

Abstract
A combined approach for targeting RNA with novel, biologically active ligands has been developed using a cyclic peptide library and in silico modeling. This approach has successfully identified novel cyclic peptide constructs that can target bTAR RNA. Subsequently, RNA/peptide interactions were effectively modeled using the HADDOCK docking program.
Keywords: RNA, Cyclic peptides, HADDOCK
Current estimates place the size of the human proteome at upwards of 107 discrete proteins.1 Under existing approaches to drug design and medicinal chemistry, only 0.005% of these proteins can be directly modulated by small molecules and are deemed druggable.2 To overcome this limitation, alternate methods to modulate protein activity are required. One promising approach would be to pursue small molecules that target RNA and modulate RNA function, consequently expanding the repertoire of targets that are available for therapeutic intervention.3–5 Herein is an account of our efforts to employ cysteine-constrained phage libraries6 to identify novel cyclic peptides that target a predetermined RNA sequence. Evidence supports that cyclic peptides often display higher affinity and stability in comparison to their linear counterparts,7 and, therefore, cyclic peptides or cyclic peptide mimics may be applicable for targeting RNA in vivo.
The well-characterized bovine immunodeficiency virus (BIV) trans-activating response element (bTAR)8 RNA was chosen to develop and evaluate our approach. Although bTAR has been targeted previously with cyclic peptides,9,10 these peptides were derived from the endogenous poly-cationic bTAR binding peptide, bTat. Other groups have used cyclic peptides to target RNA;11 however these peptides are also poly-cationic based. Methodologies that generate cyclic peptides that are not based upon either endogenous binding sequences or limited to poly-cationic interactions to drive RNA binding may also have utility as templates for the design of RNA binding ligands. Furthermore, for deriving cyclic peptide mimics that target pre-determined RNA sequences, techniques that immediately generate a cyclic peptide template for further structure-based design would have obvious inherent benefits over strategies that provide linear peptides as design templates. Most notable is that the active conformation is ensured in a cyclic peptide format and a structure-based design effort would not have to be undertaken to ensure that the active conformation of the linear peptide is preserved within a cyclic peptide format.
Previous studies employing phage display have selected linear peptides capable of binding pre-determined RNA sequences that were not poly-cationic in nature.12–14 However, to the best of our knowledge, cysteine-constrained phage libraries had not been used to generate cyclic peptide motifs that targeted a pre-determined RNA sequence. We generated novel cyclic peptide motifs that targeted bTAR using a comprehensive, cysteine-constrained phage library that contained 7 random amino acids (library diversity 1.28 × 109) flanked by two invariant cysteine residues (that oxidatively cyclize upon phage assembly) on the end of the phage pIII coat protein. The phage library was screened against a biotinylated 28 nucleotide bTAR sequence (5′-Biotin-GCUUCGUGUAGCUCAUUAGCUCCGAGCC-3′). Binding phage were selected by employing a strategy that utilized both positive and negative selection rounds. In a positive selection round, the phage library was incubated with the biotinylated bTAR motif and binding phage were captured with streptavidin-coated magnetic beads. In a negative selection round, the enriched phage library was incubated with the biotinylated, randomer RNA oligonucleotide N1 (5′-Biotin-GCAAUGUUGACGACAGUUAGAUUAGAGA-3′) to remove poly-cationic peptides that bound in a non-sequence specific fashion. In this latter case, negative binding phage were captured with streptavidin-coated magnetic beads, and the magnetic beads were collected and discarded. The supernatant was then directly placed into a positive selection cycle. A total of five rounds of selections were completed with one round of negative selection, round 2.
Two cyclic peptide motifs that were chosen for further biophysical characterization of binding with bTAR RNA are depicted in Figure 1. These constructs correspond to two phage inserts occurring multiple times in our sequencing pool that contained a tryptophan residue that could be exploited for fluorescence quenching experiments. As with previous reports that have documented the use of phage libraries to target RNA motifs,12–14 neither of these peptides are poly-cationic. Taking advantage of the inherent fluorescence of the tryptophan residue in each peptide, fluorescence quenching was used to determine the Kd of each peptide as shown in Figure 2. Peptide 1 bound with a Kd of 7.9 ± 0.5 μM to bTAR, while peptide 2 bound with a Kd of 9.1 ± 1.2 μM to bTAR. These affinities are similar to longer, linear peptides that have been selected to bind to other, un-related RNA sequences.12–14
Figure 1.

Cysteine-constrained peptides selected to bind bTAR.
Figure 2.
