

Abstract
Activation of the nuclear factor κB (NF-κB) signaling pathway may be associated with the development of cardiac hypertrophy and transition to heart failure (HF). The transgenic mouse, Myo-Tg, develops hypertrophy and HF as a result of overexpression of myotrophin in the heart associated with elevated level of NF-κB activity. Using this mouse model and an NF-κB-targeted gene array, we first determined the components of NF-κB signaling cascade and the NF-κB-linked genes that are expressed during the progression to cardiac hypertrophy and HF. Secondly, we explored the effects of inhibition of NF-κB signaling events by using gene knock-down approach: RNA interference (RNAi) through delivery of a short hairpin RNA (shRNA) against NF-κB-p65 using a lentiviral vector (L-sh-p65). When the shRNA was delivered directly into hearts of 10-week old Myo-Tg mice, a significant regression of cardiac hypertrophy, associated with significant reduction in NF-κB activation and atrial natriuretic factor (ANF) expression. Our data suggest, for the first time, that inhibition of NF-κB using direct gene delivery of sh-p65-RNA results in regression of cardiac hypertrophy. This data validates NF-κB as a therapeutic target to prevent hypertrophy/HF.
Keywords: myotrophin, NF-κB, RNA interference, transgenic mice, hypertrophy, heart failure
INTRODUCTION
Cardiac hypertrophy and heart failure (HF) are the major cause for the morbidity and mortality in humans (1). Hypertrophy is attributed to a number of diverse stimuli, including physiological, mechanical, hormonal, and even genetic influences (2). The underlying molecular mechanisms are still not clearly understood. NF-κB is a ubiquitous inducible transcription factor that activates a number of genes known to play roles in various cardiovascular disease states (3-7), in the pathogenesis of cardiac remodeling, and HF (8-12). Recently, we have shown that NF-κB activation occurs during the development of cardiac hypertrophy in spontaneously hypertensive rats (SHR) and also in failing human hearts (13, 14).
In a majority of cells, NF-κB resides in the cytoplasm in an inactive form bound to inhibitory proteins called IκBs (IκBα, IκBβ, and IκBε). The phosphorylation and subsequent proteolytic degradation of IκB proteins are key steps in the activation of NF-κB, its translocation into the nucleus, and its subsequent stimulation of expression of various inflammatory genes (4, 15, 16). This process is mediated by a high-molecular-mass kinase complex that includes IκB kinase α and β (IKKα and IKKβ) (17-19).
The signaling cascade that mediates the development of hypertrophy and its culmination in HF has not been clearly defined. Previously, we have shown that factors other than blood pressure are responsible for the development of cardiac hypertrophy (20). We identified a factor, myotrophin (a 12-kDa soluble protein), from the hypertrophied hearts of humans and spontaneously hypertensive rats that stimulated protein synthesis in cardiac myocytes, inducing myocardial growth (21-24). The signaling mechanism for myotrophin-induced myocyte growth in vitro, required activation of NF-κB (25). Recently, we developed a transgenic mouse model called Myo-Tg; in these mice, myotrophin is overexpressed, specifically in the heart, through use of an α-myosin heavy chain promoter. In Myo-Tg mice, overexpression of myotrophin in the myocardium initiates hypertrophy and there is a gradual progression from chronic hypertrophy to HF over a span of 9 months (26). Myo-Tg mice exhibit left ventricular hypertrophy, multiple focal fibrosis, myocytes necrosis, pleural effusion, and compromised cardiac function that closely mimics the symptoms of human HF (26). Myo-Tg mice thus provide us with a suitable model to study the molecular events during the progression of hypertrophy and its transition to HF.
The RNA interference (RNAi) system has been used extensively for silencing the translation of an active gene and has recently been applied to mammalian cells using small interfering RNA (siRNA) technology (27-29). The endogenous mediators of sequence-specific mRNA degradation are 21- and 22-nt siRNA duplexes generated from longer double-stranded RNAs by the ribonuclease III activity of the evolutionary-conserved Dicer enzyme (27, 30). At present it is not known whether this technique can be effectively applied in both in vitro and in vivo systems to study the mechanism of cardiac hypertrophy and HF.
We hypothesize that blockade of NF-κB could be of great therapeutic benefit in cardiac hypertrophy and HF. In this study, we report our findings on the NF-κB signaling cascade and NF-κB-targeted gene expression profiling both in vitro and in vivo using isolated adult and neonatal myocytes from Myo-Tg mice. We have also examined the effect of inhibition of the NF-κB system by using an RNAi-based inhibitor. The RNAi inhibitor (sh-p65) is a short hairpin RNA (shRNA) that is complementary to p65 delivered directly into the myocardium of Myo-Tg mice using a lentiviral (L) vector.
RESULTS
A) Activation of the NF-κB signaling cascade during progression of cardiac hypertrophy in Myo-Tg mice
a) NF-κB binding activity and upregulation of IκB protein and IκB kinase
To investigate whether myotrophin overexpression activates NF-κB in Myo-Tg mice during the progression to HF, we performed an electrophoretic mobility shift assay (EMSA) using nuclear extracts from the hearts of age and sex-matched wild-type (WT) and Myo-Tg mice at 4-, 16-, and 36-week of age. Our data (Fig. 1A) showed that binding activity was more pronounced in 36-week old mice (4.4-fold increase compared to WT) than in animals 4 and 16 weeks old (2.8- and 3.6-fold, respectively; p < 0.001). The specific binding of NF-κB was confirmed by the addition of a 100-fold molar excess of unlabeled NF-κB DNA, which abolished the binding and by antibody supershift assays which showed a significant shift of NF-κB binding complexes (Fig. 1A). In addition, we determined the NF-κB binding complex in other organs of Myo-Tg mice that include lungs, brain, liver and kidney. Our data showed that the NF-κB activity was found only in the heart but not the other organs suggesting it’s specificity to the heart only (1B). Furthermore, western blot analysis showed nuclear translocation of NF-κB as early as 4 weeks of age and was further increased at 36 weeks of age. Histone protein was used as an internal loading control (Fig. 1C). We also determined the localization of active p65 protein in the myocardium and observed a robust expression at the end stage of hypertrophy (Fig. 1D). We confirmed our findings by isolating adult myocytes from WT and Myo-Tg mice at 16 weeks of age. Our data showed a significant activation of p65 NF-κB protein (green stain) in Myo-Tg adult myocytes (Fig. 1E) and an enhanced NF-κB activation at 16 weeks of Myo-Tg mice (Fig. 1F).
Figure 1.

NF-κB activation during progression of hypertrophy in Myo-Tg mice hearts. (A) Nuclear protein was extracted from the hearts of 4-, 16-, and 36-week-old WT and Myo-Tg mice. Binding reactions were performed with an NF-κB oligonucleotide labeled with 32P-dATP. The complex formation was eliminated with excess unlabeled NF-κB oligonucleotide. The complex formation was confirmed by supershift analysis using p65 antibody. NE: Nuclear extract from 16 week Myo-Tg mice. (B) Nuclear protein was extracted from lung, brain, kidney, heart and liver separately and binding reactions were performed as described in (A). (C) Western blots profile of NF-κB p65 protein into the nucleus. Histone antibody was used as an internal nuclear protein loading control. (D) Expression of p65 active protein in the heart section of both WT and Myo-Tg mice and were photographed with an Olympus photomicroscope at 20 X magnification. This figure is representative of six different mice in each group (WT and Myo-Tg). (E). Confocal images of p65 protein from adult myocytes taken from WT and Myo-Tg mice at 16 weeks of age and were stained with p65 polyclonal antibody followed by FITC-conjugate (green), alpha-sarcomeric actinin monoclonal antibody for myocyte staining (red), followed by Texas Red-conjugate and DAPI (blue) for nuclear staining. Immunofluorescent NF-κB p65 staining was observed using a laser scanning confocal microscope and 63 X, 0.7 oil immersion objective lens. The figure is a representative of 3 different mice. The arrow indicates the translocation of NF-κB-p65 orotein into the nucleus. (F) EMSA analysis using nuclear extract from adult myocytes from 16-week old Myo-Tg mice.
