

Abstract
α1-Adrenoceptors and extracellular signal-regulated kinases 1 and 2 (ERK1/2) regulate salivary secretion. However, whether α1-adrenoceptors couple to ERK1/2 activation and the specific α1-adrenoceptor subtypes involved in salivary glands is unknown. Western blotting of ERK1/2 phosphorylation showed phenylephrine activated ERK1/2 by 2-3-fold in submandibular gland slices and 3-4-fold in submandibular acinar (SMG-C10) cells with an EC50 of 2.7 ± 2 μM. ERK1/2 activation was blocked by either prazosin or HEAT, indicating α1-adrenoceptors stimulate ERK1/2 in native glands and SMG-C10 cells. Inhibition of [125I]HEAT binding by 5-methylurapidil (selective for α1A over α1B/α1D), but not BMY 7378 (selective for α1D over α1A/α1B), was biphasic and best-fit by a two-site binding model with KiH and KiL values for 5-methylurapidil of 0.64 ± 0.3 and 91 ± 7 nM, respectively, in SMG-C10 membranes. From these binding data, we obtained subtype-selective concentrations of 5-methylurapidil to determine the α1-adrenoceptor subtype/s activating ERK1/2 in SMG-C10 cells. 5-methylurapidil (20 nM) did not affect phenylephrine- or A-61603- (α1A-selective agonist) induced ERK1/2 activation; whereas, 30 μM chloroethylclonidine (α1B-selective antagonist) inhibited ERK1/2 activation by phenylephrine, indicating α1B-adrenoceptors, but not α1A-adrenoceptors, activate ERK1/2 in submandibular cells. We also examined α1-adrenoceptor location and dependence on cholesterol-rich microdomains for activating ERK1/2. Sucrose density gradient centrifugation showed 71 ± 3% of α1-adrenoceptor binding sites were in plasma membranes. Cholesterol-disrupting agents filipin and methyl-β-cyclodextrin inhibited phenylephrine-stimulated ERK1/2. These results show only α1B-adrenoceptors activate ERK1/2 and suggest subtype-specific ERK1/2 signaling by α1B-adrenoceptors may be determined by localization to cholesterol-rich microdomains in submandibular cells.
Keywords: α1-adrenoceptor subtypes, extracellular signal-regulated kinase, submandibular gland acinar cells
1. Introduction
The major salivary glands of humans and other mammals include the sublingual, parotid and submandibular glands, which produce fluid, electrolytes, proteins and mucins that ultimately comprise saliva in the oral cavity. Saliva serves to protect the oral mucosa and teeth, lubricate the throat for easy swallowing, and inhibit microbial overgrowth of the oral cavity. The importance of saliva is clearly demonstrated by the deterioration of the mucous membranes of the mouth and throat, leading at times to serious complications in individuals suffering with dysfunctional salivary glands caused by radiation therapy for head neck cancers (Vissink et al., 2003). For the oral cavity to function properly, not only must sufficient amounts of saliva be produced, but also the components, i.e. mucins and proteins, of saliva must be secreted in appropriate proportions to maintain oral homeostasis (Kaplan and Baum, 1993).
A significant proportion of daily saliva production originates in the acinar cells of the submandibular gland. The secretory response of submandibular acinar cells is partially regulated by the sympathetic division of the autonomic nervous system. Stimulation of sympathetic nerves causes release of noradrenaline, which activates α1-adrenoceptors on submandibular acinar cells, eliciting a viscous secretion high in mucins (Quissell and Barzen, 1980). Classical signaling pathways coupling α1-adrenoceptors to secretion involve the formation of inositol 1,4,5-trisphosphate, which then stimulates an elevation of intracellular free calcium. Subsequent activation of Ca2+-dependent Cl− channels results in Cl− efflux that is the driving force for fluid secretion (Martinez and Reed, 1988). In addition, elevated intracellular free calcium also modulates exocytotic secretion of proteins and mucins (Quissell et al., 1992).
More recently, the extracellular signal-regulated kinases 1 and 2 (ERK1/2), which are members of the mitogen-activated protein (MAP) kinase superfamily, have also been found to have a role in the signal transduction pathways that control secretion. For example, ERK1/2 affects the expression of salivary proteins (Jung et al., 2000), and salivary gland ion (Zentner et al., 1998) and water (Hoffert et al., 2000) channels. Additionally, ERK1/2 modulates the activity of salivary Na+-K+-ATPase (Plourde and Soltoff, 2006) and salivary mucin secretion (Slomiany and Slomiany, 2004). Taken together, these results suggest an important role for ERK1/2 in regulating the production and composition of saliva. However, whether α1-adrenoceptors are an important trigger for activation of ERK1/2 in submandibular acinar cells is unknown.
α1-Adrenoceptors are not a homogenous population of receptors, but consist of three structurally and pharmacologically distinct subtypes referred to as the α1A-, α1B-, and α1D-adrenoceptors (Hieble et al., 1995). All three of the α1-adrenoceptor subtypes have been shown to couple to ERK1/2 activation in a variety of cell types (Zhong and Minneman, 1999; Hague et al., 2002; Jiao et al., 2002), although the physiological functions of this pathway are not well understood. Recently, Huang et al. (2007) reported in cardiomyocytes that α1-adrenoceptors activate an anti-apoptotic, cell survival pathway by stimulation of ERK 1/2. We have reported that both the native submandibular gland and an immortalized acinar cell line (SMG-C10) cloned from submandibular glands similarly express both the α1A- and α1B-adrenoceptor subtypes (Bockman et al., 2004). Like native acinar cells, SMG-C10 cells are polarized epithelia, express functional adrenergic, muscarinic and purinergic receptors that couple to an elevation in intracellular calcium, and share other morphological characteristics unique to secretory epithelia (Quissell et al., 1997; Liu et al., 2000). Taken in their entirety, these reports show that the SMG-C10 cell line is an important new tool for examining the role of α1A- and α1B-adrenoceptor subtypes in regulating submandibular gland acinar cell functions.
The expression of multiple α1-adrenoceptor subtypes in submandibular acinar cells suggests the potential for these individual subtypes to couple to different signaling pathways and physiological functions. For example, in other cell types from tissues that natively express multiple α1-adrenoceptor subtypes, the α1A subtype specifically couples to phosphatidylinositol turnover, while the α1B subtype activates the MAP kinase pathway (Wenham et al., 1997; McWhinney et al., 2000). It is now recognized that this α1-adrenoceptor subtype-specific signaling may be determined by subtype-specific localization in intracellular compartments or cholesterol-rich microdomains of the plasma membrane such as caveolae (Mackenzie et al., 2000; McWhinney et al., 2000).
The first aim of the present study was to investigate whether or not α1-adrenoceptors couple to the activation of ERK1/2 in the native submandibular gland and SMG-C10 acinar cell line. The second aim of this study was to determine if the α1A-, the α1B- or both adrenoceptor subtypes mediate ERK1/2 activation in SMG-C10 acinar cells. The third aim was to identify the subcellular location of α1-adrenoceptors and their dependence on cholesterol-rich microdomains for activating ERK1/2 in SMG-C10 acinar cells. These studies provide new information on the signaling pathways that regulate salivary gland contribution to homeostasis in the oral cavity.
2.Materials and Methods
2.1 Drugs and Reagents
The drugs and reagents used were obtained from the following sources: 8-[2-[4-(methoxyphenyl)-1-piperazinyl]-8-azaspiro[4,5]decane-7,9-dione (BMY 7378) dihydrochloride, chloroethylclonidine dihydrochloride, cholesterol, filipin, 5-methylurapidil, phenylephrine hydrochloride, prazosin hydrochloride, d,l-propranolol hydrochloride, 2-(2-methoxy-3H-1,4-benzodioxin-2-yl)-4,5-dihydro-1H-imidazole (RX 821002) hydrochloride and methyl-β-cyclodextrin (Sigma-Aldrich, St. Louis, MO); 2-(beta-(4-hydroxyphenyl)-ethylaminomethyl)-tetralone (HEAT) hydrochloride and N-[5-(4,5-dihydro-1H-imidazol-2yl)-2-hydroxy-5,6,7,8-tetrahydro-naphthalen-1-yl]methane sulphonamide (A-61603) hydrobromide (Tocris Cookson, Ballwin, MO); rabbit polyclonal phospho-p44/42 MAP kinase (Thr202/Tyr204) and p44/42 MAP kinase antibodies (Cell Signaling Technology, Beverly, MA); horseradish peroxidase-conjugated anti-rabbit IgG (Santa Cruz Biotechnology, Santa Cruz, CA); [7-methoxy-3H]prazosin ([3H]prazosin) (70-87 Ci/mmol) and [125I]HEAT (PerkinElmer Life and Analytical Sciences, Boston, MA). All drugs were dissolved in distilled water and then diluted in the appropriate buffer unless otherwise indicated.
