

Abstract
COPD is one of the leading causes of death worldwide and the age-adjusted mortality for this disease has risen significantly over the past thirty years. Current pharmacological treatments do not effectively address the inflammatory and apoptotic mechanisms that are critical in the development of this disease. Thus, despite therapy, patients typically experience a continued deterioration of their clinical status. Markers of oxidative stress are increased in the lungs of COPD patients and epidemiologic and animal studies indicate that antioxidants can protect the lungs from the damaging effects of cigarette smoke. To date, however, clinical trials of antioxidants for COPD have yielded disappointing results. This review discusses the pharmokinetic factors that limit the use of exogenous antioxidants as a treatment for this disease. In addition, it addresses strategies to overcome these limitations so that the beneficial properties of antioxidants can be translated into effective therapies for COPD patients.
Keywords: Antioxidants, Chronic Obstructive Pulmonary Disease, Emphysema, Inflammation, Cigarette Smoke, Bioavailability
1. Limitations of current pharmacotherapeutic agents in the treatment of COPD
Chronic Obstructive pulmonary disease (COPD) affects 13.5 million Americans(1) and now ranks as the fourth leading cause of death nationwide. While great strides have been made in the treatment of cardiovascular diseases, the age-adjusted mortality for chronic obstructive pulmonary disease (COPD) has actually risen 71% over the past thirty years(2). Indeed, estimates predict that COPD will be the third leading cause of death and the fifth leading cause of disability worldwide by the year 2020(3, 4). In order to improve these trends in the future, it is imperative that pharmacologic agents be developed that can halt or reverse the pathophysiologic changes that occur in this disease. Current guidelines for COPD management advocate the use of inhaled β2-agonists, inhaled anticholinergics and inhaled corticosteroids for symptomatic management(5). Long acting bronchodilators including the anti-cholinergic tiotropium, given once daily, and the β2-agonists salmeterol and formoterol given twice daily all improve lung function, quality of life and reduce the time to first exacerbation when compared to placebo(6-8). Likewise, several clinical trials have shown that inhaled steroids significantly reduce the rate of exacerbations in COPD(9, 10). Importantly, a recent study demonstrated that combination therapy with salmeterol and fluticasone proprionate reduced lymphocytic inflammation in the lung tissue of COPD patients(11). Despite these beneficial effects, neither bronchodilators nor inhaled steroids are able to alter the rate of decline of lung function(12) or improve survival(9) for this disease. This may be due in part to the inability of these drugs to fully reverse the inflammatory changes that occur in this disease(13, 14). Furthermore, these agents do not alter the oxidative or apoptotic processes that are so critical to the pathogenesis of this disease(15, 16). As a result, COPD patients experience an unabated progression of their disease that decreases their quality of life and increases their risk of premature death.
Cigarette smoke exposes the lung to extreme levels of oxidative stress(17). These smoke-derived oxidants damage epithelial cells of the lower respiratory tract by causing direct injury to membrane lipids, proteins, carbohydrates and DNA. The importance of oxidative stress has been confirmed by several studies that have identified the presence of markers of free radical damage in patients with COPD. Increased levels of 8-hydroxy-deoxyguansoine were detected in the urine of COPD patients(18) and elevated levels of 3-nitrotyrosine(19) and lung lipid peroxidation products(20) were noted in the airway cells and epithelium of COPD patients and these markers demonstrated a strong correlation with disease severity as measured by FEV1. Cigarette smoke exposure induced the expression of IL-1β, IL-8 and GM-CSF in human bronchial epithelial cells via the activation of both the NF-κB and MAPK pathways(21). Importantly, the smoke-mediated induction of MAPK and NF-κB signaling in these cells was blocked by the administration of the antioxidant epigallocatechin gallate (EGCG)(22). This data indicates that redox factors have a vitally important role in modulating intracellular signaling events that regulate the inflammatory responses to cigarette smoke exposure. In addition to its inflammatory effects, oxidative stress promotes alveolar cell apoptosis and emphysema formation by blocking the binding of vascular endothelial cell growth factor to its receptor(23). Thus, the oxidant/anti-oxidant balance in the lung has critical effects on the inflammatory and apoptotic responses that are involved in this disease. Unfortunately, neither bronchodilators nor inhaled steroids are able to effectively combat the oxidative stress that occurs in the lungs of patients with COPD(24). Indeed, this may account for the inability of these agents to significantly modulate the clinical course of this disease(25).