Myotrophin protein was determined at initiation (4 weeks), progression (16 weeks) and transition (36 weeks) phases of cardiac hypertrophy. Our data showed a significant increased in myotrophin protein level in Myo-Tg mice compared to WT littermates (Fig. 2A). Phosphorylation of IκBα was detected by as early as 4 weeks of age and attained a maximum at 36 weeks (Fig. 2B). A low level of IκBα phosphorylation was found to be present in all WT mice, probably because of low-level NF-κB activation they exhibited. Cardiac IκBα levels were significantly higher (1.8-, 2.6- and 3.5-fold respectively, p < 0.001) than in the corresponding WT littermates (Fig. 2C). Our data also revealed a linear increase in the pIκBα/IκBα ratio (1.2, 1.3, and 1.54) at 4, 16, and 36 weeks, respectively. In contrast, IκBβ levels did not differ between WT and Myo-Tg mouse hearts (Fig. 2D). Actin antibody was used as an internal protein loading control (Fig. 2E). To explore whether the activation of NF-κB in Myo-Tg mice is mediated through IKKβ, we determined the IKKβ activity in Myo-Tg mice and WT mice hearts (Fig. 2F). IKKβ activity was detected in all Myo-Tg mouse hearts and was very low in WT hearts. In extracts from Myo-Tg mice, a 4.8-fold increase in IKKβ activity was observed at 16 weeks and a 2.9-fold increase (p < 0.001) at 36 weeks, compared with 4-week-old Myo-Tg mice.
Figure 2.
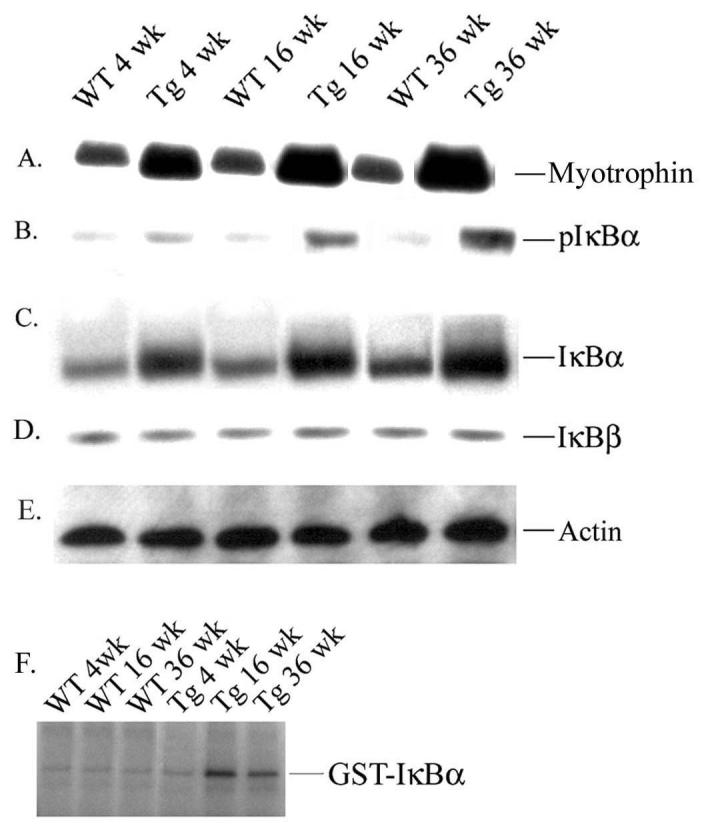
Expression of myotrophin, IκBα proteins and IKKβ activity in the hearts of Myo-Tg mice. Cytoplasmic protein extracts were made from both WT and Myo-Tg mouse hearts at 4, 16, and 36 weeks of age. Tissue extracts (10 μg) were analyzed for (A) myotrophin and tissue extracts (50 μg) were analyzed for (B) IκBα phosphorylation, (C) the intracellular level of total IκBα protein content, and (D) IκBβ level using myotrophin, phospho-IκBα, IκBα, and IκBβ antibody as probes. (E) Actin protein was used as an internal loading control. Results are presented as the mean SEM and represent six different mice in each group (WT and Myo-Tg) (p < 0.001 compared with the WT mice). (F) Tissue extracts (500 μg) were immuno-precipitated with IKKβ antibody and kinase activity was determined with GST-IκBα as a substrate. These results are presented as the mean SEM and represent six different mice (p < 0.001 compared with the WT mice).
b) Status of transcriptional level of IκBα and p65 and other NF-κB-dependent genes
We investigated the level of mRNA expression of myotrophin, IκBα and p65 by northern blot analysis in Myo-Tg and WT mice during progression of cardiac hypertrophy. Myotrophin mRNA level was significantly induced at 4-,16- and 36 weeks of age in all Myo-Tg mice compared to WT littermates (Fig 3 A). IκBα mRNA levels increased 2.5-, 3.6-, and 3.8-fold (p < 0.001) in Myo-Tg mice; p65 mRNA levels in 4-, 16- and 36-weeks-old Myo-Tg mice also increased by 1.8-, 2.9-, and 3.2-fold (p < 0.001) respectively, compared with WT mice (Fig. 3 B-D). 18 S RNA was used as an internal loading control (Fig. 3 B and 3 E).
Figure 3.

Transcriptional analysis of myotrophin, IκBα, p65, and NF-κB-dependent genes during progression of cardiac hypertrophy. Total RNA was extracted from hearts of 4-, 16-, and 36-week old WT and Myo-Tg mice. mRNA expression was determined using (A) Myotrophin (C) IκBα and (D) p65 cDNAs labeled with 32P-dCTP as a probes. (B and E) 18S rRNA probe was used as a loading control. Results are presented as the mean SEM and represent five different mice (p < 0.001 compared with the WT mice). (F). NF-κB-dependent gene expression was performed by northern blot using (a) TNFα, (b) ANF, (c) IL-1β, (d) IL-6, (e) c-myc cDNAs as a probes; 18S rRNA was used as an internal loading control. These results are presented as the mean SEM and represent three different mice (p < 0.001 compared with the WT mice).
To determine the expression profile of NF-κB-regulated inflammatory gene expression in Myo-Tg mice, we performed a northern blot analysis. Genes studied were tumor necrosis factor-α (TNFα), interleukin (IL)-1β, IL-6, atrial natriuretic factor (ANF), and c-myc, which are already known to play a critical role in HF. The results are summarized in Fig. 3F (a to f). Compared with WT mice, Myo-Tg mice hearts showed a significant increase in mRNA levels for the inflammatory cytokines (TNFα, IL-1β, and IL-6) as well as ANF and c-myc (cardiac hypertrophy marker genes). TNFα, ANF, and IL-6 showed maximum expression (4.8-, 5.2-, and 5.6-fold, respectively, compared to WT mice, p < 0.001) at the end stage of hypertrophy (Fig. 3F a, b, and e).
To gain insight into the NF-κB-target gene activation at 4 and 36 weeks of age, we used the TranSignal™ mouse NF-κB Target Gene Array System. The complete expression profile of NF-κB-targeted gene array at 4 and 36 weeks of age for both Myo-Tg and WT mice is shown in Appendix I (see supplement). We used p < 0.001 as the critical level (Bonferroni’s correction). Genes found to be upregulated consistently during the onset of hypertrophy (fold value ≥2.4 compared to WT) were IL-m, IL-10 and IL-11, alox-12, Bcl 21, cyclin D1 and D3, CSF-2, CSF-3, Fcer2a, HAS-1, IκBα, IGBP-2, mnSOD, NF-κB-2 Ptx3, PTGIS-2, and TNFα. Genes found to be upregulated at 36 weeks of age (fold value ≥2.4 compared to WT) were alox-12, AHRR, ADORA-1, apoC3, bcl2a1a, BLR-1, cyclin D1 and D3, CSF-2 and CSF-3, HMGN-1, GRO-1, HMG-14, Hmox-1, has-1, ICAM-1, IRF-2, IL-10, IL-11, IL-6, LOX-1, LAMB-2, MAD3 (IκBα), MadCam-1, myc, NF-κB-1 and 2, olr-1, PENK-1, PDGFβ, pax8, PTGIS, TNFα, and VEGFc. Gene’s upregulated only during the progression from hypertrophy to HF were AHRR, ADORA-1, bcl2a1a, BLR-1, GSTP-1, GRO-1, HMGN-1, fasL, IRF-2, ICAM-1, LAMB-2, NF-κB-1, MadCAM-1, myc, PDGF, and rel.
