

Abstract
The objective of this study was to determine whether heme oxygenase-1 (HO-1) or heme metabolites exert cytoprotective effects on interleukin-18-mediated endothelial cell (EC) death. Treatment with IL-18 increased NF-κB activation, PTEN induction, suppressed Akt activation, and stimulated EC death. While ectopic expression of p65 enhanced PTEN transcription, adenoviral transduction of dnIκB-α, dnp65, or dnIKKβ was inhibitory. Furthermore, IL-18 suppressed HO-1 mRNA expression via enhanced mRNA degradation. Overexpression of HO-1, treatment with HO-1 inducer hemin or the CO donor Cobalt (III) protoporphyrin IX all reversed IL-18-mediated NF-κB activation, PTEN induction, Akt suppression, and EC death. Furthermore, hemin induced HO-1 expression, and HO-1 knockdown, HO-1 inhibition, or CO scavengers all reversed the pro-survival effects of hemin. In addition, the CO donors CORM-1 and CORM-3 and the heme metabolites biliverdin and bilirubin attenuated IL-18-induced EC death via a similar signaling pathway. IL-18 induced p38α MAPK activation, and suppressed p38β isoform expression. While p38α knockdown attenuated, p38β knockdown potentiated IL-18-mediated EC death. Hemin and HO-1 reversed IL-18-mediated p38α induction, and restored p38β levels. These results demonstrate that IL-18 suppresses HO-1 expression and induces EC death. HO-1 overexpression, HO-1 induction, or treatment with heme metabolites all reverse IL-18-mediated p38α MAPK and NF-κB activation, PTEN induction, Akt suppression, and EC death. Thus, HO-1 inducers and CO donors may have the therapeutic potential to effectively block IL-18 signaling and reduce IL-18-dependent vascular injury and inflammation.
Keywords: Interleukin-18, cell death, heme oxygenase, carbon monoxide, biliverdin, bilirubin, NF-κB, PTEN
Introduction
Interleukin (IL)-18 is a proinflammatory cytokine which signals via the IL-18R, a heterodimer comprised of the ligand-binding IL-18Rα and the signal transducing IL-18Rβ subunits. Increased levels of IL-18 have been found in various autoimmune and inflammatory diseases including patients with heart failure and coronary artery disease where its levels correlate with disease severity [1–6]. In these patients, a concomitant increase in IL-18 binding protein (IL-18BP) is observed [6] IL-18BP is a naturally occurring inhibitor of IL-18, is detected in the plasma of healthy subjects at 10- to 20-fold molar excess over IL-18 [7], and is decreased in patients with myocardial ischemic injury and dilated cardiomyopathy. Interestingly, IL-18 shows greater affinity for IL-18BP than for IL-18R, and its binding to IL-18BP is essentially irreversible [8]. Therefore, IL-18BP or IL-18 neutralizing antibodies may potentially be employed to block IL-18 signaling and reduce inflammation and tissue injury.
IL-18 is a pleiotropic cytokine that promotes inflammation, hypertrophy, mitogenesis, and apoptosis, depending on the cell type [9–11]. As a pro-apoptotic cytokine, IL-18 induces endothelial cell death via activation of both the intrinsic and extrinsic pro-apoptotic signaling pathways [11]. In these cells, IL-18 suppresses the pro-survival factor Akt and stimulates NF-κB-dependent PTEN induction and cell death [12]. Overexpression or induction of hemeoxygenaese-1 (HO-1) has been demonstrated to be cytoprotective. HO-1 is one of two isoforms of hemeoxygenases that catalyze the conversion of heme to free iron, biliverdin and carbon monoxide (CO). Biliverdin is then converted to bilirubin by biliverdin reductase (BVR). Though biliverdin and bilirubin have been considered toxic waste products in the past, recent reports indicate that these byproducts possess potent anti-oxidant and anti-inflammatory properties [13–17]. Similarly, CO affects various signaling pathways critical to cell death, proliferation, and inflammation [17].
Of the two isoforms, HO-1 is inducible, and HO-2 is constitutively expressed. HO-1 is induced by various noxious stimuli that include endotoxin, oxidative stress, cytokines, and hemin, where its induction exerts antioxidant and cytoprotective effects [17]. Since IL-18 induces endothelial cell death [11,12], we hypothesized that HO-1 induction or HO-1 overexpression may block IL-18-mediated endothelial cell death. Furthermore, given that IL-18 suppresses Akt and induces PTEN in an NF-κB-dependent manner, we further hypothesized that HO-1 may inhibit cell death via suppression of NF-κB-PTEN signaling and restoration of Akt activation.
Material and methods
Recombinant human IL-18, IL-18 neutralizing antibodies, control IgG, IL-18BPa/Fc chimera, and Fc were purchased from R&D Systems (Minneapolis, MN). Recombinant IL-18 BPa/Fc, a disulfide-linked homodimeric protein, transcribed from a DNA sequence encoding amino acid residues 1–192 of human interleukin 18 binding protein and fused to the Fc region of human IgG1 via a polypeptide linker. The efficacy of IL-18BPa/Fc for blocking IL-18 mediated gene regulation has been previously demonstrated both in vivo and in vitro [12,18]. IL-18 neutralizing antibodies and control IgG were used at 10 µg/ml, and IL-18BPa/Fc and Fc were used at a concentration of 200 ng/ml. Antibodies against PTEN, actin, and p38β (sc-6187) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-p38 MAPK (#9212), phospho-p38 MAPK, p38α MAPK (#2371), and phospho-p65 (Ser276) antibodies were obtained from Cell Signaling Technology (Beverly, MA). Anti-p38α MAPK antibodies were rabbit mAb and detect endogenous levels of total p38α MAPK protein, and do not cross-react with other p38 MAP kinase isoforms such as β, γ, or δ. Anti-human HO-1 monoclonal antibodies were purchased from Stressgen (Victoria, Canada). Radiochemicals were obtained from Amersham Biosciences (Piscataway, NJ). SB202190 (a potent and selective inhibitor of p38 MAPK inhibitor; 30 µM for 30 min in DMSO) and DMSO were from EMD Biosciences (San Diego, CA). Hemoglobin (10 µM), hemin (ferriprotoporphyrin IX chloride; 2.5 µM for 24 h), tricarbonyldichlororuthenium (II) dimer (CORM-1), αTubulin antibodies and other chemicals were purchased from Sigma-Aldrich. CORM-1 is a first generation CO-releasing molecule. It was used at 10 µM concentration. At this concentration CORM-1 did not affect cell viability (trypan blue dye exclusion). Only prolonged treatment with high concentrations of CORM-1 was shown to be toxic [19]. CORM-3 (tricarbonylchloro (glycinate)ruthenium (II) ([Ru(CO)3Cl(glycinate)]) and inactive CORM-3 (iCORM-3) were previously described [20,21]. CORM-3 is a second generation CORM, and is an effective CO donor in vitro, ex vivo and in vitro, and does not affect cell viability for up to 50 µM [19,20]. It was stored in dark-colored vials at −20°C, and was prepared fresh on the day it was used. iCORM-3 was prepared by dissolving CORM_3 in saline and allowed to sit at room temperature for 18 h to liberate CO [21]. Both CORM-3 and iCORM-3 were dissolved in saline and used at 10 µM. Cobalt (III) protoporphyrin IX (CoPPIX; #ALX-430-076-M025; 10 µM for 24 h) and zinc (II) protoporphyrin IX (ZnPPIX; # ALX-430-049-M025; 10 µM for 24 h) were purchased from AXXORA, LLC (San Diego, CA). While CoPPIX acts a CO donor, ZnPPIX serves as an inhibitor. ZnPPIX has also been shown to irreversibly attenuate Ca2+ currents [22], inhibit hematopoisis in rabbit and human bone marrow [23], and non-selectively inhibit NOS enzymes and soluble guanylate cyclase [24]. Both CoPPIX and ZnPPIX were dissolved in 0.2M NaOH, neutralized with 1M HCl, adjusted to 1 mg/ml concentration, filter sterilized and stored at −80°C until further use. Bilirubin and biliverdin dihydrochloride (10 µM; ICN Biomedicals) were prepared similar to CoPPIX, and stored at −80°C. Bilirubin exerts toxic effects at high concentrations. But at lower concentrations it exerts anti-oxidant effects efficiently scavenging peroxyl radicals [25], and both biliverdin and bilurubin have been shown to inhibit p38 MAPK activation [26].
Cell culture
Non-transformed human cardiac microvascular endothelial cells (EC) were obtained from ScienCell Research Laboratories (San Diego, CA) and cultured as previously described [11,12]. The cells were grown in EC medium (ECM) supplied by the manufacturer and supplemented with 5% serum (complete medium). At 70–80% confluency, the complete medium was replaced with medium containing 0.5% BSA. After overnight incubation to achieve quiescence, recombinant human (rh) IL-18 (R&D Systems) was added for the indicated time periods. Specificity of IL-18 was verified by preincubating IL-18 with anti-IL-18 neutralizing antibodies or IL-18BPa/Fc chimera for 1 h at 37 °C and 14 h at 4 °C before addition. Normal human IgG and Fc served as controls. Cells were harvested, snap-frozen, and stored at −80°C.
Transient cell transfections and reporter assays
A 4.5 kb of the 5’-flanking region of human HO-1 was amplified from human genomic DNA using the sense primer 5′-ggt acc TTG GGC TTG TCT TCC TTG CT-3′ and the antisense primer 5′-ctc gag CATCCGGCCGGTGCTGGGCTCGT-3′ and cloned into the pCR2.1-TOPO vector, digested with Kpn I and Xho I, and subcloned into the pGL3-Basic reporter vector (pHO-1-Luc) as previously described [27]. EC were transiently transfected with pHO-1-Luc (2 µg), and 24 h later, treated with IL-18 for 12 hours. The Renilla-luciferase reporter plasmid (100ng; pRL-TK vector; Promega, Madison, WI) was used as an internal control. The NF-κB-driven luciferase reporter plasmid (pNF-κB-Luc), the control plasmid pEGFP-Luc, and the pPTEN-Luc reporter vector were previously described [12].
The PTEN promoter contains two putative NF-κB binding sites (NF-κB-1, −1450 to −1441, GGGTATTCCC; NF-κB-2: −1574 to −1565, GGGAATCTCT). To verify whether one or both of these NF-κB sites play a role in IL-18-mediated PTEN induction, we generated deletion constructs lacking NF-κB-1 (sense, 5’-ggtacc AAATCTCTGCGAACGATTGTGA-3’; antisense, 5’-aagctt GTCTGGG AGCCTGTGG-3’) or both NF-κB-1 and NF-κB-2 (sense, 5’-ggtaccTACACTGAGCAGCGTGGTCA-3’; antisense, 5’-aagctt GTCTGGG AGCCTGTGG-3’). EC were transiently transfected with the full length or the deletion constructs as described above. pRL-TK vector served as an internal control.
p38 MAPK isoform-specific siRNA (100 nM; p38α, sc-39116; p38β, sc-29433; Santa Cruz Biotechnology, Inc.) were transfected using Oligofectamine™ (Invitrogen). Knockdown of protein was confirmed at 48 h by Western blotting.
The transfection efficiency of EC, determined using pEGFP-N1 vector (Clontech, BD Biosciences, Palo Alto, CA), was 34.6±2.21% (mean ± SE, n=22). pcDNA3 and pGL3-Basic vectors served as controls where appropriate. Luciferase reporter data were normalized for transfection efficiency using the corresponding Renilla-luciferase values, and expressed as mean relative stimulation ± SE for a representative experiment from three separate and independent experiments with each performed in triplicate. Cells were transfected using LipofectAMINEPLUS, and the amount of DNA transfected was constant (2 µg) in all experiments. After transfection, the viability of the cells was determined using trypan blue. Twenty-four hours after transfection, the media were changed and the cells were stimulated with IL-18. Luciferase activity was determined using the Promega Biotech dual-luciferase reporter assay system and a TD 20/20 luminometer (Turner Designs, Sunnyvale, CA).