2. Redox Regulation of TNF Signaling
The binding of TNF to the TNF receptor (TNFR) has been linked to apoptosis, proliferation and the activation of NF-κB and c-Jun N-terminal kinase (JNK)(26). By affecting these key cell signaling processes, TNF is able to induce the development of smoking related lung diseases(27-29). TNF-α levels are elevated in the lungs of smokers(30) and COPD patients(31) and the absence of the TNF receptor renders mice resistant to smoke-induced inflammation(27). Moreover, animal studies have shown that mice that lack the TNF receptors R1 and R2 are protected against both elastase(32) and cigarette smoke-induced emphysema(33, 34). Though TNF is critical in the pathogenesis of COPD, the mechanisms by which cigarette smoke alters TNF signaling remain to be determined. Several studies, however, indicate that oxidants have a central role in this process(35, 36). It is estimated that each cigarette puff contains 1014 free radicals(17). These smoke-derived oxidants trigger TNF signaling by directly stimulating the receptor(36) or by activating TNF-receptor associated proteins such as RIP (Receptor Interacting Protein) and TRAF2 (TNF receptor associated factor-2)(37) (see arrows in Figure 1). In addition, reactive oxygen species cause apoptosis signaling kinase-1 (ASK-1), a MAP Kinase Kinase that is triggered by TNF(38), to dissociate from thioredoxin thus freeing it to activate JNK(39, 40). Aside from enhancing the phosphorylation of JNK, oxidants are capable of sustaining JNK signaling by inactivating MAPK phosphatases (MKPs) that return JNK to its basal state(41). Importantly, oxidants can cooperate with TNF in the activation of both NF-κB (42) and AP-1(43). This is critical since the activation of these transcription factors have been linked to cigarette smoke-induced lung inflammation(29, 44).
Figure 1. JNK Stimulation by Cigarette Smoke-Derived Oxidants.
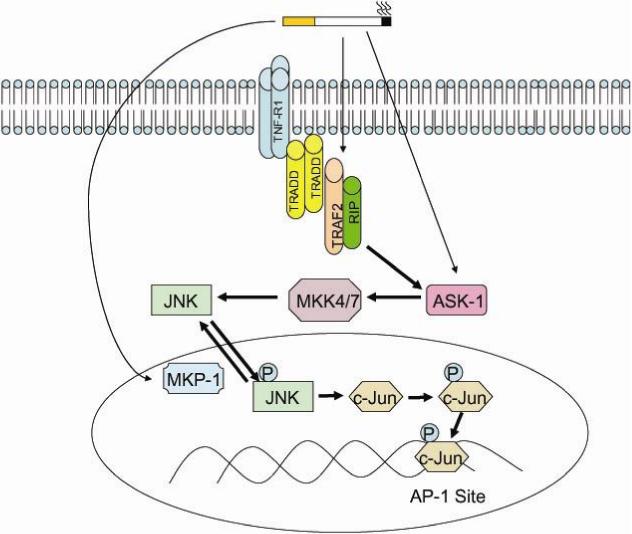
Oxidants produced by cigarette smoke activate JNK by directly stimulating TRAF2 and RIP of the TNF receptor complex. These smoke-derived oxidants also activate apoptosis signaling kinase-1 (ASK-1), an important MAPK Kinase. Furthermore, oxidants can maintain JNK in its active state by inactivating map kinase phosphatases (MKP-1) thereby preventing the dephosphorylation of JNK.
The lung has a rich network of enzymatic antioxidants to protect itself from this oxidative burden including superoxide dismutase(SOD) and glutathione peroxidase(GPX)(45) (Figure 2). SOD1 which is located in the cytosol and is the primary SOD of the lung(46) detoxifies superoxide by converting it to hydrogen peroxide. This can then be further detoxified by enzymes like GPX which convert hydrogen peroxide into water(47). Indeed, the classical GPX, GPX1, has anti-inflammatory properties in mice(48, 49) and can prevent the stress-induced activation of MAPK proteins in vivo(50).
Figure 2. Antioxidant Defenses Against Cigarette Smoke-Derived Free Radicals.

Cigarette smoke produces oxidants such as H2O2 that are able to traverse the cell membrane and it can stimulate oxidant producing enzymes on the cell surface that result in the production of superoxide and nitric oxide. In addition, the enhanced metabolic stress caused by cigarette smoke can increase superoxide production within the cell. The lung epithelium has a network of cellular antioxidants functions to detoxify superoxide and hydrogen peroxide. If this system is overwhelmed toxic radicals can accumulate and cause cell damage and death.