2.2 Cell Culture
SMG-C10 cells (generously provided by Dr. David O. Quissell, School of Dentistry, University of Colorado Health Sciences Center, Denver, CO) were seeded onto T-25 (2 × 105cells) or T-75 (8 × 105 cells) Falcon Primaria tissue culture flasks (BD Biosciences, Franklin Lakes, NJ) and grown in Dulbecco's modified Eagle's medium (DMEM)/F-12 nutrient mixture (1:1) and 2.5% fetal bovine serum (Invitrogen, Carlsbad, CA). Growth medium was supplemented with 2 mM glutamine and 4 μg/ml transferrin (Invitrogen); 0.1 μM retinoic acid, 2 nM triiodothyronine, 1 μM hydrocortisone, 5 μg/ml insulin, and 50 μg/ml gentamicin (Sigma-Aldrich); 50 ng/ml epidermal growth factor (BD Biosciences); and trace element mix (Biofluids, Rockville, MD). Cells were grown to confluence at 37°C in a humidified 95% air/5% CO2incubator and used for experiments between passages 23 and 29. Each individual experiment was performed using cells from a different passage number that were cultured on a different day.
2.3 ERK1/2 Assay
Male Sprague-Dawley rats (140-190 g) were pretreated for 24 h with reserpine (3 mg/kg, i.p.) to deplete noradrenaline from sympathetic nerves in the gland slices and prevent endogenous noradrenaline from activating ERK1/2. Rats were anesthetized with pentobarbital (50 mg/kg, i.p.) and then exsanguinated by cutting the abdominal aorta. Submandibular glands were removed and placed in ice-cold HEPES-buffered Krebs' solution (HBK), pH 7.4, containing 125 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 5 mM NaHCO3, 1.25 mM NaH2PO4, 11.1 mM dextrose, and 15 mM HEPES; trimmed of visible fat, fascia, lymph nodes, blood vessels and ducts; and then minced with iris scissors. The gland slices were washed by centrifugation at 100g for 5 min at 4°C, and the supernatant was discarded. The gland slices were resuspended in HBK and then equilibrated at 37°C for 30 min in T-25 tissue culture flasks. Confluent SMG-C10 cultures, maintained in DMEM/F12 lacking serum and growth factors for 18 h before experimentation, were washed free of medium three times and then equilibrated at 37°C for 30 min in HBK.
ERK1/2 was measured in submandibular gland slices or SMG-C10 cells treated with either HBK alone (basal) or agonist in HBK for 7 min. Where it was appropriate, cells or gland slices were incubated with a competitive receptor antagonist or an inhibitor for 30 min before agonist addition. In some experiments, cells were pretreated with the irreversible α1B-adrenoceptor antagonist, chloroethylclonidine, as previously described (Bockman et al., 2004). Briefly, cells were incubated with 30 μM chloroethylclonidine for 12 min at 37 °C. Chloroethylclonidine treatment was terminated by a 3-fold dilution with ice-cold HBK and then followed with three successive washings over a 15-min period to remove any unbound drug before agonist addition.
2.4 Western Blot Analysis of ERK1/2
ERK1/2 activity was measured using a modification of the method described by Hague et al. (2002). ERK1/2 activity was stopped by rapid aspiration of the incubation solution followed with addition of ice-cold lysis buffer containing 137 mM NaCl, 20 mM Tris HCl, 1 mM CaCl2, 1 mM MgCl2, 1% Nonidet P-40, 1 mM EDTA, 1 μM aprotinin, 100 μM phenylmethylsulfonyl fluoride, and 10 nM okadaic acid (Sigma-Aldrich). Samples were homogenized twice in ice-cold lysis buffer using a Janke and Kunkel S25N-8G element attached to an Ultra-Turrax T25 dispersing instrument (Janke and Kunkel, Staufen, Germany) at 22,000 rpm for 10 s and centrifuged at 12,000g for 15 min. Following recovery of the supernatants and determination of their protein content with the Coomassie Plus Better Bradford Assay Kit (Pierce, Rockford, IL), samples were stored at −20°C for later use.
Sample extracts (20 μg protein) were boiled for 5 min in loading buffer (pH 6.8) containing 60 mM Tris HCl, 25% glycerol, 2% sodium dodecyl sulfate, 14.4 mM 2-mercaptoethanol, and 0.1% bromophenol blue, and then resolved by electrophoresis on Ready Gel 4-15% Tris-HCl/polyacrylamide gels (Bio-Rad, Hercules, CA) using a Mini-PROTEAN 3 system (Bio-Rad). Following electrophoresis, the gel contents were transferred to NitroPure 0.45 micron nitrocellulose membranes (GE Osmonics, Minnetonka, MN) for immunoblotting. Phosphorylated ERK1/2 and total (phosphorylation-state independent) ERK1/2 were detected by incubating the nitrocellulose membranes with phospho-p44/42 (1:2,000) and p44/42 (1:5,000) MAP kinase antibodies, respectively, overnight at 4°C. The membranes were then incubated with horseradish peroxidase-conjugated anti-rabbit IgG diluted to 1:15,000 and treated with SuperSignal West Pico reagent (Pierce) to visualize the protein bands for ERK1/2 by chemiluminescence detection with Hyperfilm ECL (Amersham Biosciences, Piscataway, NJ).
2.5 Radioligand Binding Assay
Cells were washed twice with phosphate-buffered saline and removed from T-75 tissue culture flasks with a rubber policeman. Cells were homogenized twice in 10 volumes of ice-cold 50 mM Tris buffer (pH 7.4) using a Janke and Kunkel S25N-8G element attached to an Ultra-Turrax T25 dispersing instrument (Janke and Kunkel) at 22,000 rpm for 10 s. The homogenate was centrifuged at 30,000g for 15 min, and the supernatant was discarded. The membrane pellet was resuspended in Tris buffer, washed twice more by centrifugation, and stored at −80°C for later use.
Membrane pellets were resuspended by homogenization in 50 mM Tris buffer. Total [125I]HEAT binding was determined using duplicate tubes containing 300 μl of membrane suspension, 100 μl of 250 pM [125I]HEAT, and 100 μl of various concentrations of unlabeled drugs. Nonspecific binding was measured as 250 pM [125I]HEAT binding in the presence of 10 μM phentolamine. After a 30-min incubation in a shaking water bath at 37°C, membrane suspensions were filtered through GF/B glass fiber filter strips (Whatman, Maidstone, UK) using a 48-sample cell harvester (Brandel, Gaithersburg, MD). Tubes and filters were washed three times with ice-cold Tris buffer, and radioactivity retained on the filters was counted using a Beckman Gamma 5500 gamma counter (Beckman Coulter, Fullerton, CA). Specific binding was calculated as the difference between total and nonspecific binding. The protein concentration was determined by the method of Lowry et al. (1951) using bovine serum albumin as the standard.
2.6 Sucrose Density Gradient Centrifugation
Sucrose density gradient fractionation was carried out using a modification of the method described by Wang et al. (1997). Cells were washed and then lysed in ice-cold, hypotonic 1 mM Tris buffer (pH 7.4) containing 2 mM EDTA. The lysate was layered on top of a discontinuous sucrose density gradient composed of 1.7 ml of 15% sucrose (w/v), 5.0 ml of 30% sucrose, and 2.5 ml of 60% sucrose in 20 mM Tris buffer (pH 7.4). Following centrifugation at 100,000g for 65 min at 4°C, 1 ml fractions were collected from the top of the tubes, and [3H]prazosin binding (2.5 nM) to membranes in each fraction was determined as described above for [125I]HEAT binding, except that trapped radioactivity was counted by liquid scintillation spectroscopy.
2.7 Data Analysis
In Western blot experiments, the bands for both the 44- and 42-kDa proteins representing ERK1 and ERK2, respectively, were scanned and quantified by densitometry (Molecular Analyst, Bio-Rad). The densities of both bands for phosphorylated ERK1/2 were divided by band densities for total ERK1/2 to normalize for variable protein loading. Changes in the phosphorylation level of ERK1/2 were interpreted as indicative of changes in the activation of ERK1/2. The effect of various inhibitors on activation of ERK1/2 was compared statistically to the maximal ERK1/2 activation caused by phenylephrine using the one-sample t test. In some experiments, half-maximal effective concentrations (EC50 values) for agonist-induced ERK1/2 activation were calculated from concentration-response curves by nonlinear regression of all points on the curve. EC50 values were compared statistically using the Student's t test. In all cases, values are given as means ± S.E.M. and the 95% confidence level was used to conclude that there was a significant difference between groups.
