

Abstract
A three-part tandem mass spectrometric strategy that entails MSn analysis and a post-column LC-MS cobalt complexation method is developed to identify flavonoid monoglucuronide metabolites synthesized using the 1A1 isozyme of human UDP-glucuronosyltransferase (UGT). Ten flavonoid aglycons were used as substrates, spanning the subclasses of flavones, flavonols and flavanones. The products were characterized by LC-MS and LC-MSn, with post-column cobalt complexation employed to pinpoint the specific sites of conjugation. The dissociation of complexes of the form [Co(II) (flavonoid glucuronide – H) (4,7-diphenyl-1,10-phenanthroline)2]+ allowed identification of the products and differentiation of isomers. The correlation between glycosylation site and elution order is used to provide additional structural confirmation. Flavonoids lacking a 3′ hydroxyl group were glucuronidated only at position 7, while those containing this functionality also formed 3′-O-glucuronides and sometimes 4′-O-glucuronides, thus supporting the conclusion that the presence or absence of the 3′-OH group is the major determinant of the regioselectivity of glucuronidation. Moreover, the specific distribution of multiple glucuronide products (7-O, 3′-O, 4′-0) is governed by the subclass of flavonoid.
Introduction
Much progress has been made in the understanding of flavonoid metabolism over the past decade. The currently-accepted paradigm involves the consumption of flavonoid glycosides in plant-based food products, deglucosylation in the small intestine by β-glucosidase or lactose phloridzin hydrolase, and rapid metabolism by Phase I and (especially) Phase II enzymes [1–3]. Glucuronidation and sulfation are important metabolic routes for most flavonoids, while methylation or hydroxylation may also occur depending on the structure of the molecule in question [2, 3]. There has also been a report of glutathione-related metabolites in human urine [4]. As a result of these rapid conjugation reactions, neither the original flavonoid glycosides (except anthocyanins) nor the aglycon forms (except catechins) are found in plasma [5–8]. Early reports of unmodified flavonoid glycosides circulating in the bloodstream [9–11] were likely mistaken identifications of flavonoid glucuronides, which have similar chromatographic and ultraviolet (UV) spectroscopic characteristics [7, 12]. Flavonoids that fail to be absorbed in the small intestine may be broken down by microflora in the large intestine [1–3]. This process may release the free aglycons, which can then be absorbed and metabolized, but mostly results in the release of small phenolic acids, which are expelled in the urine [1–3]. Quantitative in vivo studies generally show that only a small percentage of consumed flavonoid glycosides is recovered in urine as conjugated phase II metabolites [13–15]. Walle et al. used 14C-labelled quercetin to show that up to 81% of the administered dose ultimately is exhaled in the form of carbon dioxide [16]. There remains considerable interest in the conjugated metabolites as they may retain some of the bioactivity of the original molecules [5, 17].
In spite of breakthroughs in the field of flavonoid metabolism, much is still unknown about the precise structure of Phase II flavonoid metabolites, particularly in terms of the conjugation positions. Most reports do not supply this information, identifying observed metabolites imprecisely as, for example, unspecified quercetin glucuronides. A recent review [5] listed all conjugated metabolites that have been identified in human in vivo studies, but the very short list included conjugates of only a few flavonoid aglycons. One reason for the dearth of such information is a lack of sensitive and specific analytical methods. While there are several approaches currently available, there are problems associated with each. The standard method for structural determination of organic molecules is nuclear magnetic resonance (NMR) spectroscopy, but its lack of sensitivity does not lend itself to analysis of low-concentration metabolites, especially when scaling up is prohibitive (such as in vivo bioavailability, pharmacokinetic, and mechanistic studies). Thus few metabolism studies have employed this technique [18, 19]. An alternative method using UV spectroscopy [20] to determine the conjugation position of flavonoids has been used only rarely for metabolites [17], probably due to the complex set of experiments with numerous UV shift reagents required to make this determination. A third strategy requires synthesis of suspected metabolites and comparison of the chemical properties of these synthetic compounds to those of the observed metabolites. While this approach is sometimes employed to identify flavonoid metabolites [13, 21, 22], it is too laborious for routine use.
We have developed several methods for identifying the conjugation positions and saccharide identities of flavonoid glycosides using tandem mass spectrometry [23–29]. These methods exploit the natural chelating ability of flavonoids to form metal complexes in solution, which can then be analyzed in a mass spectrometer for structural identification. Upon collision-induced dissociation (CID), characteristic fragmentation patterns are observed that allow the conjugation positions to be determined. These fragmentation patterns are consistent enough to provide positive identification of unknown compounds without the need for standards. For example, a manganese complexation method [26] that distinguished five different glucosylation positions as well as glucoside/galactoside isomers was used to identify kaempferol 3-O-glucoside, kaempferol 3-O-galactoside, isorhamnetin 3-O-glucoside and isorhamnetin 3-O-galactoside in a Silphium albiflorum extract [30].
Most recently, we reported a metal complexation/tandem mass spectrometry method for differentiating the conjugation positions of monoglucuronidated flavonols, flavones and flavanones [31]. The metal complexation strategy entailed formation of complexes of the type [Co(II) (FG - H) (4,7-dpphen)2]+ where (FG - H) is one molecule of deprotonated flavonoid glucuronide and (4,7-dpphen)2 are two molecules of an auxiliary ligand, 4,7-diphenyl-1,10-phenanthroline. Upon CID, this complex yielded product ions corresponding to the loss of a glucuronic acid moiety, of an auxiliary ligand, and of the aglycon portion of the flavonoid, in various combinations and ratios that correlated with the position of glucuronidation. A four-way differentiation of the 3-O-, 7-O-, 3′-O- and 4′-O-glucuronides of quercetin was demonstrated. This previous investigation focused on a limited set of flavonoid glucuronides, but we speculated that the versatility and sensitivity of the method would allow facile adaptation to other flavonoid metabolites, such as the ones produced enzymatically in the present study.
The purpose of the current work is twofold. The first goal is to explore the wider applicability of the described metal complexation methods to flavonoid metabolism studies. Second, we report the regiospecificity of flavonoid glucuronidation by the 1A1 isozyme of human UDP-glucuronosyltransferase (UGT1A1). While there have been several studies on the glucuronidating activity of various isozymes of UGT using flavonoids as substrates [32–38], fewer have explored the regioselectivity of such reactions [19, 39]. This report represents the first comprehensive assessment of the impact of the structural features of flavonoids on the enzymatic formation of flavonoid glucuronides. The glucuronidated derivatives of ten flavonols, flavones and flavanones (structures of the flavonoid aglycons are shown in Scheme 1) provide a diverse set of analytes to challenge the performance of the new method. The correlation between flavonoid conjugation sites and reversed-phase high-performance liquid chromatography (HPLC) retention time is also discussed and used as an additional tool in elucidating the structures of the enzymatically synthesized products.
Scheme 1.

Structures of flavonoid aglycons used as substrates for UGT1A1.
Experimental
Materials
UGT1A1 isozyme (human, recombinant), UDP-glucuronic acid (UDPGA) trisodium salt, 4,7-diphenyl-1,10-phenanthroline (4,7-dpphen), cobalt(II) bromide, acacetin, myricetin, hesperetin, naringenin and quercetin were purchased from Sigma-Aldrich (St. Louis, MO). Eriodictyol, kaempferol, luteolin, luteolin 7-O-glucoside and luteolin 4′-O-glucoside were purchased from Indofine (Hillsborough, NJ). Apigenin, homoeriodictyol, quercetin 3-O-glucoside and quercetin 4′-O-glucoside were purchased from Extrasynthèse (Genay, France). Quercetin 7-O-glucoside was purchased from Apin (Abingdon, UK). Quercetin 3-O-glucuronide, quercetin 7-O-glucuronide, quercetin 3′-O-glucuronide and quercetin 4′-O-glucuronide were synthesized [22, 31] by Paul Needs and Paul Kroon at the Institute of Food Research (Norwich, UK). Potassium phosphate was purchased from Thermo Fisher Scientific, Inc. (Waltham, MA).
Enzymatic Synthesis of Flavonoid Glucuronides
The method for synthesizing the flavonoid glucuronides was adapted from one described by Plumb et al. [40]. The UGT1A1, having an estimated enzyme concentration of 12 mg/mL, was divided into 25 μL aliquots which were stored at −80 °C until use. Flavonoids were prepared as 10 mM solutions in methanol, which were employed despite turbidity of a few solutions. Synthesis occurred in a microcentrifuge tube, to which were added 2 mM aqueous UDPGA (65 μL), 20 mM potassium phosphate buffer pH 7.0 (378.75 μL) and 10 mM flavonoid (6.25 μL). The reaction was initiated by adding a 25 μL aliquot of UGT1A1 to the tube. The tubes were incubated in a 37 °C water bath (VWR Model 1227, West Chester, PA) with gentle agitation for 6 hours. A few reactions were repeated with 1 hour incubation times, but these gave similar results to the 6 hour incubations, so these data will not be specifically presented here. Reactions were stopped by adding 1.5 mL acetone. The tubes were centrifuged for 10 minutes at 16000 g, and the supernatants were removed and blown with nitrogen to evaporate the acetone. The remaining supernatant was refrigerated until analysis.