TNF Signaling and Apoptosis in COPD
Aside from promoting inflammation, another important effect of oxidant-mediated TNF stimulation is the induction of apoptotic responses in the lung(51). TNF-α receptor binding initiates two intracellular events that have divergent effects on cell survival and proliferation (see Figure 3). Upon stimulation, TNFR1 recruits TNFR1-associated death domain (TRADD) which then forms two signaling complexes. Complex I (pro-survival) consists of TNFR1, TRADD, Receptor Interacting Protein (RIP) and TNF receptor associated factor 2 (TRAF2). This complex leads to the rapid activation of NF-κB which antagonizes apoptotic responses(52) and promotes cellular proliferation(53, 54). NF-κB confers resistance to TNF toxicity by stimulating the expression of Gadd45β which suppresses sustained JNK activation(55). This is critical since prolonged JNK activation will promote apoptosis by activating caspase-8(56-58) and inhibiting the anti-apoptotic proteins Bcl-2 and Bcl-x(59, 60). On the other hand, complex II (pro-death), which is comprised of TRADD, RIP, FAS-associated death domain (FADD) and caspase-8, activates caspase-8 which promotes apoptosis through the activation of a cascade of cellular caspases(61, 62). Ultimately, the effect of TNF on cell survival and proliferation is determined by the balance of these signaling complexes. This balance is influenced by the cell type and the co-stimulatory factors to which the cell is exposed. Indeed, hepatocytes that normally proliferate following TNF-α stimulation under basal conditions(63) will undergo cell death when co-treated with ethanol(64). COPD patients have an increase in apoptosis(16, 65, 66) and cellular proliferation(16) in their lung. It has been theorized that an imbalance in apoptosis and proliferation could result in a net loss of lung cells and tissue(16). Indeed, Calabrese and colleagues demonstrated that alveolar epithelial cell apoptosis increased disproportionately to alveolar cell proliferation in emphysema(67). Tuder and colleagues have shown that the SOD mimetic M40419 can prevent apoptosis and emphysema formation which is induced in the lungs of rats by VEGF receptor blockade(23). Furthermore, deficiencies in antioxidant responses have been linked to apoptosis and emphysema formation in mice following cigarette smoke exposure(68). Thus, these findings suggest that antioxidants can influence disease development by altering the apoptotic and proliferative responses that are mediated by TNF in response to cigarette smoke exposure in the lung.
Figure 3. TNF Regulation of Cell Death and Survival.

TNF binding to TNFR1 stimulates two signaling complexes with opposing effects on cell survival. Complex I (pro-survival) triggers the NF-κB pathway which up regulates the expression of XIAP, a protease that inhibits apoptosis by blocking the caspase signaling cascade. On the other hand, complex II (pro-death) promotes apoptosis by stimulating Fas associated death domain (FADD). This induces apoptosis by stimulating cytochrome c release from the mitochondria and by activating caspases-3, -8 and -10. Cell fate is influenced by the balance of these signaling effects.
4. Impact of Antioxidants on Lung Health
Evidence from several epidemiologic studies supports the assertion that antioxidants can preserve lung health and prevent the development of COPD symptoms. The Second National Health and Nutrition Examination Survey (NHANES II) found that dietary and serum levels of vitamin C inversely correlated with chronic respiratory symptoms(69) and a study by Hu and colleagues showed that dietary vitamin C intake was positively associated with lung function(70). These reports were complemented by NHANES III which surveyed the lung health effects of dietary and serum levels of vitamin C, vitamin E, β-carotene and selenium in 18,162 subjects. NHANES III found that dietary consumption of vitamin E and carotene was positively associated with lung function as determined by FEV1(71) although only vitamin E consumption continued to show a beneficial effect after stratifying for smoking status. For the serum studies, each antioxidant evaluated showed a positive correlation with FEV1 though again the effects were highly dependent on the smoking history of the individual. Importantly, vitamin E and selenium had the strongest positive relationship with FEV1 amongst smokers while the beneficial effects of β-carotene were completely eliminated in heavy smokers. The protective effects of vitamin E were further confirmed in a cross sectional analysis of 1,616 subjects in western New York(72). Multiple linear regression analyses of this cohort found a strong correlation between FEV1 and serum vitamin E levels after accounting for smoking status. In fact, a reduction of one standard deviation in serum vitamin E levels was equivalent to the negative effects of 1 to 2 years of aging in the lung. Together, these results signify that antioxidants maintain lung function and deter the development of COPD symptoms. In addition, the findings indicate that certain antioxidants (i.e. vitamin E) may be more effective at counteracting the damaging effects of cigarette smoke-derived oxidants.
Flavanoids are a class of polyphenolic compounds with potent anti-inflammatory and antioxidative effects that are present in high quantities in solid fruits and teas(73). The MORGEN study evaluated the impact of these compounds on FEV1 and COPD symptoms in a cohort of 13,651 subjects(74). Total flavanoid intake was associated with improved lung function and decreased cough and breathlessness. When the flavanoids were divided into subclasses (i.e. catechins, flavanols and flavones), it was evident that the majority of the favorable effects were associated with catechin consumption. Indeed, after adjusting for smoking status, subjects with the highest catechin intake had an FEV1 that was 130 ml greater than those in the lowest quintile. Furthermore, high catechin intake nearly decreased by half the symptoms of cough, phlegm and breathlessness in the study subjects. In contrast, flavanol and flavone intake was independently associated with chronic cough only. Consistent with NHANES III, the MORGEN study demonstrated that antioxidant consumption had a strong positive correlation with lung health as assessed by FEV1 and respiratory symptoms. Moreover, the beneficial effects of these compounds varied depending on the class of antioxidants. This variation is likely due to the intrinsic potency and bioavailability of the various antioxidant classes. Indeed, bioavailability will have a critical influence on the beneficial effects of antioxidants in the lung.