Competition binding data were analyzed using a nonlinear least-squares curve-fitting program (GraphPad Prism; GraphPad Software, San Diego, CA) to determine IC50 values. Ki values were calculated from IC50 values by the method of Cheng and Prusoff (1973). The F test was used to determine whether or not the binding data fit best to a 1- or 2-site binding model. A value of P < 0.05 was used to conclude that the two-site model fit the data best.
3. Results
3.1 Phenylephrine-Stimulated ERK1/2 Activation in Submandibular Gland Acinar Cells
To determine whether or not the α1-adrenoceptor selective agonist phenylephrine activates ERK1/2 in submandibular acinar cells, we examined the time-course for phenylephrine-stimulated ERK1/2 phosphorylation in slices and cells from the submandibular gland. Figure 1A shows a line graph of mean ERK1/2 responses (n = 3-4) in SMG-C10 cells exposed to 10 μM phenylephrine for various times. Phenylephrine elicited a transient activation of ERK1/2 that peaked between 5 - 7 min and returned to baseline between 20 - 60 min. A similar time-course for phenylephrine-induced ERK1/2 stimulation was also obtained in submandibular gland slices, where 10 μM phenylephrine stimulated ERK1/2 by 3.4-fold over basal, with maximal stimulation at 5-10 min.
Figure 1.
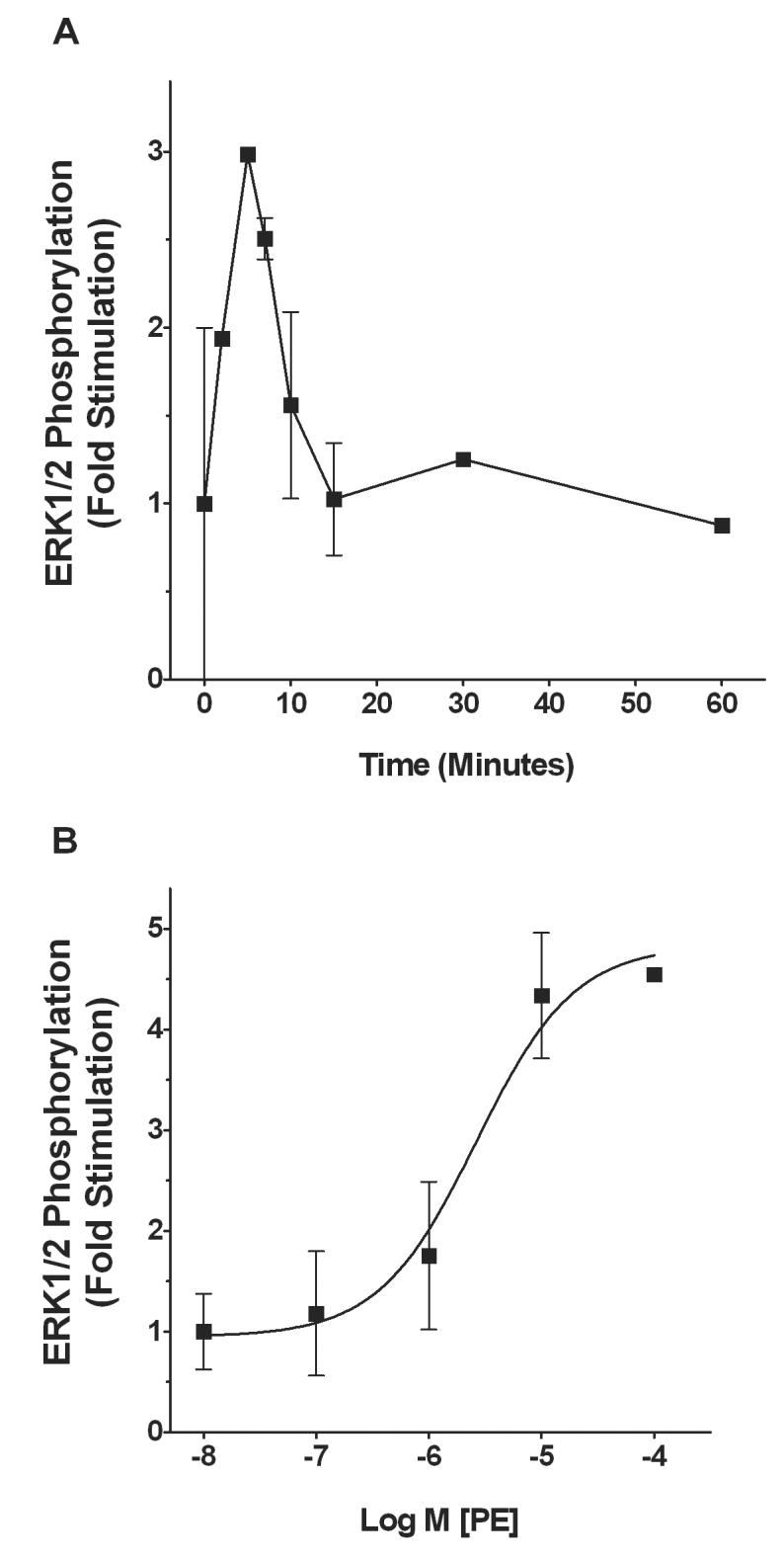
Time course and concentration-response curve for phenylephrine (PE) stimulation of ERK1/2 phosphorylation in SMG-C10 cells. (A) Cells were stimulated with 10 μM PE and ERK1/2 phosphorylation measured by Western blotting at time points ranging from 10s - 60 min. The data are fold stimulation over basal ERK1/2 activation in the absence of PE and are normalized to maximal ERK1/2 stimulation. (B) Mean concentration-response curve for PE stimulation of ERK1/2 using a 7 min exposure time. Each point on the concentration-response curve is the fold stimulation over basal ERK1/2 activation in the absence of PE normalized to maximal ERK1/2 stimulation. All data are the means ± S.E.M. of 3-4 individual experiments, each using different cell cultures.
The ERK1/2 responses to phenylephrine were also concentration-dependent. Figure 1B illustrates a mean concentration-response curve for phenylephrine-stimulated ERK1/2 activation at 7 min in SMG-C10 cells. The maximally effective concentration of phenylephrine in stimulating ERK1/2 was 10 μM, which increased phosphorylation of ERK1/2 by 4.5 ± 0.8-fold relative to basal levels. The mean EC50 value for phenylephrine was 2.7 ± 2 μM (n = 3-4). In submandibular gland slices, 10 μM phenylephrine also elicited maximum ERK1/2 activation that was 1.9 ± 0.3-fold over basal (n = 3).
3.2 α1-Adrenoceptors Mediate Phenylephrine-Stimulated ERK1/2 Activation
To determine the adrenoceptor type mediating phenylephrine-induced ERK1/2 activation, we measured ERK1/2 phosphorylation in phenylephrine-stimulated gland slices and SMG-C10 cells incubated with various adrenoceptor antagonists that selectively block either α1-, α2- or β-adrenoceptors. Figure 2 shows mean bar graphs of ERK1/2 responses in submandibular gland slices (Fig. 2A) and SMG-C10 cells (Fig. 2B) in the absence and presence of either prazosin or HEAT, which are both α1-adrenoceptor selective antagonists. In gland slices, either 100 nM prazosin or 100 nM HEAT completely inhibited phenylephrine-stimulated ERK1/2 activation (p < 0.05, n = 4-5) to basal levels measured in unstimulated slices (Fig. 2A).
Figure 2.