LC-MSn analysis
Liquid chromatography took place on a Waters Alliance 2695 HPLC system (Milford, MA). The stationary phase was a Waters Symmetry C18 column, 2.1 × 50 mm, 3.5 μm particle size, with a guard column. Typical injection volumes were 15 to 30 μL for negative ion mode analyses, and 30 to 45 μL for positive ion mode. The mobile phases were 0.33% formic acid in water (A) and 0.33% formic acid in methanol (B). A typical gradient began at 35% B, increased to 50% B over 25 minutes, then increased to 95% B over 2 minutes, with a constant flow rate of 0.1 mL/min. For the naringenin, eriodictyol and homoeriodictyol reaction products, the first part of the gradient increased from 30% B to 45% B in 25 minutes; while for acacetin the first part of the gradient increased from 40% B to 55% B in 25 minutes. The column effluent was sent directly to the mass spectrometer.
All mass spectrometry was performed on an LCQ Duo (Thermo Electron, Waltham, MA) quadrupole ion trap with electrospray ionization (ESI). Samples were analyzed in both negative and positive ion modes. In both polarities, a spray voltage of 5.0 kV was used, the heated capillary temperature was 200 °C, and the automatic gain control was set to a target of 2 × 107 ions with a maximum injection time of 500 msec and 5 microscan averaging. All other mass spectrometer parameters were set as needed to obtain optimal signal. Negative ion mode was used to search for flavonoid glucuronides and to confirm the identities of the aglycon portion of these molecules. Positive ion mode was used for analysis of metal complexes, which were formed by post-column addition of a methanolic solution of 5 μM CoBr2 and 4,7-dpphen. These reagents were infused at a rate of 20 μL/min controlled by a syringe pump, and added to the column effluent via a tee between the column and the mass spectrometer. Due to the fragility of the metal complexes, an isolation width of 4 Da was used [41]. Because varying the collision energy will change the appearance of fragment ion spectra, a constant normalized collision energy [42] of 35% (of 5 Vp-p) was used to dissociate the complexes.
HPLC retention time analysis
Retention time was used as a source of supplementary data for the identification of the flavonoid glucuronides. In order to determine trends in retention order, flavonoid glycosides and glucuronides were injected individually, and the retention times were measured by a Waters 486 UV detector. All other chromatographic equipment was the same as used to analyze the enzymatic synthesis products. The gradient employed began at 15% B, increased to 70% B over 12 minutes, then increased to 95% B over 2 minutes, with a flow rate of 0.3 mL/min. Unlike the slow, specific gradients used to separate the reaction products, this gradient allows the quick separation of a wider variety of flavonoid derivatives.
Results and Discussion
For identification of the flavonoid glucuronides, the two key steps include the determination of the aglycon skeleton and the location of the site of conjugation. In our approach, the aglycon skeleton is elucidated from MSn spectra of the deprotonated flavonoid glucuronide with spectral comparison to standard flavonoid aglycons. The site of conjugation is pinpointed based on interpretation of the MS/MS spectra of metal complexes of the type [Co(II) (FG - H) (4,7-dpphen)2]+. As discussed in an earlier report[31], the characteristic signs of 7-O-glucuronidation from this type of metal complex include significant losses of the glucuronic acid moiety (-GlcA) and of the auxiliary ligand (-Aux), both individually and concurrently, as well as the loss of the flavonoid aglycon (-Agl), which is a diagnostic fragment for the 7-O-glucuronidation position. The metal complexes of B-ring-glucuronides typically display significant amounts of the –Aux and –(Aux + GlcA) fragment ions upon CID, but yield only minor amounts of the -GlcA fragment ion [31]. Upon CID, metal complexes of flavonol 3-O-glucuronides produce the –(Aux + GlcA) fragment ion predominantly [31]. Supplemental Figure 1 shows proposed structures for these general types of product ions.
Single-product reactions
After the enzymatic incubation of each of the ten flavonoids, the initial screening employed LC-MS with negative ion mode analysis. The total ion chromatograms were searched for masses corresponding to the aglycon (unreacted starting material), the monoglucuronidated flavonoid (aglycon + 176) and the diglucuronidated flavonoid (aglycon + 176 + 176). Unreacted aglycon was observed in most cases, but no diglucuronidated products were produced in any of the reactions. All of the reactions produced monoglucuronidated products, with five flavonoids generating a single product, and the other five producing either two or three products. This section concerns the former group, consisting of apigenin, naringenin, acacetin, kaempferol and homoeriodictyol.
Although most studies involving the identification of flavonoid conjugates by mass spectrometry do not employ MSn for verification of the flavonoid skeleton, we have recently encountered a urinary metabolite with the correct mass and initial fragments for a quercetin glucuronide sulfate, but which required MS4 analysis to prove that it was not a quercetin derivative at all [43]. Hence confirming the identity of the aglycon portion is an important step in identifying these molecules and is essential for preventing false positives. The MS/MS spectra of each of the monoglucuronidated products showed the characteristic loss of a glucuronic acid residue, −176 Da. An additional isolation/dissociation step (MS3) was used to fragment the aglycon portions of these molecules. The resulting spectra are shown in Figure 1. Because the flavonoid aglycons yield a variety of product ions, the structures were easily confirmed by comparing the MS/MS spectra of the aglycon standards, which were nearly identical to the spectra shown. Acacetin, like many methoxylated flavonoid aglycons, yields only a single fragment ion due to the loss of a methyl radical, which is not a diagnostic dissociation pathway [44, 45]. Hence, the MS4 spectrum of the acacetin monoglucuronide is shown in Figure 1 rather than the MS3 spectrum, and it was compared to the MS3 spectrum of the acacetin standard (because there is no glucuronic acid moiety to remove, one less stage of fragmentation is required). Some of the diagnostic dissociation routes include retro Diels-Alder pathways that lead to the 1,3A− ion (m/z 151) and small molecule losses such as –H2O, -CO and –CO2 [46, 47].
Figure 1.
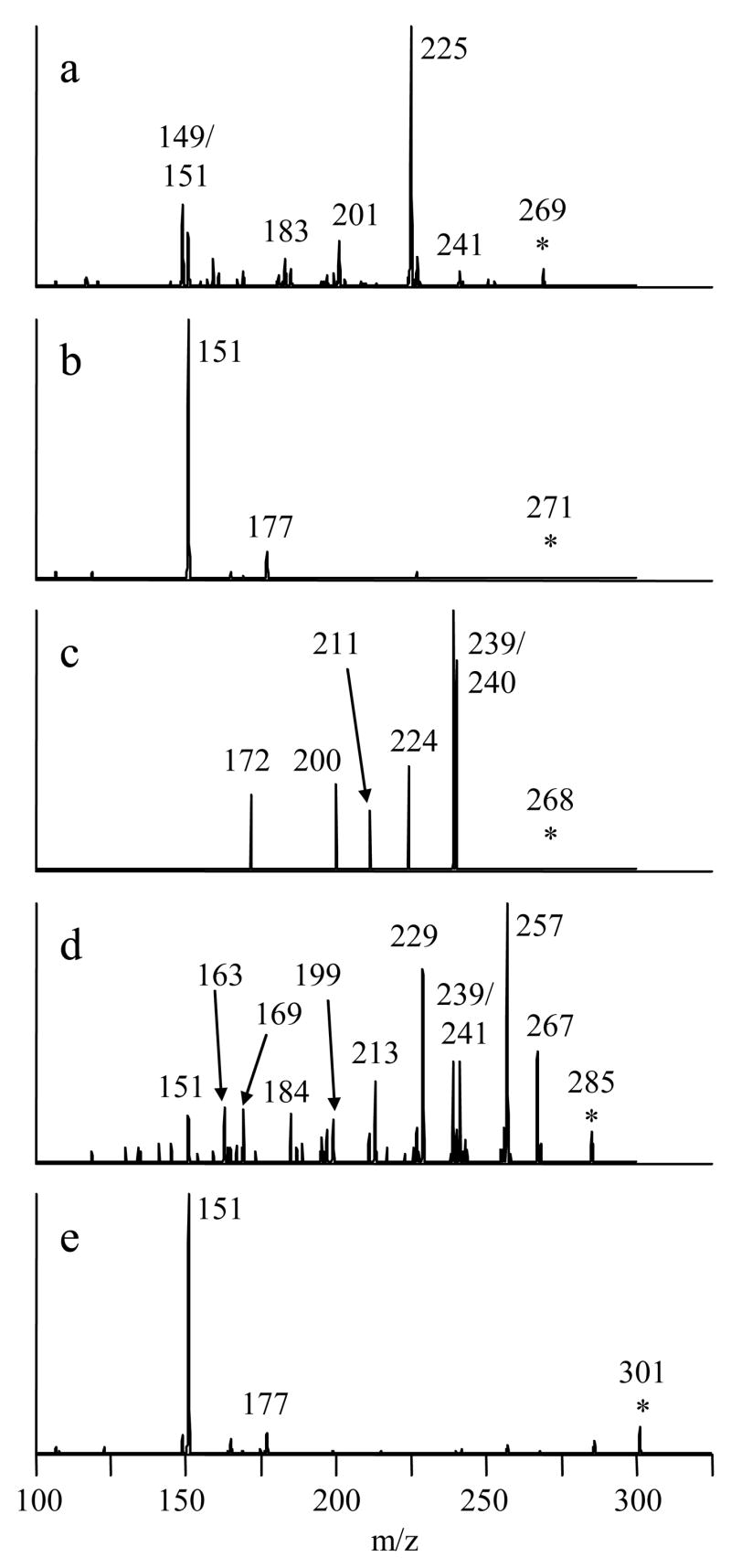
MSn spectra used to confirm the identities of the aglycon skeletons of deprotonated flavonoid monoglucuronides. a) apigenin monoglucuronide, 445→269→ b) naringenin monoglucuronide, 447→271→ c) acacetin monoglucuronide, 459→283→ 268→ d) kaempferol monoglucuronide, 461→285→ e) homoeriodictyol monoglucuronide, 477→301→.