B) Effect of transduction of L-sh-p65 gene on myotrophin-induced myocytes growth To evaluate the role of NF-κB in myotrophin-induced myocytes growth, we took RNAi approach
Neonatal rat myocytes were transduced with lentivirus plasmid containing sh-p65 and EGFP, (LRV-U6-p65-CMV-EGFP), mentioned as (L-sh-p65) in the text, in the presence or absence of myotrophin, and NF-κB activation was determined by EMSA. Our data showed a significant inhibition of NF-κB activity in presence of myotrophin in L-sh-p65 transduced cells (Fig. 4A) (68.4% reduction compared to myotrophin-stimulated neonatal myocytes, p < 0.001) whereas in non-transduced cells NF-κB activation was observed. Further confirmation of NF-κB binding was determined by western blotting in which p65 translocation was significantly inhibited in L-sh-p65 transfected cells (Fig. 4B). Histone was used as an internal nuclear protein loading control (Fig 4B lower panel).
Figure 4.

RNA interference suppresses NF-κB-p65 activation, myocyte growth, and ANF expression in neonatal myocytes. (A) Cells were transduced with L-sh-p65 at a multiplicity of infection (MOI) of 30 for 48 hours and were stimulated with myotrophin. EMSA was performed as described in Fig 1. (B) Western blot analysis using NF-κB-p65 antibody as a probe in L-sh-p65-transduced neonatal myocytes. L-EGFP was used as a control. Histone antibody was used as an internal nuclear protein loading control. (C). Myocyte growth was measured by [3H]-leucine incorporation into myocyte protein. L-sh-p65 was transduced into neonatal myocytes in presence and absence of myotrophin. Cells were also transduced with scramble sequences separately in presence and absence of myotrophin. Results are presented as the mean ± SEM and represent set of three different experiments in neonatal myocytes (p < 0.001 compared with unstimulated cells). (D) ANF expression was measured by using ANF cDNA as a probe in L-sh-p65-transduced neonatal myocytes in the presence or absence of myotrophin. Note: in all experiments, a scrambled sequence did not show any effect.
To determine the role of NF-κB in myocyte growth, we measured [3H]-leucine incorporation into myocyte protein. Our data demonstrated that myotrophin-induced enhanced protein synthesis was significantly inhibited in L-sh-p65-transduced myocytes (76.6% ± 3.8 vs. 31.1% ± 5.8 over control; p < 0.001) suggesting a potential role NF-κB in myocyte growth (Fig. 4C). The scramble sequence did not show any inhibitory effect on myotrophin-induced 3H leucinene incorporation. Furthermore, ANF expression was significantly attenuated in L-sh-p65-transduced myocytes (168.8 ± 5.8 vs. 48.6 ± 4.3) compared to myotrophin-stimulated myocytes (Fig. 4D). The scrambled sequence did not show any effect on ANF expression.
C) Effect of L-sh-p65 treatment on cardiac mass, and function and NF-κB signaling components in Myo-Tg mice
L-sh-p65 lentiviral vectors were directly injected into the myocardium of two groups of 10 weeks old mice (n=6) over a period of 6 weeks and we determined the cardiac mass remodeling and expression of EGFP to confirm the localization into the myocardium. As shown in Fig. 5A and B, L-sh-p65-treated Myo-Tg mice show a significant attenuation of heart weight to body weight ratio in comparison to a mice with a sham procedure (ratio of 9.6 ± 0.05 vs. 6.8 ± 0.33, p = 0.002) and EGFP expression was found in the myocardium as shown in Fig 5C, which was absent in WT mice. Echocardiographic data from 6-week sh-p65-treated Myo-Tg mice showed an improvement of cardiac function compared to untreated Myo-Tg mice. The left atrial area was marginally decreased in sh-p65-treated animals. When compared with the untreated group, sh-p65-treated mice showed a significant improvement of ejection fraction (25.82 ± 1.16 vs. 39.57 ± 1.9; p < 0.06) (Fig. 5 D). Similarly, Doppler myocardial tissue velocities and fractional shortening were marginally increased in sh-p65-treated group (data not shown). Thus, all three systolic parameters showed a change in the same direction with improvement in animals with inhibited NF-κB activation.
Figure 5.

Attenuation of cardiac mass and inhibition of NF-κB activation cascade in Myo-Tg mice treated with L-sh-p65. (A) Typical appearance of heart size in untreated Myo-Tg and L-sh-p65-transduced Myo-Tg mice. (B) Heart weight:body weight ratio in L-sh-p65-transduced Myo-Tg mice. Values represent mean ± SE and p = 0.002 compared with untreated Myo-Tg mice (n=6). (C) Confocal image showing the expression of EGFP in L-sh-p65-transduced Myo-Tg mice. Magnification is 40 X. (D) Representative M-mode tracings of the left ventricle obtained in a Myo-Tg (Sham) and sh-p65-Tg-treated mice. Arrows indicate endocardial borders in diastole (broken arrows) and systole (full arrows). Improved LV fractional shortening and decreased left ventricle internal dimension are evident in the L-sh-p65-treated mouse. IVS, interventricular septum; LV, left ventricle cavity; PW, posterior wall. (E) EMSA was performed using nuclear extract from WT, Myo-Tg, and L-sh-p65-treated Myo-Tg mice as described in Fig. 1A. (E) Quantification of EMSA using an arbitrary density unit (10 μg/NE). NE: nuclear extract. These results are presented as the mean ± SE and represent six different mice (p = 0.002 compared with the sham mice, two-way ANOVA). (G) p65 nuclear protein translocation was demonstrated by western blot analysis p65 antibody as described in Fig. 1B. (H) IκBα protein levels were determined by western blot analysis using IκBα antibody. (I) mRNA expression profiling of IκBα, p65, ANF, and TNFα in Myo-Tg and L-sh-p65-transduced Myo-Tg mice (two separate animals) using specific cDNAs as probes.
To establish the cause-and-effect relationship of NF-κB activation and cardiac remodeling, we analyzed the NF-κB signaling components. Direct delivery of the NF-κB-targeted shRNA into the hearts of Myo-Tg mice resulted in significant reduction in NF-κB activity (208. 7.1 ± 7.1 vs 132.3 ± 11.0; 35.7% compared to sham; p = 0.002,) and inhibition of translocation of p65 protein into the nucleus but there was no effect on WT mice (Fig. 5E, F and G). The amount of total IκBα cytosolic protein was analyzed by immunoblot analysis. L-sh-p65 treatments significantly reduced IκBα levels in Myo-Tg mice (39.2% compared to sham; p =0.002) (Fig 5H). We further determined the mRNA levels of IκBα and p65. L-sh-p65-treated Myo-Tg mice showed a significant reduction of both IκBα and p65 (49.6% and 52% reduction compared to sham, respectively; p= 0.002) (Fig. 5I). Our studies also revealed the down-regulation of ANF and TNFα (32.3%, 36.4%, respectively, p< 0.001) (Fig 5I).
DISCUSSION
The novel and most significant finding in this study is that activation of the NF-κB signaling cascade is necessary for the initiation of hypertrophy and plays an important role in the progression of hypertrophy to HF. We analyzed for the first time NF-κB-targeted gene expression during the onset of hypertrophy and its transition to HF. Furthermore, to establish a cause and effect relationship between NF-κB activation and remodeling of cardiac mass, we demonstrated that either by knocking down the NF-κB-p65 gene using lentivirus-mediated siRNA delivery or by treating Myo-Tg mice with a NF-κB inhibitor, PDTC (data not shown) lead to attenuation of cardiac mass with improved cardiac functions. Lentiviral delivery of the shRNA was the first demonstration of direct gene delivery and activity of siRNA in the mouse heart. This multifaceted study may provide new ideas for drug development in the treatment of human HF. Here, we reported five lines of evidence that substantiate these findings.
First, in Myo-Tg mice, NF-κB was activated during the initiation phase of hypertrophy (4 weeks) and became most pronounced in the myocardium at 36 weeks, i.e. the end stage of HF (Fig. 1A, C and D). This observation was substantiated by localization of active NF-κB-p65 protein in the myocytes from 16-week-old Myo-Tg mice (Fig. 1E and F) thus emphasizing the important role of this transcription factor in the process.
