64)<1] (fig. 2b).
Fig. 2.
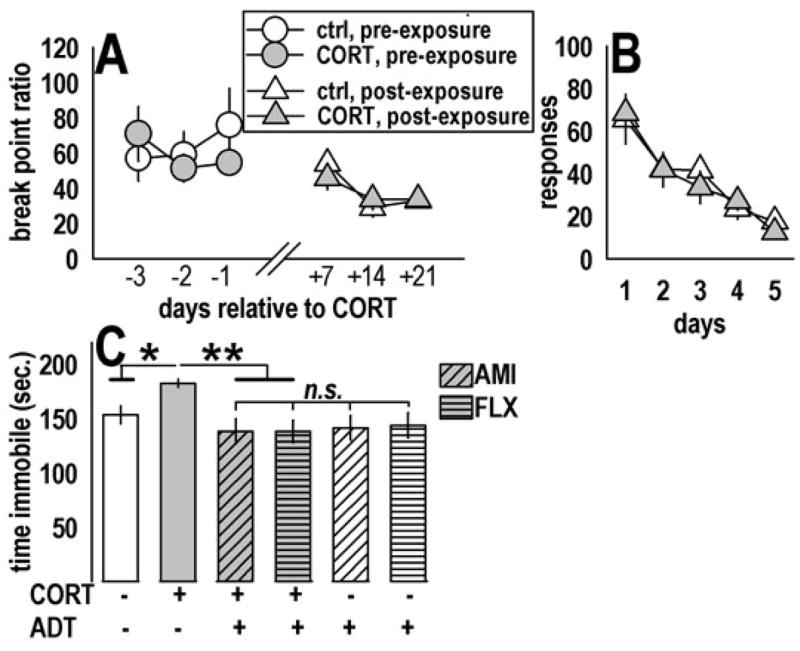
Prior CORT exposure did not influence operant responding in PR-experienced mice or under non-reinforcement (extinction) conditions, but increased immobility in the TST. (a) CORT-exposed mice with previous experience with the PR task were resistant to CORT’s effects on subsequent PR responding. These data provide evidence that CORT specifically increases sensitivity to changing reinforcement contingencies from the relatively non-taxing schedule of reinforcement used during training to the taxing PR schedule. (b) Prior CORT exposure also did not alter responding in the complete absence of reinforcement; as such, the observed decrease in PR responding after CORT (fig. 1d) cannot be attributed to increased sensitivity to extinction conditions. Rather, decreased responding likely reflects decreased motivation to acquire an outcome, a hallmark symptom of depression. (c) Prior CORT exposure increased immobility (“behavioral despair”) in the TST; importantly, chronic oral administration of both AMI (diagonal lines) and FLX (horizontal lines) restored responding, suggesting CORT produces an ADT-sensitive depressive-like response in this test. AMI and FLX alone did not further decrease immobility, perhaps because ADTs were not on-board during testing. Feelings of helplessness and despair are also core symptoms of depression. Bars and symbols represent means (±SEM) per treatment group (*p≤0.05, **p≤0.001).
TST
Analysis of total immobility time in the TST revealed main effects of ADTs [F(2,52)=4.9, p=0.01), and ADT-treated mice did not differ from each other (ps≥0.5). When analyzed together, an interaction of CORT and ADT was detected [F(1,51)=3.8, p=0.05] (fig. 2c). Post-hoc analyses revealed CORT-exposed mice had higher immobility scores than other groups (vs. control p=0.04; vs. subsequent ADT p=0.001).
Western blotting
pERK1/2 was analyzed in multiple cortico-limbic regions after CORT and AMI. CORTxAMI interaction effects were detected in the dentate gyrus [F(1,1)=4.6, p=0.04] and dorsal striatum [F(1,1)=5.8, p=0.02]: Prior CORT suppressed pERK1/2 in the dentate gyrus, and this deficit was reversed by AMI, such that AMI-treated samples were significantly increased compared to CORT-exposed samples (ps≤0.01), while AMI-only and control groups did not differ (p=0.9) (fig. 3a). In the dorsal striatum, post-hoc tests revealed a main effect of CORT (p=0.004) and selective decrease in pERK1/2 in CORT-exposed animals subsequently treated with AMI (p=0.001), with a trend for the same effect when CORT and CORT+AMI samples were compared (p=0.08) (fig. 3b). Interestingly, restriction of our analysis to pERK1, but not pERK2, also revealed a main effect of CORT [F(1,1)=5.9, p=0.02] (not shown).