The metal complexation methods described in the Experimental section were used to elucidate the location of the glucuronic acid moiety of these monoglucuronidated flavonoids. In particular, complexes of the type [Co(II) (FG - H) (4,7-dpphen)2]+ self-assembled in the LC effluent (in which CoBr2 and 4,7-diphenyl-1,10-phenanthroline were introduced post-column), were transported to the gas phase by ESI, and analyzed by tandem mass spectrometry. The CID mass spectra of these complexes are shown in Figure 2. All five of the complexes show evidence of glucuronidation at the 7 position, based on the diagnostic pathways summarized above (e.g. -Aux, -GlcA, -Agl), and thus all products are identified as the 7-O-glucuronides of the relevant flavonoid.
Figure 2.
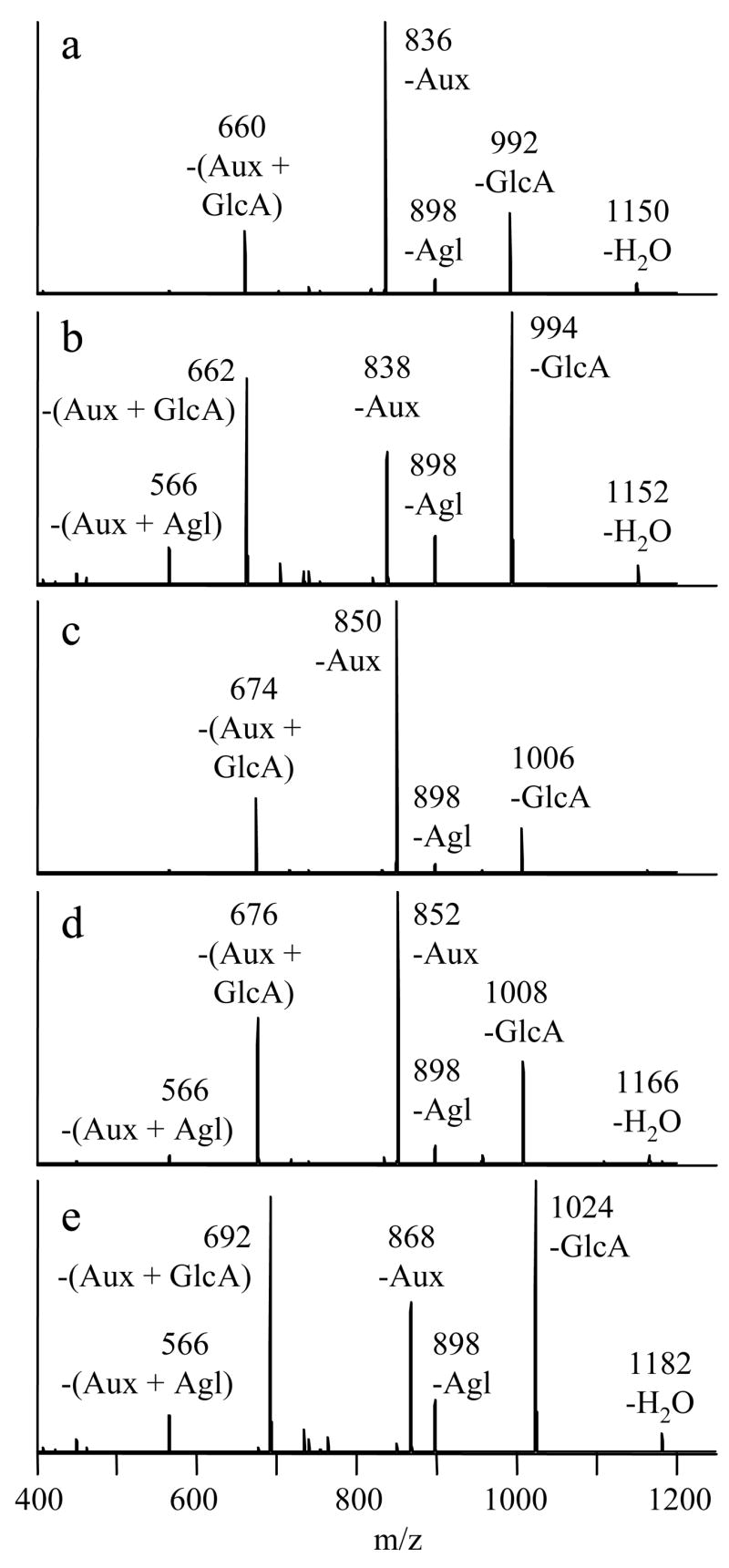
CID mass spectra of [Co(II) (FG-H) (4,7-dpphen)2]+ complexes of flavonoid monoglucuronides. a) FG=apigenin, 1168→ b) FG=naringenin, 1170→ c) FG=acacetin, 1182→ d) FG=kaempferol, 1184→ e) FG=homoeriodictyol, 1200→.
The fragmentation patterns also show some dependence on the class of flavonoid in the complex. Complexes involving flavanone (naringenin and homoeriodictyol) 7-O-glucuronides produce greater relative abundances of the diagnostic ion due to loss of the aglycon moiety (-Agl), as well as those due to loss of a glucuronic acid residue (–GlcA) and loss of the auxiliary ligand in conjunction with the elimination of a glucuronic acid residue –(Aux + GlcA). Additionally, they produce a significant fragment ion resulting from the loss of both the auxiliary ligand and the aglycon group, –(Aux + Agl). The relative abundances of these ions are lower for the complex involving the flavonol (kaempferol) 7-O-glucuronide, and lowest for those complexes involving flavone (apigenin and acacetin) 7-O-glucuronides. The analogous complex involving baicalin (a flavone 7-O-glucuronide) was reported to show similarly low abundances of these fragment ions [31].
The remaining potential glucuronidation sites should also be considered in case of ambiguous fragment ion signatures from the metal complexes. The –(Agl) and –(GlcA) ions do not feature prominently as dissociation products of flavonoid B-ring-glucuronide complexes, so the glucuronidation at the 3′ and 4′ positions is ruled out. Flavonoid 5-O-conjugates are purported to be unfavorable products due to hydrogen bonding to the 4-keto group [3, 47]. Nonetheless, an enzymatic synthesis of flavonoid 5-O-glucuronides has been reported [48, 49]. Our work has suggested that the fragmentation of metal complexes containing flavonoid 5-O-glucuronides mimics that of the 3-O-glucuronides (unpublished data). None of the spectra in Figure 2 display the –(Aux + GlcA) ion as the only abundant fragment, so both the 3-O- and 5-O-glycosylation positions are ruled out, further corroborating the initial identification of the five unknown compounds as 7-O-glucuronides.
HPLC retention time analysis
The enzymatic incubations of the other five flavonoids (luteolin, eriodictyol, quercetin, hesperetin, and myricetin) all produced more than one glucuronide derivative. The presence of multiple products means that retention time analysis can be used to assist in compound identification. Since this information plays a crucial role in identifying some of the glucuronidated products, a discussion of this approach is warranted before presenting the remaining mass spectral data.
There are several known trends regarding flavonoid structure and reversed-phase HPLC retention times. Since saccharides are more polar than flavonoid aglycons, flavonoid diglycosides elute before flavonoid monoglycosides, which elute before flavonoid aglycons [50]. It has also been reported that the retention time order is flavanones < flavonols < flavones, for compounds that otherwise have the same substitution patterns [50]. Furthermore, studies have shown that the identities of the saccharides influence HPLC mobility in predictable ways. For example, when all else is equal, a 7-rutinoside elutes before a 7-neohesperidoside, and a 3-galactoside elutes before a 3-glucoside [50]. Such trends are upheld in numerous articles which report reversed-phase HPLC retention times, regardless of the exact chromatographic method employed [30, 51–55].