Given the evidence from these epidemiologic studies, researchers have sought to determine whether antioxidant administration would improve disease outcomes in COPD. The agent most commonly used for these studies is n-acetylcysteine (NAC), a known precursor of glutathione(75, 76). The advantage of NAC is that it has recognized antioxidant properties, an established safety profile and is relatively easy to tolerate in its oral form(77, 78). Several controlled trials have shown that prolonged treatment with NAC significantly improved pulmonary symptoms and decreased the frequency and severity of disease exacerbations(79, 80). On the other hand, a recent clinical trial determined that NAC treatment did not modify the clinical course of acute exacerbations of COPD(81). Furthermore, the BRONCUS study, which evaluated the effect of NAC on COPD progression, found that it did not alter the yearly rate of decline of FEV1(82). Subanalyses, however, did show that NAC lowered the rate of exacerbation in patients who were not taking inhaled steroids and decreased air trapping as measured by the FRC. Nevertheless, the negative result for the primary outcome of the BRONCUS study has dampened the enthusiasm for the use of antioxidants for this disease.
5. The Lung Bioavailability of Exogenous Antioxidants
The ability of a compound to act as a free radical scavenger is related to its standard one-electron reduction potential (E°’), a measure of the reactivity of an antioxidant as a hydrogen or electron donor under standard conditions. A lower E°’ signifies that it requires less energy for an agent to serve as a donor thus indicating that it has more potent antioxidant activity. Dietary and endogenous antioxidants have differing reduction potentials. Ascorbate (Vitamin C) has a very low reduction potential and thus a high antioxidant capacity (280 mV) while α-tocopherol (480 mV), Epigallocatechin gallate (430 mV) and flavanoids (510 mV) have comparable levels of antioxidant activity(83, 84). Interestingly, the antioxidant potency of all these compounds is significantly greater than glutathione (920 mV)(83), which is a key antioxidant present in the epithelial lining fluid of the lung(85). However, it is important to note that the physiologic role an antioxidant plays depends not only on its reduction potential but also on its concentration. Although glutathione has a low reduction potential, its concentration in the lung is much higher than ascorbate or α-tocopherol(86). Thus, it contributes significantly to the free radical scavenging capacity of the lung(87). Indeed, the ability of an antioxidant to achieve relevant concentrations within the plasma or lung tissue is at least as important as its potency in determining its protective effects from the damaging consequences of cigarette smoke-derived oxidants.
Ultimately, the beneficial effects of an antioxidant will be determined by its unique pharmokinetic properties. For this reason, it is critical that the bioavailability of an antioxidant be carefully examined prior to evaluating its potential health effects. Though several antioxidant compounds exert potent anti-inflammatory and anti-apoptotic effects in vitro, these effects occur at concentrations that are often impossible to achieve in vivo. As an example, in cultured lung epithelial cells, NAC administration prevented chromatin remodeling by blocking the oxidant-mediated decrease in HDAC activity(88). This is critical since chromatin remodeling and altered HDAC activity are key factors in the pathogenesis of the human disease(89). However, the positive effects that were observed in this study were obtained at NAC concentrations that are impossible to achieve in the human lung. In fact, the treatment regimen of NAC utilized in the BRONCUS study does not generate sustained increases in plasma or lung tissue concentrations of cysteine or glutathione(90). Thus, the dose used in the BRONCUS study would be insufficient to increase the antioxidant capacity of the lungs and alter key signaling events that regulate the development of the disease.
Poor lung bioavailability is an important factor that has limited the use of antioxidant supplementation as a treatment for COPD. There are several formidable challenges that affect the bioavailability of dietary and exogenous antioxidants including the release from food structure (bioaccessibility), the passage from the gut lumen into the body (absorption) and host metabolism. For example, carotenoids such as lycopene and β-carotene are lipophilic compounds that function as photoprotective pigments in plants(91). While within the leaf structure these antioxidants are protected; however, their anti-oxidative properties can be compromised by light and heat exposure during food processing and preparation(92). Moreover, carotenoids and other plant-derived antioxidants such as α-tocopherol are hydrophobic and require the close proximity of dietary lipids in order to be accessible for absorption(93). When fats are present in the gut lumen, the enterocyte can package these antioxidants in chylomicrons, which are then able to enter the circulation(94). However, if these lipophilic antioxidants are consumed apart from a fat-rich meal, then the amount actually absorbed can be negligible.