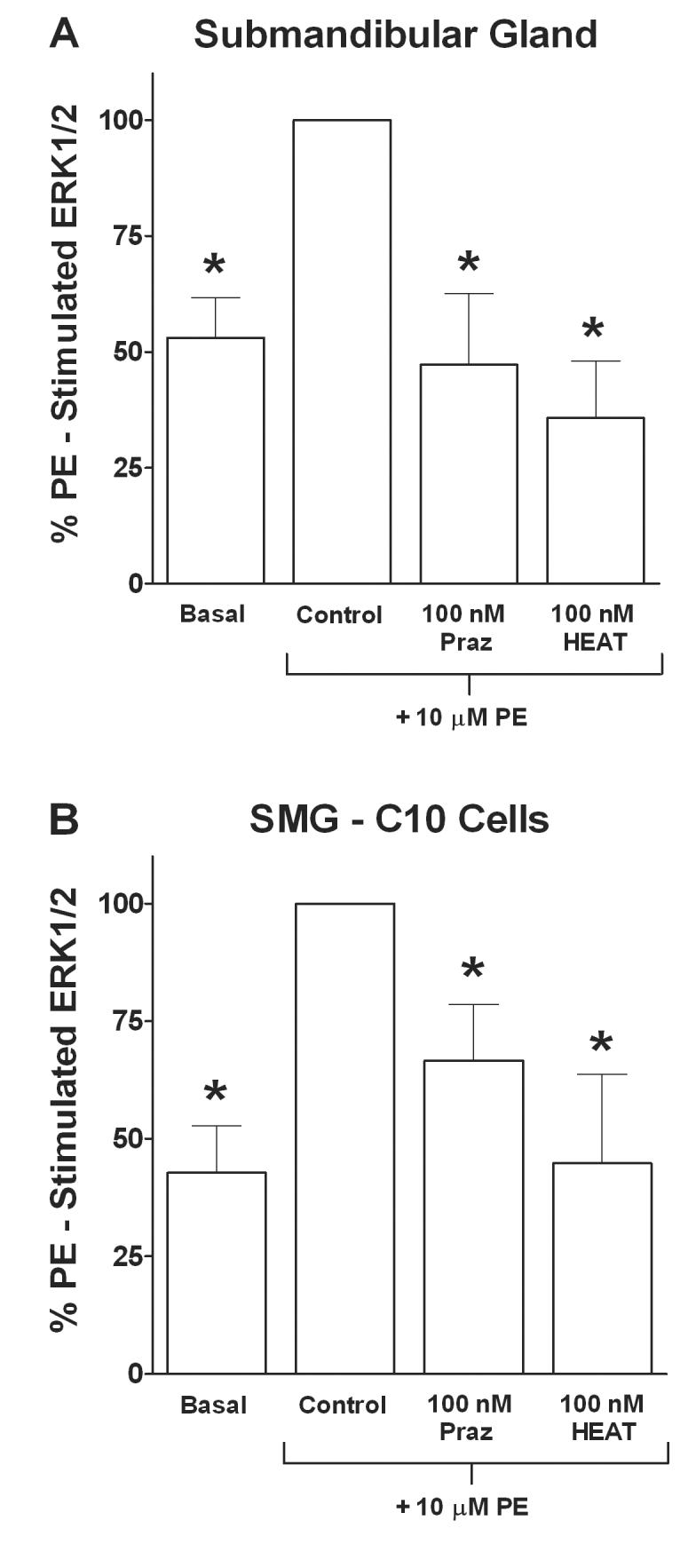
Effects of the α1-adrenoceptor antagonists prazosin (Praz) and HEAT on phenylephrine (PE) stimulated ERK1/2 in submandibular gland slices or SMG-C10 cells. Slices or cells were incubated with 100 nM of an antagonist for 30 min followed by addition of 10 μM PE for 7 min. Data are plotted as % of 10 μM PE stimulated ERK1/2 in the absence of antagonist (Control). (A) Bar graph of data using submandibular gland slices, showing significant inhibition of PE stimulated ERK1/2 to basal levels by the α1-adrenoceptor antagonists Praz and HEAT. (B) Bar graph of data using SMG-C10 cells, showing significant inhibition by Praz and HEAT of PE stimulation of ERK1/2. All data are the means ± S.E.M. of 4-5 individual experiments, each using submandibular gland slices taken from a different animal or using different SMG-C10 cell cultures. *Significantly different from Control (10 μM PE alone), p < 0.05.
A similar pharmacological profile for antagonist inhibition of ERK1/2 responses to phenylephrine was obtained in SMG-C10 cells. In cells, both 100 nM prazosin and 100 nM HEAT significantly inhibited phenylephrine-stimulated ERK1/2 activation (p < 0.05, n = 5) by 60 ± 20% and 100 ± 30%, respectively (Fig. 2B). In addition, neither 100 nM of the α2-adrenoceptor selective antagonist RX 821002 (9 ± 14% inhibition) nor 100 nM of the β-adrenoceptor selective antagonist propranolol (21 ± 6% inhibition) had a significant effect on phenylephrine-induced ERK1/2 activation (p > 0.05, n = 4). Taken together, these results suggest α1-adrenoceptors, but not α2- or β-adrenoceptors, mediate phenylephrine-induced ERK1/2 activation in submandibular gland cells.
3.3 Radioligand Binding Characterization of α1-Adrenoceptor Subtypes
The α1-adrenoceptors present in membranes from SMG-C10 cells were directly identified in radioligand binding experiments with [125I]HEAT. Figure 3 illustrates mean competition curves for the inhibition of [125I]HEAT binding by α1-adrenoceptor subtypenonselective (prazosin) and selective (5-methylurapidil and BMY 7378) antagonists. From these curves, we obtained Hill coefficients and affinities (Ki values) for drugs to determine the α1-adrenoceptor subtypes expressed in SMG-C10 cell membranes (Table 1A). Competition curves for prazosin inhibition of [125I]HEAT binding were monophasic with a mean Hill coefficient of -0.75 ± 0.1, and fit best by a one-site binding model (p > 0.05) with a mean Ki value of 0.33 ± 0.1 nM. In contrast, competition curves for the inhibition of [125I]HEAT binding by 5-methylurapidil were biphasic and characterized by relatively shallow slopes with a mean Hill coefficient of -0.58 ± 0.1. Additionally, competition binding curves for 5-methylurapidil were fit best by a two-site binding model (p < 0.05) with 21 ± 4% high-affinity binding sites and 79 ± 5% low-affinity binding sites, suggesting the presence of a heterogeneous population of α1-adrenoceptor subtypes (Table 1A). 5-methylurapidil has highest affinity for the α1A-adrenoceptor subtype and lower but equal affinities for the α1B- and α1D-adrenoceptors. Thus, the high (KiH)- and low ( KiL)-affinity values for 5-methylurapidil of 0.64 ± 0.3 nM and 91 ± 7 nM, respectively, indicate the presence of the α1A-adrenoceptor subtype and either the α1B-or α1D-adrenoceptor subtypes. We then generated competition-binding curves for BMY 7378, which has highest affinity, i.e. approximately 1 nM, for the α1D-adrenoceptor subtype and lower but equal affinities for the α1A- and α1B-adrenoceptors. Competition curves for BMY 7378 inhibition of [125I]HEAT binding were monophasic, relatively steep (Hill coefficient = −0.87 ± 0.2), and fit best by a one-site binding model (p > 0.05) with a Ki value of 122 ± 27 nM (Table 1A), indicating the lack of the α1D-adrenoceptor subtype in submandibular acinar cells.
Figure 3.

Mean competition curves showing α1-adrenoceptor antagonist inhibition of [125I]HEAT binding in membranes from SMG-C10 cells. For each concentration of antagonist, [125I]HEAT binding is expressed as percent of specific binding in the absence of drugs. The competition curves were fit to one- and two-site binding models and mean Ki values (Table 1) were determined from the curve fits. Competition curves for the non-subtype selective antagonist prazosin and the α1D-subtype selective antagonist BMY 7378 fit best to a one-site binding model. In contrast, competition curves for 5-methylurapidil (5-MU) fit significantly better to a two-site model (p < 0.05 using an F test; Table 1) indicating that both α1A- and α1B-adrenoceptors are present in SMG-C10 cells. Each curve is the mean ± S.E.M. of 4-5 individual experiments, each using different SMG-C10 cell cultures.
TABLE 1.
Affinity values, and percentage and subcellular location of binding sites for α1-adrenoceptor drugs in submandibular gland acinar cells (SMG-C10).
| A | |||||||
|---|---|---|---|---|---|---|---|
| Drug | Ki (nM) | nH | KiH (nM) | % High | KiL (nM) | % Low | n |
| Prazosin | 0.33 ± 0.1 | −0.75 ± 0.1 | 4 | ||||
| BMY 7378 | 122 ± 27 | −0.87 ± 0.2 | 4 | ||||
| 5-methylurapidila | −0.58 ± 0.1 | 0.64 ± 0.3 | 21 ± 4 | 91 ± 7 | 79 ± 5 | 5 | |
| B | |||||||
| % [3H]Prazosin Binding | 19 ± 2 | 71 ± 3 | |||||
| Sucrose Density Gradient Fraction Number | 3 – 5b | 9 – 10c | |||||
A. The values are mean ± S.E.M.; n, number of separate SMG-C10 cell cultures. Ki, is affinity in nM of drugs for inhibiting specific [125I]HEAT binding; nH, Hill coefficient. Affinity values from binding data that fit best to a two-site binding model are expressed as KiH (high-affinity binding site) and KiL (low-affinity binding site). % High, percentage of high-affinity binding sites; % Low, percentage of low affinity binding sites. B. % [3H]Prazosin Binding, percentage of specific [3H]prazosin binding sites in subcellular fractions separated by sucrose density gradient centrifugation. Values are mean ± S.E.M. of five experiments using separate SMG-C10 cell cultures.