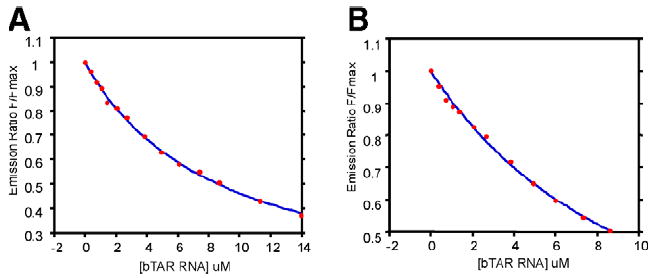
Fluorescence quenching profiles for peptide 1 with (A) bTAR RNA and (B) peptide 2 with bTAR RNA.
Preliminary specificity experiments of peptide 1 and peptide 2 for the bTAR oligonucleotide were also performed. Figure 3A and 3B depicts binding of the two cyclic peptide constructs with non-biotinylated Negative Randomer, N1, used during selection. Although N1 is comparable in length to the bTAR target, it is devoid of any secondary structure. Peptide 1 displayed a slight selectivity with a Kd of 15.9 ± 3.2 μM compared to binding bTAR with a Kd of 7.9 ± 0.5 μM, however peptide 2 bound N1 with a Kd analogous to bTAR, at 9.1 ± 1.2 μM. Peptide 1 was subsequently tested with a second Negative RNA control, N2, designed to mimic the secondary structure of bTAR RNA but with a different primary sequence, depicted in Figure 3C. Fluorescence quenching indicates that although peptide 1 has higher affinity for N2 than N1 with a Kd of 11.8 ± 0.5 μM, peptide 1 is capable of differentiating between secondary structure analogues and binding in a sequence specific manner.
Figure 3.
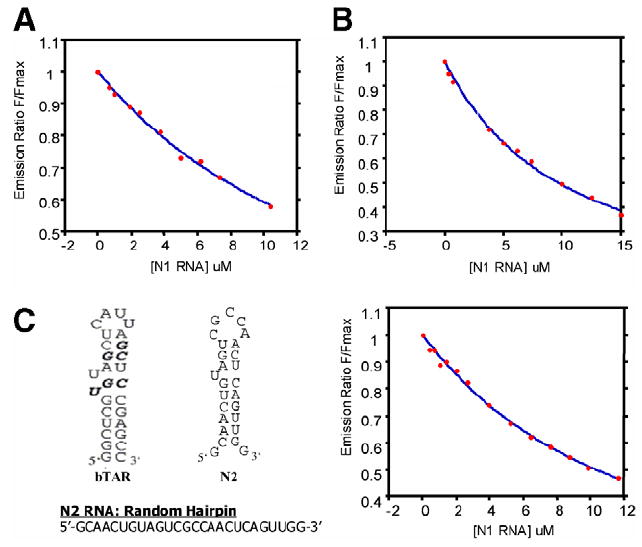
Fluorescence quenching profiles (QPs) for peptide 1 and peptide 2 with negative controls. (A) Peptide 1 with N1; (B) peptide 2 with N1; (C) bTAR sequence (bTat binding indicated in bold) compared to the N2 sequence, secondary structure, and QPs profile with peptide 1.
Preliminary modeling of each peptide/RNA complex was accomplished with HADDOCK.15 The HADDOCK docking program generates the lowest energy structure via a three-stage process: an initial rigid-body docking assignment, followed by a semi-flexible assignment, and finally the inclusion of water in the calculation (details are provided in the Supporting Information, general information about the HADDOCK program can be found at: http://www.nmr.chem.uu.nl/haddock/). The lowest energy structure for each complex is depicted in Figure 4. HADDOCK predicted that while both di-sulfide linkages provide a necessary covalent linkage to generate the cyclic peptide architecture, they do not participate in H-bond donor/acceptor interactions with the oligonucleotide. Therefore, the di-sulfide bridge should be amenable to replacement with a redox stable surrogate with little or no loss in affinity.
Figure 4.