Once dietary or supplemental antioxidants are absorbed, they are carried via the portal vein to the liver where they undergo extensive metabolism by phase II enzymes. These enzyme modifications can dramatically alter the antioxidant potency and half life of these compounds. Resveratrol (red wine) and EGCG (green tea) are dietary compounds that are readily absorbed into the bloodstream after ingestion and are known to exert protective antioxidant effects on lung epithelial cells in culture(95, 96). However, these agents undergo extensive phase II modifications that severely limit their bioavailability. In fact, resveratrol experiences such rapid sulfation and glucuronidation by the liver that plasma levels in humans are non-detectable within minutes after consumption(97). EGCG also goes through extensive phase II metabolism including glucuronidation and methylation(98, 99). These modifications prevent tissue penetration and accelerate clearance from the plasma(99). Thus, although intravenous administration is able to achieve high levels of free EGCG in the lung and plasma of rodents(100), this effect is transient as liver and lung tissue enzymes rapidly transform EGCG into its inactive form. Furthermore, multi-drug resistance-related proteins (Mrp) 1 and 2 actively pump EGCG out of the intracellular compartment of cells which limits its ability to alter redox regulated intracellular signaling events(101). Similarly, α-tocopherol supplements are well absorbed in the plasma and become distributed in the tissues. However, studies with radiolabeled α-tocopherol show that the increase in plasma levels induced by the supplement are offset by a concomitant decrease in pre-existing α-tocopherol(102). Thus, despite aggressive supplementation, total levels of this vitamin remain largely unaltered in the plasma. From these studies it is clear that the pharmokinetic processes that control antioxidant absorption, activity and clearance are quite complex. An enhanced understanding of the factors regulating antioxidant homeostasis will be needed in order to develop strategies to generate clinically relevant increases in the antioxidant capacity in the lung.
6. Future Directions in Antioxidant Therapy
Cigarette smoke exposes the lung to extreme oxidative stress(103) and the accumulation of free radical damage is linked to the development of COPD(19). Epidemiological studies indicate that antioxidant administration decreases the incidence and severity of COPD(104); however, controlled, clinical trials of antioxidants have yielded largely disappointing results to date(82). Our laboratory has recently published that the transgenic expression of human superoxide dismutase-1 (SOD) prevents cigarette smoke-induced inflammation and emphysema formation in mice(105). These findings directly establish the ability of antioxidants to alter the pathogenic responses that are central to the development of this disease. Before these findings can be translated into effective therapies for patients, issues of bioavailability need to be carefully addressed. SOD transgenic mice exhibit a four-fold increase in superoxide dismutase-1 activity at baseline. Currently, there are no compounds that upon administration will generate similar enhancements of antioxidant activity in the lung. Thus, realizing the therapeutic promise of antioxidants will require significant advances in our understanding of their pharmokinetics coupled with new and innovative strategies of drug delivery. Importantly, in our transgenic model, the enhanced expression of SOD predated the exposure to cigarette smoke. Thus, these results demonstrate that augmenting SOD activity can prevent disease formation; however, it remains to be determined whether SOD or other antioxidants would be able to reverse the damaging effects of chronic cigarette smoke exposure in the lung.
As discussed above, dietary and host factors severely limit the absorption, concentration and half-life of exogenously administered antioxidants. An improved understanding of these factors would enable researchers to design potent antioxidants that would be better able to maintain sustained concentrations within the lung tissue compartment. This could involve creating new antioxidants that are resistant to phase II modification or altering existing antioxidants so that they would retain their potency and bioavailability even after enzymatic modification. Indeed, the novel membrane permeable free radical scavenger Tempol has shown promise in reducing lung inflammation in response to shock(106). Additionally, the development of improved inhaled delivery techniques would allow clinically relevant concentrations of antioxidants to be deposited in the lung while avoiding the first pass metabolism that occurs during systemic absorption(107). Current inhalational devices deliver the majority of the drug to large and medium size airways of the lung(108). This ignores the small airways and alveolar regions which are key sites in the pathogenesis of the human disease(109). Improvements in distal lung deposition would be required prior to using this technology to deliver effective antioxidant treatment for patients. This may be possible in the near future as there are promising inhalational techniques that appear to produce uniform distribution in the distal lung(110). Future antioxidant strategies may also seek to counteract the cigarette smoke-mediated induction of oxidant forming enzymes such as NADPH oxidase(111) and xanthine oxidase(112) that are present within the lung epithelium. Indeed, these enzymes can contribute significantly to the oxidative injury that is mediated by cigarette smoke exposure (see Figure 2). In addition, if the exact mechanisms of the effects of antioxidants were established, it would enable researchers to identify the regulatory proteins that are modulated by antioxidant activity in the lung. Once these target proteins are determined, pharmacologic strategies could be developed to alter their levels or activity. This targeted approach could reproduce the benefits of antioxidants while overcoming the present pharmacodynamic issues that have hindered the efficacy of antioxidant treatments. The combined approaches that are outlined here will lead to the development of effective treatment strategies for this disease.