We have observed that similar generalizations can be made in cases in which both the flavonoid portion and the glycosidic portion are the same, with only the glycosylation site differing between compounds. Table 1 lists the retention times for three groups of such isomers. In all three groups, the 7-O-glycoside elutes before the 4′-O-glycoside. When the 3-O-glycoside is included, it elutes between these two isomers. Finally, quercetin 3′-O-glucuronide elutes after quercetin 4′-O-glucuronide. Thus the retention times of such isomers can be ordered based on glycosylation site as 7 < 3 < 4′ < 3′. This elution order for quercetin monoglucuronides has been reported previously [12, 19, 56]. Regarding the rarer 5-O-glycosides, Grayer et al. reported that the 5-O-glucosides of luteolin and apigenin elute before their 7-O-glucoside analogs [57]. Harborne and Boardley report the retention time of quercetin 5-O-glucoside as lower than that of quercetin 3-O-glucoside [58]. We have also observed a case in which the 5-O-glucuronide, the 7-O-glucuronide and the 3-O-glucuronide of a flavonol eluted in that order (unpublished results). The evidence points towards a general overall elution order (by position of conjugation) of 5 < 7 < 3 < 4′ < 3′, at least for flavonoid glucosides and glucuronides.
Table 1.
HPLC retention times of selected isomeric flavonoid glycosides
| aglycon | linkage | saccharidea | retention time (min) |
|---|---|---|---|
| quercetin | 7-O- | Glc | 8.19 |
| quercetin | 3-O- | Glc | 9.04 |
| quercetin | 4′-O- | Glc | 9.82 |
| luteolin | 7-O- | Glc | 8.78 |
| luteolin | 4′-O- | Glc | 10.09 |
| quercetin | 7-O- | GlcA | 8.72 |
| quercetin | 3-O- | GlcA | 9.28 |
| quercetin | 4′-O- | GlcA | 10.46 |
| quercetin | 3′-O- | GlcA | 11.32 |
Glc=glucose, GlcA=glucuronic acid
The significance of deciphering this elution rule is that it may be used to confirm the identities of flavonoid glucosides or glucuronides that have been elucidated by tandem mass spectrometric methods. Moreover, an otherwise unidentifiable isomer may be assigned by retention time if the some of the other isomers have been identified. Both strategies will be applied to the identification of compounds from reactions that yielded multiple products.
Multi-product reactions
The same LC-MS negative ion mode screening strategy described earlier was also pursued for the reaction products of luteolin, eriodictyol, quercetin, hesperetin and myricetin. Two or three different glucuronides were identified in each elution profile based on the diagnostic loss of a glucuronic acid moiety (−176 Da) in the CID mass spectra. Once identified as glucuronides, these products were subjected to MS3 analysis to yield the aglycon-specific fragmentation patterns shown in Figure 3. The monoglucuronidated flavonoid isomers in each reaction mixture yielded almost identical MS3 profiles, though only the results from the earliest-eluting isomer are shown. All of these spectra matched the MS/MS spectra obtained from standards of the flavonoid aglycons.
Figure 3.
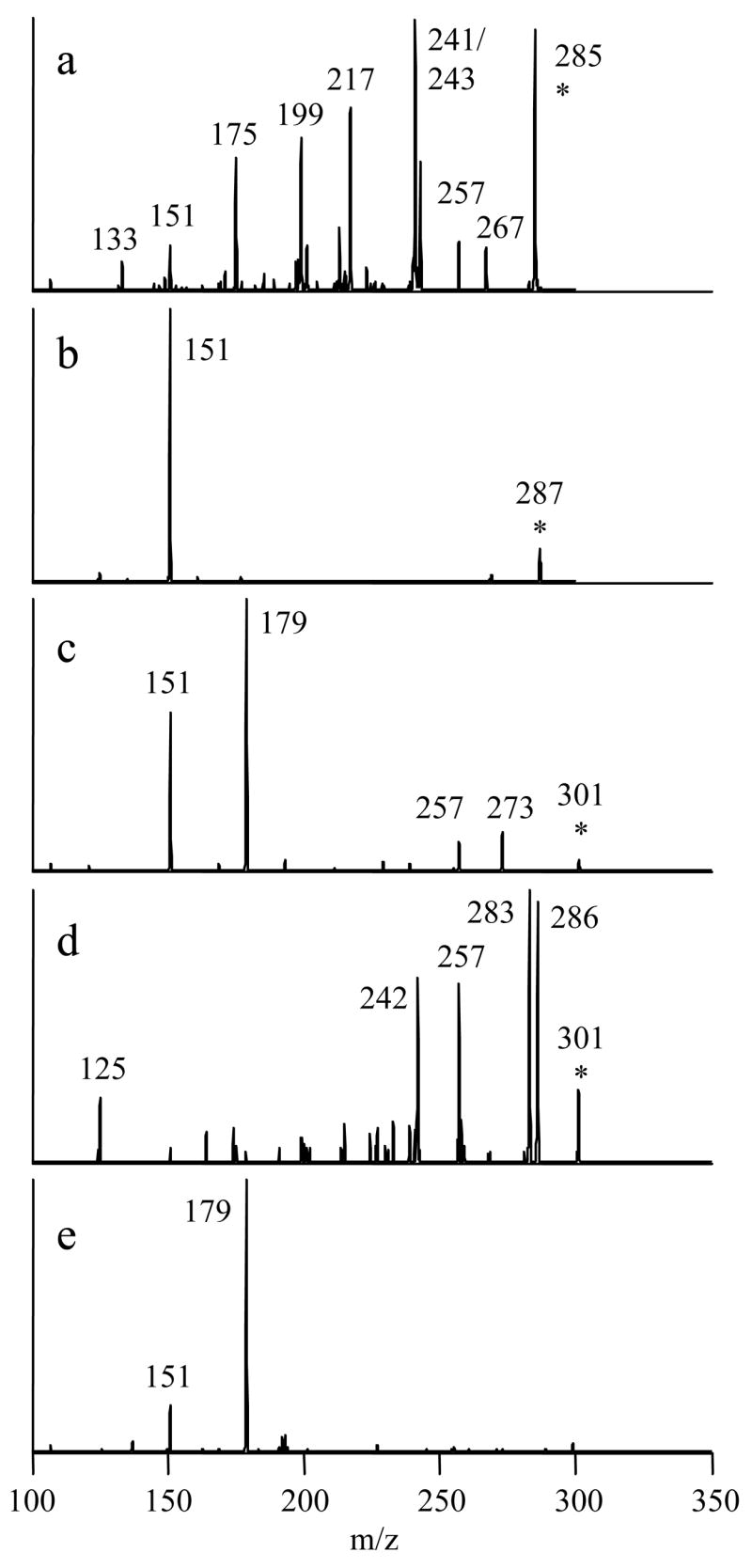
MSn spectra used to confirm the identities of the aglycon skeletons of deprotonated flavonoid monoglucuronides. a) luteolin monoglucuronide, 461→285→ b) eriodictyol monoglucuronide, 463→287→ c) quercetin monoglucuronide, 477→301→ 268→ d) hesperetin monoglucuronide, 477→301→ e) myricetin monoglucuronide, 493→317→.
After confirming that the reaction products were monoglucuronides and identifying the aglycon structures, CID of the [Co(II) (FG-H) (4,7-dpphen)2]+ complexes was used to identify the glucuronidation positions in a manner similar to that described above. The MS/MS fragmentation patterns of the earliest-eluting isomer from each mixture are shown in Figure 4, and the characteristic array of fragments indicate 7-O-glucuronidation in each case. The flavanone (eriodictyol and hesperetin) 7-O-glucuronide complexes again yield the highest abundance of the –Agl and several other fragment ions, while the flavone (luteolin) 7-O-glucuronide complex shows the lowest abundances of these fragment ions. The relative yield of 7-O-glucuronides was very low in the quercetin and myricetin reaction mixtures based on the intensities of these components in the chromatographic profiles, and 75 μL injections were required to obtain good mass spectra. The low signal intensity of myricetin 7-O-glucuronide resulted in a very noisy MS/MS spectrum from the complex, especially in the high mass range. The diagnostic –Agl fragment ion is indistinguishable from the noise. The identification can still be made on the basis of three fragment ions: -Aux, -GlcA and –(Aux + GlcA). Typically, only metal complexes of 7-O-glucuronides yield high abundances of all three of these ions, though flavonoid class also affects the abundances.
Figure 4.
CID mass spectra of [Co(II) (FG-H) (4,7-dpphen)2]+ complexes of earliest eluting flavonoid monoglucuronides from multi-product reactions. a) FG=luteolin, 1184→ b) FG=eriodictyol, 1186→ c) FG=quercetin, 1200→ d) FG=hesperetin, 1200→ e) FG=myricetin, 1216→.