Fig. 3.
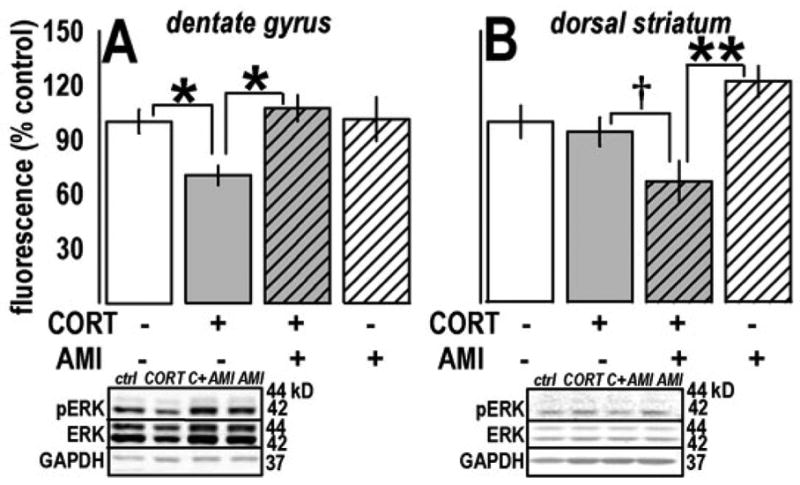
ADT and CORT chronically regulated pERK1/2 in dentate gyrus and dorsal striatum. (a) Prior CORT exposure suppressed ERK1/2 phosphorylation in the dentate gyrus, consistent with evidence from targets both upstream and downstream and with behavioral consequences of CORT. As with PR responding and immobility, subsequent AMI reversed the deficit. (b) In the dorsal striatum, a main effect of CORT exposure and a CORTxAMI interaction effect were observed. Post-hoc comparisons revealed a selective down-regulation of pERK1/2 in CORT-exposed animals subsequently treated with AMI. Analysis of pERK1 alone revealed the same effect (not shown). Bars represent means (±SEM) per treatment group (**p≤0.001, *p≤0.05, †p=0.08). Representative blots are displayed in the same order as the experimental groups in the plots above. “C+AMI” refers to the CORT+AMI group.
Regulation of pERK2 after CORT/AMI was observed in other regions. In CA1/CA3-rich samples, we detected a main effect of AMI [F(1,1)=10.1, p=0.003] and a trend for a CORTxAMI interaction [F(1,1)=3.8, p=0.06] (fig. 4a). In the amygdala, a main effect of AMI [F(1,1)=5.0, p=0.03] and a CORTxAMI interaction were detected [F(1,1)=4.4, p=0.04, post-hoc ps<0.04], with increased pERK2 in the CORT+AMI group (fig. 4b). pERK2 was unchanged in the mPFC [CORTxAMI interaction F(1,1)=1.2, p=0.3] (fig. 4c). We detected no regulation of pERK1 (all ps>0.1).
Fig. 4.

AMI increased pERK2 in CA1/CA3 and amygdala samples. (a) AMI increased pERK2, but not pERK1, in CA1/CA3 hippocampal samples. (b) Analysis of pERK1/2 in amygdala samples revealed significant up-regulation of pERK2 after AMI treatment, as well as a CORTxAMI interaction in this region. (c) pERK1/2 was unchanged in the mPFC. Bars represent means (±SEM) per treatment group (*p≤0.05 as indicated). Representative blots are displayed in the same order as the experimental groups in the plots above. “C+AMI” refers to the CORT+AMI group.
A timecourse analysis of the dentate gyrus revealed that, 14 days after CORT, pERK1/2 in otherwise naïve animals was decreased [F(2,2)=4.6, p=0.03, post-hoc ps≤0.02] compared to control and 1-day samples (fig. 5a). Because decreased pERK1/2 may result from decreased BDNF binding, we analyzed expression and phosphorylation of its receptor. TrkB immunoreactivity showed a trend for a time effect [F(2,2)=3.2, p=0.07] with high variability in the 14-day group. Comparison of 1-day CORT-exposed against 1-day control mice or all control mice revealed a down-regulation in that group (ps=0.01) but not the 14-day group. By contrast, phosphorylated trkB was decreased along the same timecourse as pERK1/2 [F(2,2)=4, p=0.04; 14-day vs. control p=0.04] (fig. 5b), while serum CORT was unchanged [F(2,2)<1] (fig. 5c).