Competition binding curves fit best by nonlinear least-squares curve-fitting regression to a two-site binding model (p < 0.05) using the F test.
Fractions containing intracellular vesicles.
Fractions containing plasma membranes.
3.4 Functional Characterization of the α1-Adrenoceptor Subtype Causing ERK1/2 Activation
We determined the α1-adrenoceptor subtype that mediates ERK1/2 activation by incubating SMG-C10 cells with receptor subtype-selective concentrations of the competitive antagonist 5-methylurapidil (α1A-selective) or irreversible antagonist chloroethylclonidine (selective for alkylating α1B-adrenoceptors), and then stimulating with phenylephrine (Fig. 4A). At a concentration that we determined by radioligand binding to be α1A-selective in SMG-C10 cells, i.e. thirty times the KiH (Fig. 3; Table 1A), 5-methylurapidil (20 nM) had no significant effect (p > 0.05) on phenylephrine-stimulated ERK1/2 activation (Fig. 4A). Similarly, an α1D-adrenoceptor-selective concentration of BMY 7378 (10 nM) had little effect on phenylephrine-stimulated ERK1/2 activation (8% inhibition). In contrast, 30 μM chloroethylclonidine for 12 min, a treatment that we previously showed to inactivate only the α1B-adrenoceptor subtype in SMG-C10 cells (Bockman et al., 2004), significantly inhibited (p < 0.05 ) phenylephrine-stimulated ERK1/2 activation (Fig. 4A). These results suggest that the α1B-adrenoceptor is the predominant α1-adrenoceptor subtype mediating phenylephrine-stimulated ERK1/2 activation in submandibular acinar cells.
Figure 4.
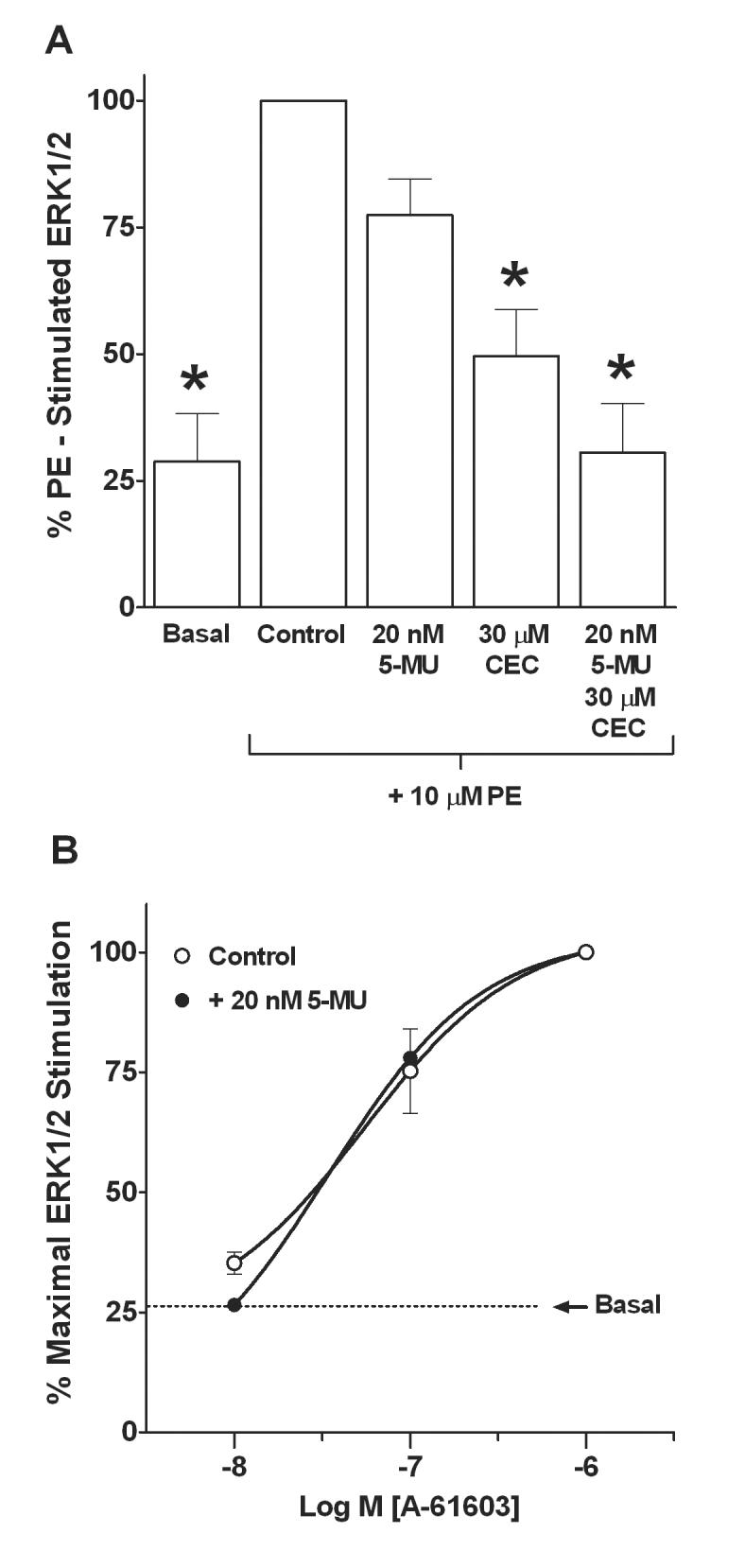
Effects of the α1-adrenoceptor subtype selective antagonists 5-methylurapidil (5-MU) and chloroethylclonidine (CEC) on phenylephrine (PE) or A-61603 stimulated ERK1/2 in SMG-C10 cells. (A) Bar graph of α1-adrenoceptor subtype selective antagonist effects on PE stimulated ERK1/2. Cells were incubated with either 5-MU for 30 min, CEC for 12 min, or 5-MU for 30 min followed by 5-MU plus CEC for 12 min. Samples treated with the irreversible antagonist CEC were extensively washed to remove non-bound antagonists. Cells were then treated with 10 μM PE for 7 min to stimulate ERK1/2. Data are plotted as % of Control (10 μM PE stimulated) ERK1/2. PE stimulated ERK1/2 was only inhibited by treatment with the α1B-subtype selective antagonist CEC. (B) Mean concentration-response curves for A-61603 stimulation of ERK1/2 in the absence and the presence of an α1A-adrenoceptor blocking concentration of 5-MU. SMG-C10 cells were incubated without or with 5-MU for 30 min and then stimulated for 7 min with increasing concentrations of A-61603. Each point on the concentration-response curve is the percentage of the maximal ERK1/2 stimulation produced by A-61603. The dashed line is the mean basal level of ERK1/2 activation in the absence of drugs. Only higher concentrations of A-61603 that stimulate α1B-adrenoceptors activated ERK1/2 and this stimulation was not affected by α1A-adrenoceptor blockade by 5-MU. All data are means ± S.E.M. of 4-5 individual experiments, each using different SMG-C10 cell cultures. *Significantly different from Control (10 μM PE alone), p < 0.05.
Because the selectivity of chloroethylclonidine for inactivating the α1B-adrenoceptor subtype is dependent on experimental conditions, a protection experiment was performed to confirm the results using chloroethylclonidine. SMG-C10 cells were first incubated with 20 nM 5-methylurapidil for 30 min to bind selectively to 100% of the α1A-adrenoceptor population, protecting it from possible inactivation by chloroethylclonidine. In the presence of 5-methylurapidil, cells were next treated with 30 μM chloroethylclonidine for 12 min to inactivate the unprotected α1B-adrenoceptors. Then cells were washed free of 5-methylurapidil, leaving only α1A-adrenoceptors available for agonist stimulation. We then tested whether or not the α1A-adrenoceptor subtype contributes to the ERK1/2 response to phenylephrine in SMG-C10 cells. Figure 4A illustrates that phenylephrine stimulation of α1A-adrenoceptors did not activate ERK1/2, suggesting ERK 1/2 responses to phenylephrine are mediated solely by the α1B-adrenoceptor subtype in SMG-C10 cells.