Second, Myo-Tg mouse hearts showed a gradual increase in the phosphorylation of IκBα along with a significant increase in total cytosolic content of IκBα protein, but not IκBβ protein, compared to WT mouse hearts (Fig. 2B, C, and D). These findings provided insight into the role of IκBα proteins during HF. The enhanced level of IκBα may be explained by its rapid turnover and auto-feedback mechanism. Because IκBα promoter region contains an NF-κB binding site, IκBα is quickly re-synthesized, bound to the NF-κB DNA element in the nucleus and transactivated. This auto-regulatory feedback circuit maintains a transient expression of different NF-κB responsive genes that has been illustrated in other cell types under other stimuli (31, 32). Based on this concept, we suggest that increased levels of IκBα may be beneficial as it releases NF-κB from DNA binding and eliminates further NF-κB activation. The multi-subunit IκB kinase (IKK) is responsible for the phosphorylation of IκBα, which appears to be an important phenomenon in NF-κB activation (18, 19, 33). IKKβ also primarily accounts for the activation of NF-κB in response to inflammatory stimuli, whereas IKKα is essential for keratinocyte differentiation (34-36). Recent studies have demonstrated that a multi-subunit IKK complex, which contains two interacting catalytic subunits, IKKα and IKKβ, and a regulatory subunit, IKKγ, mediates specific phosphorylation of IκBα at Ser 32 and Ser 36, resulting in NF-κB activation (18, 19, 37). The hearts of Myo-Tg mice showed a significant increase in IKKβ activity at 16 weeks, followed by a gradual decrease (Fig. 2F), suggesting that enhanced IKKβ activity leads to IκBα phosphorylation and subsequent translocation of NF-κB. It is difficult to explain why IKKβ activity is peaked at 16 weeks of age but it could be due to the same auto-feedback mechanism (37, 38) and that reduces IKKβ activity during HF.
Myo-Tg mouse hearts also showed upregulation of IκBα and p65 mRNA compared with those of WT mice (Fig. 3 C and D). Upregulation of IκBα mRNA may also be explained by the presence of NF-κB binding sites in IκBα gene, as described above. Recent studies have suggested the existence of an auto-regulatory feedback mechanism in NF-κB/IκBα activation (5,39) leading us to speculate that IκBα controls NF-κB activation and activated NF-κB in turn regulates expression of the IκBα gene.
Third, Myo-Tg mouse hearts showed upregulation of several NF-κB-dependent genes (TNFα, IL-1β, IL-6, c-myc, and ANF) during progression of cardiac hypertrophy (Fig. 3F). Regulation of these genes may therefore depend on activation of NF-κB as the promoter regions of these genes contain NF-κB binding sites (6). In addition, for the first time, we report the participation of NF-κB-targeted genes as a causal factor during the onset of hypertrophy and its progression to HF by using NF-κB-target gene array. The expression of NF-κB target genes depends not only on the activity of NF-κB itself, but also on a number of other factors, including NF-κB’s interaction with other transcription factors, and the level of expression may vary under different physiological conditions. It will be intriguing to analyze these genes at their protein level as they may represent novel cardiac-specific genes and may potentially provide keys to unlocking the underlying mechanisms of HF. Investigations to elucidate the function of these genes are currently underway and may eventually define these genes as a new therapeutic target for increased myocardial protection (5, 38, 40).
Fourth, most importantly, to correlate an association between alteration between cardiac mass and NF-κB activation, we have utilized RNAi technology both in vitro and in vivo. Knocking down of NF-κB-p65 gene in neonatal myocytes showed a significant inhibition of myocyte growth and ANF expression (Fig. 4). A significant reduction in cardiac mass associated with improved cardiac function and an attenuation of the NF-κB activation cascade were observed when shRNA-p65 was introduced in a lentiviral expression vector directly into the hearts of Myo-Tg mice (Fig. 5). This is the first report of in vivo demonstration of the effect of knowing down of NF-κB-p65 in cardiac remodeling. RNAi is mediated by the RNA-induced protein complex (RISC), guided by siRNA to achieve the specific recognition of homologous mRNA sequences and subsequent degradation of the mRNA by nucleases [(41, 42). Our studies also suggest that sh-p65 gene delivery is not toxic and the effects appear to be specific and last over several weeks, a period of time sufficient to analyze the phenotypic changes. We observed only a partial inhibition of the protein, which can be explained by various means (e.g., dose and kinetics, persistence of protein after transfection, design of the molecule, and most importantly the limitation of the RISC complex).
It has been reported that cardiac-specific overexpression of an IκBα triple mutant (S32A, S36A and Y42F) completely blocked cytokine-induced NF-κB activation in response to TNFα-induced cardiomyopathy (43, 44). Data from studies on these 3M mice suggest that the serine and tyrosine phosphorylation pathways are differentially activated in different cardiac pathophysiological processes and NF-κB plays an important role in this process. Recently, a genetic breed was made between TNFα transgenic mice and p50 knock out mice to assess whether the cardiotoxic effects of proinflammatory cytokines were mediated through NF-κB (45, 46) and revealed that blockade of NF-κB activation attenuated myocardial inflammation and improved cardiac function in male TNFα transgenic mice. All these results corroborate the findings of our report.
Cumulatively, our data indicate that NF-κB and its known signaling cascade play key roles in the initiation of hypertrophy and transition to HF in the Myo-Tg mouse model. Delivery of the L-sh-NF-κB-p65 gene into Myo-Tg mouse hearts inhibited the NF-κB signaling cascade, attenuated cardiac mass, and improved cardiac function. The data from this study suggest that inhibition of NF-κB will offer a therapeutic benefit in prevention of hypertrophy/HF.
MATERIALS AND METHODS
Generation of transgenic mice overexpressing myotrophin in the heart
The generation of our Myo-Tg mice has been described previously (26). The studies were conducted with the approval of The Cleveland Clinic Foundation’s Institutional Review Board. Age and sex-matched wild type (WT) mice were used for comparison with Myo-Tg mice. We considered 4 weeks of age as the onset of hypertrophy [heart wt: body wt = 6.2 ± 0.3 (Myo-Tg) vs. 4.2 ± 0.14 (WT); p < 0.05], 16 weeks as the progression [heart wt: body wt = 8.8 ± 0.25 (Myo-Tg) vs. 5.0 ± 0.12 (WT); p < 0.05] and 36 weeks as a transition phase of hypertrophy to HF [heart wt: body wt = 12.3 ± 0.025 (Myo-Tg) vs. 4.5 ± 0.1(WT); p < 0.05].
Extraction of cytoplasmic, nuclear protein, western blotting and northern blotting
Nuclear and cytoplasmic extracts were made according to the method described by Dignam et al. (47). Western blot analysis was performed as described previously (14). Total RNA was isolated from the ventricle of WT and Myo-Tg mice according to the protocol of Chomczynsky and Sacchi, 1987 (48) and northern blotting was performed as described previously (25).
Electrophoretic mobility shift assay (EMSA), IKKβ activity and histological analysis
An EMSA was performed using a double-stranded NF-κB oligonucleotide as a probe, as described previously (25), (14). The sequence of NF-κB probe used in this study was 5′AGT TGA GGG GAC TTT CCC AGG C3′. Left ventricular tissue from age-matched WT and Myo-Tg were homogenized and IKKβ activity was determined as described previously (14). Sections were then photographed with an Olympus photomicroscope at 20 X magnification.
NF-κB-target gene array analysis
The NF-κB-target gene array was performed using the TranSignal mouse NF-κB Target Gene Array kit from Panomics, Inc. (Redwood City, CA) as described previously (14).