Fig. 5.
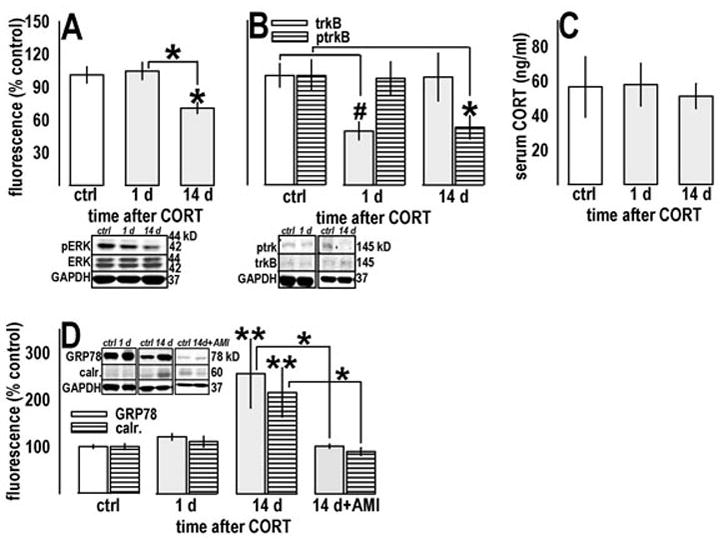
Prior CORT exposure chronically regulated pERK1/2, trkB, ptrkB, and heat-shock proteins associated with depression in humans in the dentate gyrus. (a) In behaviorally naïve mice, prior CORT decreased pERK1/2 14, but not 1 day, after exposure. (b) By contrast, prior CORT decreased trkB receptor expression (open bars) 1, but not 14, days after exposure; nonetheless, trkB phosphorylation levels (horizontal stripes) mirrored pERK1/2 expression, suggesting decreased pERK1/2 is a consequence of decreased trkB receptor binding. (c) These decreases were detected in the absence of increased endogenous CORT, as measured in blood serum. These findings are consistent with the observation that many individuals with depressive symptoms do not experience hypercortisolemia. (d) Finally, decreased pERK1/2 and ptrkB coincided with increases in GRP78 (open bars) and calr. (horizontal stripes), related heat-shock proteins associated with depression in humans, but not previously studied in murine models of depression. Both targets were sensitive to chronic AMI administration in a subsequent experiment. Control samples did not differ in any experiment and have been combined. Bars represent means (±SEM) per treatment group (**p≤0.001, *p≤0.05 compared to control samples unless otherwise indicated, #p<0.05 in a t-test comparison against same-day and all control samples). “d” refers to days subsequent to CORT exposure.
We also analyzed GRP78 and calr., heat-shock proteins associated with cellular insult. Both paralleled pERK1/2 and ptrkB expression patterns, with day 14 differing from control samples and 1-day samples [GRP78: F(3,3)=6.5, p=0.001, post-hoc ps≤0.009; calr.: F(3,3)=6.7, p=0.001, post-hoc ps≤0.007], as well as samples from animals subsequently treated with AMI, which did not differ from controls (ps≥0.7) (fig. 5d).
Discussion
Our main findings implicate ERK1/2 phosphorylation events in both ADT efficacy and the chronic depressive-like state and can be summarized as follows: 1) Prior CORT exposure selectively decreased responding in a PR task. CORT did not affect non-reinforced responding (extinction) or break point ratios in PR-experienced mice, suggesting CORT specifically impairs responding when the PR response contingency is novel and not previously learned. The deficit was reversed by AMI treatment; decreased PR responding after CORT may thus model motivational loss in depression. 2) CORT also increased immobility in the TST, a test of “behavioral despair.” Chemically distinct ADTs decreased immobility after CORT, further suggesting oral CORT exposure produces an ADT-sensitive depressive-like behavioral phenotype. 3) Post-mortem analysis revealed a selective, long-lasting, and AMI-sensitive decrease in pERK1/2 in the dentate gyrus that coincided with decreased trkB phosphorylation. 4) In the dorsal striatum, CORT and subsequent AMI decreased pERK1/2, an effect that could be attributed to regulation of the ERK1 isoform. 5) AMI increased ERK2 phosphorylation in CA1/CA3 and amygdala. Together, these data demonstrate that prior CORT exposure carries face, predictive, and construct validity as a depression model and disrupts ERK1/2 phosphorylation in the dentate gyrus, while AMI normalizes phosphorylation levels in this region and concomitantly regulates phosphorylation events in other regions.