Adenoviral transduction
Adenovirus expressing catalytically inactive mutant PTEN (Ad-ciPTEN), wild type NF-κBp65, dominant negative (dn) p65, dnIKKβ, phosphorylation-deficient IκB-α (S32A/S36A; dnIκB-α), and GFP were used as previously described [12]. Adenovirus expressing HO-1 was used as previously described [28]. EC were grown to 60–70% confluency in complete medium. The medium was replaced with PBS, and the cells were infected at 100 multiplicity of infection (MOI) for 1h at 22° C. The transfection medium was replaced with media containing 0.5% BSA. After 24 h the cells were treated with IL-18.
Electrophoretic mobility shift (EMSA), ELISA, reporter assays
NF-κB DNA-binding activity in nuclear protein extracts was analyzed by EMSA [9–12]. The following double-stranded oligonucleotides were used: consensus, 5′-AGTTGAGGGGACTTTCCCAGGC-3′; mutant, 5′-AGTTGAGGCGACTTTCCCAGGC-3′ (Santa Cruz Biotechnology, Santa Cruz, CA). Absence of protein extract and competition with 100-fold molar excess of unlabeled or corresponding mutant oligonucleotides served as controls. Subunit composition of NF-κB was determined by ELISA (Active Motif, Carlsbad, CA) [12]. NF-κB activation was confirmed in transient transfection assays using an NF-κB-reporter vector (pNF-κB-Luc). pEGFP-Luc served as a control in these experiments.
mRNA analysis
Total RNA isolation, enrichment for poly(A)+ RNA, Northern blotting, and PTEN cDNA were previously described [12]. 28S rRNA was used as an internal control. PTEN expression was targeted by siRNA (PTENsiRNA#1, 5′-AAGAGGAUGGAUUCGACUUAGACUU-3′; PTENsiRNA#2, 5′-AUCGUUAGCAGAAACAAAAGGAGAU-3′) as described previously [12]. Knockdown of PTEN was confirmed at 72 h by Western blotting. Mismatch siRNA were used as controls (mismatch siRNA#1, 5′-AAGAGGAUGGUAUCGACUUAGACUU-3′; mismatch siRNA#2, 5′-AUCGUGAGCACAAAGAAAAUGAGAU-3′). HO-1 expression was analyzed by real time quantitative PCR (RT-qPCR) as previously described [29], using an ABI Prism 7900 Sequence Detection System (Applied Biosystems) and QuantiTect™ SYBR Green PCR kit (Qiagen, Valencia, CA). The sequence-specific primers used in these experiments were previously described (HO-1, X06985, 265 bp; sense, 5’-CCAGCGGGCCAGCAACAAAGTGC-3’, antisense, AAGCCTTCAGTGCCCACGGTAAGG-3; GAPDH, sense, 5′-TTGTTGCCATCAATGACCC-3’; antisense, 5′-CTTCCCGTTCTCAGCCTTG-3’). Omission of template or reverse transcriptase served as internal controls. Absolute standard curves of HO-1 and GAPDH were constructed from the results of parallel PCRs performed on serial dilutions of standard DNA that were originally generated by in vitro transcription. Input cDNA concentrations were normalized to GAPDH mRNA levels. HO-1 expression was targeted by siRNA that targets nucleotides 612 to 630 of the HO-1 mRNA coding sequence (5′-rGACUGCGUUCCUGCUCAACdTdT-3′ and 5′-rGUUGAGCAGGAACGCAGUCdTdT-3′; 50 nM).
HO-1 mRNA stability and transcription after IL-18 treatment were analyzed by actinomycin D treatment and nuclear run-on assays, respectively, as described previously [30].
Protein analysis
Isolation of whole cell homogenates, electrophoresis, electroblotting, and Western blotting using the ECL chemiluminescence detection system were previously described [9–12]. Mitochondrial and cytoplasmic fractions were prepared using the Mitochondrial Fractionation Kit (Active Motif). Cytochrome c levels were measured colorimetrically (FunctionELISA Cytochrome c kit, Active Motif) [12]. p38 MAPK activity in whole cell homogenates was determined by immunocomplex kinase assays using a commercially available colorimetric assay kit (#9820; p38 MAP Kinase Assay Kit; Cell Signaling Technology, Inc.). The assay is based immunoprecipitation of p38 MAP kinase by immobilized phospho-p38 MAPK (Thr180/Tyr182) monoclonal antibody followed by an in vitro kinase assay using ATF-2 as a substrate. ATF-2 phosphorylation is detected by Western blotting using phospho-ATF-2 (Thr71) antibody.
Cell death detection
Following 48h incubation in ECM+0.5% BSA, EC were treated with IL−18 (100ng/ml) for 24 h. Detached cells were collected and added to the scraped adherent cells, and analyzed for apoptosis using the annexin V–FITC apoptosis detection kit (Oncogene Research Products, San Diego, CA) [11,12]. The cells were analyzed by flow cytometry at an excitation wavelength of 488 nm. The FITC signal was detected at 518 nm, and that of PI at 620 nm.
Statistical analysis
Results are expressed as mean ± standard error (±SE). For statistical analysis we used ANOVA followed by an appropriate post hoc multiple comparison test (Tukey method). Data were considered statistically significant at P < 0.05.
Results
IL-18 induces NF-κB activation in EC
Interleukin-18 is a pro-apoptotic cytokine that induces EC death via activation of the NF-κB-dependent PTEN signaling pathway [12]. Confirming these results, IL-18 induced NF-κB DNA binding activity in EC, and treatment with anti-IL-18 neutralizing antibodies or IL-18BPa/Fc chimera prior to IL-18 treatment blunted this response (Fig. 1A). IL-18 stimulation of NF-κB-dependent reporter gene activity confirmed the EMSA results (Fig. 1B). Once again, IL-18 neutralizing antibodies and IL-18BPa/FC chimera blunted NF-κB reporter activity (Fig. 1B). ELISA revealed significant increases in nuclear p65 and p50 levels following IL-18 treatment (Fig. 1C). Furthermore, IL-18 stimulated p65 phosphorylation (Fig. 1D). These results indicate that IL-18 is a potent inducer of NF-κB activation in EC, and both p65 and p50 contribute to IL-18-mediated NF-κB activation (Fig. 1).
Figure 1. IL-18 induces NF-κB activation in EC.
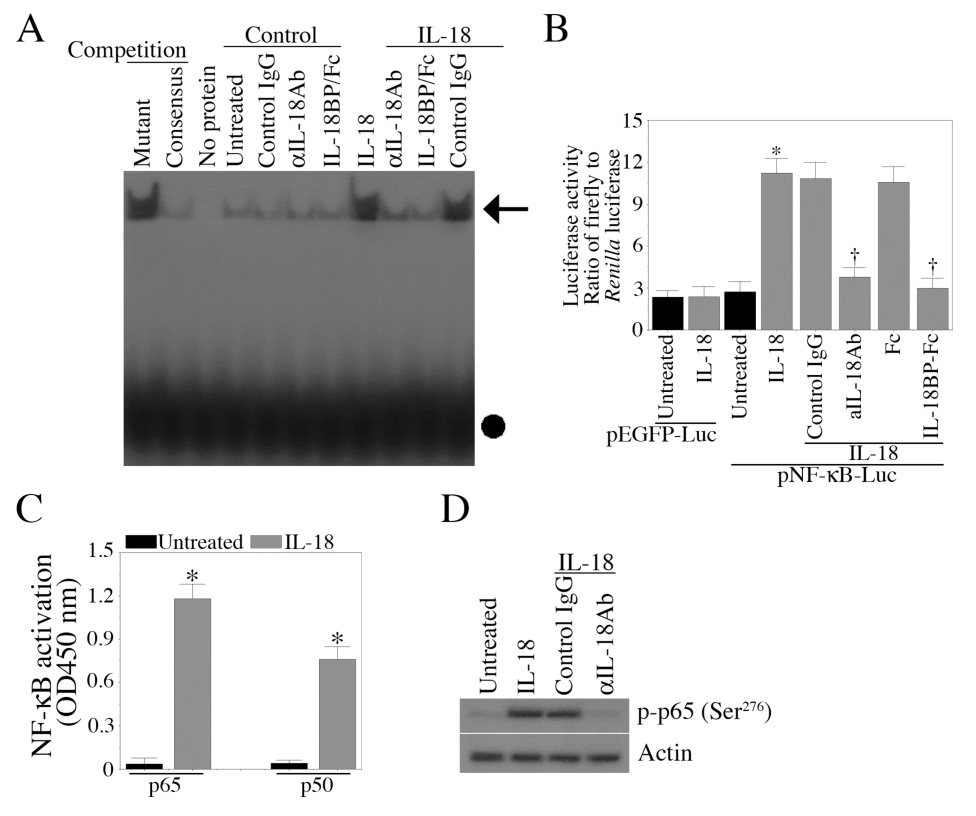
A, IL-18 induces NF-κB DNA binding activity. Quiescent EC were treated with IL-18 (100 ng/ml for 1 h), and NF-κB DNA binding activity was assessed by EMSA using nuclear protein extracts. Specificity of IL-18 was verified by preincubating the cells with IL-18 neutralizing antibodies (10 µg/ml for 1 h) or IL-18BP/Fc chimera (200 ng/ml). Normal IgG (10 µg/ml) and Fc (200 ng/ml) served as controls. These experiments were performed at least 3 times. B, IL-18 stimulates NF-κB-driven reporter gene activity. EC transiently transfected with pNF-κB-Luc vector, and 24 h later treated with IL-18 (n=3). Co-transfection with Renilla luciferase vector was used to compensate for variations in transfection efficiency. pEGFP-Luc served as a control. Firefly and Renilla luciferase activities were determined after 7 h. C, IL-18 activates p65 and p50 subunits of NF-κB. Quiescent EC treated as in A were analyzed for NF-κB p50 and p65 subunits in the nuclear protein extracts by ELISA (n=3). D, IL-18 induces p65 phosphorylation. Quiescent EC were treated with IL-18 for 20 min, and analyzed for p65 phosphorylation by Western blotting using cell lysates (n=3). A, arrow denotes NF-κB-specific DNA-protein complexes, the solid circle represents unincorporated labeled probe. B, *p<0.001 Vs. Untreated, †p<0.01 Vs. IL-18; C, *p<0.001 Vs. respective Untreated.
IL-18 stimulates PTEN transcription
Activation of NF-κB leads to suppression or activation of PTEN expression depending on the stimulus and the cell type [31,32]. We previously demonstrated that IL-18 stimulates PTEN induction in EC via NF-κB-activation [12]. The results shown in Fig. 2 indicate that IL-18 induces PTEN mRNA expression (Fig. 2A) and protein levels (Fig. 2B). Furthermore, IL-18 stimulated PTEN promoter-reporter activity (Fig.2C), an effect that was blunted following pretreatment with IL-18 neutralizing antibodies or IL-18BPa/Fc chimera. IL-18-mediated PTEN promoter reporter activity was attenuated by the adenoviral transduction of dnIκBα, dnp65 or dnIKKβ (Fig. 2D), indicating that IL-18 mediates PTEN expression via NF-κB activation. The PTEN promoter has two putative NF-κB binding sites located at −1574/−1565 (NF-κB-1) and −1450/−1441 (NF-κB-2) [33–35]. Therefore, we generated deletion constructs lacking one or both NF-κB-binding sites. EC were transiently transfected with the full length or deletion constructs and treated with IL-18 after 24 h. Results in Fig. 2E show that transfection with the full length promoter resulted in a significant increase in basal PTEN reporter activity. While deletion of NF-κB-1 reduced, deletion of both sites significantly attenuated basal promoter reporter activity. These effects were more pronounced after IL-18 treatment. While IL-18 stimulated PTEN promoter activity, deletion construct lacking NF-κB-1 site showed reduced activity (Fig. 2E). Deletion of both the sites, however, inhibited IL-18-mediated PTEN reporter activity (Fig. 2E). These results indicate that (i) IL-18 is a potent inducer of PTEN expression in EC, (ii) induces PTEN expression via NF-κB, and (iii) both NF-κB sites contribute to IL-18-mediated PTEN transcription (Fig. 2).