Design and construction of L-sh-p65
The shRNAs were designed such that the antisense strand came before the sense strand during transcription. The sense sequence was followed by the loop sequence 5′TTC AAG AGA3′ and then by the antisense sequence to form a hairpin. These small oligonucleotides had Eco RI and Sal I overhangs to allow for ligation into the Sal I/Eco RI sites immediately after the U6 promoter of the lentiviral plasmid, LRV-U6-CMV-EGFP (L). Clones were verified by sequencing. The empty vector was used as a control plasmid in this study. A scrambled sequence of the same length was also used as a control. The sequences for the two strands of the construct for L-sh-p65 were as shown:
Sense
5′[p] TCGAC GCC TAT CCC TTT ACG TCTT TTC AAG AGA AAG TAA AGG GAT AGG GC TTT TTTG 3′
Antisense
5′ [p] AATTC AAA AAA GCC CTA TCC CTT TAC GTC TT TCT CTT GAA AAG ACG TAA AGG GAT AGG GCG 3′
Viral production and purification
Preparation, purification of the lentiviral particle and transfection were done as described previously (49). In brief, the lentivirus particles were typically made by transfecting 293-T cells with the LRV-U6-CMV-EGFP plasmid containing either sh-p65 (L-sh-p65) or scramble insert. Viral supernatants were harvested 48 and 72 h post-transfection and filter sterilized through 0.45μ. Using this system, we achieved optimal yields of vector of high transducing efficiency (106-107 transducing units (TU)/mL and close to 104 TU/ng of p24 core protein). The virus particles were further concentrated by spinning 150 ml of viral supernatant at 30,000 rpm for 2 h in an ultracentrifuge and resuspending in 150 μl of serum free Dulbecco’s modified Eagles’s medium F12 (DMEM-F12) media. The final concentration of viral particles are 109-1010/ml.
Lentiviral transduction
Twenty-four hours after plating, rat neonatal cardiomyocytes were transduced (in triplicates) separately in six-well laminin coated plates with 30 MOI of L-sh-p65 and LRV-U6-CMV-EGFP (control) viral particles in presence of 8 μg/ml of polybrene. We use 2 ml of viral supernatant, which contain 2 × 107-108 viral particles for each transduction experiment. After 48 hours, the efficiency of the transduction was measured by monitoring EGFP expression under fluorescence microscope. The transduction efficiency was found to be 80-90% after two successive infections.
Immuno-cytochemical analysis
Adult mouse myocytes were washed in DMEM-F-12 containing 10% FBS two times and plated on laminin coated six-well plates for over night. Immuno-cytochemical and confocal analysis were performed as described before (17).
Preparation of cardiac neonatal myocytes and measurement of protein synthesis
Neonatal rat myocytes using 3-day old rat pups were prepared as described previously (25). To determine the rate of protein synthesis in myotrophin-induced myocyte growth, bioassays were performed as described previously (25). In brief, after transduction, cells were stimulated with myotrophin for 16-18 hours followed by 10μCi of 3H leucine for two hours. Cells were TCA precipitated and [3H] leucine incorporation was measured.
Delivery of siRNA using the lentiviral vector
Animals (WT and Myo-Tg mice) were anesthetized using 0.1 mL of cocktail of ketamine and xylene (1 mL ketamine, 0.9 mL xylene, and 1.5 mL of PBS), ventilated, and subjected to a lateral thoracotomy for direct visualization. We gave two injections (40 μL each) containing sh-p65 gene (4 × 106-107) in each of the anterior-septal and posterior-lateral walls and one injection near the apex of the heart. There were 5-8 mice per treatment group. At the end of a 6-week treatment period, the animals were sacrificed, their hearts were removed, and the following parameters were determined: a) cardiac mass, b) body weight, c) NF-κB activation, d) p65 gene and protein expression, and e) ANF expression.
Determination of cardiac function data collection and analysis
Echocardiography was performed using a Vivid 7 echocardiography machine (GE Medical, Milwaukee, WI). LV mass and velocity of the posterior wall were calculated by bullet equation (50) and tissue Doppler echocardiography (TDE) mode (51). In brief, mice were assessed in a conscious state in a supine position. Data were analyzed using Echopac PC (GE Medical, Milwaukee, WI), M-mode echocardiography, 2-dimensional echocardiography (frame rate >160 fps) using a 14MHz epicardial linear transducer. Data were digitized and stored in a proprietary format for further analysis. Left atrial area was measured from the basal short-axis view of the heart that maximized left atrial appendage size. LV end-diastolic and end-systolic diameters (LV-EDD and LV-ESD) measured from M-mode data in a standard manner. Fractional shortenings (FSh) were calculated as Fsh = 1 - LV ESD/ LV EDD where the diameters are measured from M-mode data. LV end-diastolic and end-systolic volumes were measured from the parasternal short- and long-axis view by bullet equation. LV mass was calculated from 2D echocardiography data by bullet equation also. We reconstructed the velocity of the posterior wall of the left ventricle using parasternal short-axis view at the papillary muscle level recorded in TDE mode. The region of interest for the analysis was set to 1×1 mm. At least 6 beats were averaged to obtain peak myocardial tissue systolic velocity of the posterior wall.
Statistical Analysis
Results are expressed as mean ± S.E and the differences between groups were by paired Student’s t test. Differences were considered significant at p < 0.001. Data were also analyzed by two-way analysis of variance (ANOVA) using GraphPad Prism software (GraphPad Software, Inc., San Diego, USA) for L-sh-p65 transduced animals. For NF-κB-target gene array analysis, genes are arranged in order by the t-statistic, i.e. from largest to smallest standardized difference in mean. We used 0.001 as the critical level (Bonferroni’s correction).
Supplementary Material
ACKNOWLEDGEMENTS