The two most widely expressed ERK isoforms—1 and 2—have historically been considered to play complementary roles in synaptic plasticity and gene transcription. For example, hippocampal LTP requires ERK1/2 (26); however, LTP induction preferentially activates ERK2 (27), suggesting the isoforms have some non-overlapping functions. Consistent with this hypothesis, ERK1 inhibits striatal LTP (28, but see 29). Here, AMI treatment broadly regulated pERK1/2 in a fashion expected to support LTP-like processes, with increased pERK2 in hippocampus and amygdala and restoration of pERK1/2 in dentate gyrus after CORT, but decreased pERK1/2 signal in the striatum, an effect that could be largely accounted for by pERK1.
Based on behavioral responding in the PR task, reduced dentate gyrus and striatal ERK1/2 phosphorylation might be hypothesized to exert opposite effects on motivated responding. Specifically, the initial decrease in responding after CORT was associated with decreased pERK1/2 in the dentate gyrus, while the restoration of responding by AMI treatment was associated with decreased pERK1/2 in the dorsal striatum. Likely for this reason, peripheral administration of a MEK inhibitor that broadly blunts phosphorylation of both ERK isoforms did not mimic CORT when injected before or immediately after the session in otherwise drug-naïve mice (data not shown). By contrast, CORT and AMI produced regionally-selective alterations in pERK1/2 and robustly altered responding in a manner consistent with motivational deficit and its restoration, respectively.
The neurotrophic hypothesis of ADT efficacy has been broadly attributed to both chemical and non-chemical ADTs, particularly as it pertains to the dentate gyrus (e.g.,30), however, the link between ADT efficacy and enhanced ERK1/2 phosphorylation has been difficult to verify. One explanation may be related to the observation that ERK1/2 phosphorylation is a rapid and dynamic event associated with environmental stimuli (31), providing a narrow time window in which to capture enhanced phosphorylation. Here, sacrificing animals at a biologically significant time—in the food-restricted state and when mice would have been acquiring food reinforcers—revealed pERK1/2 up-regulation after AMI consistent with reported increased phosphorylation of the target transcription factor, CREB, by distinct ADTs (32,7).
CORT persistently regulates pERK1/2
Stress-related neuronal insult is widely hypothesized to be long-lasting, with some supporting evidence from animal models. For example, decreased hippocampal CREB phosphorylation is a persistent consequence of stress (5), and decreased pCREB may emerge during and/or after chronic stress (c.f.,33). Our finding that pERK1/2 is decreased 14, but not 1, day after CORT is consistent with this hypothesis. The finding may reflect long-term neural insult to cells in this region, so we analyzed the same tissue for markers of cellular damage, GRP78 and calr., heat-shock proteins that have also been associated with depression (23) and bipolar disorder (24) in humans. As with pERK1/2, GRP78 and calr. were unchanged immediately after CORT exposure, but were robustly regulated at a later time point, emphasizing the importance of analyzing biochemical targets related to depression immediately and long after stress-related manipulations in rodents, as targets of interest may be sensitive to glucocorticoid/stress exposure in a time-dependent manner.
This point is of particular relevance to depression, in which previous life stressors may induce, contribute to, or exacerbate major depressive episodes without immediately disrupting mood. Nevertheless, much of what is believed to be known about the neurobiological factors of depression, including the potential importance of neurotrophin- and neuroplasticity-related signaling, is based upon our understanding of (often acute) actions of standard ADTs in otherwise naïve rodents. Despite methodological differences between experimenter-administered CORT and widely embraced chronic mild stress (CMS) models, both methods induce diverse depressive-like behaviors in the rodent and offer the opportunity to evaluate effects of chronically elevated glucocorticoid exposure on behavior and biochemical targets, and whether ADTs reverse or interact with these consequences. The CORT method may also, however, offer a solution to CMS protocol variability and reported difficulties in data replication (e.g.,34,35). While no stress-related depression model can fully recapitulate the heterogeneity of depressive disorders, a model that produces a chronic, varied behavioral sequela sensitive to chronic ADT may offer an advance for understanding the neurobiology of the disease.