Figure 2. IL-18 induces PTEN expression.
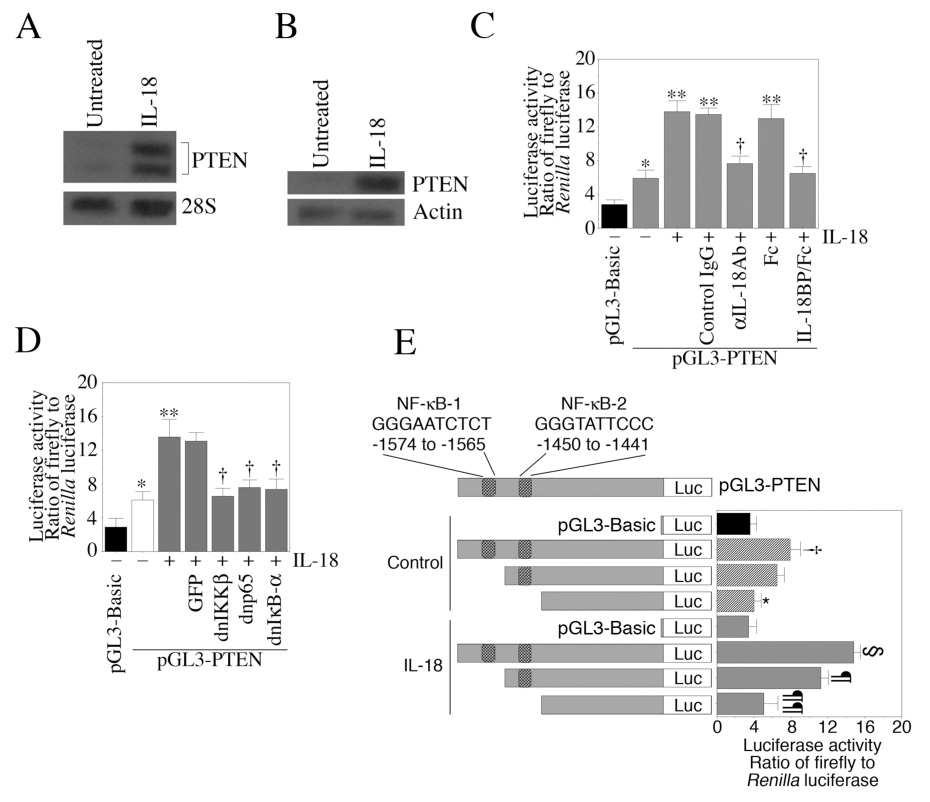
A, IL-18 induces PTEN mRNA expression. Quiescent EC were treated with IL-18 for 2 h, and PTEN mRNA expression was analyzed by Northern blotting (n=3). 28S rRNA served as an internal control. B, IL-18 induces PTEN protein expression. Quiescent EC were treated with IL-18 or 12 h. PTEN protein levels were analyzed in cleared cell lysates by Western blotting (n=3). C, IL-18 stimulates PTEN promoter-reporter activity. EC were transiently transfected with pGL3-PTEN for 24 h, and then treated with IL-18 (100 ng/ml) for 7 h (n=8). pGL3-Basic served as a vector control. EC were co-transfected with pRL-TK vector (100 ng) to normalize for variations in transfection efficiency. Specificity of IL-18 was verified by incubating cells with anti-IL-18 neutralizing antibodies or IL-18BP/Fc chimera 1 h prior to IL-18 addition. D, IL-18-induced PTEN promoter activity is blunted by dnIKKβ, dnp65 and dnIκB-α. EC transiently transfected with pGL3-PTEN were transduced with adenoviral dnIKKβ, dnp65 or dnIκB-α for 24 h, and then treated with IL-18 for 7 h (n=6). GFP served as a control. E, Deletion of NF-κB sites blunts IL-18-mediated PTEN promoter-reporter activity. EC transiently transfected for 24 h with the full length PTEN promoter reporter vector (shown in upper panel) or deletion constructs lacking one or both NF-κB sites were treated IL-18 for 7 h (n=8). pGL3-Basic served as a control. EC were co-transfected with pRL-TK vector to normalize for variations in transfection efficiency. C, *p<0.05, **p<0.01 Vs. pGL3-Basic, †p<0.05 Vs. IL-18; D, *p<0.05, **p<0.001 Vs. pGL3-Basic, †p<0.05 Vs. IL-18 alone; E, †p<0.05 Vs. empty vector, *p<0.05 Vs. control pGL3-PTEN, §p<0.01 Vs. control pGL3-PTEN or IL-18-treated pGL3-Basic, ¶, p<0.05 Vs. IL-18-treated pGL-PTEN, ¶¶p<0.001 Vs. IL-18-treated pGL3-PTEN.
IL-18 induces EC death via NF-κB- and PTEN-dependent mechanisms
IL-18 induces NF-κB activation in EC (Fig.1). Furthermore, IL-18 induces NF-κB-dependent PTEN expression (Fig.3D). Therefore, we examined whether IL-18-mediated EC death is dependent on NF-κB and PTEN. Cell death was analyzed using Annexin V-FITC/PI staining by flow cytometry. Conforming our previous results [12], results in Fig. 3A show that IL-18 induces significant death in EC at 24 h (results from six independent experiments are summarized in panel B). The pro-apoptotic effects of IL-18 were further confirmed in panel C which shows increased release of mitochondrial cytochrome c into the cytoplasm (Fig. 3C). While adenoviral transduction of dnIKKβ, dnp65 or dnIκB-α attenuated IL-18-induced EC death (Fig. 3D), overexpression of constitutively inactive PTEN reversed IL-18-mediated cell death (Fig. 3E). These results indicate that IL-18 induces EC death in an NF-κB-PTEN-dependent manner (Fig. 3).
Figure 3. IL-18 induces endothelial cells death.
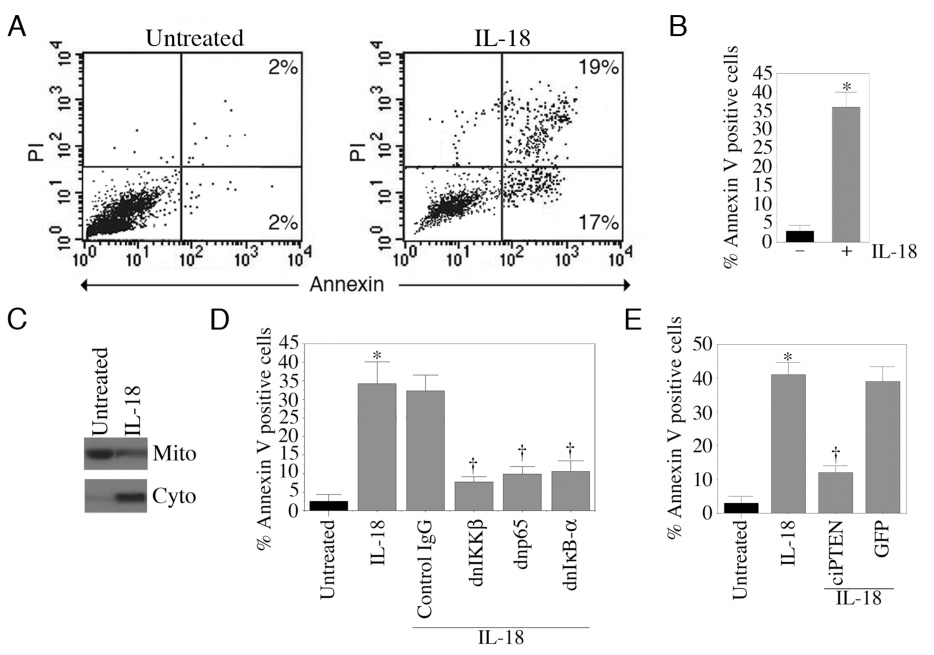
A, IL-18 induces EC death. Quiescent EC were treated with IL-18. After 24 h, cells were harvested, stained with Annexin V-FITC/PI, and analyzed by flow cytometry in untreated controls (left hand panel) and following IL-18 treatment (right hand panel). Results from six independent experiments are summarized in panel B. C, IL-18 induces cytochrome c release from mitochondria to cytoplasm. Quiescent EC were treated with IL-18 for 12 h (n=3). Mitochondrial and cytoplasmic extracts were analyzed for cytochrome c levels by Western blotting. D, NF-κB inhibition blunts IL-18-mediated EC death. EC were treated as in Fig. 2D were analyzed for cell death at 24 h post-IL-18 treatment (n=6). E, Constitutively inactive PTEN inhibits IL-18-mediated EC death. EC were transduced with adenoviral ciPTEN for 24 h, and then treated with IL-18 for an additional 24 h (n=6). Cell death was analyzed as in A. B, *p<0.001 Vs. Untreated; D, *p<0.001 Vs. Untreated, †p<0.001 Vs. IL-18; E, *p<0.001 Vs. Untreated, †p<0.001 Vs. IL-18.
IL-18 inhibits HO-1 mRNA expression
HO-1 is a stress-response gene that cleaves the α-mesocarbon of heme yielding equimolar concentrations of CO, free iron and biliverdin [16]. Both CO and biliverdin exert anti-apoptotic and anti-oxidant effects (16). Cytokines play a critical role in various cardiovascular diseases. Cytokines have been shown to modulate HO-1 expression in a cell type-dependent manner. TNF-α, a proinflammatory and pro-apoptotic cytokine, has been shown to modulate HO-1 expression. In a recent study, Kirino et al. have demonstrated that TNF-α suppresses HO-1 expression in human peripheral monocytes via increased mRNA degradation [27]. Since reduced HO-1 expression is associated with increased cell death, we investigated whether IL-18 modulates HO-1 expression in EC. Results in Fig. 4A show that EC express HO-1 mRNA under basal conditions, and IL-18 treatment inhibited its expression. Decreased mRNA levels reflect reduced transcription and/or increased mRNA degradation. Therefore, we studied the effects of IL-18 on HO-1 transcription by nuclear run-on assay and mRNA stability by actinomycin D pulse. Our results show that IL-18 failed to modulate HO-1 transcription and HO-1 promoter reporter (pHO-1-Luc) activity (data not shown). However, IL-18 decreased stability of HO-1 mRNA (Fig. 4B), demonstrating that IL-18 suppresses HO-1 expression via enhanced mRNA degradation (Fig. 4).
Figure 4. IL-18 attenuates HO-1 expression.
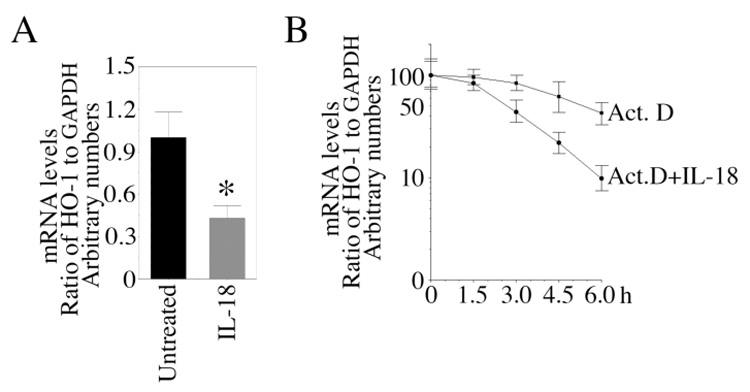
A, IL-18 inhibits HO-1 mRNA expression. Quiescent EC were treated with IL-18 for 24 h. HO-1 mRNA expression was analyzed by RT-qPCR (n=6). GAPDH served as internal control. HO-1 mRNA expression in untreated controls was considered as 1.B, IL-18 decreases stability of HO-1 mRNA. EC were treated with IL-18 for 12 h, followed by Actinomycin D (5µg/ml) for 1.5, 3, 4.5 and 6 h. HO-1 and GAPDH mRNA expressions were analyzed by RT-qPCR, and results from 3 independent experiments is shown as a ratio of HO-1 to GAPDH. A, *p<0.01 Vs. Untreated.