Acknowledgments
Sources of Funding: The Flight Attendant Medical Research Institute (FAMRI), The Alpha-1 Anti-trypsin Foundation and NIH (1R01HL086936-01)
Abbreviations
- COPD
Chronic Obstructive Pulmonary Disease
- FEV1
Forced Expiratory Volume in one Second
- IL-1β
Interleukin-1-beta
- GM-CSF
Granulocyte Monocyte Colony Stimulating Factor
- NF-κB
Nuclear Factor Kappa B
- MAPK
Mitogen Activated Protein Kinase
- EGCG
Epigallocatechin Gallate
- TNF
Tumor Necrosis Factor
- JNK
c-Jun N Terminal Kinase
- RIP
Receptor Interacting Protein
- TRAF
Tumor Necrosis Factor Receptor Associated Factor
- ASK
Apoptosis Signaling Kinase
- MKP
MAPK Phosphatases
- SOD
Superoxide Dismutase
- GPX
Glutathione Peroxidase
- TNFR
Tumor Necrosis Factor Receptor
- TRADD
Tumor Necrosis Factor Receptor Associated Death Domain
- FADD
FAS Associated Death Domain
- XIAP
Human X-Chromosome-Linked Inhibitor of Apoptosis Protein
- NHANES II
National Health and Nutrition Examination Survey
- NAC
N-acetylcysteine
- HDAC
Histone Deacetylase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Coultas DB, Mapel D, Gagnon R, Lydick E. The health impact of undiagnosed airflow obstruction in a national sample of United States adults. Am J Respir Crit Care Med. 2001;164:372–377. doi: 10.1164/ajrccm.164.3.2004029. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Ward E, Hao Y, Thun M. Trends in the leading causes of death in the United States, 1970−2002. JAMA. 2005;294:1255–1259. doi: 10.1001/jama.294.10.1255. [DOI] [PubMed] [Google Scholar]
- 3.Michaud CM, Murray CJ, Bloom BR. Burden of disease--implications for future research. Jama. 2001;285:535–539. doi: 10.1001/jama.285.5.535. [DOI] [PubMed] [Google Scholar]
- 4.Sullivan S, Ramsey S, Lee T. The economic burden of COPD. Chest. 2000;117(supp):5S–9S. doi: 10.1378/chest.117.2_suppl.5s. [DOI] [PubMed] [Google Scholar]
- 5.Pauwels RA, Buist AS, Calverley PM, Jenkins CR, Hurd SS. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summary. Am J Respir Crit Care Med. 2001;163:1256–1276. doi: 10.1164/ajrccm.163.5.2101039. [DOI] [PubMed] [Google Scholar]
- 6.Tashkin DP, Cooper CB. The role of long-acting bronchodilators in the management of stable COPD. Chest. 2004;125:249–259. doi: 10.1378/chest.125.1.249. [DOI] [PubMed] [Google Scholar]
- 7.Donohue JF, Menjoge S, Kesten S. Tolerance to bronchodilating effects of salmeterol in COPD. Respir Med. 2003;97:1014–1020. doi: 10.1016/s0954-6111(03)00131-8. [DOI] [PubMed] [Google Scholar]
- 8.Brusasco V, Hodder R, Miravitlles M, Korducki L, Towse L, Kesten S. Health outcomes following treatment for six months with once daily tiotropium compared with twice daily salmeterol in patients with COPD. Thorax. 2003;58:399–404. doi: 10.1136/thorax.58.5.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calverley PM, Anderson JA, Celli B, Ferguson GT, Jenkins C, Jones PW, Yates JC, Vestbo J. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med. 2007;356:775–789. doi: 10.1056/NEJMoa063070. [DOI] [PubMed] [Google Scholar]
- 10.Alsaeedi A, Sin DD, McAlister FA. The effects of inhaled corticosteroids in chronic obstructive pulmonary disease: a systematic review of randomized placebo-controlled trials. Am J Med. 2002;113:59–65. doi: 10.1016/s0002-9343(02)01143-9. [DOI] [PubMed] [Google Scholar]
- 11.Barnes NC, Qiu YS, Pavord ID, Parker D, Davis PA, Zhu J, Johnson M, Thomson NC, Jeffery PK. Antiinflammatory effects of salmeterol/fluticasone propionate in chronic obstructive lung disease. Am J Respir Crit Care Med. 2006;173:736–743. doi: 10.1164/rccm.200508-1321OC. [DOI] [PubMed] [Google Scholar]
- 12.Vestbo J, Sorensen T, Lange P, Brix A, Torre P, Viskum K. Long-term effect of inhaled budesonide in mild and moderate chronic obstructive pulmonary disease: a randomised controlled trial. Lancet. 1999;353:1819–1823. doi: 10.1016/s0140-6736(98)10019-3. [DOI] [PubMed] [Google Scholar]
- 13.Culpitt SV, Maziak W, Loukidis S, Nightingale JA, Matthews JL, Barnes PJ. Effect of high dose inhaled steroid on cells, cytokines, and proteases in induced sputum in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1999;160:1635–1639. doi: 10.1164/ajrccm.160.5.9811058. [DOI] [PubMed] [Google Scholar]