Other considerations
While both stress and CORT decrease hippocampal BDNF expression during, immediately after, and long after exposure (3,36,37,4,38), neither chronic CORT nor dexamethasone, a synthetic glucocorticoid, are reported to alter mRNA expression of catalytic trkB, the receptor for BDNF (39,4,40). Nevertheless, we report decreased trkB immediately after CORT exposure. Treatment regiments in previous experiments lasted 5–10 days, raising the possibility that only prolonged exposure (20 days here) decreases trkB expression. Notably, ptrkB and pERK1/2 were down-regulated by CORT on the same timecourse, suggesting decreased ERK1/2 phosphorylation may be attributed to decreased trkB binding. By contrast, decreased receptor expression appears to be a more immediate consequence of chronic glucocorticoid binding. Nonetheless, both are likely targets of ADTs.
The lack of prefrontal ERK1/2 regulation is surprising, given evidence that chemical ADTs, electroconvulsant seizure, and stress influence kinase expression and/or phosphorylation in this region (33,15,16,41). In previous studies, tissue was harvested within 24 hours of manipulation; here, tissue was harvested after an extended wash-out period. Therefore, ADT/stress-related prefrontal ERK regulation may be transient, in contrast to the dentate gyrus, where both CORT and AMI persistently altered pERK1/2. Future studies should further characterize prefrontal ERK regulation.
Long-term HPA axis hyper-activity has well-established adverse effects on neural plasticity and mood (42,43). We hypothesize decreased hippocampal pERK1/2 after CORT reflects decreased LTP in the dentate gyrus (44), rendering the CORT-exposed animal less sensitive to LTP “reinforcement” by motivational drive (45,46,47) and conferring risk for further cognitive/motivational deficit (48). Additionally, pERK1/2 expression patterns after CORT and AMI could reflect intracellular events associated with decreased neurogenesis and its enhancement by ADTs (c.f.,49). In this case, AMI alone would be expected to increase pERK1/2 above control levels. Although we did not observe this effect, future studies could address the possibility that CORT- and/or AMI-induced alterations in neurogenesis contribute to responding in the PR task and TST.
Conclusions
Evidence suggests that ADTs robustly promote transcriptional events in the hippocampus related to neurotrophin expression and binding, but the majority of studies have been conducted in naïve rodents, despite the possibility that the long-term molecular targets of stress (a major risk factor for depression) and ADTs do not overlap. We reveal opposite effects of CORT and AMI on ERK1/2 signaling in the dentate gyrus, but while we provide evidence that CORT and AMI share an intracellular target in this region, our data suggest that persistent regulatory effects may in fact differ in other regions. Elucidating how AMI and other ADTs act on pERK1/2 in region- and isoform-specific manners should aid in understanding the neurotrophic- and neuroplasticity-related contributions to ADT action and the development of novel targets for treating depression.
Acknowledgments
This work was supported by PHS MH 25642 (JRT) and a Sigma Xi grant-in-aid award (SLG). The authors would like to thank Erika Andrade and Drs. Angus Nairn and Dilja Krueger for excellent advice and support.
Footnotes
The authors declare they have no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Duman RS, Heninger GR, Nestler EJ. A molecular and cellular theory of depression. Arch Gen Psychiatry. 1997;54:597–606. doi: 10.1001/archpsyc.1997.01830190015002. [DOI] [PubMed] [Google Scholar]
- 2.Chen AC-H, Shirayama Y, Shin K-H, Neve RL, Duman RS. Expression of cAMP response element binding protein (CREB) in hippocampus produces an antidepressant effect. Biol Psychiatry. 2001;49:753–762. doi: 10.1016/s0006-3223(00)01114-8. [DOI] [PubMed] [Google Scholar]