Adenoviral transduction of HO-1 blocks IL-18-mediated EC death
We have demonstrated that IL-18 suppresses HO-1 expression (Fig. 4) and induces EC death (Fig. 3). We next investigated whether HO-1 overexpression blunts IL-18-mediated EC death. Results in Fig. 5A show that adenoviral transduction of wtHO-1 significantly attenuated IL-18-mediated EC death. Western blotting revealed efficient HO-1 protein expression in EC trasduced with Ad.HO-1 (Fig. 5A, right side panel). Further investigation revealed that HO-1 overexpression blunts IL-18-mediated NF-κB activation (Fig. 5B) and PTEN protein levels (Fig. 5C; results from three independent experiments are summarized in the bottom panel). Importantly, HO-1 overexpression restored activation levels of the pro-survival factor Akt as evidenced by increased phospho-Akt levels (Fig. 5D; results from three independent experiments are summarized in the bottom panel). However, total Akt levels were not affected by HO-1 overexpression (Fig. 5D). These results demonstrate that HO-1 overexpression exerts prosurvival effects by suppressing IL-18-mediated NF-κB activation and PTEN induction, and restoring phospho-Akt levels (Fig. 5).
Figure 5. HO-1 blocks IL-18-mediated EC death.
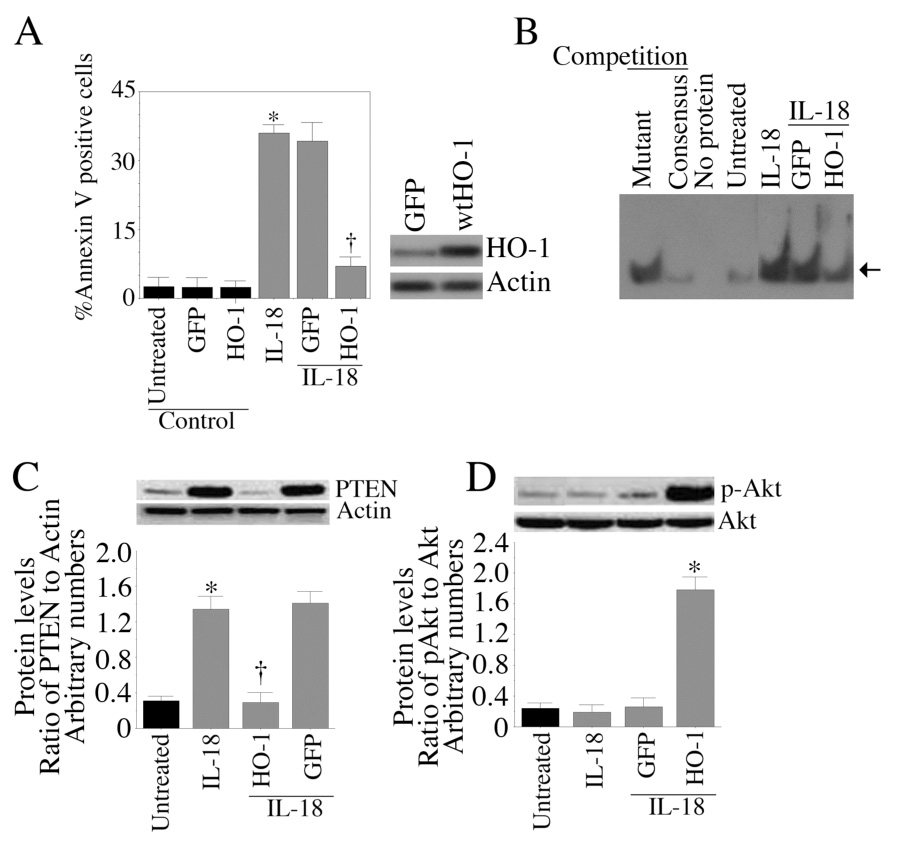
A, EC were transduced with Ad.HO-1 for 24 h (HO-1 expression was analyzed by Western blotting, right side panel, (n=3)) and then treated with IL-18 (100 ng/ml) for an additional 24 h. Cell death was analyzed by Annexin V-FITC/PI staining and flow cytometry (n=3). B, Adenoviral transduction of HO-1 attenuates IL-18-mediated NF-κB activation. EC were transduced with Ad.HO-1 for 24 h, and then treated with IL-18 for one hour (n=3). Nuclear protein was extracted and analyzed for NF-κB DNA binding activity by EMSA. C, HO-1 blunts IL-18-mediated PTEN expression. EC transduced with Ad.HO-1 were treated with IL-18 for 12 h. PTEN protein levels in cleared cell lysates were analyzed by Western blotting. Actin served as a control. Densitometric analysis from three independent experiments is shown in the lower panel as a ratio of PTEN to corresponding Actin levels. D, HO-1 reverses IL-18-mediated inhibition of Akt activation. EC treated as in C for 1 h were analyzed for Akt activation by Western blotting using activation-specific antibodies. Densitometric analysis from three independent experiments is summarized in the lower panel. Arrow in B denotes NF-κB-specific DNA-protein complexes. A, *p<0.001 Vs. Untreated, †p<001 Vs. IL-18; C, *p<0.001 Vs. Untreated, †p<0.01 Vs. IL-18; D, *p<0.001 Vs. Vs. IL-18 or IL-18-treated GFP-transfected cells.
Hemin and CO-donors blunt IL-18-mediated NF-κB activation, PTEN transcription, and cell death
We have demonstrated that HO-1 overexpression suppresses IL-18-mediated EC death via activation of Akt and suppression of NF-κB-PTEN signaling (Fig. 5). We next investigated whether the HO-1 substrate hemin and the CO donor CoPPIX mimic the HO-1 cytoprotective effects. The results in Fig. 6A show that indeed treatment with the HO-1 inducer hemin or the CO donor CoPPIX significantly attenuated IL-18-mediated EC death. Both hemin and CoPPIX blunted IL-18-mediated NF-κB reporter activity (Fig. 6B). Similar to CoPPIX, two other CO donors CORM-1 and CORM-3 attenuated IL-18-mediated EC death (Fig. 6C) and NF-κB reporter activity (Fig. 6D). However, treatment with the inactive form of CORM-3, iCORM-3 failed to confer any protection (Fig. 6C). Of note, CORM-3 appeared to be more cytoprotective than CORM-1 (Fig. 6C). Furthermore, hemin, CoPPIX, CORM-1 and CORM-3 all reversed IL-18-mediated PTEN induction (Fig. 6E) and suppression of Akt activation (Fig. 6F). Together, these results indicate that hemin and CO donors mimic HO-1 effects, and reverse IL-18-induced endothelial cell death (Fig. 6).
Figure 6. Hemin and CO donors attenuate IL-18-mediated EC death.
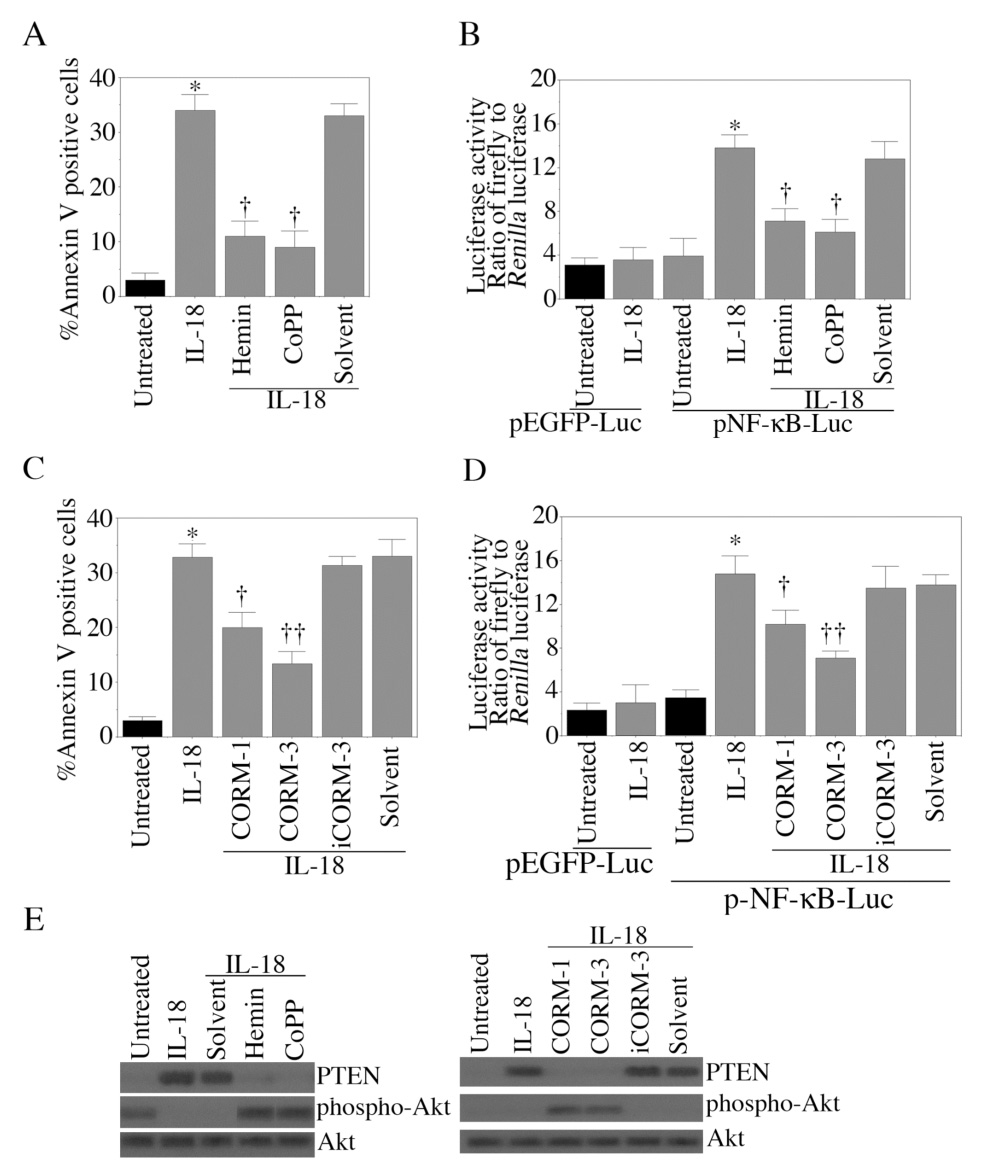
A, Hemin and CoPPIX inhibit IL-18-mediated EC death. Quiescent EC were treated with hemin (2.5µM for 24 h) or CoPPIX (10 µM for 24 h) prior to IL-18 addition. 24 h later, cell death was assessed by Annexin V-FITC/PI staining and flow cytometry. ZnPPIX served as a control. Results from 6 independent experiments are shown. B, Hemin and CoPPIX blunt IL-18-mediated NF-κB activation. EC transiently transfected with NF-κB reporter vector were treated with IL-18 with and without hemin, CoPPIX, ZnPPIX or their solvent control. pEGFP transfection served as a control. pRLTK vector was used to normalize for variations in transfection efficiency. Luciferase activities were determined 7 h post-treatment. These experiments were performed 8 times. C, CO donors blunt IL-18-mediated EC death. Quiescent EC were treated with CORM-1 or CORM-3 (10 µM) prior to IL-18 addition. iCORM-3 (10 µM) served as a control. After 24 h, cell death was analyzed by Annexin V-FITC/PI staining and flow cytometry (n=6). D, CO donors inhibit IL-18-mediated NF-κB activation. EC were treated with CO donors and IL-18 as in B. Luciferase activities were determined after 7 h. These experiments were performed 8 times. E, CO donors (left side panel, Hemin and CoPPIX; right side panel, CORM-1, CORM-3) reverse IL-18-mediated PTEN induction and Akt suppression. Quiescent EC were treated with IL-18 and/or CO donors for 12 h. Akt, phospho-Akt and PTEN levels were analyzed by Western blotting (n=3). A, *p<0.001, **p<0.01 Vs. Untreated, †p<0.01 Vs. IL-18; B, *p<0.001 Vs. Untreated, †p<0.05 Vs. IL-18; C, *p<0.001 Vs. Untreated, †p<0.05 Vs. IL-18; D, *p<0.001 Vs. Untreated, †p<0.05 Vs. IL-18.