This study was supported in part by National Institutes of Health grant HL R01 47794 to Dr. S. Sen, American Heart Association (Ohio Valley Affiliate) Beginning Grant-in-Aid 0565226B (to S.G.), and NIH R01 HL66251 (to Q.W.). The authors acknowledge Dr. A. Brasier for IκBα cDNA, Dr. S. Ghosh for p65 cDNA, Ms. Linda Vargo (Imaging Facility and Histology Core at The Cleveland Clinic Foundation for her expert technical assistance in immunohistology), and the expert secretarial help of Lori Sims, and the editing assistance of Christine Kassuba.
Appendix 1: NF-κB targeted gene expression profile at 4 and 36 weeks Myo-Tg mice
NF-κB targeted gene expressed at 4 weeks Tg mice (n= 4)
| Gene | Group | Mean | SE(±) | P-value | Tg/WT |
|---|---|---|---|---|---|
| AGER | WT Tg |
6.25 20.25. |
1.10 2.13 |
<0.001 | 3.24 |
| AGP-1 | WT Tg |
62.75 89.5. |
1.43 2.21 |
<0.001 | 1.42 |
| Alox-12 | WT Tg |
10.25 25.0 |
1.49 1.08 |
<0.001 | 2.43 |
| ApoC3 | WT Tg |
44.75 110.25 |
1.65 4.21 |
<0.001 | 2.46 |
| Bcl2a1a | WT Tg |
5.25 10.75 |
0.85 1.10 |
<0.001 | 2.04 |
| Bcl21 | WT Tg |
14.75 59.0 |
1.65 2.64 |
<0.001 | 4.0 |
| Bdkrb | WT Tg |
12.5 56.5 |
1.70 2.50 |
<0.001 | 4.52 |
| Blr-1 | WT Tg |
7.0 16.75 |
0.91 1.31 |
<0.001 | 2.39 |
| Ccnd-1 | WT Tg |
11.0 33.5 |
1.29 1.93 |
<0.001 | 3.04 |
| Ccnd-3 | WT Tg |
29.5 116.25 |
1.93 2.01 |
<0.001 | 3.94 |
| CD 69 | WT Tg |
8.5 23.5 |
1.04 1.70 |
<0.001 | 2.76 |
| CSF-1 | WT Tg |
17.0 28.5 |
0.91 1.44 |
<0.001 | 1.67 |
| CSF-2 | WT Tg |
12.5 32.5 |
1.19 1.04 |
<0.001 | 2.6 |
| CSF-3 | WT Tg |
28.75 98.75 |
1.65 2.56 |
<0.001 | 3.5 |
| Fcer2a | WT Tg |
31.25 73.75 |
1.10 2.28 |
<0.001 | 2.36 |
| FTH | WT Tg |
14.25 38.0 |
0.85 1.08 |
<0.001 | 2.66 |
| GSTP-1 | WT Tg |
118.0 181.5 |
2.73 2.21 |
<0.001 | 1.53 |
| H2T23 | WT Tg |
61.0 136.25 |
1.82 2.28 |
<0.001 | 2.23 |
| HAS-1 | WT Tg |
20.5 78.25 |
1.70 2.21 |
<0.001 | 3.81 |
| HMGN-1 | WT Tg |
85.25 150.75 |
1.25 1.93 |
<0.001 | 1.76 |
| IGBP-2 | WT Tg |
15.25 54.25 |
1.25 1.65 |
<0.001 | 3.55 |
| IL-1β | WT Tg |
11.75 23.75 |
0.85 1.65 |
<0.001 | 2.02 |
| IL-6 | WT Tg |
16.25 37.0 |
0.85 1.08 |
<0.001 | 2.27 |
| IL-10 | WT Tg |
18.75 79.75 |
1.10 2.13 |
<0.001 | 4.25 |
| IL-11 | WT Tg |
33.25 111.5 |
2.42 2.59 |
<0.001 | 3.35 |
| IL-m | WT Tg |
23.75 69.0 |
1.37 1.47 |
<0.001 | 2.90 |
| IRF-2 | WT Tg |
101.5 143.5 |
1.7 2.39 |
<0.001 | 1.41 |
| LAMB-2 | WT Tg |
63.5 122.5 |
1.19 2.66 |
<0.001 | 1.92 |
| LTA | WT Tg |
18.75 25.25 |
1.03 1.75 |
<0.003 | 1.34 |
| Lyzs | WT Tg |
25.5 39.5 |
2.53 2.59 |
<0.001 | 1.54 |
| Mmp-3 | WT Tg |
7.5 14.5 |
0.64 0.95 |
<0.001 | 1.93 |
| mnSOD | WT Tg |
6.5 33.25 |
0.64 2.13 |
<0.001 | 5.11 |
| MSX-1 | WT Tg |
11.0 22.5 |
0.70 1.32 |
<0.001 | 2.04 |
| NF-κB-2 | WT Tg |
10.5 73.0 |
0.64 2.64 |
<0.001 | 6.85 |
| IκBα | WT Tg |
11.5 38.25 |
1.04 1.93 |
<0.001 | 3.32 |
| PDGFβ | WT Tg |
51.25 94.25 |
1.10 2.13 |
<0.001 | 1.83 |
| PENK-1 | WT Tg |
26.75 49.25 |
2.28 1.70 |
<0.001 | 1.84 |
| PTGIS-2 | WT Tg |
14.5 36.5 |
0.64 1.19 |
<0.001 | 2.51 |
| Ptx-3 | WT Tg |
15.5 86.25 |
1.32 1.65 |
<0.001 | 5.56 |
| PAX8 | WT Tg |
46.5 86.0 |
1.7 1.29 |
<0.001 | 1.84 |
| TNFα | WT Tg |
15.5 39.75 |
0.64 1.65 |
<0.001 | 2.56 |
NF-κB targeted gene expressed at 36 weeks Tg mice (n= 4)
| Gene | Group | Mean | SE (±) | P-value | Tg/WT |
|---|---|---|---|---|---|
| AHRR | WT Tg |
5.75 29.0 |
0.85 3.3 |
<0.001 | 5.08 |
| ALOX-12 | WT Tg |
13.5 30.6 |
1.19 7.71 |
<0.05 | 2.75 |
| ApoC3 | WT Tg |
30.0 84.5 |
3.87 4.11 |
<0.001 | 2.83 |
| A1AT | WT Tg |
33.5 67.5 |
1.04 1.55 |
<0.001 | 2.01 |
| ADORA1 | WT Tg |
30.0 89.0 |
0.91 3.02 |
<0.001 | 2.96 |
| AGER | WT Tg |
28.5 61.0 |
1.04 1.47 |
<0.001 | 2.14 |
| AGT | WT Tg |
6.75 15.0 |
0.85 1.22 |
<0.001 | 2.23 |
| BcL2a | WT Tg |
93.5 178.7 |
1.55 4.81 |
<0.001 | 1.91 |
| BcL2a1a | WT Tg |
33.5 92.7 |
1.19 1.75 |
<0.01 | 2.76 |
| BGN | WT Tg |
12.0 25.2 |
1.08 2.46 |
<0.001 | 2.10 |
| BLR-1 | WT Tg |
12.7 58.0 |
1.37 2.48 |
<0.001 | 4.56 |
| Ccnd-1 | WT Tg |
13.25 70.25 |
1.37 2.01 |
<0.001 | 4.37 |
| Ccnd-3 | WT Tg |
10.25 39.75 |
1.49 2.28 |
<0.001 | 3.89 |
| CD80 | WT Tg |
12.0 26.25 |
1.08 2.01 |
<0.001 | 2.10 |
| CD69 | WT Tg |
34.5 97.5 |
1.04 2.66 |
<0.001 | 2.82 |
| CSF-1 | WT Tg |
5.25 9.5 |
0.85 2.78 |
N.S | 1.82 |
| CSF-2 | WT Tg |
12.5 70.2 |
3.06 2.28 |
<0.001 | 5.62 |
| CSF-3 | WT Tg |
18.25 75.7 |
1.10 2.56 |
<0.001 | 4.16 |
| Fcer2a | WT Tg |
23.0 88.25 |
1.6 3.37 |
<0.001 | 3.83 |
| F8 | WT Tg |
5.75 22.75 |
1.25 2.13 |
<0.001 | 3.99 |
| GRO-1 | WT Tg |
32.25 135.75 |
0.94 2.86 |
<0.001 | 4.21 |
| GSTP-1 | WT Tg |
53.5 154.25 |
1.70 2.68 |
<0.001 | 2.88 |
| H2T23 | WT Tg |
46.25 108.75 |
1.54 1.49 |
<0.001 | 2.34 |
| HMGN-1 | WT Tg |
60.25 187.5 |
1.10 3.12 |
<0.001 | 3.16 |
| FB | WT Tg |
7.25 22.0 |
0.62 1.58 |
<0.001 | 3.40 |
| FasL | WT Tg |
6.0 17.0 |
1.08 1.58 |
<0.001 | 2.83 |
| FTH | WT Tg |
3.75 13.5 |
0.85 1.32 |
<0.001 | 3.64 |
| GAL-3 | WT Tg |
4.0 9.5 |
0.91 1.19 |
<0.003 | 2.35 |
| Hmox | WT Tg |
3.0 6.25 |
0.40 1.03 |
<0.01 | 2.08 |
| Gly96 | WT Tg |
6.75 19.5 |
1.10 1.55 |
<0.001 | 2.91 |
| HAS-1 | WT Tg |
22.75 71.25 |
1.49 1.79 |
<0.001 | 3.13 |
| ICAM-1 | WT Tg |
4.75 10.25 |
0.85 1.49 |
<0.003 | 2.18 |
| IGFBP-1 | WT Tg |
17.5 35.5 |
1.32 1.5 |
<0.001 | 2.02 |
| IGFBP-2 | WT Tg |
15.2 32.5 |
1.31 1.93 |
<0.001 | 2.13 |
| InfB | WT Tg |
6.25 13.75 |
1.10 1.37 |
<0.001 | 2.21 |
| InfG | WT Tg |
6.75 14.75 |
0.47 1.49 |
<0.001 | 2.30 |
| IL-11 | WT Tg |
19.75 65.0 |
1.03 3.80 |
<0.001 | 3.29 |
| IL-15 | WT Tg |
5.0 9.5 |
0.70 1.19 |
<0.003 | 1.90 |
| IL-1β | WT Tg |
13.0 20.0 |
0.91 1.68 |
<0.003 | 1.53 |
| IL-m | WT Tg |
7.5 16.0 |
1.02 1.2.08 |
<0.003 | 2.13 |
| IL-2 | WT Tg |
2.75 5.75 |
0.47 0.85 |
<0.003 | 2.12 |
| IL-6 | WT Tg |
33.75 69.25 |
0.85 1.37 |
<0.001 | 2.05 |
| IL-10 | WT Tg |
29.0 61.75 |
1.08 1.25 |
<0.001 | 2.12 |
| IRF-2 | WT Tg |
53.0 156.7 |
1.29 2.28 |
<0.001 | 2.95 |
| LAMB-2 | WT Tg |
32.75 70.0 |
0.85 0.91 |
<0.001 | 2.14 |
| LTA | WT Tg |
9.0 14.25 |
1.08 1.65 |
<0.001 | 1.58 |
| Lyzs | WT Tg |
35.2 36.0 |
2.86 1.47 |
N.S | 1.02 |
| MMP-3 | WT Tg |
14.25 21.25 |
1.10 2.49 |
<0.05 | 1.49 |
| Myc | WT Tg |
11.25 33.5 |
0.85 2.1 |
<0.001 | 2.99 |
| NF-κB-1 | WT Tg |
12.75 75.0 |
0.85 1.08 |
<0.001 | 5.90 |
| NF-κB-2 | WT Tg |
30.5 75.0 |
6.25 2.73 |
<0.001 | 2.45 |
| IκBα | WT Tg |
19.25 36.0 |
0.85 1.08 |
<0.001 | 1.87 |
| MadCAM-1 | WT Tg |
6.25 30.75 |
1.10 2.42 |
<0.01 | 4.95 |