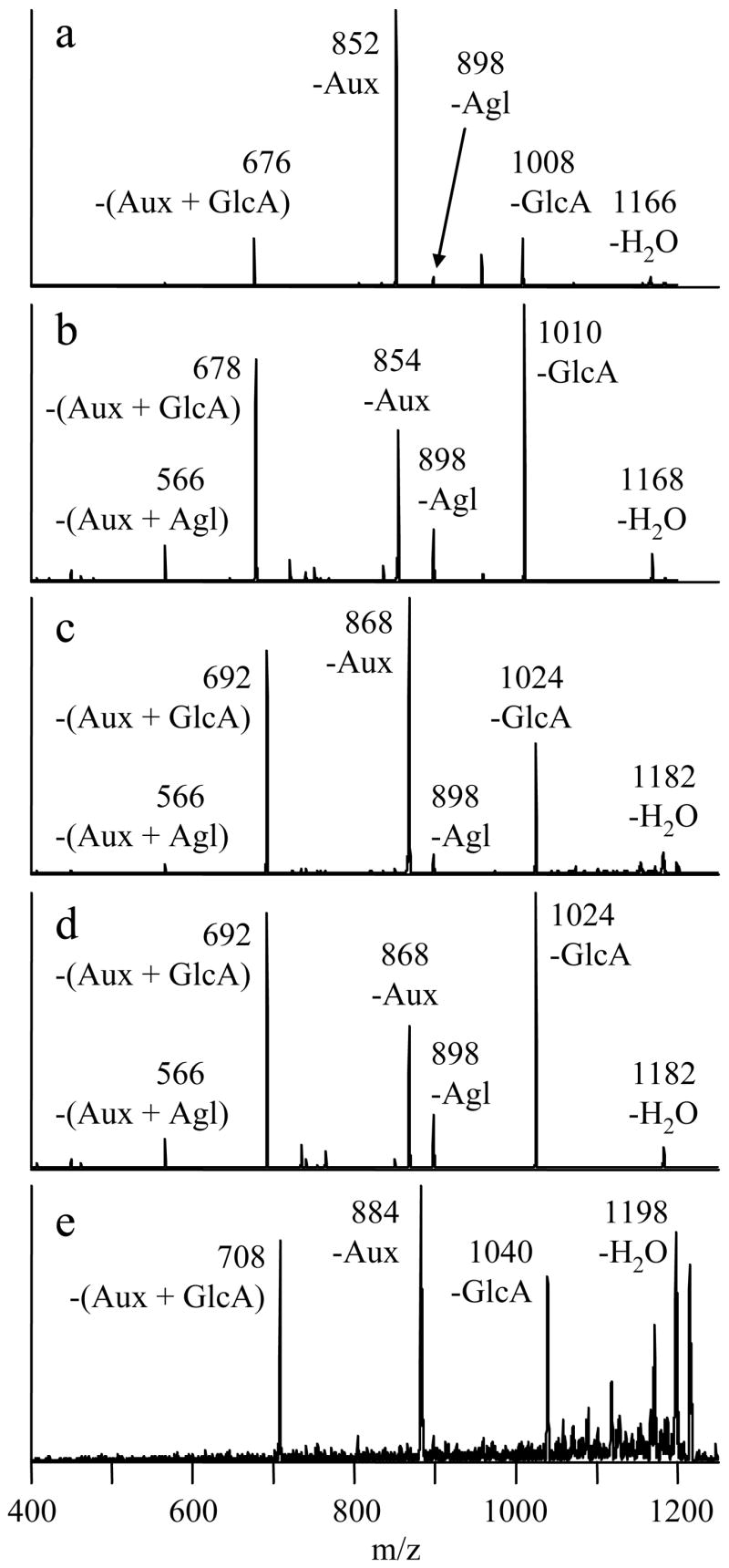
The enzymatic reactions involving eriodictyol and hesperetin yielded only one product in addition to the 7-O-glucuronide, while the remaining flavonoids produced two additional products. All of these products eluted after the respective 7-O-glucuronides. The CID mass spectra from the metal complexes of the latest-eluting isomers in each mixture are shown in Figure 5. These fragmentation patterns are indicative of B-ring-glucuronidation. As stated earlier, the two characteristic fragmentation pathways of complexes containing B-ring-glucuronides are –Aux and –(Aux + GlcA). The fragment ion abundances from these B-ring-glucuronide metal complexes appear to be partially dependent on the class of the flavonoid, as noted earlier for the 7-O-glucuronide complexes. While all of the spectra show both of the characteristic fragment ions of B-ring-glucuronides, the metal complex of the flavone (luteolin) B-ring-glucuronide strongly favors the loss of the auxiliary ligand, while the complexes of the flavanone (eriodictyol and hesperetin) B-ring-glucuronides preferentially yield the –(Aux + GlcA) ion. The flavonol (quercetin and myricetin) B-ring-glucuronide complexes display intermediate behavior, yielding similar amounts of each of these two fragment ions. Hence, knowledge of the flavonoid class helps in the interpretation of the fragmentation patterns of these metal complexes.
Figure 5.
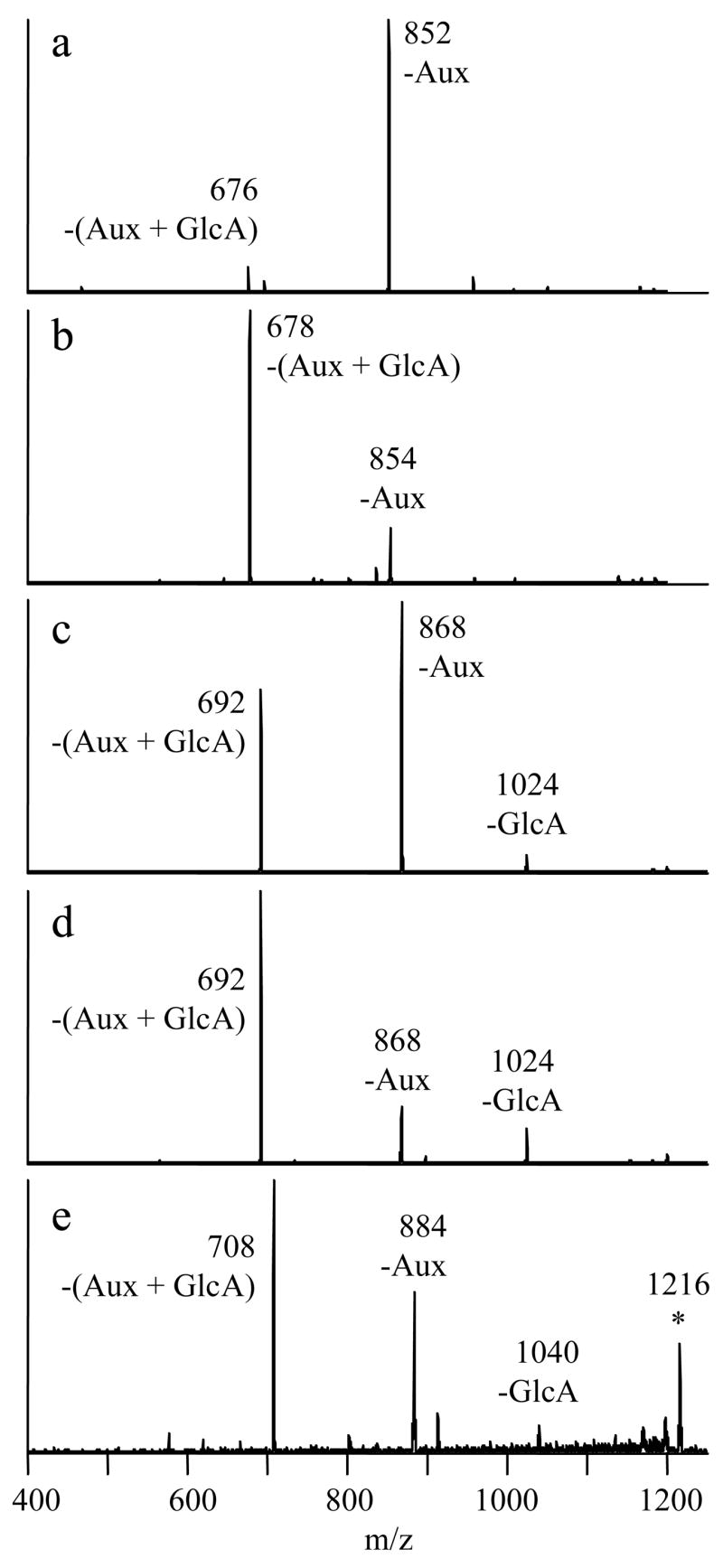
CID mass spectra of [Co(II) (FG-H) (4,7-dpphen)2]+ complexes of latest eluting flavonoid monoglucuronides from multi-product reactions. a) FG=luteolin, 1184→ b) FG=eriodictyol, 1186→ c) FG=quercetin, 1200→ d) FG=hesperetin, 1200→ e) FG=myricetin, 1216→.