HO-1 knockdown, suppression, and hemoglobin reverse hemin’s pro-survival effects
In addition to being a substrate for HO-1, hemin also induces HO-1 expression [36]. Therefore, we investigated whether hemin induces HO-1 expression in EC, and whether knockdown of HO-1 abrogates the pro-survival effects of hemin. Our results show that hemin induces HO-1 mRNA expression in a time-dependent manner, with maximal induction detected at 24 h (Fig. 7A). Furthermore, siRNA-mediated HO-1 knockdown blunted the pro-survival effects of hemin (Fig. 7B; knockdown of HO-1 was confirmed by Western blotting, Fig. 7C). Similarly, the HO-1 inhibitor ZnPPIX (Fig. 7B) and the CO scavenger hemoglobin (Fig. 7D) effectively reversed the cytoprotective effects of hemin. Together these results demonstrate that HO-1 knockdown or suppression reverses hemin’s cytoprotective effects (Fig. 7).
Figure 7. Hemin blocks IL-18-mediated EC death via HO-1 induction.
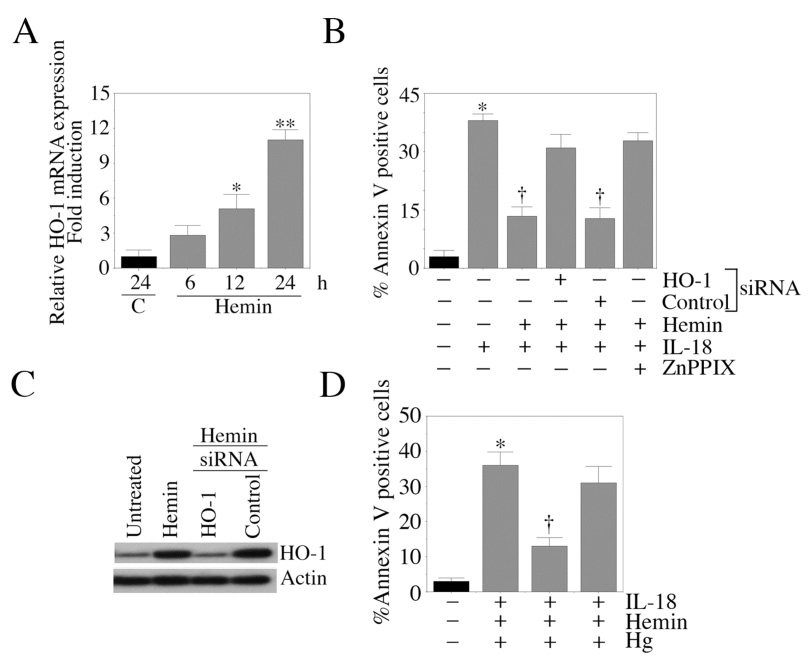
A, Hemin induces HO-1 expression. Quiescent EC were treated with hemin (10 µM). Cells were harvested at the indicated time periods, and analyzed for HO-1 mRNA expression by RT-qPCR (n=6). B, HO-1 knockdown blocks hemin-mediated rescue of IL-18-dependent EC death. EC were treated with HO-1 siRNA for 48 h, and then treated with hemin and IL-18 for an additional 24 h. Cell death was analyzed by Annexin V-FITC/PI staining and flow cytometry (n=6). C, HO-1 knockdown is confirmed by Western blotting (n=3). EC treated as in C were analyzed for HO-1 knockdown by Western blotting at 48 h post-HO-1 siRNA treatment. siRNA that does not target any gene in the mammalian genome served as a control. D, Hemoglobin (Hg) reverses hemin-mediated rescue of IL-18-depndent EC death. Quiescent EC were treated with hemin and Hg prior to IL-18 addition. After 24 h, cell death was analyzed by Annexin V-FITC/PI staining and flow cytometry (n=6). A, *p<0.01, **p<0.001 Vs. Untreated; B, *p<0.001 Vs. Untreated, †p<0.01 Vs. IL-18 alone; D, *p<0.001 Vs. Untreated, †p<0.01 Vs. IL-18.
Biliverdin and bilirubin inhibit IL-18-mediated EC death
HO catalyzes the conversion heme to biliverdin. Biliverdin is subsequently converted to bilirubin by biliverdin reductase. Both biliverdin and bilirubin exert strong antioxidant and anti-inflammatory effects [37]. Therefore, we investigated whether exposure to these heme metabolites modulate IL-18-mediated EC death. The results in Fig. 8A show that cell death was blunted following treatment with either biliverdin or bilirubin. Furthermore, biliverdin and bilirubin reversed IL-18-induced NF-κB reporter activity (Fig. 8B), PTEN induction, and suppression of Akt phosphorylation (Fig. 8C). These results demonstrate that while hemoglobin reverses the pro-survival effects of hemin (Fig. 7D), its metabolites confer cytoprotection by inhibiting IL-18-induced endothelial cell death (Fig. 8).
Figure 8. The hemoglobin degradation products bilirubin and biliverdin attenuate IL-18-mediated EC death.
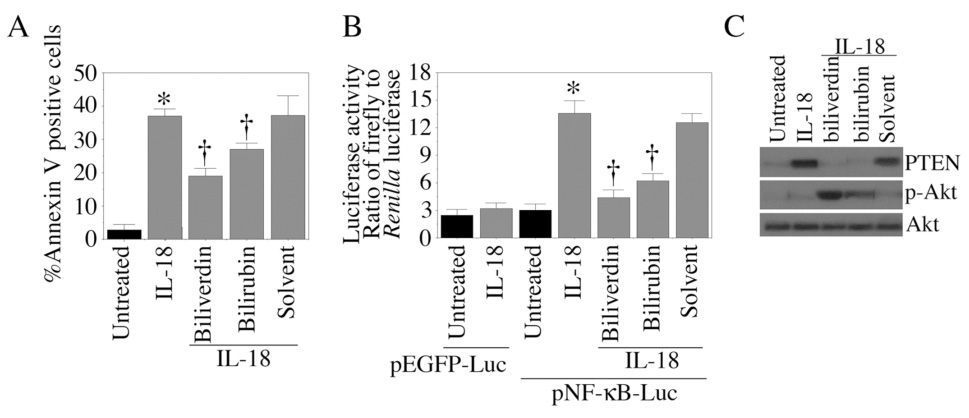
A, Bilirubin and biliverdin blunt IL-18 induced EC death. Quiescent EC were treated with IL-18 and/or bilirubin or biliverdin (10 µM) for 24 h. Cell death was analyzed by Annexin V-FITC/PI staining and flow cytometry. These experiments were performed at least 6 times. B, Bilirubin and biliverdin blunt IL-18-mediated NF-κB-dependent reporter gene activity. EC transiently transfected with the NF-κB reporter vector were treated with IL-18 and/or bilirubin or biliverdin. Luciferase activities were analyzed 7 h post-treatment (n=6). C, Bilirubin and biliverdin reverse IL-18-mediated PTEN induction and Akt suppression. Quiescent EC were treated with IL-18 and/or bilirubin or biliverdin for 1 (Akt) or 12 h (PTEN). PTEN, phospho-Akt, and Akt levels were assessed by Western blotting (n=3). A, B, *p<0.001 Vs. Untreated, †p<0.05 Vs. IL-18.
IL-18 differentially regulates p38 MAPK isoforms
p38 MAPK is a mitogen-activated serine-threonine protein kinase whose activation plays a role in diverse cellular activities including cell death. We have previously demonstrated that IL-18 stimulates p38 MAPK activation in EC, and inhibition of p38 MAPK by SB203580 significantly attenuated IL-18-dependent p65 phosphorylation, NF-κB activation, and PTEN promoter activity [12]. While the p38α isoform promotes cell death, the p38α isoform exerts anti-apoptotic effects. Since HO-1 exerts anti-apoptotic effects via degradation of p38α MAPK (38), we investigated whether IL-18 differentially regulates p38 MAPK isoforms in EC, and determined whether knockdown of p38α will abrogate IL-18-mediated EC death. Results show that IL-18 induces p38 MAPK phosphorylation (Fig. 9A) and kinase activity (Fig. 9B), effects that were inhibitable by SB202190, thus confirming our previous observations [12]. Using isoform-specific antibodies and Western blotting, we further demonstrated that IL-18, while inducing p38α, inhibits p38β expression. However, levels of the control protein αTubulin were not changed. A representative autoradiogram from three independent experiments is shown in Fig. 9C with the corresponding densitometric values summarized in Fig. 9D. These results demonstrate that IL-18 differentially regulates p38 MAPK isoforms, i.e. IL-18 induced p38α while suppressing p38β.
Figure 9. IL-18 stimulates p38 MAPK activation.
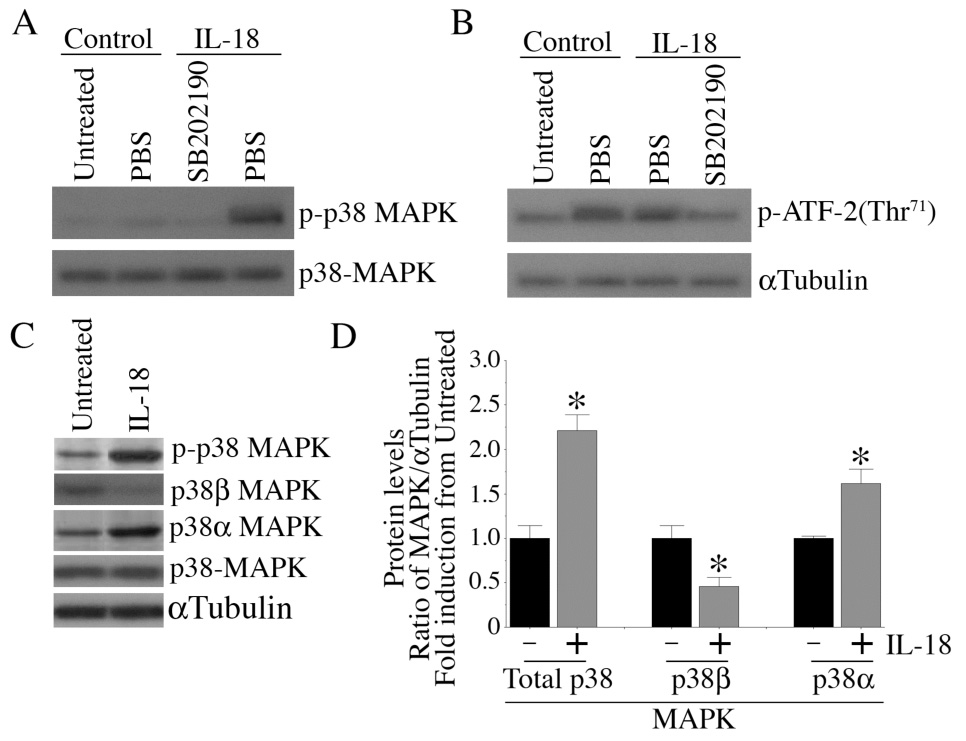
A, IL-18 induces p38 MAPK phosphorylation. Quiescent EC were treated with SB202190 in DMSO (30 µM for 30 min) prior to IL-18 addition (100 ng/ml for 1 h). Total protein was extracted and cleared cell lysates were analyzed for total and p-p38 MAPK by Western blotting using activation-specific antibodies. A representative of three independent experiments is shown. B, IL-18 stimulates p38 MAPK enzyme activity. EC treated as in A were analyzed for p38 MAPK activity using a commercially available colorimetric assay kit (n=3). ATF-2 served as a substrate. C, IL-18 differentially regulates p38 MAPK isoforms. Quiescent EC were treated with IL-18 for 1 h, and cleared cell lysates were analyzed for total p38 MAPK and p38 MAPK isoforms (p38α, and p38β) by Western blotting using isoform-specific antibodies. µTubulin served as a control. Autoradiographic signals from three independent experiments were semiquantified by videoimage analysis and the results are presented in panel D as a ratio of MAPK to αTubulin. D, *p at least <0.05 Vs. respective Untreated.