| NOS | WT Tg |
12.25 19.25 |
1.31 1.37 |
<0.001 | 1.57 |
| NpYR | WT Tg |
32.75 40.0 |
1.54 2.34 |
<0.001 | 1.22 |
| Olr-1 | WT Tg |
46.5 67.75 |
2.9 1.37 |
<0.001 | 1.45 |
| PAFR | WT Tg |
13.5 21.75 |
1.04 1.65 |
<0.001 | 1.60 |
| PDGFβ | WT Tg |
40.0 126.75 |
1.29 0.85 |
<0.001 | 3.16 |
| PENK-1 | WT Tg |
33.0 143.75 |
1.08 5.90 |
<0.001 | 4.35 |
| PTGIS | WT Tg |
41.0 140.75 |
1.08 1.75 |
<0.001 | 3.43 |
| PTGIS2 | WT Tg |
43.0 90.25 |
1.82 1.49 |
<0.001 | 2.09 |
| Ptx-2 | WT Tg |
33.25 65.75 |
2.05 1.65 |
<0.001 | 1.98 |
| Rel | WT Tg |
27.25 70.75 |
1.49 2.92 |
<0.001 | 2.60 |
| Saa-1 | WT Tg |
34.0 67.0 |
1.08 2.19 |
<0.001 | 1.97 |
| Scya-2 | WT Tg |
28.25 37.50 |
1.10 1.93 |
<0.003 | 1.32 |
| TNC | WT Tg |
24.0 36.25 |
1.29 1.54 |
<0.001 | 1.51 |
| TNF | WT Tg |
18.0 73.25 |
0.81 4.32 |
<0.001 | 4.06 |
| UPAR | WT Tg |
35.25 49.75 |
1.65 3.59 |
<0.003 | 1.41 |
| VEGFc | WT Tg |
40.25 63.75 |
0.85 2.13 |
<0.001 | 1.58 |
| Vim | WT Tg |
33.7 42.5 |
1.31 2.21 |
<0.003 | 1.26 |
| Wt1 | WT Tg |
50.5 75.0 |
1.04 1.68 |
<0.001 | 1.48 |
| GAPDH | WT Tg |
97.2 97.7 |
0.47 0.62 |
1.00 |
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med. 1990;322:1561–1566. doi: 10.1056/NEJM199005313222203. [DOI] [PubMed] [Google Scholar]
- 2.Kent RL, Mann DL, Cooper G. Signals for cardiac muscle hypertrophy in hypertension. J Cardiovasc. Pharmacol. 1991;17(Suppl 2):S7–13. doi: 10.1097/00005344-199117002-00003. [DOI] [PubMed] [Google Scholar]
- 3.Baldwin AS., Jr. The NF-κB and I-κB proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 4.May MJ, Ghosh S. Signal transduction through NF-kappa B. Immunol Today. 1998;19:80–88. doi: 10.1016/s0167-5699(97)01197-3. [DOI] [PubMed] [Google Scholar]
- 5.Chen F, Castranova V, Shi X, Demers LMA. New insights into the role of nuclear factor-κB, a ubiquitous transcription factor in the initiation of diseases. Clin. Chem. 1999;45:7–17. [PubMed] [Google Scholar]
- 6.Blackwell TS, Christman JW. The role of nuclear factor-kappa B in cytokine gene regulation. Am. J Respir. Cell Mol Biol. 1997;17:3–9. doi: 10.1165/ajrcmb.17.1.f132. [DOI] [PubMed] [Google Scholar]
- 7.Bourcier T, Sukhova G, Libby P. The nuclear factor kappa-B signaling pathway participates in dysregulation of vascular smooth muscle cells in vitro and in human atherosclerosis.PG. J Biol Chem. 1997;272 doi: 10.1074/jbc.272.25.15817. [DOI] [PubMed] [Google Scholar]
- 8.Ritchie ME. Nuclear factor-kappaB is selectively and markedly activated in humans with unstable angina pectoris. Circulation. 1998;98:1707–1713. doi: 10.1161/01.cir.98.17.1707. [DOI] [PubMed] [Google Scholar]
- 9.Kubota T, McTiernan CF, Frye CS, Slawson SE, Lemster BH, Koretsky AP, Demetris AJ, Feldman AM. Dilated cardiomyopathy in transgenic mice with cardiac-specific overexpression of tumor necrosis factor-alpha. Circ. Res. 1997;81:627–635. doi: 10.1161/01.res.81.4.627. [DOI] [PubMed] [Google Scholar]
- 10.Wong SC, Fukuchi M, Melnyk P, Rodger I, Giaid A. Induction of cyclooxygenase-2 and activation of nuclear factor-kappaB in myocardium of patients with congestive heart failure. Circulation. 1998;98:100–103. doi: 10.1161/01.cir.98.2.100. [DOI] [PubMed] [Google Scholar]
- 11.Kalra D, Baumgarten G, Dibbs Z, Seta Y, Sivasubramanian N, Mann DL. Nitric oxide provokes tumor necrosis factor-alpha expression in adult feline myocardium through a cGMP-dependent pathway. Circulation. 2000;102:1302–1307. doi: 10.1161/01.cir.102.11.1302. [DOI] [PubMed] [Google Scholar]
- 12.Li Y, Ha T, Gao X, Kelley J, Williams DL, Browder IW, Kao RL, Li C. NF-κB activation is required for the development of cardiac hypertrophy in vivo. Am. J. Physiol. Heart Circ. Physiol. 2004;287:H1712–H1720. doi: 10.1152/ajpheart.00124.2004. [DOI] [PubMed] [Google Scholar]
- 13.Gupta S, Young D, Sen S. Inhibition of NF-kappaB induces regression of cardiac hypertrophy, independent of blood pressure control, in spontaneously hypertensive rats. Am. J. Physiol Heart Circ. Physiol. 2005;289:H20–H29. doi: 10.1152/ajpheart.00082.2005. [DOI] [PubMed] [Google Scholar]
- 14.Gupta S, Sen S. Role of the NF-kappaB signaling cascade and NF-kappaB-targeted genes in failing human hearts. J. Mol. Med. 2005;83:993–1004. doi: 10.1007/s00109-005-0691-z. [DOI] [PubMed] [Google Scholar]
- 15.Baeuerle PA, Baltimore D. NF-kappa B: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 16.Beg AA, Finco TS, Nantermet PV, Baldwin AS., Jr. Tumor necrosis factor and interleukin-1 lead to phosphorylation and loss of I kappa B alpha: a mechanism for NF-kappa B activation. Mol Cell Biol. 1993;13:3301–3310. doi: 10.1128/mcb.13.6.3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IκB kinase that activates the transcription factor NF-κB. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 18.Zandi E, Karin M. Bridging the gap: composition, regulation, and physiological function of the IkappaB kinase complex. Mol Cell Biol. 1999;19:4547–4551. doi: 10.1128/mcb.19.7.4547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zandi E, Rothwarf DM, Delhase M, Hayakawa M, Karin M. The IκB kinase complex (IKK) contains two kinase subunits, IKKα and IKKβ, necessary for IκB phosphorylation and NF-κB activation. Cell. 1997;91:243–252. doi: 10.1016/s0092-8674(00)80406-7. [DOI] [PubMed] [Google Scholar]
- 20.Sen S, Tarazi RC, Khairallah PA, Bumpus FM. Cardiac hypertrophy in spontaneously hypertensive rats. Circ. Res. 1974;35:775–781. doi: 10.1161/01.res.35.5.775. [DOI] [PubMed] [Google Scholar]
- 21.Sil P, Misono K, Sen S. Myotrophin in human cardiomyopathic heart. Circ. Res. 1993;73:98–108. doi: 10.1161/01.res.73.1.98. [DOI] [PubMed] [Google Scholar]
- 22.Sen S, Kundu G, Mekhail N, Castel J, Misono K, Healy B. Myotrophin: purification of a novel peptide from spontaneously hypertensive rat heart that influences myocardial growth. J. Biol. Chem. 1990;265:16635–16643. [PubMed] [Google Scholar]
- 23.Mukherjee DP, McTiernan CF, Sen S. Myotrophin induces early response genes and enhances cardiac gene expression. Hypertension. 1993;21:142–148. doi: 10.1161/01.hyp.21.2.142. [DOI] [PubMed] [Google Scholar]
- 24.Sil P, Mukherjee DP, Sen S. Quantification of myotrophin from spontaneously hypertensive and normal rat hearts. Circ. Res. 1995;76:1020–1027. doi: 10.1161/01.res.76.6.1020. [DOI] [PubMed] [Google Scholar]