Some of the spectra also contain small amounts of the –GlcA fragment ion, which is usually associated with 7-O-glucuronidation. For the hesperetin glucuronide data in Figure 5d, this is likely a contribution from the earlier-eluting hesperetin 7-O-glucuronide, as the two compounds were only partially separated. However, the -GlcA fragment ion appears to be an intrinsic part of the spectrum for the complexes of the quercetin and myricetin B-ring-glucuronides because there are no co-eluting isomers in these cases. A reasonable guideline is that the MS/MS of flavone 7-O-glucuronide complexes displays at least 15% relative abundance of the –GlcA fragment ion (Figures 2a, 2c and 4a), but this fragment ion is absent from the MS/MS of flavone B-ring-glucuronide complexes (Figure 5a). For flavonols, the same product ion appears with ~50% relative abundance for the 7-O-glucuronides (Figures 2d, 4c and 4e), but with ~10% relative abundance for B-ring-glucuronides (Figures 5c and 5e). Since product ion distributions are dependent on collision energy [31], these guidelines are only applicable to the conditions used in this set of experiments.
The collision energy used in the current set of experiments was specifically chosen to take advantage of a subtle difference in the fragmentation of the complexes of quercetin 4′-O-glucuronide and quercetin 3′-O-glucuronide [31]. When a collision voltage in the range of 1.5–1.8 V was used [31], it was noted that a greater abundance of the –Aux fragment ion relative to the –(Aux + GlcA) fragment ion, as occurs in Figure 5c, is indicative of quercetin 3′-O-glucuronide. If the relative abundances are reversed, this is characteristic of quercetin 4′-O-glucuronide; and this was observed for an isomeric compound in the quercetin reaction mixture that eluted just prior to quercetin 3′-O-glucuronide (data not shown). The retention time guidelines also support the identification of these two products as quercetin 4′-O-glucuronide and quercetin 3′-O-glucuronide, in order of elution.
There are no known rules for differentiating the 3′-O- and 4′-O-glucuronides of flavonoids other than quercetin. Hence other means are required to precisely identify the remaining B-ring-glucuronides. The hesperetin B-ring-glucuronide is easiest to assign. Hesperetin has only one B ring hydroxyl group, so the unknown compound must be hesperetin 3′-O-glucuronide. Like quercetin, both the luteolin and myricetin reaction mixtures also display two glucuronide species that elute after the 7-O-glucuronide. As luteolin has two B ring hydroxyl groups, the two unknowns are assigned as luteolin 4′-O-glucuronide and luteolin 3′-O-glucuronide, in order of elution. These assignments were possible despite the failure of luteolin 4′-O-glucuronide to form significant amounts of the metal complex in question, for reasons that remain unknown. Both late-eluting myricetin glucuronides form complexes which dissociate to provide the same two major fragment ions shown in Figure 5e. Myricetin has three B ring hydroxyl groups but two are equivalent, so the two isomers are identified as myricetin 4′-O-glucuronide and myricetin 3′-O-glucuronide, in order of elution. The eriodictyol derivative is the most difficult to assign because there are two B ring hydroxyl groups but only one B-ring-glucuronide is formed. While it is impossible to precisely identify this compound based on the available data, it is hypothesized to be eriodictyol 3′-O-glucuronide, not eriodictyol 4′-O-glucuronide, for reasons that will be explained in the next section.
An estimate of the relative product yields of these multi-product reactions was made based on the peak areas of the initial negative ion mode MS scans from the chromatographic profiles (Table 2). Some of our results can be compared with data presented by Boersma et al. [19], who reported product distributions for reactions of luteolin and quercetin with UGT1A1 based on HPLC, LC-MS and NMR spectroscopy. Their observed product distribution was 21:29:50 for the 7-O-, 3′-O- and 4′-O-glucuronides of luteolin, which is similar to our results (10:30:60). The distribution for quercetin was 6:85:9, which also parallels our data (trace:90:10). As in the current study, quercetin 3-O-glucuronide was not observed as a reaction product.
Table 2.
Estimated product distributions from multi-product reactions
| aglycon | % 7-O-GlcA | % 3′-O-GlcA | % 4′-O-GlcA |
|---|---|---|---|
| eriodictyol | 40 | 60a | — |
| hesperetin | 70 | 30 | — |
| luteolin | 10 | 30 | 60 |
| quercetin | trace | 90 | 10 |
| myricetin | trace | 70 | 30 |
All values were rounded to the nearest 10%.
the compound may possibly be eriodictyol 4′-O-glucuronide
Structure-activity relationships
The flavonoids that produce a single monoglucuronidated product and those which produce multiple products constitute two distinct groups, distinguished by the presence or the absence of a 3′ hydroxyl group. The sole reaction product for compounds that lack this structural feature is the 7-O-glucuronide. The regioselectivity of UGT1A1 shifts toward the flavonoid B ring when a 3′ hydroxyl group is present, but the extent to which this happens depends on other structural features of the flavonoid. The two flavanones with a 3′ hydroxyl group, eriodictyol and hesperetin, form only one product in addition to the 7-O-glucuronide. The additional product of hesperetin was identified as the 3′-O-glucuronide, but the additional product of eriodictyol could not be conclusively determined. However, the important activating properties of the 3′ hydroxyl group suggest that eriodictyol 3′-O-glucuronide is the product that is formed. For both eriodictyol and hesperetin, there remains a significant yield of the 7-O-glucuronide (relative yields of 40% and 70%, respectively). Luteolin is the only flavone in the study that features a 3′ hydroxyl group. There is a larger shift toward B-ring-glucuronidation as luteolin 7-O-glucuronide accounts for only about 10% of the product. For the flavonols containing the 3′ hydroxyl group (quercetin and myricetin), only trace amounts of their 7-O-glucuronides were formed. Hence, while the presence or absence of the 3′ hydroxyl group is the major determinant of the regioselectivity of glucuronidation, there are secondary effects due to flavonoid class.
The structural features that govern the regioselectivity of the glucuronidation reaction (the presence or absence of the 3′ hydroxyl group, the C2–C3 double bond, and the 3 hydroxyl group) are also important factors in the antioxidant capability of flavonoids, and may act synergistically in that capacity as well [59]. The same structural features have also been cited as important determinants of the antiproliferative effects of flavonoids against several cancer cell lines [60]. It is interesting to note that while the presence of the ortho-catechol group (i.e. the 3′ and 4′ hydroxyl groups) is often claimed to be a source of beneficial flavonoid activity, our work shows a tendency for this group to be preferentially metabolized by UGT1A1, which would potentially mitigate the bioactivity of these molecules. On the other hand, the B-ring-glucuronides of quercetin retain the inhibitory effects of the aglycon against xanthine oxidase and lipoxygenase better than when quercetin is conjugated at other positions [17].
Conclusions
A three-part tandem mass spectrometry strategy that entails MS/MS analysis of deprotonated metabolites to pinpoint those that are glucuronides, then MS3 analysis to elucidate the specific aglycon structure, then MS/MS analysis of metal complexes of the type [Co(II) (FG-H) (4,7-dpphen)2]+ allows confident identification of enzymatically-synthesized flavonoid glucuronides, including the conjugation site. This strategy was used to determine the regioselectivity of UGT1A1 toward flavonol, flavone and flavanone substrates. The 7 position of the flavonoid is the sole site of glucuronidation except when a 3′ hydroxyl group is present. A 3′ hydroxyl group promotes the formation of B-ring-glucuronides in addition to 7-O-glucuronides. The flavonoid class determines the product distribution in such cases. This result represents a significant advance in understanding flavonoid metabolism. Studies involving other UGT isozymes are planned. Furthermore, we have demonstrated the success of metal complexation methods in the identification of new flavonoid monoglucuruonides with the assistance of HPLC retention time analysis. Of the eighteen flavonoid monoglucuronides produced, only one remains ambiguous (the eriodictyol B-ring-glucuronide). Insights regarding the effect of flavonoid class on the fragmentation of the metal complexes will prove useful for future applications of this method.