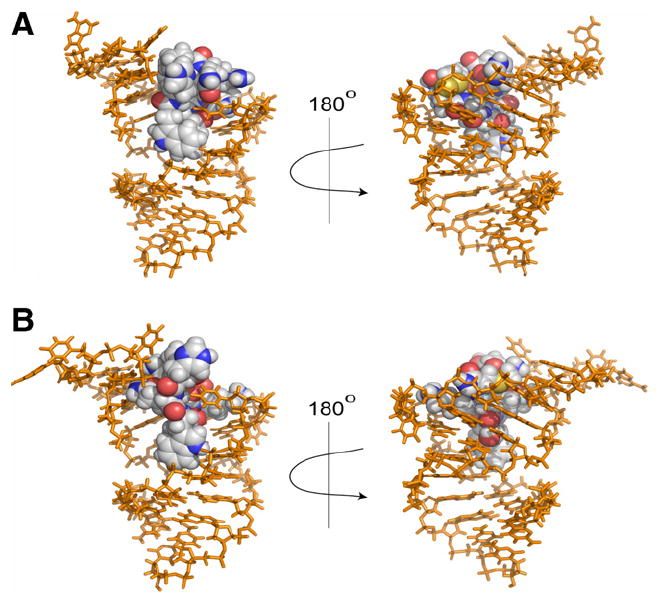
Predicted structures of RNA/peptide binding complexes. Peptide shown as spheres. (A) Cyclic peptide 1. (B) Cyclic peptide 2.
In conclusion, we have demonstrated that it is possible to generate novel cyclic peptide motifs that bind with relatively high affinity to a predetermined RNA target by use of cysteine-constrained phage libraries. These peptides are not poly-cationic and may be recognizing RNA through specific RNA/peptide interactions that may be exploitable for future structure-based design efforts. Given the flexibility of phage methodology, it should be possible to use this approach to generate novel, cyclic peptide scaffolds for any predetermined RNA motif. Furthermore, this approach is not limited to or reliant upon a known binding motif. Application of this approach and adaptation of HADDOCK to structure-based refinement of both affinity and specificity of cyclic peptides that target pre-determined RNA sequences is currently underway.
Acknowledgments
We greatly acknowledge financial support from North Carolina State University, the NIH (GM55769, J.C.), and the Kenan Institute for Engineering, Technology and Science (J.C.). We also thank Professors David Shultz (NCSU Chemistry), Tatyana Smirnova (NCSU Chemistry), and Keith Weninger (NCSU Physics) for computational and fluorescence assistance.
References and notes
- 1.Anderson N, Anderson N. Mol Cell Proteomics. 2002;1:845. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
- 2.Renfrey S, Featherstone J. Nat Rev Drug Disc. 2002;1:175. doi: 10.1038/nrd766. [DOI] [PubMed] [Google Scholar]
- 3.Tor Y. Chembiochem. 2003;4:998. doi: 10.1002/cbic.200300680. [DOI] [PubMed] [Google Scholar]
- 4.Hermann T. Biopolymers. 2003;70:4. doi: 10.1002/bip.10410. [DOI] [PubMed] [Google Scholar]
- 5.Cheng AC, Calabro V, Frankel AD. Curr Opin Struct Biol. 2001;11:478. doi: 10.1016/s0959-440x(00)00236-0. [DOI] [PubMed] [Google Scholar]
- 6.Mclafferty MA, Kent RB, Ladner RC, Markland W. Gene. 1993;128:29. doi: 10.1016/0378-1119(93)90149-w. [DOI] [PubMed] [Google Scholar]
- 7.Davies J. J Pep Sci. 2003;9:471. doi: 10.1002/psc.491. [DOI] [PubMed] [Google Scholar]
- 8.Gonda MA, Luther DG, Fong SE, Tobin GJ. Virus Res. 1994;32:155. doi: 10.1016/0168-1702(94)90040-x. [DOI] [PubMed] [Google Scholar]
- 9.Runyon S, Puglisi J. J Am Chem Soc. 2003;125:15704. doi: 10.1021/ja036344h. [DOI] [PubMed] [Google Scholar]
- 10.Athanassiou Z, Dias R, Moehle K. J Am Chem Soc. 2004;126:6906. doi: 10.1021/ja0497680. [DOI] [PubMed] [Google Scholar]
- 11.Tamilarasu N, Huq I, Rana TM. Bioorg Med Chem Lett. 2000;10:971. doi: 10.1016/s0960-894x(00)00140-2. [DOI] [PubMed] [Google Scholar]
- 12.Frugier M, Schimmel P. Proc Natl Acad Sci. 1997;94:11291. doi: 10.1073/pnas.94.21.11291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mucha P, Szyk A, Rekowski P, Weiss PA, Agris PF. Biochemistry. 2001;40:14191. doi: 10.1021/bi010978o. [DOI] [PubMed] [Google Scholar]
- 14.Agris PF, Marchbank MT, Newman W, Guenther R, Ingram P, Swallow J, Mucha P, Szyk A, Rekowski P, Peletskaya E, Deutscher SL. J Protein Chem. 1999;18:425. doi: 10.1023/a:1020688609121. [DOI] [PubMed] [Google Scholar]
- 15.Dominguez C, Boelens R, Bonvin AMJJ. J Am Chem Soc. 2003;125:1731. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]