- 14.Loppow D, Schleiss MB, Kanniess F, Taube C, Jorres RA, Magnussen H. In patients with chronic bronchitis a four week trial with inhaled steroids does not attenuate airway inflammation. Respir Med. 2001;95:115–121. doi: 10.1053/rmed.2000.0960. [DOI] [PubMed] [Google Scholar]
- 15.Zheng T, Kang MJ, Crothers K, Zhu Z, Liu W, Lee CG, Rabach LA, Chapman HA, Homer RJ, Aldous D, et al. Role of cathepsin S-dependent epithelial cell apoptosis in IFN-gamma-induced alveolar remodeling and pulmonary emphysema. J Immunol. 2005;174:8106–8115. doi: 10.4049/jimmunol.174.12.8106. [DOI] [PubMed] [Google Scholar]
- 16.Imai K, Mercer BA, Schulman LL, Sonett JR, D'Armiento JM. Correlation of lung surface area to apoptosis and proliferation in human emphysema. Eur Respir J. 2005;25:250–258. doi: 10.1183/09031936.05.00023704. [DOI] [PubMed] [Google Scholar]
- 17.Pryor WA, Prier DG, Church DF. Electron-spin resonance study of mainstream and sidestream cigarette smoke: nature of the free radicals in gas-phase smoke and in cigarette tar. Environ Health Perspect. 1983;47:345–355. doi: 10.1289/ehp.8347345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Igishi T, Hitsuda Y, Kato K, Sako T, Burioka N, Yasuda K, Sano H, Shigeoka Y, Nakanishi H, Shimizu E. Elevated urinary 8-hydroxydeoxyguanosine, a biomarker of oxidative stress, and lack of association with antioxidant vitamins in chronic obstructive pulmonary disease. Respirology. 2003;8:455–460. doi: 10.1046/j.1440-1843.2003.00490.x. [DOI] [PubMed] [Google Scholar]
- 19.Ichinose M, Sugiura H, Yamagata S, Koari A, Shirato K. Increase in Reactive Nitrogen Species Production in Chronic Obstructive Pulmonary Disease Airways. Am J Respir Crit Care Med. 2000;162:701–706. doi: 10.1164/ajrccm.162.2.9908132. [DOI] [PubMed] [Google Scholar]
- 20.Rahman I, van Schadewijk AA, Crowther AJ, Hiemstra PS, Stolk J, MacNee W, De Boer WI. 4-Hydroxy-2-nonenal, a specific lipid peroxidation product, is elevated in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2002;166:490–495. doi: 10.1164/rccm.2110101. [DOI] [PubMed] [Google Scholar]
- 21.Hellermann GR, Nagy SB, Kong X, Lockey RF, Mohapatra SS. Mechanism of cigarette smoke condensate-induced acute inflammatory response in human bronchial epithelial cells. Respir Res. 2002;3:22. doi: 10.1186/rr172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Syed DN, Afaq F, Kweon MH, Hadi N, Bhatia N, Spiegelman VS, Mukhtar H. Green tea polyphenol EGCG suppresses cigarette smoke condensate-induced NF-kappaB activation in normal human bronchial epithelial cells. Oncogene. 2007;26:673–682. doi: 10.1038/sj.onc.1209829. [DOI] [PubMed] [Google Scholar]
- 23.Tuder RM, Zhen L, Cho CY, Taraseviciene-Stewart L, Kasahara Y, Salvemini D, Voelkel NF, Flores S. Oxidative Stress and Apoptosis Interact and Cause Emphysema Due to Vascular Endothelial Growth Factor Receptor Blockade. Am J. Resp. Cell Mol. Biol. 2003;29:88–97. doi: 10.1165/rcmb.2002-0228OC. [DOI] [PubMed] [Google Scholar]
- 24.Verhoeven GT, Wijkhuijs AJ, Hooijkaas H, Hoogsteden HC, Sluiter W. Effect of an inhaled glucocorticoid on reactive oxygen species production by bronchoalveolar lavage cells from smoking COPD patients. Mediators Inflamm. 2000;9:109–113. doi: 10.1080/096293500411578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barnes PJ. Corticosteroid resistance in airway disease. Proc Am Thorac Soc. 2004;1:264–268. doi: 10.1513/pats.200402-014MS. [DOI] [PubMed] [Google Scholar]
- 26.Dempsey PW, Doyle SE, He JQ, Cheng G. The signaling adaptors and pathways activated by TNF superfamily. Cytokine Growth Factor Rev. 2003;14:193–209. doi: 10.1016/s1359-6101(03)00021-2. [DOI] [PubMed] [Google Scholar]
- 27.Churg A, Dai J, Tai H, Xie C, Wright JL. Tumor necrosis factor-alpha is central to acute cigarette smoke-induced inflammation and connective tissue breakdown. Am J Respir Crit Care Med. 2002;166:849–854. doi: 10.1164/rccm.200202-097OC. [DOI] [PubMed] [Google Scholar]
- 28.Churg A, Wang RD, Tai H, Wang X, Xie C, Dai J, Shapiro SD, Wright JL. Macrophage metalloelastase mediates acute cigarette smoke-induced inflammation via tumor necrosis factor-alpha release. Am J Respir Crit Care Med. 2003;167:1083–1089. doi: 10.1164/rccm.200212-1396OC. [DOI] [PubMed] [Google Scholar]