IL-18-mediated EC death is blunted by p38α MAPK knockdown
We have demonstrated that IL-18 differentially regulates p38 MAPK isoforms (Fig.9). However it is not known whether hemin or HO-1 expression blunts IL-18-mediated EC death via inhibition of p38α. Results in Fig. 10A show that treatment with hemin (2.5 µM for 24 h) resulted in increased p38β with concomitant down regulation of p38α expression. These experiments were repeated three times with results summarized in Fig. 10B. Since forced expression of HO-1 blunted IL-18-mediated EC death (Fig.5A), we investigated whether HO-1 overexpression results in differential activation of p38 MAPK isoforms. Results in Fig. 10C show that HO-1 overexpression resulted in a modest increase in p38β expression in the absence of IL-18 stimulation. Confirming our earlier results (Fig. 9), IL-18 induced p38α and inhibited p38β expression, and these effects were reversed following HO-1 overexpression (Fig. 10C). These experiments were repeated three times with results summarized in Fig. 10D. Since IL-18 inhibited p38β and induced p38β expression, we next investigated whether knockdown of p38α would inhibit IL-18-mediated EC death. EC were treated with p38α or p38β isoform-specific siRNA for 48, and then treated with IL-18 for an additional 24 h. Results in Fig. 10E demonstrate that while knockdown of p38α attenuates, knockdown of p38α potentiates IL-18-mediated EC death. Knockdown of p38α and p38β was confirmed by Western blotting (Fig. 10F). Knockdown of p38β did not induce cell death under basal conditions. Together, these results indicate that (i) hemin or HO-1 overexpression differentially regulates p38 MAPK isoforms, i.e. inhibits p38α and induces p38β expression, and (ii) while knockdown of p38α inhibits, knockdown of p38β potentiates IL-18-mediated EC death (Fig. 10).
Figure 10. Hemin and HO-1 differentially regulate p38 MAPK isoforms.
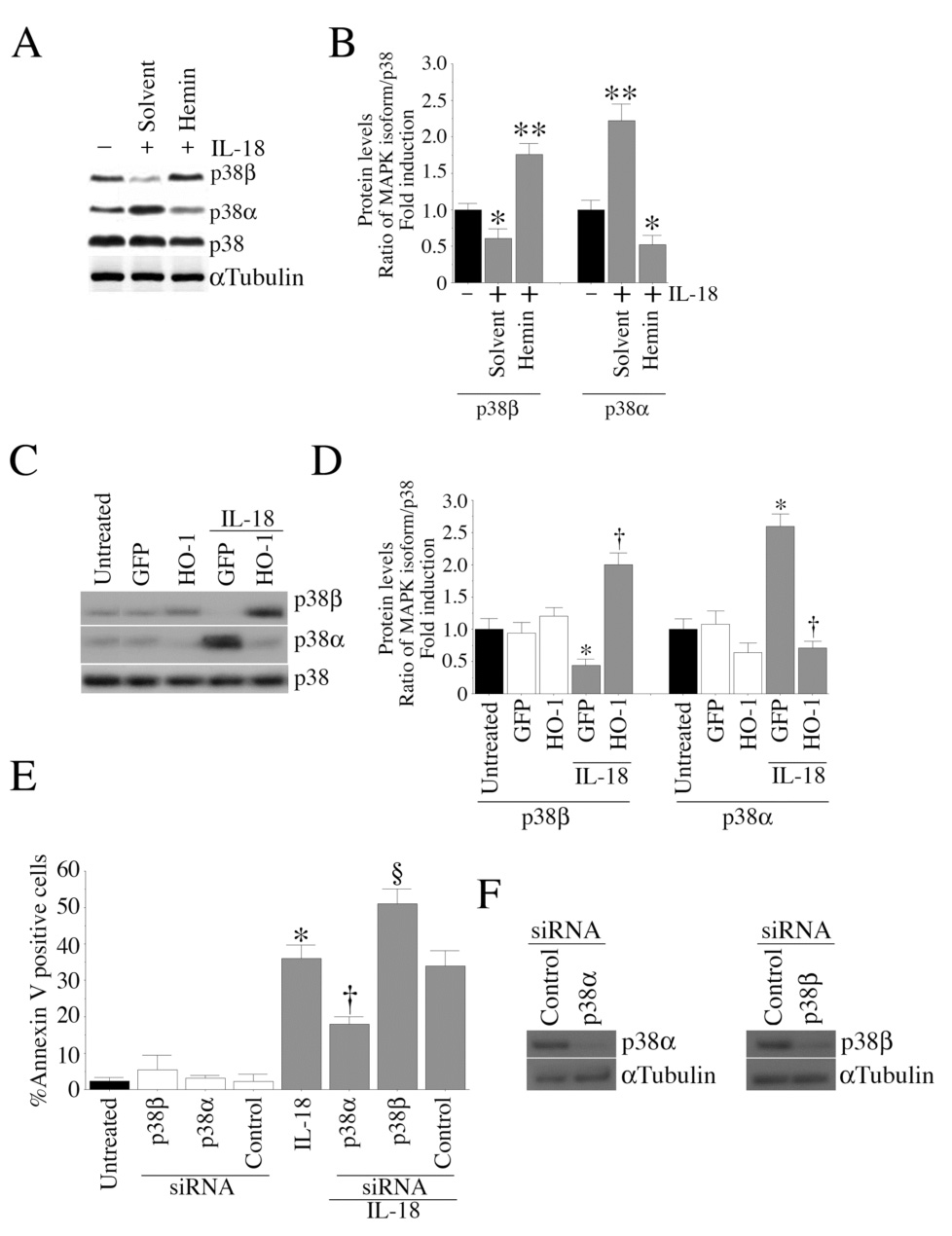
A, Hemin differentially regulates p38 MAPK isoforms. Quiescent EC were treated with hemin (10 µM for 24 h), and analyzed for total p38 MAPK and p38 isoforms by Western blotting. B, The autoradiographic signals in panel A were semiquantified by videoimage analysis, and presented as a ratio of p38 MAPK isoforms to that of total p38 MAPK from three independent experiments. C, Ectopic expression of HO-1 reverses IL-18 mediated suppression in p38β MAPK levels. EC transduced with Ad-HO-1 or Ad.GFP for 24 h were treated with IL-18. One hour later, cleared cell lysates were analyzed for total p38 and p38 isoforms by Western blotting. D, The autoradiographic signals in panel C were semiquantified by videoimage analysis, and results are presented as a ratio of p38 MAPK isoforms to that of total p38 MAPK from three independent experiments. E, Knockdown of p38β isoform potentiates IL-18-mediated EC death. EC were treated with p38 MAPK isoform-specific siRNA (100 nM for 48 h) and then treated with IL-18. After 24 h, cell death was analyzed by Annexin V-FITC/PI staining and flow cytometry (n=6). F, Knockdown of p38 MAPK isoforms was confirmed by Western blotting (n=3). αTubulin served as a control. B, *p<0.05 Vs. respective Untreated or solvent, **p<0.001 Vs. Untreated; D, *p<0.01 Vs. respective Untreated, †p<0.001 Vs. IL-18-treated GFP; E, *p<0.001 Vs. Untreated, †p<0.01 Vs. IL-18, §p<0.05 Vs. IL-18.
Discussion
We demonstrate that HO-1 overexpression or HO-1 induction inhibits IL-18-mediated endothelial cell death. The CO donor CoPPIX and the heme metabolites biliverdin and bilirubin mimic HO-1 induction and inhibit IL-18-mediated endothelial cell death. Furthermore, HO-1 overexpression, HO-1 induction, CO donors, as well as the heme metabolites all inhibit cell death via a similar signaling pathway; they suppress IL-18-mediated NF-κB activation and PTEN induction, and reverse IL-18-mediated suppression of Akt activity. Thus, HO-1 inducers and CO donors have the therapeutic potential to effectively block IL-18 signaling and reduce IL-18-dependent vascular injury and inflammation.
Endothelial cells play a critical role in vascular homeostasis. In a healthy vessel endothelial cells play an anti-thrombotic and anti-inflammatory role [39]. However, their dysfunction and death contribute to the development and progression of atherosclerosis [39]. Endothelial cell apoptosis is observed in atherosclerotic plaques localized mainly to areas of macrophage infiltration [40]. Activated macrophages secrete various proinflammatory cytokines. These cytokines induce adhesion molecule expression on endothelial cells leading to platelet adhesion and immune cell recruitment. Cytokines also induce other proinflammatory mediators (e.g. prostaglandin E2) resulting in endothelial cell dysfunction and death [41]. This leads to plaque instability, rupture, and thrombus formation. Oxidized LDL in the vessel wall as well as systemic and locally expressed CRP have all been shown to exert proinflammatory effects. Recently, CRP has been shown to induce IL-18 expression in endothelial cells [42]. Since endothelial cells express IL-18 receptors and their expression levels are increased in atherosclerotic lesions [43], locally generated IL-18 likely contributes to endothelial cell dysfunction. In addition, systemic IL-18 levels are increased in acute coronary syndromes [1], and show a positive correlation with intima-media thickening. It is plausible that systemic IL-18 may enhance endothelial cell dysfunction and death.
IL-18 induces endothelial cell death via activation of NF-κB [11,12]. Activation of NF-κB proceeds following the phosphorylation and degradation of the inhibitory subunit IκB-α. Since HO-1 attenuates IL-18-mediated NF-κB activation, it is possible that HO-1 attenuates NF-κB activation via inhibition of IκB-α degradation. In support of this hypothesis, Sarady et al. have demonstrated that HO-1 overexpression in a murine macrophage cell line reduced LPS-mediated GM-CSF expression by inhibiting IκB-α degradation [44]. In contrast, Brouard, et al. have demonstrated that HO-1-derived CO requires the activation of NF-κB to protect endothelial cells from TNF-α-mediated apoptosis (45). These conflicting observations suggest that the role of NF-κB in HO-1-mediated cell survival is cell- and stimulus-specific. Our data show that, similar to HO-1 overexpression, treatment with the HO-1 inducer hemin, CO donors or heme metabolites promoted cell survival by suppressing IL-18-mediated NF-κB activation. In contrast, the CO scavenger hemoglobin reversed the pro-survival effects of hemin. These observations suggest that CO may attenuate IL-18-mediated NF-κB activation by inhibiting IκB-α phosphorylation and degradation. Since IL-18 induced the expression of pro-apoptotic PTEN in an NF-κB-dependent manner, it is possible that the pro-survival effects of HO-1 in endothelial cells are mediated, at least in part, via suppression of NF-κB-PTEN signaling.