Supplementary Material
Acknowledgments
This work was supported by the Welch Foundation (F1155) and the NIH (R01-GM63512). We thank Paul Needs and Paul Kroon for the donation of synthetic quercetin glucuronides, and Michelle Lane, Erik Wilder and Kally O’Reilly of the UT-Austin Dept. of Human Ecology (ML, EW) and the Institute for Cellular and Molecular Biology (KO) for invaluable advice and assistance in the enzymatic synthesis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Manach C, Donovan JL. Pharmacokinetics and Metabolism of Dietary Flavonoids in Humans. Free Radic Res. 2004;38:771–785. doi: 10.1080/10715760410001727858. [DOI] [PubMed] [Google Scholar]
- 2.Williamson G, Day AJ, Plumb GW, Couteau D. Human Metabolic Pathways of Dietary Flavonoids and Cinnamates. Biochem Soc Trans. 2000;28:16–22. doi: 10.1042/bst0280016. [DOI] [PubMed] [Google Scholar]
- 3.Prasain JK, Wang CC, Barnes S. Mass Spectrometric Methods for the Determination of Flavonoids in Biological Samples. Free Radic Biol Med. 2004;37:1324–1350. doi: 10.1016/j.freeradbiomed.2004.07.026. [DOI] [PubMed] [Google Scholar]
- 4.Hong YJ, Mitchell AE. Identification of Glutathione-Related Quercetin Metabolites in Humans. Chem Res Toxicol. 2006;19:1525–1532. doi: 10.1021/tx0601758. [DOI] [PubMed] [Google Scholar]
- 5.Williamson G, Barron D, Shimoi K, Terao J. In Vitro Biological Properties of Flavonoid Conjugates Found in Vivo. Free Radic Res. 2005;39:457–469. doi: 10.1080/10715760500053610. [DOI] [PubMed] [Google Scholar]
- 6.Wittig J, Herderich M, Graefe EU, Veit M. Identification of Quercetin Glucuronides in Human Plasma by High-Performance Liquid Chromatography-Tandem Mass Spectrometry. J Chromatogr B. 2001;753:237–243. doi: 10.1016/s0378-4347(00)00549-1. [DOI] [PubMed] [Google Scholar]
- 7.Day AJ, Williamson G. Biomarkers for Exposure to Dietary Flavonoids: A Review of the Current Evidence for Identification of Quercetin Glycosides in Plasma. Br J Nutr. 2001;86:S105–S110. doi: 10.1079/bjn2001342. [DOI] [PubMed] [Google Scholar]
- 8.Sesink ALA, O’leary KA, Hollman PCH. Quercetin Gluouronides but Not Glucosides Are Present in Human Plasma after Consumption of Quercetin-3-Glucoside or Quercetin-4′-Glucoside. J Nutr. 2001;131:1938–1941. doi: 10.1093/jn/131.7.1938. [DOI] [PubMed] [Google Scholar]
- 9.Paganga G, Rice-Evans CA. The Identification of Flavonoids as Glycosides in Human Plasma. FEBS Lett. 1997;401:78–82. doi: 10.1016/s0014-5793(96)01442-1. [DOI] [PubMed] [Google Scholar]
- 10.Aziz AA, Edwards CA, Lean MEJ, Crozier A. Absorption and Excretion of Conjugated Flavonols, Including Quercetin-4′-O-Beta-Glucoside and Isorhamnetin-4′-O-Beta-Glucoside by Human Volunteers after the Consumption of Onions. Free Radic Res. 1998;29:257–269. doi: 10.1080/10715769800300291. [DOI] [PubMed] [Google Scholar]
- 11.Boyle SP, Dobson VL, Duthie SJ, Kyle JAM, Collins AR. Absorption and DNA Protective Effects of Flavonoid Glycosides from an Onion Meal. Eur J Nutr. 2000;39:213–223. doi: 10.1007/s003940070014. [DOI] [PubMed] [Google Scholar]
- 12.Day AJ, Mellon F, Barron D, Sarrazin G, Morgan MRA, Williamson G. Human Metabolism of Dietary Flavonoids: Identification of Plasma Metabolites of Quercetin. Free Radic Res. 2001;35:941–952. doi: 10.1080/10715760100301441. [DOI] [PubMed] [Google Scholar]
- 13.Mullen W, Edwards CA, Crozier A. Absorption, Excretion and Metabolite Profiling of Methyl-, Glucuronyl-, Glucosyl- and Sulpho-Conjugates of Quercetin in Human Plasma and Urine after Ingestion of Onions. Br J Nutr. 2006;96:107–116. doi: 10.1079/bjn20061809. [DOI] [PubMed] [Google Scholar]
- 14.Manach C, Morand C, Gil-Izquierdo A, Bouteloup-Demange C, Remesy C. Bioavailability in Humans of the Flavanones Hesperidin and Narirutin after the Ingestion of Two Doses of Orange Juice. Eur J Clin Nutr. 2003;57:235–242. doi: 10.1038/sj.ejcn.1601547. [DOI] [PubMed] [Google Scholar]
- 15.Mullen W, Graf BA, Caldwell ST, Hartley RC, Duthie GG, Edwards CA, Lean MEJ, Crozier A. Determination of Flavonol Metabolites in Plasma and Tissues of Rats by HPLC-Radiocounting and Tandem Mass Spectrometry Following Oral Ingestion of [2-C-14]Quercetin-4′ Glucoside. J Agric Food Chem. 2002;50:6902–6909. doi: 10.1021/jf020598p. [DOI] [PubMed] [Google Scholar]
- 16.Walle T, Walle UK, Halushka PV. Carbon Dioxide Is the Major Metabolite of Quercetin in Humans. J Nutr. 2001;131:2648–2652. doi: 10.1093/jn/131.10.2648. [DOI] [PubMed] [Google Scholar]
- 17.Day AJ, Bao YP, Morgan MRA, Williamson G. Conjugation Position of Quercetin Glucuronides and Effect on Biological Activity. Free Radic Biol Med. 2000;29:1234–1243. doi: 10.1016/s0891-5849(00)00416-0. [DOI] [PubMed] [Google Scholar]
- 18.Abe K, Katayama H, Suzuki A, Yumioka E. Biological Fate of Orally Administrede Naringin and Naringenin in Rats. Japan J Pharmacogn. 1993;47:402. [Google Scholar]
- 19.Boersma MG, Van Der Woude H, Bogaards J, Boeren S, Vervoort J, Cnubben NHP, Van Iersel MLPS, Van Bladeren PJ, Rietjens IMCM. Regioselectivity of Phase II Metabolism of Luteolin and Quercetin by UDP-Glucuronosyl Transferases. Chem Res Toxicol. 2002;15:662–670. doi: 10.1021/tx0101705. [DOI] [PubMed] [Google Scholar]
- 20.Mabry TJ, Markham KR, Thomas MB. The Systematic Identification of Flavonoids. Springer-Verlag; New York: 1970. pp. 35–229. [Google Scholar]
- 21.Matsumoto H, Ikoma Y, Sugiura M, Yano M, Hasegawa Y. Identification and Quantification of the Conjugated Metabolites Derived from Orally Administered Hesperidin in Rat Plasma. J Agric Food Chem. 2004;52:6653–6659. doi: 10.1021/jf0491411. [DOI] [PubMed] [Google Scholar]
- 22.Needs PW, Kroon PA. Convenient Syntheses of Metabolically Important Quercetin Glucuronides and Sulfates. Tetrahedron. 2006;62:6862–6868. [Google Scholar]
- 23.Satterfield M, Brodbelt JS. Structural Characterization of Flavonoid Glycosides by Collisionally Activated Dissociation of Metal Complexes. J Am Soc Mass Spectrom. 2001;12:537–549. doi: 10.1016/S1044-0305(01)00236-7. [DOI] [PubMed] [Google Scholar]
- 24.Pikulski M, Brodbelt JS. Differentiation of Flavonoid Glycoside Isomers by Using Metal Complexation and Electrospray Ionization Mass Spectrometry. J Am Soc Mass Spectrom. 2003;14:1437–1453. doi: 10.1016/j.jasms.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 25.Davis BD, Brodbelt JS. Determination of the Glycosylation Site of Flavonoid Monoglucosides by Metal Complexation and Tandem Mass Spectrometry. J Am Soc Mass Spectrom. 2004;15:1287–1299. doi: 10.1016/j.jasms.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 26.Davis BD, Brodbelt JS. LC-MSn Methods for Saccharide Characterization of Monoglycosyl Flavonoids Using Postcolumn Manganese Complexation. Anal Chem. 2005;77:1883–1890. doi: 10.1021/ac048374o. [DOI] [PubMed] [Google Scholar]
- 27.Zhang JM, Brodbelt JS. Silver Complexation and Tandem Mass Spectrometry for Differentiation of Isomeric Flavonoid Diglycosides. Anal Chem. 2005;77:1761–1770. doi: 10.1021/ac048818g. [DOI] [PubMed] [Google Scholar]
- 28.Zhang JM, Wang JM, Brodbelt JS. Characterization of Flavonoids by Aluminum Complexation and Collisionally Activated Dissociation. J Mass Spectrom. 2005;40:350–363. doi: 10.1002/jms.793. [DOI] [PubMed] [Google Scholar]
- 29.Pikulski M, Aguilar A, Brodbelt JS. Tunable Transition Metal-Ligand Complexation for Enhanced Elucidation of Flavonoid Diglycosides by Electrospray Ionization Mass Spectrometry. J Am Soc Mass Spectrom. 2007;18:422–431. doi: 10.1016/j.jasms.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 30.Wojcinska M, Williams J, Mabry TJ, Ahmed AA, Davis BD, Toth G, El-Sayed NH, Matlawska I, Clevinger J. Flavonol Triglycosides from the Leaves of Silphium Albiflorum. Natural Product Communications. 2006;1:941–948. [Google Scholar]