- 3.Smith MA, Makino S, Kvetnansky R, Post RM. Stress and glucocorticoids affect the expression of brain-derived neurotrophic factor and neurotrophin-3 mRNAs in the hippocampus. J Neurosci. 1995;15:1767–1777. doi: 10.1523/JNEUROSCI.15-03-01768.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nibuya M, Takahashi M, Russell DS, Duman RS. Repeated stress increases catalytic TrkB mRNA in rat hippocampus. Neurosci Lett. 1999;267:81–84. doi: 10.1016/s0304-3940(99)00335-3. [DOI] [PubMed] [Google Scholar]
- 5.Laifenfeld D, Kerry R, Grauer E, Klein E, Ben-Shachar D. Antidepressant and prolonged stress in rats modulate CAM-L1, laminin, and pCREB, implicated in neuronal plasticity. Neurobiol Dis. 2005;20:432–441. doi: 10.1016/j.nbd.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 6.Blendy J. The role of CREB in depression and antidepressant treatment. Biol Psychiatry. 2006;59:1144–1150. doi: 10.1016/j.biopsych.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 7.Thome J, Sakai N, Shin K, Steffen C, Zhang YJ, Impey S, et al. cAMP response element-mediated gene transcription is upregulated by chronic antidepressant treatment. J Neurosci. 2000;20:4030–4036. doi: 10.1523/JNEUROSCI.20-11-04030.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mazzucchelli C, Brambilla R. Ras-related and MAPK signaling in neuronal plasticity and memory formation. Cell Mol Life Sci. 2000;57:604–611. doi: 10.1007/PL00000722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rodrigues SM, Schafe GE, LeDoux JE. Molecular mechanisms underlying emotional learning and memory in the lateral amygdala. Neuron. 2004;44:75–91. doi: 10.1016/j.neuron.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 10.Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C, Sweatt JD. The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of Hippocampus. J Neurosci. 1999;19:4337–4348. doi: 10.1523/JNEUROSCI.19-11-04337.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sweatt JD. The neuronal MAP kinase cascade: A biochemical signal integration system subserving synaptic plasticity and memory. J Neurochem. 2001;76:1–10. doi: 10.1046/j.1471-4159.2001.00054.x. [DOI] [PubMed] [Google Scholar]
- 12.Shirayama Y, Chen AC-H, Nakagawa S, Russell DS, Duman RS. Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J Neurosci. 2002;22:3251–3261. doi: 10.1523/JNEUROSCI.22-08-03251.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duman CH, Schlesinger L, Kodama M, Russell DS, Duman RS. A role for MAP kinase signaling in behavioral models of depression and antidepressant treatment. Biol Psychiatry. 2007;61:661–670. doi: 10.1016/j.biopsych.2006.05.047. [DOI] [PubMed] [Google Scholar]
- 14.Hisaoka K, Nishida A, Koda T, Miyata M, Zensho H, Morinobu A, et al. Antidepressant drug treatments induce glial cell line-derived neurotrophic factor (GDNF) synthesis and release in rat C6 glioblastoma cells. J Neurochem. 2001;79:25–34. doi: 10.1046/j.1471-4159.2001.00531.x. [DOI] [PubMed] [Google Scholar]
- 15.Tiraboschi E, Tardito D, Kasahara J, Moraschi S, Pruneri P, Gennarelli M, et al. Selective phosphorylation of nuclear CREB by fluoxetine is linked to activiation of CaM kinase IV and MAP kinase cascades. Neuropsychopharmacology. 2004;29:1831–1840. doi: 10.1038/sj.npp.1300488. [DOI] [PubMed] [Google Scholar]
- 16.Fumagalli F, Molteni R, Calabrese F, Frasca A, Racagni G, Riva MA. Chronic fluoxetine administration inhibits extracellular signal-related kinase 1/2 phosphorylation in rat brain. J Neurochem. 2005;93:1551–1560. doi: 10.1111/j.1471-4159.2005.03149.x. [DOI] [PubMed] [Google Scholar]
- 17.Dwivedi Y, Rizavi HS, Roberts RC, Conley RC, Tamminga CA, Pandey GN. Reduced activation and expression of ERK1/2 MAP kinase in the post-mortem brain of depressed suicide subjects. J Neurochem. 2001;77:916–928. doi: 10.1046/j.1471-4159.2001.00300.x. [DOI] [PubMed] [Google Scholar]