Recently, Silva et al. have demonstrated that HO-1 exerts anti-apoptotic effects in bovine aortic endothelial cells by suppressing p38α MAPK expression [38]. These authors demonstrated that HO-1-generated CO was responsible for this down-regulation and the observed reduction in cell death. However, although HO-1 expression resulted in reduced p38 protein levels, neither p38α activity nor p38β levels were affected [38]. In contrast, our results demonstrate that treatment with IL-18 increases p38α and suppresses p38β levels. Furthermore, treatment with hemin or HO-1 overexpression reversed this phenomenon, and enhanced p38β expression. These results were further confirmed in studies using siRNA-mediated isoform-specific knockdown. While knockdown of p38α inhibited, knockdown of p38β potentiated IL-18-mediated endothelial cell death. Since the CO donors CoPPIX, CORM-1 and CORM-3 were all capable of inhibiting IL-18-mediated endothelial cell death, it is plausible that CO donors may act via a similar signaling pathway involving activation of p38β and suppression of p38α.
Our results demonstrate that IL-18 suppresses activation of the pro-survival factor Akt. Treatment with IL-18 suppressed Akt phosphorylation in a time-dependent manner, while significantly increasing the expression of the proapoptotic factor PTEN. These effects were reversed following HO-1 overexpression or HO-1 induction by hemin, and are mimicked by CO donors and heme metabolites. Treatment with these agents restored phospho-Akt levels and suppressed PTEN. However, the mechanism by which these agents restored phospho-Akt is not known and warrants further study. It is possible that HO-1 may have regulated Akt activation via two different mechanisms. Brunt et al. have demonstrated functional codependence of HO-1 and Akt in HO-1 treated hydrogen peroxide-induced smooth muscle cell death [46]. Though HO-1 is regulated primarily at the transcriptional level, Akt-induced HO-1 phosphorylation resulted in increased HO-1 activity. These results suggest that Akt regulates HO-1 expression post-transcriptionally [47]. HO-1 phosphorylation also increased its binding affinity for cytochrome P450 reductase and BVR [47]. Whether such interactions occur in IL-18-treated endothelial cells transduced with HO-1 or treated with HO-1 inducers or CO donors is not known. Our results indicate that IL-18 suppresses Akt activation in a time-dependent manner while simultaneously increasing pro-apoptotic PTEN expression. It is possible that forced expression of HO-1 or HO-1 induction prior to IL-18 might increase HO-1-Akt interactions leading to suppressed IL-18-induced cell death.
We have previously demonstrated that IL-18 activates both the intrinsic and the extrinsic pro-apoptotic signaling pathways that converge at caspase-3 activation [11]. HO-1 has been shown to blunt cell death by regulating multiple apoptotic signaling pathways. In monocytes, hemin-induced HO-1 activity significantly inhibited serum withdrawal and dexamethasone-induced cell death [48]. While these stimuli resulted in caspase-3 activation, the cytoprotective effects of HO-1 were caspase-3-independent. In contrast, the anti-apoptotic effect of CO is associated with lowered caspase-3 activity in hepatocytes [49]. It is not known whether HO-1 suppresses IL-18-mediated endothelial cell death by a mechanism involving caspase-3.
Our results also demonstrate that the heme metabolites biliverdin and bilirubin blunt IL-18-mediated endothelial cell death. It has been previously reported that bilirubin administration was vasoprotective in a rat model of vascular balloon injury [50], similar to that seen with HO-1 overexpression, suggesting that one component of the cytoprotective effects of HO-1 is likely mediated by bilirubin and/or biliverdin. For example, prior studies have shown that administration of bilirubin inhibited JNK phosphorylation, c-Jun phosphorylation, and endothelial cell apoptosis [51]. Since IL-18 activates JNK [52], it is conceivable that bilirubin and biliverdin may have inhibited endothelial cell death via suppression of IL-18-induced JNK activation.
The protective effects of CO against the deleterious effects of cytokines and pro-oxidants may result from enhanced expression of anti-apoptotic genes. We have previously shown that IL-18, while increasing the expression of proapoptotic bcl-XS, inhibits the expression of antiapoptotic bcl-XL in endothelial cells [11]. HO-1 overexpression or induction, and CO donors might alter this ratio in favor of bcl-XL thus inhibiting cell death. In streptozotocin-induced diabetic kidney, HO-1 induction increased bcl-XL expression, but failed to modulate bax expression [26]. Similarly, CoPPIX has been shown to induce bcl-XL expression [53]. It is thus possible that CO donors or HO-1 may exert cytoprotective effects in endothelial cells by stimulating bcl-XL expression.
In summary, our results show that HO-1 either overexpressed or induced, as well as treatment with CO donors or heme metabolites significantly inhibit IL-18-mediated endothelial cell death by attenuating p38α MAPK and NF-κB activation and PTEN expression, and reversing the suppression of Akt phosphorylation. Since myocardial ischemic injury and inflammation and coronary artery diseases are characterized by enhanced IL-18 expression and early endothelial cell death, administration of HO-1 inducers or HO-1 metabolites may have therapeutic potential in limiting IL-18-dependent tissue injury and inflammation.
Acknowledgments
Grant support: NIH RO1 HL-68020
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References Cited
- 1.Mallat Z, Henry P, Fressonnet R, Alouani S, Scoazec A, Beaufils P, Chvatchko Y, Tedgui A. Increased plasma concentrations of interleukin-18 in acute coronary syndromes. Heart. 2002;88:467–469. doi: 10.1136/heart.88.5.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yamagami H, Kitagawa K, Hoshi T, Furukado S, Hougaku H, Nagai Y, Hori M. Associations of serum IL-18 levels with carotid intima-media thickness. Arterioscler Thromb Vasc Biol. 2005;25:1458–1462. doi: 10.1161/01.ATV.0000168417.52486.56. [DOI] [PubMed] [Google Scholar]
- 3.Suchanek H, Mysliwska J, Siebert J, Wieckiewicz J, Hak L, Szyndler K, Kartanowicz D. High serum interleukin-18 concentrations in patients with coronary artery disease and type 2 diabetes mellitus. Eur Cytokine Netw. 2005;16:177–185. [PubMed] [Google Scholar]
- 4.Seta Y, Kanda T, Tanaka T, Arai M, Sekiguchi K, Yokoyama T, Kurimoto M, Tamura J, Kurabayashi M. Interleukin-18 in patients with congestive heart failure: induction of atrial natriuretic peptide gene expression. Res Commun Mol Pathol Pharmacol. 2000;108:87–95. [PubMed] [Google Scholar]
- 5.Pomerantz BJ, Reznikov LL, Harken AH, Dinarello CA. Inhibition of caspase 1 reduces human myocardial ischemic dysfunction via inhibition of IL-18 and IL-1 beta. Proc Natl Acad Sci U S A. 2001;98:2871–2876. doi: 10.1073/pnas.041611398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mallat Z, Heymes C, Corbaz A, Logeart D, Alouani S, Cohen-Solal A, Seidler T, Hasenfuss G, Chvatchko Y, Shah AM, Tedgui A. Evidence for altered interleukin 18 (IL)-18 pathway in human heart failure. FASEB J. 2004;18:1752–1754. doi: 10.1096/fj.04-2426fje. [DOI] [PubMed] [Google Scholar]
- 7.Novick D, Schwartsburd B, Pinkus R, Suissa D, Belzer I, Sthoeger Z, Keane WF, Chvatchko Y, Kim SH, Fantuzzi G, Dinarello CA, Rubinstein M. A novel IL-18BP ELISA shows elevated serum IL-18BP in sepsis and extensive decrease of free IL-18. Cytokine. 2001;14:334–342. doi: 10.1006/cyto.2001.0914. [DOI] [PubMed] [Google Scholar]
- 8.Novick D, Kim SH, Fantuzzi G, Reznikov LL, Dinarello CA, Rubinstein M. Interleukin-18 binding protein: a novel modulator of the Th1 cytokine response. Immunity. 1999;10:127–136. doi: 10.1016/s1074-7613(00)80013-8. [DOI] [PubMed] [Google Scholar]
- 9.Chandrasekar B, Mummidi S, Mahimainathan L, Patel DN, Bailey SR, Imam SZ, Greene WC, Valente AJ. Interleukin-18-induced human coronary artery smooth muscle cell migration is dependent on NF-kappaB- and AP-1-mediated matrix metalloproteinase-9 expression and is inhibited by atorvastatin. J Biol Chem. 2006;281:15099–15109. doi: 10.1074/jbc.M600200200. [DOI] [PubMed] [Google Scholar]
- 10.Chandrasekar B, Mummidi S, Claycomb WC, Mestril R, Nemer M. Interleukin-18 is a pro-hypertrophic cytokine that acts through a phosphatidylinositol 3-kinase-phosphoinositide-dependent kinase-1-Akt-GATA4 signaling pathway in cardiomyocytes. J Biol Chem. 2005;280:4553–4567. doi: 10.1074/jbc.M411787200. [DOI] [PubMed] [Google Scholar]
- 11.Chandrasekar B, Vemula K, Surabhi RM, Li-Weber M, Owen-Schaub LB, Jensen LE, Mummidi S. Activation of intrinsic and extrinsic proapoptotic signaling pathways in interleukin-18-mediated human cardiac endothelial cell death. J Biol Chem. 2004;279:20221–20233. doi: 10.1074/jbc.M313980200. [DOI] [PubMed] [Google Scholar]
- 12.Chandrasekar B, Valente AJ, Freeman GL, Mahimainathan L, Mummidi S. Interleukin-18 induces human cardiac endothelial cell death via a novel signaling pathway involving NF-κB-dependent PTEN activation. Biochem Biophys Res Commun. 2006;339:956–963. doi: 10.1016/j.bbrc.2005.11.100. [DOI] [PubMed] [Google Scholar]
- 13.Clark JE, Foresti R, Sarathchandra P, Kaur H, Green CJ, Motterlini R. Heme oxygenase-1-derived bilirubin ameliorates postischemic myocardial dysfunction. Am J Physiol Heart Circ Physiol. 2000;278:H643–H651. doi: 10.1152/ajpheart.2000.278.2.H643. [DOI] [PubMed] [Google Scholar]
- 14.Guo Y, Stein AB, Wu WJ, Tan W, Zhu X, Li QH, Dawn B, Motterlini R, Bolli R. Administration of a CO-releasing molecule at the time of reperfusion reduces infarct size in vivo. Am J Physiol Heart Circ Physiol. 2004;286:H1649–H1653. doi: 10.1152/ajpheart.00971.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Otterbein LE, Bach FH, Alam J, Soares M, Tao Lu H, Wysk M, Davis RJ, Flavell RA, Choi AM. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med. 2000;6:422–428. doi: 10.1038/74680. [DOI] [PubMed] [Google Scholar]
- 16.Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev. 2006;86:583–650. doi: 10.1152/physrev.00011.2005. [DOI] [PubMed] [Google Scholar]