In addition, Figure 4B illustrates concentration-response curves for the α1A-adrenoceptor-selective agonist A-61603 in causing ERK1/2 activation in the absence and presence of an α1A-selective concentration (20 nM) of 5-methylurapidil predicted from our radioligand binding experiments to saturate 100% of the α1A-receptors in SMG-C10 cells. In the absence and presence of 5-methylurapidil, the EC50 values for A-61603-stimulated ERK1/2 activation were 52 ± 5 nM (n = 4) and 33 ± 4 nM (n = 4), respectively, and were not significantly different (p > 0.05) from one another. Taken together, these results suggest the α1B- but not the α1A-adrenoceptor subtype mediates ERK1/2 activation in submandibular acinar cells.
3.5 α1-Adrenoceptor Subcellular Location and Cholesterol Dependence of ERK1/2 Activation
We examined the subcellular location of α1-adrenoceptors in SMG-C10 cells by first separating subcellular fractions from the total cell lysate using sucrose density gradient centrifugation and then measuring [3H]prazosin binding in membrane fractions to determine the distribution of α1-adrenoceptors. Figure 5A illustrates a mean line graph of the gradient profile depicting the percentage of specific [3H]prazosin binding sites in each membrane fraction. The majority (71 ± 3 %) of the overall α1-adrenoceptor binding sites were located in the heavy plasma membrane fractions 9-10; whereas, the light vesicle fractions 3-5 expressed a smaller percentage (19 ± 2 %) of overall α1-adrenoceptor binding sites (Table 1B).
Figure 5.

Subcellular location of α1-adrenoceptors and effects of cholesterol depletion/disruption by filipin (Fil) or methyl-β-cyclodextrin (mβCD) on phenylephrine (PE) stimulated ERK1/2 in SMG-C10 cells. (A) Sucrose density gradient fractionation of α1-adrenoceptors. Sucrose density gradient centrifugation was used to separate subcellular fractions and the α1-adrenoceptors present in those fractions were determined using [3H]Prazosin binding. Data are the % specific [3H]Prazosin bound, which is total bound minus nonspecific bound in each fraction. Fraction numbers are 1 ml volumes isolated from the sucrose gradient following centrifugation. The majority of specific [3H]Prazosin binding was in heavy plasma membrane fractions 9-10 with a lesser percentage in light vesicle fractions 3-5. Points are means ± S.E.M. of 5 individual experiments performed in duplicate, each using different SMG-C10 cell cultures. (B) Bar graph of the effects of Fil and mβCD on PE stimulated ERK1/2. Cells were incubated with Fil, the vehicle DMSO, mβCD, or mβCD + cholesterol (Chol) for 30 min and then stimulated for 7 min with PE. The cholesterol depletors/disruptors Fil and mβCD inhibited PE stimulated ERK1/2, while cholesterol prevented the inhibition caused by mβCD. Data are plotted as percent of Control (10 μM PE stimulated) ERK1/2. Data are means ± S.E.M. of 5 experiments, each using different SMG-C10 cell cultures. *Significantly different from Control (10 μM PE alone), p < 0.05.
The plasma membrane of submandibular gland cells contains cholesterol-enriched microdomains, such as caveolae, which concentrate G protein-coupled receptors and their effector molecules important in regulating secretion (Brazer et al., 2003). Thus, we investigated the membrane cholesterol dependence of α1B-adrenoceptor-mediated ERK1/2 activation in submandibular acinar cells with methyl-β-cyclodextrin and filipin, both of which are used to deplete cholesterol or to otherwise disrupt cholesterol-rich membrane structure and signaling. For these studies, phenylephrine-induced ERK1/2 activation was measured in SMG-C10 cells incubated with 6 μg/ml filipin or 10 mM methyl-β-cyclodextrin (Fig. 5B). Both filipin and methyl-β-cyclodextrin significantly inhibited (p < 0.05, n = 6) phenylephrine-stimulated ERK1/2 activation by 87 ± 30% and 64 ± 20%, respectively, compared to unstimulated basal levels. We also incubated cells with methyl-β-cyclodextrin in the presence of 2 mM cholesterol (Fig. 5B). Exogenous cholesterol prevents methyl-β-cyclodextrin from extracting membrane cholesterol and thus serves as a control for confirming the specificity of methyl-β-cyclodextrin's action. Figure 5B shows methyl-β-cyclodextrin and cholesterol together had no effect (p > 0.05) on phenylephrine-induced ERK1/2 activation in SMG-C10 cells, indicating the effects of methyl-β-cyclodextrin are due to depletion of membrane cholesterol. These data show that in SMG-C10 cells, phenylephrine-stimulated ERK1/2 activation is cholesterol-dependent and may occur in cholesterol-rich plasma membrane domains.
4.Discussion
The submandibular gland is a major producer of the fluids, electrolytes and mucins that form saliva. In the submandibular gland, α1-adrenoceptors are coupled through the Gq/11 protein to an elevation in intracellular free calcium, which activates Ca2+-dependent chloride channels to cause fluid secretion and which modulates exocytotic secretion of mucins (Martinez and Reed, 1988; Quissel et al., 1992). The mitogen-activated protein kinases, ERK1/2, may also have an important role in the stimulus-secretion coupling pathway in salivary glands (Zentner et al., 1998, Hoffert et al., 2000; Jung et al., 2000; Slomiany and Slomiany, 2004; Plourde and Soltoff, 2006). However, whether ERK1/2 contributes to α1-adrenoceptor-stimulated secretion or is activated by α1-adrenoceptors in submandibular glands is currently unknown. Moreover, the submandibular gland expresses a heterogeneous α1-adrenoceptor population comprised of the α1A- and α1B-adrenoceptor subtypes. Thus, it is also important to know whether the α1A-, the α1B- or both adrenoceptor subtypes mediate ERK1/2 activation in submandibular acinar cells. In the present study, we determined whether or not α1-adrenoceptors couple to the activation of ERK1/2 and identified the α1-adrenoceptor subtype/s mediating ERK1/2 activation in submandibular gland acinar cells. The contemporary view that G protein-coupled receptors and their effector molecules, such as ERK1/2, are closely associated with cholesterol-rich plasma membrane microdomains suggests the importance of evaluating the role of these compartments in regulating ERK1/2 activation by α1-adrenoceptors. Thus, we also examined the subcellular location of α1-adrenoceptors and their dependence on cholesterol-rich microdomains for activating ERK1/2 in submandibular acinar cells.
The submandibular gland is comprised mainly of acinar cells, which are responsible for generating the secretory product. SMG-C10 cells are secretory epithelia of acinar origin and exhibit many of the phenotypical characteristics of fully differentiated native submandibular acinar cells that make them a useful system for studying receptor-regulated secretion (Quissell et al., 1997; Liu et al., 2000). In salivary gland research, well differentiated acinar cell lines are scarce, and excepting SMG-C10 cells, there are none that express both the α1A- and α1B-adrenoceptor subtypes comparable to those in native submandibular acinar cells (Bockman et al., 2004), establishing SMG-C10 cells as a model for examining α1-adrenoceptor-mediated secretion. In our study, selective α1-adrenoceptor blockade with either prazosin or HEAT inhibited phenylephrine-stimulated ERK1/2 activation to basal levels, indicating that α1-adrenoceptors mediate the ERK1/2 response to phenylephrine in submandibular glands and SMG-C10 cells. In the current study, we also found that the magnitude of the ERK1/2 response to phenylephrine was two-fold greater in SMG-C10 cells than in submandibular glands, and thus we were better able to quantify the concentration-response relationship for phenylephrine-induced ERK1/2 activation in SMG-C10 cells than in native submandibular glands. Consequently, we then determined whether the α1A-, the α1B- or both adrenoceptor subtypes mediate ERK1/2 activation in SMG-C10 cells.