- 18.Dwivedi Y, Rizavi HS, Conley RR, Pandey GN. ERK MAP kinase signaling in postmortem brain of suicide subjects: differential regulation of upstream Raf kinases Raf-1 and B-Raf. Mol Psychiatry. 2006;11:86–98. doi: 10.1038/sj.mp.4001744. [DOI] [PubMed] [Google Scholar]
- 19.Baldwin AE, Sadeghian K, Holahan MR, Kelley AE. Appetitive instrumental learning is impaired by inhibition of cAMP-dependent protein kinase within the nucleus accumbens. Neurobiol Learn Mem. 2002;77:44–62. doi: 10.1006/nlme.2000.4002. [DOI] [PubMed] [Google Scholar]
- 20.Caldarone BJ, Karthigeyan K, Harrist A, Hunsberger JG, Witmack E, King SL, et al. Sex differences in response to oral amitriptyline in three animal models of depression in C57BL/6J mice. Psychopharmacology. 2003;170:94–101. doi: 10.1007/s00213-003-1518-7. [DOI] [PubMed] [Google Scholar]
- 21.Hodos W. Progressive ratio as a measure of reward strength. Science. 1961;134:943–944. doi: 10.1126/science.134.3483.943. [DOI] [PubMed] [Google Scholar]
- 22.Cryan JF, Mombereau C, Vassout A. The tail suspension test as a model for assessing antidepressant activity: Review of pharmacology and genetic studies in mice. Neurosci Biobehav Rev. 2005;29:571–625. doi: 10.1016/j.neubiorev.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 23.Brown C, Wang J-F, MacQueen G, Young LT. Increased temporal cortex ER stress proteins in depressed subjects who died by suicide. Neuropsychopharmacology. 2000;22:327–332. doi: 10.1016/S0893-133X(99)00091-3. [DOI] [PubMed] [Google Scholar]
- 24.Kakiuchi C, Ishiwata M, Nanko S, Kunugi H, Minabe Y, Nakamura K, et al. Functional polymorphisms of HSPA5: Possible association with bipolar disorder. Biochem Biophys Res Commun. 2005;336:1136–1143. doi: 10.1016/j.bbrc.2005.08.248. [DOI] [PubMed] [Google Scholar]
- 25.Rousse I, Beaulieu S, Barden N, Rochford J. Test-specific effects of amitriptyline administration on behavioral deficits in transgenic mice with impaired glucocorticoid receptor function. Dev Brain Dysfunction. 1997;10:418–431. [Google Scholar]
- 26.English JD, Sweatt JD. A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J Biol Chem. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- 27.English JD, Sweatt JD. Activation of p42 mitogen-activated protein kinase in hippocampal long term potentiation. J Biol Chem. 1996;271:24329–24332. doi: 10.1074/jbc.271.40.24329. [DOI] [PubMed] [Google Scholar]
- 28.Mazzucchelli C, Vantaggiato C, Ciamei A, Fasano S, Pakhotin DP, Frezel W, et al. Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron. 2002:807–820. doi: 10.1016/s0896-6273(02)00716-x. [DOI] [PubMed] [Google Scholar]
- 29.Selcher JC, Nekrasova T, Paylor R, Landreth GE, Sweatt JD. Mice lacking the ERK1 isoform of MAP kinase are unimpaired in emotional learning. Learning and Memory. 2001;8:11–19. doi: 10.1101/lm.37001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duman RS, Monteggia LM. A neurotrophin model for stress-related mood disorders. Biol Psychiatry. 2006;59:1116–1127. doi: 10.1016/j.biopsych.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 31.Davis S, Vanhoutte P, Pages C, Caboche J, Laroche S. The MAPK/ERK cascade targets both Elk-1 and cAMP response element binding protein to control long-term potentiation-dependent gene expression in the dentate gyrus in vivo. J Neurosci. 2000;20:4563–4572. doi: 10.1523/JNEUROSCI.20-12-04563.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nibuya M, Nester EJ, Duman RS. Chronic antidepressant administration increases the expression of cAMP response element binding protein (CREB) in rat hippocampus. J Neurosci. 1996;16:2365–2372. doi: 10.1523/JNEUROSCI.16-07-02365.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trentani A, Kuipers SD, Ter Horst GJ, Den Boer JA. Selective chronic stress-induced in vivo ERK1/2 hyperphosphorylation in medial prefrontocortical dendrites: implications for stress-related cortical pathology. Eur J Neurosci. 2002;15:1681–1691. doi: 10.1046/j.1460-9568.2002.02000.x. [DOI] [PubMed] [Google Scholar]