- 17.Immenschuh S, Schroder H. Heme oxygenase-1 and cardiovascular disease. Histol Histopathol. 2006;21:679–685. doi: 10.14670/HH-21.679. [DOI] [PubMed] [Google Scholar]
- 18.Sivakumar PV, Westrich GM, Kanaly S, Garka K, Born TL, Derry JM, Viney JL. Interleukin 18 is a primary mediator of the inflammation associated with dextran sulphate sodium induced colitis: blocking interleukin 18 attenuates intestinal damage. Gut. 2002;50:812–820. doi: 10.1136/gut.50.6.812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Motterlini R, Clark JE, Foresti R, Sarathchandra P, Mann BE, Green CJ. Carbon monoxide-releasing molecules: characterization of biochemical and vascular activities. Circ Res. 2002;90:E17–E24. doi: 10.1161/hh0202.104530. [DOI] [PubMed] [Google Scholar]
- 20.Clark JE, Naughton P, Shurey S, Green CJ, Johnson TR, Mann BE, Foresti R, Motterlini R. Cardioprotective actions by a water-soluble carbon monoxide-releasing molecule. Circ Res. 2003;93:e2–e8. doi: 10.1161/01.RES.0000084381.86567.08. [DOI] [PubMed] [Google Scholar]
- 21.Vera T, Henegar JR, Drummond HA, Rimoldi JM, Stec DE. Protective effect of carbon monoxide-releasing compounds in ischemia-induced acute renal failure. J Am Soc Nephrol. 2005;16:950–958. doi: 10.1681/ASN.2004090736. [DOI] [PubMed] [Google Scholar]
- 22.Linden DJ, Narasimhan K, Gurfel D. Protoporphyrins modulate voltage-gated Ca current in AtT-20 pituitary cells. J Neurophysiol. 1993;70:2673–2677. doi: 10.1152/jn.1993.70.6.2673. [DOI] [PubMed] [Google Scholar]
- 23.Lutton JD, Abraham NG, Drummond GS, Levere RD, Kappas A. Zinc porphyrins: potent inhibitors of hematopoieses in animal and human bone marrow. Proc Natl Acad Sci U S A. 1997;94:1432–1436. doi: 10.1073/pnas.94.4.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zakhary R, Gaine SP, Dinerman JL, Ruat M, Flavahan NA, Snyder SH. Heme oxygenase 2: endothelial and neuronal localization and role in endothelium-dependent relaxation. Proc Natl Acad Sci U S A. 1996;93:795–798. doi: 10.1073/pnas.93.2.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science. 1987;235:1043–1046. doi: 10.1126/science.3029864. [DOI] [PubMed] [Google Scholar]
- 26.Kruger AL, Peterson SJ, Schwartzman ML, Fusco H, McClung JA, Weiss M, Shenouda S, Goodman AI, Goligorsky MS, Kappas A, Abraham NG. Up-regulation of heme oxygenase provides vascular protection in an animal model of diabetes through its antioxidant and antiapoptotic effects. J Pharmacol Exp Ther. 2006;319:1144–1152. doi: 10.1124/jpet.106.107482. [DOI] [PubMed] [Google Scholar]
- 27.Kirino Y, Takeno M, Murakami S, Kobayashi M, Kobayashi H, Miura K, Ideguchi H, Ohno S, Ueda A, Ishigatsubo Y. Tumor necrosis factor alpha acceleration of inflammatory responses by down-regulating heme oxygenase 1 in human peripheral monocytes. Arthritis Rheum. 2007;56:464–475. doi: 10.1002/art.22370. [DOI] [PubMed] [Google Scholar]
- 28.Teng ZP, Chen J, Chau LY, Galunic N, Regan RF. Adenoviral transfer of the heme oxygenase-1 gene protects striatal astrocytes from heme-mediated oxidative injury. Neurobiol Dis. 2004;17:179–187. doi: 10.1016/j.nbd.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 29.Colombrita C, Lombardo G, Scapagnini G, Abraham NG. Heme oxygenase-1 expression levels are cell cycle dependent. Biochem Biophys Res Commun. 2003;308:1001–1008. doi: 10.1016/s0006-291x(03)01509-2. [DOI] [PubMed] [Google Scholar]
- 30.Chandrasekar B, Melby PC, Sarau HM, Raveendran M, Perla RP, Marelli-Berg FM, Dulin NO, Singh IS. Chemokine-cytokine cross-talk. The ELR+ CXC chemokine LIX (CXCL5) amplifies a proinflammatory cytokine response via a phosphatidylinositol 3-kinase-NF-κB pathway. J Biol Chem. 2003;278:4675–4686. doi: 10.1074/jbc.M207006200. [DOI] [PubMed] [Google Scholar]
- 31.Gu J, Tamura M, Pankov R, Danen EH, Takino T, Matsumoto K, Yamada KM. Shc and FAK differentially regulate cell motility and directionality modulated by PTEN. J Cell Biol. 1999;146:389–403. doi: 10.1083/jcb.146.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamada KM, Araki M. Tumor suppressor PTEN: modulator of cell signaling, growth, migration and apoptosis. J Cell Sci. 2001;114:2375–2382. doi: 10.1242/jcs.114.13.2375. [DOI] [PubMed] [Google Scholar]
- 33.Kim S, Domon-Dell C, Kang J, Chung DH, Freund JN, Evers BM. Down-regulation of the tumor suppressor PTEN by the tumor necrosis factor-α/nuclear factor-κB (NF-κB)-inducing kinase/NFκB pathway is linked to a default IκB-α autoregulatory loop. J Biol Chem. 2004;279:4285–4291. doi: 10.1074/jbc.M308383200. [DOI] [PubMed] [Google Scholar]
- 34.Vasudevan KM, Gurumurthy S, Rangnekar VM. Suppression of PTEN expression by NF-κB prevents apoptosis. Mol Cell Biol. 2004;24:1007–1021. doi: 10.1128/MCB.24.3.1007-1021.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xia D, Srinivas H, Ahn YH, Sethi G, Sheng X, Yung WK, Xia Q, Chiao PJ, Kim H, Brown PH, Wistuba, Aggarwal BB, Kurie JM. Mitogen-activated protein kinase kinase-4 promotes cell survival by decreasing PTEN expression through an NF kappa B-dependent pathway. J Biol Chem. 2007;282:3507–3519. doi: 10.1074/jbc.M610141200. [DOI] [PubMed] [Google Scholar]
- 36.Yoshida T, Biro P, Cohen T, Muller RM, Shibahara S. Human heme oxygenase cDNA and induction of its mRNA by hemin. Eur J Biochem. 1988;171:457–461. doi: 10.1111/j.1432-1033.1988.tb13811.x. [DOI] [PubMed] [Google Scholar]
- 37.Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science. 1987;235:1043–1046. doi: 10.1126/science.3029864. [DOI] [PubMed] [Google Scholar]
- 38.Silva G, Cunha A, Gregoire IP, Seldon MP, Soares MP. The antiapoptotic effect of heme oxygenase-1 in endothelial cells involves the degradation of p38α MAPK isoform. J Immunol. 2006;177:1894–1903. doi: 10.4049/jimmunol.177.3.1894. [DOI] [PubMed] [Google Scholar]
- 39.Landmesser U, Hornig B, Drexler H. Endothelial function: a critical determinant in atherosclerosis? Circulation. 2004;109:II27–II33. doi: 10.1161/01.CIR.0000129501.88485.1f. [DOI] [PubMed] [Google Scholar]
- 40.Kockx MM, De Meyer GR, Muhring J, Jacob W, Bult H, Herman AG. Apoptosis and related proteins in different stages of human atherosclerotic plaques. Circulation. 1998;97:2307–2315. doi: 10.1161/01.cir.97.23.2307. [DOI] [PubMed] [Google Scholar]
- 41.Dimmeler S, Hermann C, Zeiher AM. Apoptosis of endothelial cells. Contribution to the pathophysiology of atherosclerosis? Eur Cytokine Netw. 1998;9:697–698. [PubMed] [Google Scholar]
- 42.Yamaoka-Tojo M, Tojo T, Masuda T, Machida Y, Kitano Y, Kurosawa T, Izumi T. C-reactive protein-induced production of interleukin-18 in human endothelial cells: a mechanism of orchestrating cytokine cascade in acute coronary syndrome. Heart Vessels. 2003;18:183–187. doi: 10.1007/s00380-003-0719-7. [DOI] [PubMed] [Google Scholar]
- 43.Mallat Z, Corbaz A, Scoazec A, Besnard S, Leseche G, Chvatchko Y, Tedgui A. Expression of interleukin-18 in human atherosclerotic plaques and relation to plaque instability. Circulation. 2001;104:1598–1603. doi: 10.1161/hc3901.096721. [DOI] [PubMed] [Google Scholar]
- 44.Sarady JK, Otterbein SL, Liu F, Otterbein LE, Choi AM. Carbon monoxide modulates endotoxin-induced production of granulocyte macrophage colony-stimulating factor in macrophages. Am J Respir Cell Mol Biol. 2002;27:739–745. doi: 10.1165/rcmb.4816. [DOI] [PubMed] [Google Scholar]
- 45.Brouard S, Berberat PO, Tobiasch E, Seldon MP, Bach FH, Soares MP. Heme oxygenase-1-derived carbon monoxide requires the activation of transcription factor NF-κB to protect endothelial cells from tumor necrosis factor-α-mediated apoptosis. J Biol Chem. 2002;277:17950–17961. doi: 10.1074/jbc.M108317200. [DOI] [PubMed] [Google Scholar]
- 46.Brunt KR, Fenrich KK, Kiani G, Tse MY, Pang SC, Ward CA, Melo LG. Protection of human vascular smooth muscle cells from H2O2-induced apoptosis through functional codependence between HO-1 and AKT. Arterioscler Thromb Vasc Biol. 2006;26:2027–2034. doi: 10.1161/01.ATV.0000236204.37119.8d. [DOI] [PubMed] [Google Scholar]
- 47.Salinas M, Wang J, Rosa de Sagarra M, Martin D, Rojo AI, Martin-Perez J, Ortiz de Montellano PR, Cuadrado A. Protein kinase Akt/PKB phosphorylates heme oxygenase-1 in vitro and in vivo. FEBS Lett. 2004;578:90–94. doi: 10.1016/j.febslet.2004.10.077. [DOI] [PubMed] [Google Scholar]
- 48.Lang D, Reuter S, Buzescu T, August C, Heidenreich S. Heme-induced heme oxygenase-1 (HO-1) in human monocytes inhibits apoptosis despite caspase-3 upregulation. Int Immunol. 2005;17:155–165. doi: 10.1093/intimm/dxh196. [DOI] [PubMed] [Google Scholar]
- 49.Dorman RB, Bajt ML, Farhood A, Mayes J, Jaeschke H. Heme oxygenase-1 induction in hepatocytes and non-parenchymal cells protects against liver injury during endotoxemia. Comp Hepatol. 2004;3:S42–S44. doi: 10.1186/1476-5926-2-S1-S42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ollinger R, Bilban M, Erat A, Froio A, McDaid J, Tyagi S, Csizmadia E, Graca-Souza AV, Liloia A, Soares MP, Otterbein LE, Usheva A, Yamashita K, Bach FH. Bilirubin: a natural inhibitor of vascular smooth muscle cell proliferation. Circulation. 2005;112:1030–1039. doi: 10.1161/CIRCULATIONAHA.104.528802. [DOI] [PubMed] [Google Scholar]
- 51.Nakao A, Murase N, Ho C, Toyokawa H, Billiar TR, Kanno S. Biliverdin administration prevents the formation of intimal hyperplasia induced by vascular injury. Circulation. 2005;112:587–591. doi: 10.1161/CIRCULATIONAHA.104.509778. [DOI] [PubMed] [Google Scholar]
- 52.Chandrasekar B, Mummidi S, Valente AJ, Patel DN, Bailey SR, Freeman GL, Hatano M, Tokuhisa T, Jensen LE. The pro-atherogenic cytokine interleukin-18 induces CXCL16 expression in rat aortic smooth muscle cells via MyD88, interleukin-1 receptor-associated kinase, tumor necrosis factor receptor-associated factor 6, c-Src, phosphatidylinositol 3-kinase, Akt, c-Jun N-terminal kinase, and activator protein-1 signaling. J Biol Chem. 2005;280:26263–26277. doi: 10.1074/jbc.M502586200. [DOI] [PubMed] [Google Scholar]
- 53.Di Noia MA, S. Van Driesche F, Palmieri LM, Yang S, Quan A, Goodman I, Abraham NG. Heme oxygenase-1 enhances renal mitochondrial transport carriers and cytochrome C oxidase activity in experimental diabetes. J Biol Chem. 2006;281:15687–15693. doi: 10.1074/jbc.M510595200. [DOI] [PubMed] [Google Scholar]