- 25.Gupta S, Purcell NH, Lin A, Sen S. Activation of nuclear factor-kappaB is necessary for myotrophin-induced cardiac hypertrophy. J Cell Biol. 2002;159:1019–1028. doi: 10.1083/jcb.200207149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sarkar S, Leaman DW, Gupta S, Sil P, Young D, Morerhead A, Mukherjee D, Ratliff N, Sun Y, Rayborn M, Hollyfield J, Sen S. Cardiac expression of myotrophin triggers myocardial hypertrophy and heart failure in transgenic mice: Changes in gene expression profiles during initiation of hypertrophy and during heart failure measured by DNA microarray analysis. J. Biol. Chem. 2004;279:20422–20434. doi: 10.1074/jbc.M308488200. [DOI] [PubMed] [Google Scholar]
- 27.Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 28.Harborth J, Elbashir SM, Bechert K, Tuschl T, Weber K. Identification of essential genes in cultured mammalian cells using small interfering RNAs. J. Cell Sci. 2001;114:4557–4565. doi: 10.1242/jcs.114.24.4557. [DOI] [PubMed] [Google Scholar]
- 29.Sharp PA. RNAi and double-strand RNA. Genes Dev. 1999;13:139–141. [PubMed] [Google Scholar]
- 30.Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409:363–366. doi: 10.1038/35053110. [DOI] [PubMed] [Google Scholar]
- 31.Le Bail O, Schmidt-Ullrich R, Israel A. Promoter analysis of the gene encoding the I kappa B-alpha/MAD3 inhibitor of NF-kappa B: positive regulation by members of the rel/NF-kappa B family. EMBO J. 1993;12:5043–5049. doi: 10.1002/j.1460-2075.1993.tb06197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shames BD, Meldrum DR, Selzman CH, Pulido EJ, Cain BS, Banerjee A, Harken AH, Meng X. Increased levels of myocardial IkappaB-alpha protein promote tolerance to endotoxin. Am. J Physiol. 1998;275:H1084–H1091. doi: 10.1152/ajpheart.1998.275.3.H1084. [DOI] [PubMed] [Google Scholar]
- 33.Zandi E, Chen Y, Karin M. Direct phosphorylation of IκB by IKKα and IKKβ: discrimination between free and NF-κB-bound substrate. Science. 1998;281:1360–1363. doi: 10.1126/science.281.5381.1360. [DOI] [PubMed] [Google Scholar]
- 34.Li Q, Lu Q, Hwang JY, Buscher D, Lee KF, Izpisua-Belmonte JC, Verma IM. IKK1-deficient mice exhibit abnormal development of skin and skeleton. Genes Dev. 1999;13:1322–1328. doi: 10.1101/gad.13.10.1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Q, Van Antwerp D, Mercurio F, Lee KF, Verma IM. Severe liver degeneration in mice lacking the IkappaB kinase 2 gene. Science. 1999;284:321–325. doi: 10.1126/science.284.5412.321. [DOI] [PubMed] [Google Scholar]
- 36.Li Z, Bing OH, Long X, Robinson KG, Lakatta EG. Increased cardiomyocyte apoptosis during the transition to heart failure in the spontaneously hypertensive rat. Am. J Physiol. 1997;272:H2313–H2319. doi: 10.1152/ajpheart.1997.272.5.H2313. [DOI] [PubMed] [Google Scholar]
- 37.Karin M. The beginning of the end: IkappaB kinase (IKK) and NF-kappaB activation. J Biol. Chem. 1999;274:27339–27342. doi: 10.1074/jbc.274.39.27339. [DOI] [PubMed] [Google Scholar]
- 38.Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N. Engl. J Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- 39.Li C, Kao RL, Ha T, Kelley J, Browder IW, Williams DL. Early activation of IKKbeta during in vivo myocardial ischemia. Am J Physiol Heart Circ Physiol. 2001;280:H1264–H1271. doi: 10.1152/ajpheart.2001.280.3.H1264. [DOI] [PubMed] [Google Scholar]
- 40.Valen G, Yan ZQ, Hansson GK. Nuclear factor kappa-B and the heart. J Am. Coll. Cardiol. 2001;38:307–314. doi: 10.1016/s0735-1097(01)01377-8. [DOI] [PubMed] [Google Scholar]
- 41.Hannon GJ. RNA interference. Nature. 2002;418:244–251. doi: 10.1038/418244a. [DOI] [PubMed] [Google Scholar]
- 42.McManus MT, Sharp PA. Gene silencing in mammals by small interfering RNAs. Nat. Rev. Genet. 2002;3:737–747. doi: 10.1038/nrg908. [DOI] [PubMed] [Google Scholar]
- 43.Tuschl T. Expanding small RNA interference. Nat. Biotechnol. 2002;20:446–448. doi: 10.1038/nbt0502-446. [DOI] [PubMed] [Google Scholar]
- 44.Higuchi Y, Chan TO, Brown MA, Zhang J, DeGeorge BR, Jr., Funakoshi H, Gibson G, McTiernan CF, Kubota T, Jones WK, Feldman AM. Cardioprotection afforded by NF-kappaB ablation is associated with activation of Akt in mice overexpressing TNF-alpha. Am. J. Physiol Heart Circ. Physiol. 2006;290:H590–H598. doi: 10.1152/ajpheart.00379.2005. [DOI] [PubMed] [Google Scholar]
- 45.Kawamura N, Kubota T, Kawano S, Monden Y, Feldman AM, Tsutsui H, Takeshita A, Sunagawa K. Blockade of NF-kappaB improves cardiac function and survival without affecting inflammation in TNF-alpha-induced cardiomyopathy. Cardiovasc. Res. 2005;66:520–529. doi: 10.1016/j.cardiores.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 46.Kawano S, Kubota T, Monden Y, Tsutsumi T, Inoue T, Kawamura N, Tsutsui H, Sunagawa K. Blockade of NF-{kappa}B improves cardiac function and survival after myocardial infarction. Am. J. Physiol Heart Circ. Physiol. 2006 doi: 10.1152/ajpheart.01175.2005. [DOI] [PubMed] [Google Scholar]
- 47.Dignam JD, Martin PL, Shastry BS, Roeder RG. Eukaryotic gene transcription with purified components. Methods Enzymol. 1983;101:582–598. doi: 10.1016/0076-6879(83)01039-3. [DOI] [PubMed] [Google Scholar]
- 48.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 49.Malur AG, Chattopadhyay S, Maitra RK, Banerjee AK. Inhibition of STAT 1 phosphorylation by human parainfluenza virus type 3 C protein. J. Virol. 2005;79:7877–7882. doi: 10.1128/JVI.79.12.7877-7882.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Collins KA, Korcarz CE, Shroff SG, Bednarz JE, Fentzke RC, Lin H, Leiden JM, Lang RM. Accuracy of echocardiographic estimates of left ventricular mass in mice. Am. J. Physiol Heart Circ. Physiol. 2001;280:H1954–H1962. doi: 10.1152/ajpheart.2001.280.5.H1954. [DOI] [PubMed] [Google Scholar]
- 51.Derumeaux G, Mulder P, Richard V, Chagraoui A, Nafeh C, Bauer F, Henry JP, Thuillez C. Tissue Doppler imaging differentiates physiological from pathological pressure-overload left ventricular hypertrophy in rats. Circulation. 2002;105:1602–1608. doi: 10.1161/01.cir.0000012943.91101.d7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.