- 34.Matthews K, Forbes N, Reid IC. Sucrose consumption as an hedonic measure following chronic unpredictable mild stress. Physiol Behav. 1995;57:241–248. doi: 10.1016/0031-9384(94)00286-e. [DOI] [PubMed] [Google Scholar]
- 35.Forbes NF, Stewart CA, Matthews K, Reid I. Chronic mild stress and sucrose consumption: Validity as a model of depression. Physiol Behav. 1996;60:1481–1484. doi: 10.1016/s0031-9384(96)00305-8. [DOI] [PubMed] [Google Scholar]
- 36.Schaaf MJM, Hoetelmans E, de Kloet R, Vreugdenhil E. Corticosterone regulates expression of BDNF and trkB but not NT-3 and trkC mRNA in the rat hippocampus. J Neurosci Res. 1997;48:334–341. [PubMed] [Google Scholar]
- 37.Schaaf MJM, de Jong J, de Kloet ER, Vreugdenhil E. Down-regulation of BDNF mRNA and protein in the rat hippocampus by corticosterone. Brain Res. 1998;813:112–120. doi: 10.1016/s0006-8993(98)01010-5. [DOI] [PubMed] [Google Scholar]
- 38.Prickaerts J, van den Hove DLA, Fieren FLP, Kia HK, Lenaerts I, Steckler T. Chronic corticosterone manipulations in mice affect brain cell proliferation rates, but only partly affect BDNF protein levels. Neurosci Lett. 2006;396:12–16. doi: 10.1016/j.neulet.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 39.Chao HM, McEwen BS. Glucocorticoids and the expression of mRNAs for neurotrophins, their receptors and GAP-43 in the rat hippocampus. Brain Res Mol Brain Res. 1994;26:271–276. doi: 10.1016/0169-328x(94)90099-x. [DOI] [PubMed] [Google Scholar]
- 40.Vellucci SV, Parrott RF, Mimmack ML. Chronic dexamethasone-treatment alters mineralocorticoid receptor, truncated trkB and selected glutamate receptor subunit mRNA expression in the porcine hippocampus. Neuropeptides. 2002;36:291–298. doi: 10.1016/s0143-4179(02)00048-3. [DOI] [PubMed] [Google Scholar]
- 41.Kodama M, Russell DS, Duman RS. Electroconvulsive seizures increase the expression of MAP kinase phosphatases in limbic regions of rat brain. Neuropsychopharmacology. 2005;30:360–371. doi: 10.1038/sj.npp.1300588. [DOI] [PubMed] [Google Scholar]
- 42.Sapolsky RM. The possibility of neurotoxicity in the hippocampus in major depression: A primer on neuron death. Biol Psychiatry. 2000;48:755–765. doi: 10.1016/s0006-3223(00)00971-9. [DOI] [PubMed] [Google Scholar]
- 43.Kendler KS, Karkowski LM, Prescott CA. Causal relationship between stressful life events and the onset of major depression. Am J Psychiatry. 1999;156:837–841. doi: 10.1176/ajp.156.6.837. [DOI] [PubMed] [Google Scholar]
- 44.Korz V, Frey JU. Stress-related modulation of hippocampal long-term potentiation in rats: Involvement of adrenal steroid receptors. J Neurosci. 2003;23:7281–7287. doi: 10.1523/JNEUROSCI.23-19-07281.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seidenbecher T, Balschun D, Reymann KG. Drinking after water deprivation prolongs “unsaturated” LTP in the dentate gyrus of rats. Physiol Behav. 1995;57:1001–1004. doi: 10.1016/0031-9384(94)00352-6. [DOI] [PubMed] [Google Scholar]
- 46.Seidenbecher T, Reyman KG, Balschun D. A post titanic time window for the reinforcement of long-term potentiation by appetitive and aversive stimuli. Proc Natl Acad Sci U S A. 1997;94:1494–1499. doi: 10.1073/pnas.94.4.1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Straube T, Korz V, Frey JU. Bidirectional modulation of long-term potentiation by novelty-exploration in the dentate gyrus. Neurosci Lett. 2003;344:5–8. doi: 10.1016/s0304-3940(03)00349-5. [DOI] [PubMed] [Google Scholar]
- 48.Maren S, DeCola JP, Swain RA, Fanselow MS, Thompson RF. Parallel augmentation of hippocampal long-term potentiation, theta rhythm, and contextual fear conditioning in water-deprived rats. Behav Neurosci. 1994;108:44–56. doi: 10.1037//0735-7044.108.1.44. [DOI] [PubMed] [Google Scholar]
- 49.Vollmayr B, Magdalena MM, Henn FA. Neurogenesis and depression: What animal models tell us about the link. Eur Arch Psychiatry Clin Neurosci. 2007 doi: 10.1007/s00406-007-0734-2. Pubmed ahead of print. [DOI] [PubMed] [Google Scholar]