A commonly used competitive antagonist, 5-methylurapidil exhibits high affinity and selectivity for the α1A- over the α1B-adrenoceptor subtype (Gross et al., 1988). At a concentration (20 nM) that we determined by radioligand binding to be α1A-selective in SMG-C10 cells, 5-methylurapidil did not block α1-adrenoceptor-mediated ERK 1/2 activation. 20 nM is approximately thirty times the Ki of 5-methylurapidil at the α1A-subtype in SMG-C10 cells and thus would block 100% of these receptors; yet, the ERK1/2 response to phenylephrine was unaffected, suggesting α1A-adrenoceptors do not contribute to ERK1/2 activation in submandibular acinar cells. In addition, chloroethylclonidine, which is an irreversible alkylating agent that can be used to functionally inactivate the α1B-adrenoceptor subtype (Minneman et al., 1988), inhibited α1-adrenoceptor-mediated ERK1/2 activation, suggesting α1B-adrenoceptors mediate ERK1/2 activation. Additional data supporting the role of the α1B-adrenoceptor, but not the α1A-adrenoceptor subtype, in regulating ERK1/2 in submandibular acinar cells were obtained using 20 nM of 5-methylurapidil to saturate and thus protect the α1A-receptor subtype from inactivation by chloroethylclonidine. Again, phenylephrine caused no response, indicating ERK1/2 activation is specific to the α1B-adrenoceptor subtype in these cells. These results are consistent with our data obtained using A-61603, which showed that blocking 100% of the α1A-receptor population with 20 nM 5-methylurapidil had no effect on the ERK1/2 response to A-61603. A-61603 is an agonist selective for, but not specific to, the α1A-adrenoceptor subtype (Knepper et al., 1995). Taken together, these data show that the α1B-adrenoceptor subtype mediates ERK1/2 activation in submandibular acinar cells.
SMG-C10 cells are distinctive because they endogenously express multiple α1-adrenoceptors. Most other cell lines containing α1-adrenoceptors express only the α1B subtype (Zhong and Minneman, 1999). In addition, SMG-C10 cells reflect native submandibular gland acini with respect to their similar expression of both α1A- and α1B-adrenoceptor subtypes (Bockman et al., 2004); thus, they offer a unique opportunity to examine the physiological significance of these multiple subtypes in a nontransfected intact functionome. For example, there is now evidence for physical interactions between α1-adrenoceptor subtypes resulting in functional receptor heterodimers (Finch and Graham, 2006; Hague et al., 2006). Others have suggested that native expression of multiple α1-adrenoceptor subtypes provides a mechanism for differential regulation of distinct signaling pathways and physiological functions. For instance in neonatal cardiac myocytes, which also natively express both α1A- and α1B-adrenoceptor subtypes, the α1A subtype specifically couples to phosphatidylinositol turnover, while the α1B subtype activates the MAP kinase pathway (Wenham et al., 1997; McWhinney et al., 2000). These reports suggest that α1-adrenoceptor subtypes selectively couple to different signaling pathways in a cell-type, dependent fashion. Our results, which show that only the α1B subtype is coupled to ERK1/2 activation, suggest that the α1A- and α1B-adrenoceptor subtypes may also be coupled to different signaling pathways and functions in acinar cells.
Indeed, signaling specificity as conferred by subcellular location and compartmentation of adrenoceptors has been identified in a variety of cell types (Toews et al., 2003; Steinberg, 2004), although this process has not been studied in submandibular acinar cells. We found that 70% of the α1-adrenoceptors were located in heavy plasma membrane fractions; whereas, 19% of the α1-adrenoceptors were located in the light intracellular vesicle fractions. These data are similar to the distribution of α1-adrenoceptors between plasma membrane and intracellular vesicles, respectively, previously reported in other cell types expressing endogenous or transfected α1-adrenoceptors (Toews et al., 2003; Wang et al., 2000). Interestingly, these values are similar to the percentage of α1B-adrenoceptors (79%) and α1A-adrenoceptors (21%) expressed in SMG-C10 cells (Table 1). Thus, it is possible that α1B-adrenoceptors are localized to the plasma membrane, while α1A-adrenoceptors are found at intracellular sites. This suggestion is consistent with reports by others who found that α1A-adrenoceptors are more likely to localize intracellularly; whereas, α1B-adrenoceptors are found primarily on the plasma membrane (Hirasawa et al., 1997; Mackenzie et al., 2000).
In the present study, depleting membrane cholesterol or disrupting cholesterol-rich membrane structures resulted in inhibition of α1B-adrenoceptor mediated ERK1/2 activation. These results are consistent with the recent recognition that G protein-coupled receptors and their effector molecules, such as ERK1/2, are closely associated with cholesterol-rich plasma membrane microdomains such as caveolae (Ostrom and Insel, 2004). The inhibitory effects of both methyl-β-cyclodextrin and filipin on α1-adrenoceptor stimulation of ERK1/2 activation suggest that these receptors or other components of their signaling pathway require cholesterol. A likely explanation for the cholesterol requirement is that this signaling pathway is initiated in cholesterol-rich plasma membrane microdomains, either caveolae or other types of lipid rafts. Similarly, we recently reported that α1B-adrenoceptors transfected into Chinese hamster ovary cells require cholesterol for protection from proteolytic degradation (Prinster et al., 2003). Taken together with the analysis of receptor distribution in subcellular compartments, the effect of cholesterol depletion or disruption on α1B-adrenoceptor coupling to ERK1/2 activation suggests the involvement of specific membrane localization in determining subtype-selective signaling in submandibular acinar cells.
In the current report, we found that the α1B-adrenoceptor subtype regulates ERK1/2 activation in submandibular acinar cells. However, the physiological significance of α1B-adrenoceptor mediated ERK1/2 activation in acinar cells is not completely understood. Both α1-receptors and ERK1/2 are known to contribute to the secretory response in acinar cells. For example, stimulation of α1-receptors elevates intracellular free calcium, activating Ca2+-dependent chloride channels to cause fluid and mucin secretion (Martinez and Reed, 1988; Quissell et al., 1992), and induces the trafficking of aquaporin-5 water channels to the apical plasma membrane of salivary acinar cells (Ishikawa et al., 1999). In addition, ERK1/2 regulates the expression of aquaporin-5 water channels (Hoffert et al., 2000), the activity of salivary Na+-K+-ATPase (Plourde and Soltoff, 2006), and salivary mucin secretion (Slomiany and Slomiany, 2004). Taken together, these results suggest that α1B-adrenoceptor-stimulated ERK1/2 activation may regulate the production and composition of saliva. SMG-C10 cells will be useful in clarifying how these multiple signaling pathways activated by the different α1-adrenoceptor subtypes are integrated into the final acinar cell response.
Acknowledgements
Supported, in part, by National Institute of Dental and Craniofacial Research Grant RO3-DE12530 and Health Future Foundation Faculty Development Award to C. S. Bockman.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bockman CS, Bruchas MR, Zeng W, O'Connell KA, Abel PW, Scofield MA, Dowd FJ. Submandibular gland acinar cells express multiple α1-adrenoceptor subtypes. J. Pharmacol. Exp. Ther. 2004;311:364–372. doi: 10.1124/jpet.104.066399. [DOI] [PubMed] [Google Scholar]
- Brazer SC, Singh BB, Liu X, Swaim W, Ambudkar IS. Caveolin-1 contributes to assembly of store-operated Ca2+ influx channels by regulating plasma membrane localization of TRPC1. J. Biol. Chem. 2003;278:27208–27215. doi: 10.1074/jbc.M301118200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng YC, Prusoff WH. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 percent inhibition (IC50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Finch AM, Graham RM. The α1D-adrenergic receptor: cinderella or ugly stepsister. Mol. Pharmacol. 2006;69:1–4. doi: 10.1124/mol.105.020230. [DOI] [PubMed] [Google Scholar]