- 31.Davis BD, Needs PW, Kroon PA, Brodbelt JS. Identification of Isomeric Flavonoid Glucuronides in Urine and Plasma by Metal Complexation and LC-ESI-MS/MS. J Mass Spectrom. 2006;41:911–920. doi: 10.1002/jms.1050. [DOI] [PubMed] [Google Scholar]
- 32.Ebner T, Burchell B. Substrate Specificities of Two Stably Expressed Human Liver UDP-Glucuronosyltransferases of the UGT1 Gene Family. Drug Metab Dispos. 1993;21:50–55. [PubMed] [Google Scholar]
- 33.Green MD, Oturu EM, Tephly TR. Stable Expression of a Human Liver UDP-Glucuronosyltransferase (UGT2B15) with Activity toward Steroid and Xenobiotic Substrates. Drug Metab Dispos. 1994;22:799–805. [PubMed] [Google Scholar]
- 34.King CD, Green MD, Rios GR, Coffman BL, Owens IS, Bishop WP, Tephly TR. The Glucuronidation of Exogenous and Endogenous Compounds by Stably Expressed Rat and Human UDP-Glucuronosyltransferase 1.1. Arch Biochem Biophys. 1996;332:92–100. doi: 10.1006/abbi.1996.0320. [DOI] [PubMed] [Google Scholar]
- 35.Green MD, Tephly TR. Glucuronidation of Amine Substrates by Purified and Expressed UDP-Glucuronosyltransferase Proteins. Drug Metab Dispos. 1998;26:860–867. [PubMed] [Google Scholar]
- 36.Cheng ZQ, Radominska-Pandya A, Tephly TR. Studies on the Substrate Specificity of Human Intestinal UDP-Glucuronosyltransferases 1A8 and 1A10. Drug Metb Dispos. 1999;27:1165–1170. [PubMed] [Google Scholar]
- 37.King CD, Rios GR, Green MD, Tephly TR. UDP-Glucuronosyltransferases. Curr Drug Metab. 2000;1:143–161. doi: 10.2174/1389200003339171. [DOI] [PubMed] [Google Scholar]
- 38.Vaidyanathan JB, Walle T. Glucuronidation and Sulfation of the Tea Flavonoid (-)-Epicatechin by the Human and Rat Enzymes. Drug Metab Dispos. 2002;30:897–903. doi: 10.1124/dmd.30.8.897. [DOI] [PubMed] [Google Scholar]
- 39.Zhang L, Lin G, Zuo Z. Position Preference on Glucuronidation of Mono-Hydroxylflavones in Human Intestine. Life Sci. 2006;78:2772–2780. doi: 10.1016/j.lfs.2005.10.038. [DOI] [PubMed] [Google Scholar]
- 40.Plumb GW, O’leary K, Day AJ, Williamson G. Enzymatic Synthesis of Quercetin Glucosides and Glucuronides. In: Santos-Buelga C, Williamson G, editors. Methods in Polyphenol Analysis. The Royal Society of Chemistry; Cambridge: 2003. pp. 177–186. [Google Scholar]
- 41.Mcclellan JE, Murphy JP, Mulholland JJ, Yost RA. Effects of Fragile Ions on Mass Resolution and on Isolation for Tandem Mass Spectrometry in the Quadrupole Ion Trap Mass Spectrometer. Anal Chem. 2002;74:402–412. doi: 10.1021/ac015610b. [DOI] [PubMed] [Google Scholar]
- 42.Lopez LL, Tiller PR, Senko MW, Schwartz JC. Automated Strategies for Obtaining Standardized Collisionally Induced Dissociation Spectra on a Benchtop Ion Trap Mass Spectrometer. Rapid Commun Mass Spectrom. 1999;13:663–668. [Google Scholar]
- 43.Davis BD, Kroon PA, Brodbelt JS. In Vivo Human Metabolites of Flavonols and Flavanones. 55th ASMS Conference on Mass Spectrometry and Allied Topics; Indianapolis. 2007. [Google Scholar]
- 44.Ma YL, Li QM, Van den Heuvel H, Claeys M. Characterization of Flavone and Flavonol Aglycones by Collision-Induced Dissociation Tandem Mass Spectrometry. Rapid Commun Mass Spectrom. 1997;11:1357–1364. [Google Scholar]
- 45.Justesen U. Collision-Induced Fragmentation of Deprotonated Methoxylated Flavonoids, Obtained by Electrospray Ionization Mass Spectrometry. J Mass Spectrom. 2001;36:169–178. doi: 10.1002/jms.118. [DOI] [PubMed] [Google Scholar]
- 46.Fabre N, Rustan I, De Hoffmann E, Quetin-Leclercq J. Determination of Flavone, Flavonol, and Flavanone Aglycones by Negative Ion Liquid Chromatography Electrospray Ion Trap Mass Spectrometry. J Am Soc Mass Spectrom. 2001;12:707–715. doi: 10.1016/S1044-0305(01)00226-4. [DOI] [PubMed] [Google Scholar]
- 47.Cuyckens F, Claeys M. Mass Spectrometry in the Structural Analysis of Flavonoids. J Mass Spectrom. 2004;39:1–15. doi: 10.1002/jms.585. [DOI] [PubMed] [Google Scholar]
- 48.Youdim KA, Dobbie MS, Kuhnle G, Proteggente AR, Abbott NJ, Rice-Evans C. Interaction between Flavonoids and the Blood-Brain Barrier: In Vitro Studies. J Neurochem. 2003;85:180–192. doi: 10.1046/j.1471-4159.2003.01652.x. [DOI] [PubMed] [Google Scholar]
- 49.Proteggente AR, Basu-Modak S, Kuhnle G, Gordon MJ, Youdim K, Tyrrell R, Rice-Evans CA. Hesperetin Glucuronide, a Photoprotective Agent Arising from Flavonoid Metabolism in Human Skin Fibroblasts. Photochem Photobiol. 2003;78:256–261. doi: 10.1562/0031-8655(2003)078<0256:hgapaa>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 50.Robards K, Antolovich M. Analytical Chemistry of Fruit Bioflavonoids - a Review. Analyst. 1997;122:11R–34R. [Google Scholar]
- 51.Schieber A, Keller P, Streker P, Klaiber I, Carle R. Detection of Isorhamnetin Glycosides in Extracts of Apples (Malus Domestica Cv. “Brettacher”) by HPLC-PDA and HPCL-APCI-MS/MS. Phytochem Anal. 2002;13:87–94. doi: 10.1002/pca.630. [DOI] [PubMed] [Google Scholar]
- 52.De Rijke E, Zappey H, Ariese F, Gooijer C, Brinkman UAT. Liquid Chromatography with Atmospheric Pressure Chemical Ionization and Electrospray Ionization Mass Spectrometry of Flavonoids with Triple-Quadrupole and Ion-Trap Instruments. J Chromatogr A. 2003;984:45–58. doi: 10.1016/s0021-9673(02)01868-x. [DOI] [PubMed] [Google Scholar]
- 53.Sanchez-Rabaneda F, Jauregui O, Lamuela-Raventos RM, Viladomat F, Bastida J, Codina C. Qualitative Analysis of Phenolic Compounds in Apple Pomace Using Liquid Chromatography Coupled to Mass Spectrometry in Tandem Mode. Rapid Commun Mass Spectrom. 2004;18:553–563. doi: 10.1002/rcm.1370. [DOI] [PubMed] [Google Scholar]
- 54.Dugo P, Lo Presti M, Ohman M, Fazio A, Dugo G, Mondello L. Determination of Flavonoids in Citrus Juices by Micro-HPLC-ESI/MS. J Sep Sci. 2005;28:1149–1156. doi: 10.1002/jssc.200500053. [DOI] [PubMed] [Google Scholar]
- 55.Lin LZ, Harnly JM. A Screening Method for the Identification of Glycosylated Flavonoids and Other Phenolic Compounds Using a Standard Analytical Approach for All Plant Materials. J Agric Food Chem. 2007;55:1084–1096. doi: 10.1021/jf062431s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mullen W, Boitier A, Stewart AJ, Crozier A. Flavonoid Metabolites in Human Plasma and Urine after the Consumption of Red Onions: Analysis by Liquid Chromatography with Photodiode Array and Full Scan Tandem Mass Spectrometric Detection. J Chromatogr A. 2004;1058:163–168. [PubMed] [Google Scholar]
- 57.Grayer RJ, Kite GC, Veitch NC, Eckert MR, Marin PD, Senanayake P, Paton AJ. Leaf Flavonoid Glycosides as Chemosystematic Characters in Ocimum. Biochem Syst Ecol. 2002;30:327–342. [Google Scholar]
- 58.Harborne JB, Boardley M. Use of High-Performance Liquid-Chromatography in the Separation of Flavonol Glycosides and Flavonol Sulfates. J Chromatogr. 1984;299:377–385. [Google Scholar]
- 59.Rice-Evans CA, Miller NJ, Paganga G. Structure-Antioxidant Activity Relationships of Flavonoids and Phenolic Acids. Free Radic Biol Med. 1996;20:933–956. doi: 10.1016/0891-5849(95)02227-9. [DOI] [PubMed] [Google Scholar]
- 60.Kawaii S, Tomono Y, Katase E, Ogawa K, Yano M. Antiproliferative Activity of Flavonoids on Several Cancer Cell Lines. Biosci Biotech Biochem. 1999;63:896–899. doi: 10.1271/bbb.63.896. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.