- Gross G, Hanft G, Rugevics C. 5-Methyl-urapidil discriminates between subtypes of the α1-adrenoceptor. Eur. J. Pharmacol. 1988;151:333–335. doi: 10.1016/0014-2999(88)90819-9. [DOI] [PubMed] [Google Scholar]
- Hague C, Gonzalez-Cabrera PJ, Jeffries WB, Abel PW. Relationship between α1-adrenergic receptor-induced contraction and extracellular signal-regulated kinase activation in the bovine inferior alveolar artery. J. Pharmacol. Exp. Ther. 2002;303:403–411. doi: 10.1124/jpet.102.037531. [DOI] [PubMed] [Google Scholar]
- Hague C, Lee SE, Chen Z, Prinster SC, Hall RA, Minneman KP. Heterodimers of α1B- and α1D-adrenergic receptors form a single functional entity. Mol. Pharmacol. 2006;69:45–55. doi: 10.1124/mol.105.014985. [DOI] [PubMed] [Google Scholar]
- Hieble JP, Bylund DB, Clarke DE, Eikenburg DC, Langer SZ, Lefkowitz RJ, Minneman KP, Ruffolo RR., Jr. International Union of Pharmacology. X. Recommendation for nomenclature of α1-adrenoceptors: consensus update. Pharmacol. Rev. 1995;47:267–270. [PubMed] [Google Scholar]
- Hirasawa A, Sugawara T, Awaji T, Tsumaya K, Ito H, Tsujimoto G. Subtype-specific differences in subcellular localization of α1-adrenoceptors: chlorethylclonidine preferentially alkylates the accessible cell surface α1-adrenoceptors irrespective of the subtype. Mol. Pharmacol. 1997;52:764–770. doi: 10.1124/mol.52.5.764. [DOI] [PubMed] [Google Scholar]
- Hoffert JD, Leitch V, Agre P, King LS. Hypertonic induction of aquaporin-5 expression through an ERK-dependent pathway. J. Biol. Chem. 2000;275:9070–9077. doi: 10.1074/jbc.275.12.9070. [DOI] [PubMed] [Google Scholar]
- Huang Y, Wright CD, Merkwan CL, Baye NL, Liang Q, Simpson PC, O'Connell TD. An alpha1A-adrenergic-extracellular signal-regulated kinase survival signaling pathway in cardiac myocytes. Circulation. 2007;115:763–772. doi: 10.1161/CIRCULATIONAHA.106.664862. [DOI] [PubMed] [Google Scholar]
- Ishikawa Y, Skowronski MT, Inoue N, Ishida H. α1-Adrenoceptor-induced trafficking of aquaporin-5 to the apical plasma membrane of rat parotid cells. Biochem. Biophys. Res. Commun. 1999;265:94–100. doi: 10.1006/bbrc.1999.1630. [DOI] [PubMed] [Google Scholar]
- Jiao X, Gonzalez-Cabrera PJ, Xiao L, Bradley ME, Abel PW, Jeffries WB. Tonic inhibitory role for cAMP in α1A-adrenergic receptor coupling to extracellular signal-regulated kinases 1/2. J. Pharmacol. Exp. Ther. 2002;303:247–256. doi: 10.1124/jpet.102.037747. [DOI] [PubMed] [Google Scholar]
- Jung DW, Hecht D, Ho SW, O'Connell BC, Kleinman HK, Hoffman MP. PKC and ERK1/2 regulate amylase promoter activity during differentiation of a salivary gland cell line. J. Cell. Physiol. 2000;185:215–225. doi: 10.1002/1097-4652(200011)185:2<215::AID-JCP6>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Kaplan MD, Baum BJ. The functions of saliva. Dysphagia. 1993;8:225–229. doi: 10.1007/BF01354542. [DOI] [PubMed] [Google Scholar]
- Knepper SM, Buckner SA, Brune ME, DeBernardis JF, Meyer MD, Hancock AA. A-61603, a potent α1-adrenergic receptor agonist, selective for the α1A receptor subtype. J. Pharmacol. Exp. Ther. 1995;274:97–103. [PubMed] [Google Scholar]
- Liu XB, Sun X, Mork AC, Dodds MW, Martinez JR, Zhang GH. Characterization of the calcium signaling system in the submandibular cell line SMG-C6. Proc. Soc. Exp. Biol. Med. 2000;225:211–220. doi: 10.1177/153537020022500308. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Mackenzie JF, Daly CJ, Pediani JD, McGrath JC. Quantitative imaging in live human cells reveals intracellular α1-adrenoceptor ligand-binding sites. J. Pharmacol. Exp. Ther. 2000;294:434–443. [PubMed] [Google Scholar]
- Martinez JR, Reed P. Effect of alpha-receptor stimulation on Cl transport by rat submandibular acini. J. Dent. Res. 1988;67:561–564. doi: 10.1177/00220345880670030701. [DOI] [PubMed] [Google Scholar]
- McWhinney C, Wenham D, Kanwal S, Kalman V, Hansen C, Robishaw JD. Constitutively active mutants of the α1a- and the α1b-adrenergic receptor subtypes reveal coupling to different signaling pathways and physiological responses in rat cardiac myocytes. J. Biol. Chem. 2000;275:2087–2097. doi: 10.1074/jbc.275.3.2087. [DOI] [PubMed] [Google Scholar]
- Minneman KP, Han C, Abel PW. Comparison of α1-adrenergic receptor subtypes distinguished by chlorethylclonidine and WB 4101. Mol. Pharmacol. 1988;33:509–514. [PubMed] [Google Scholar]
- Ostrom RS, Insel PA. The evolving role of lipid rafts and caveolae in G protein-coupled receptor signaling: implications for molecular pharmacology. Br. J. Pharmacol. 2004;143:235–245. doi: 10.1038/sj.bjp.0705930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plourde D, Soltoff SP. Ouabain potentiates the activation of ERK1/2 by carbachol in parotid gland epithelial cells; inhibition of ERK1/2 reduces Na+-K+-ATPase activity. Am. J. Physiol. 2006;290:C702–C710. doi: 10.1152/ajpcell.00213.2005. [DOI] [PubMed] [Google Scholar]
- Prinster SC, Schulte NA, Collins MR, Toews ML. Up-regulation of α1B-adrenergic receptors with defects in G protein coupling: ligand-induced protection from receptor instability. Mol. Pharmacol. 2003;64:1126–1135. doi: 10.1124/mol.64.5.1126. [DOI] [PubMed] [Google Scholar]
- Quissell DO, Barzen KA. Secretory response of dispersed rat submandibular cells. II. Mucin secretion. Am. J. Physiol. 1980;238:C99–C106. doi: 10.1152/ajpcell.1980.238.3.C99. [DOI] [PubMed] [Google Scholar]
- Quissell DO, Barzen KA, Gruenert DC, Redman RS, Camden JM, Turner JT. Development and characterization of SV40 immortalized rat submandibular acinar cell lines. In Vitro Cell Dev. Biol. 1997;33:164–173. doi: 10.1007/s11626-997-0137-8. [DOI] [PubMed] [Google Scholar]
- Quissell DO, Watson E, Dowd FJ. Signal transduction mechanisms in salivary gland regulated exocytosis. Crit. Rev. Oral Biol. Med. 1992;3:83–107. doi: 10.1177/10454411920030010701. [DOI] [PubMed] [Google Scholar]
- Slomiany BL, Slomiany A. Src-kinase-dependent epidermal growth factor receptor transactivation in salivary mucin secretion in response to beta-adrenergic G-protein-coupled receptor activation. Inflammopharmacology. 2004;12:233–245. doi: 10.1163/1568560042342329. [DOI] [PubMed] [Google Scholar]
- Steinberg SF. β2-Adrenergic receptor signaling complexes in cardiomyocyte caveolae/lipid rafts. J. Mol. Cell Cardiology. 2004;37:407–415. doi: 10.1016/j.yjmcc.2004.04.018. [DOI] [PubMed] [Google Scholar]
- Toews ML, Prinster SC, Schulte NA. Regulation of alpha-1B adrenergic receptor localization, function, and stability. Life Sci. 2003;74:379–389. doi: 10.1016/j.lfs.2003.09.024. [DOI] [PubMed] [Google Scholar]
- Wang J, Wang L, Zheng J, Anderson JL, Toews ML. Identification of distinct carboxyl-terminal domains mediating internalization and down-regulation of the hamster α1B-adrenergic receptor. Mol. Pharmacol. 2000;57:687–694. doi: 10.1124/mol.57.4.687. [DOI] [PubMed] [Google Scholar]
- Wang J, Zheng J, Anderson JL, Toews ML. A mutation in the hamster α1B-adrenergic receptor that differentiates two steps in the pathway of receptor internalization. Mol. Pharmacol. 1997;52:306–313. doi: 10.1124/mol.52.2.306. [DOI] [PubMed] [Google Scholar]
- Wenham D, Rahmatullah RJ, Rahmatullah M, Hansen CA, Robishaw JD. Differential coupling of α1-adrenoreceptor subtypes to phospholipase C and mitogen activated protein kinase in neonatal rat cardiac myocytes. Eur. J. Pharmacol. 1997;339:77–86. doi: 10.1016/s0014-2999(97)01359-9. [DOI] [PubMed] [Google Scholar]
- Vissink A, Jansma J, Spijkervet FK, Burlage FR, Coppes RP. Oral sequelae of head and neck radiotherapy. Crit. Rev. Oral. Biol. Med. 2003;14:199–212. doi: 10.1177/154411130301400305. [DOI] [PubMed] [Google Scholar]
- Zentner MD, Lin HH, Wen X, Kim KJ, Ann DK. The amiloride-sensitive epithelial sodium channel α-subunit is transcriptionally down-regulated in rat parotid cells by the extracellular signal-regulated protein kinase pathway. J. Biol. Chem. 1998;273:30770–30776. doi: 10.1074/jbc.273.46.30770. [DOI] [PubMed] [Google Scholar]
- Zhong H, Minneman KP. α1-Adrenoceptor subtypes. Eur. J. Pharmacol. 1999;375:261–276. doi: 10.1016/s0014-2999(99)00222-8. [DOI] [PubMed] [Google Scholar]