- 29.Vlahos R, Bozinovski S, Jones JE, Powell J, Gras J, Lilja A, Hansen MJ, Gualano RC, Irving L, Anderson GP. Differential protease, innate immunity, and NF-kappaB induction profiles during lung inflammation induced by subchronic cigarette smoke exposure in mice. Am J Physiol Lung Cell Mol Physiol. 2006;290:L931–945. doi: 10.1152/ajplung.00201.2005. [DOI] [PubMed] [Google Scholar]
- 30.Kuschner WG, D'Alessandro A, Wong H, Blanc PD. Dose-dependent cigarette smoking-related inflammatory responses in healthy adults. Eur Respir J. 1996;9:1989–1994. doi: 10.1183/09031936.96.09101989. [DOI] [PubMed] [Google Scholar]
- 31.Keatings VM, Collins PD, Scott DM, Barnes PJ. Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med. 1996;153:530–534. doi: 10.1164/ajrccm.153.2.8564092. [DOI] [PubMed] [Google Scholar]
- 32.Lucey EC, Keane J, Kuang PP, Snider GL, Goldstein RH. Severity of elastase-induced emphysema is decreased in tumor necrosis factor-alpha and interleukin-1beta receptor-deficient mice. Lab Invest. 2002;82:79–85. doi: 10.1038/labinvest.3780397. [DOI] [PubMed] [Google Scholar]
- 33.Churg A, Wang RD, Tai H, Wang X, Xie C, Wright JL. Tumor necrosis factor-alpha drives 70% of cigarette smoke-induced emphysema in the mouse. Am J Respir Crit Care Med. 2004;170:492–498. doi: 10.1164/rccm.200404-511OC. [DOI] [PubMed] [Google Scholar]
- 34.D'Hulst A,I, Bracke KR, Maes T, De Bleecker JL, Pauwels RA, Joos GF, Brusselle GG. Role of tumour necrosis factor-alpha receptor p75 in cigarette smoke-induced pulmonary inflammation and emphysema. Eur Respir J. 2006;28:102–112. doi: 10.1183/09031936.06.00059305. [DOI] [PubMed] [Google Scholar]
- 35.Han D, Hanawa N, Saberi B, Kaplowitz N. Hydrogen peroxide and redox modulation sensitize primary mouse hepatocytes to TNF-induced apoptosis. Free Radic Biol Med. 2006;41:627–639. doi: 10.1016/j.freeradbiomed.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 36.Pantano C, Shrivastava P, McElhinney B, Janssen-Heininger Y. Hydrogen peroxide signaling through tumor necrosis factor receptor 1 leads to selective activation of c-Jun N-terminal kinase. J Biol Chem. 2003;278:44091–44096. doi: 10.1074/jbc.M308487200. [DOI] [PubMed] [Google Scholar]
- 37.Shen HM, Lin Y, Choksi S, Tran J, Jin T, Chang L, Karin M, Zhang J, Liu ZG. Essential roles of receptor-interacting protein and TRAF2 in oxidative stress-induced cell death. Mol Cell Biol. 2004;24:5914–5922. doi: 10.1128/MCB.24.13.5914-5922.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nishitoh H, Saitoh M, Mochida Y, Takeda K, Nakano H, Rothe M, Miyazono K, Ichijo H. ASK1 is essential for JNK/SAPK activation by TRAF2. Mol Cell. 1998;2:389–395. doi: 10.1016/s1097-2765(00)80283-x. [DOI] [PubMed] [Google Scholar]
- 39.Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. Embo J. 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hsieh CC, Papaconstantinou J. Thioredoxin-ASK1 complex levels regulate ROS-mediated p38 MAPK pathway activity in livers of aged and long-lived Snell dwarf mice. Faseb J. 2006;20:259–268. doi: 10.1096/fj.05-4376com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 42.Janssen-Heininger YM, Macara I, Mossman BT. Cooperativity between oxidants and tumor necrosis factor in the activation of nuclear factor (NF)-kappaB: requirement of Ras/mitogen-activated protein kinases in the activation of NF-kappaB by oxidants. Am J Respir Cell Mol Biol. 1999;20:942–952. doi: 10.1165/ajrcmb.20.5.3452. [DOI] [PubMed] [Google Scholar]
- 43.Manna SK, Mukhopadhyay A, Aggarwal BB. Resveratrol suppresses TNF-induced activation of nuclear transcription factors NF-kappa B, activator protein-1, and apoptosis: potential role of reactive oxygen intermediates and lipid peroxidation. J Immunol. 2000;164:6509–6519. doi: 10.4049/jimmunol.164.12.6509. [DOI] [PubMed] [Google Scholar]
- 44.Walters MJ, Paul-Clark MJ, McMaster SK, Ito K, Adcock IM, Mitchell JA. Cigarette smoke activates human monocytes by an oxidant-AP-1 signaling pathway: implications for steroid resistance. Mol Pharmacol. 2005;68:1343–1353. doi: 10.1124/mol.105.012591. [DOI] [PubMed] [Google Scholar]
- 45.Macnee W, Rahman I. Oxidants and antioxidants as therapeutic targets in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1999;160:S58–65. doi: 10.1164/ajrccm.160.supplement_1.15. [DOI] [PubMed] [Google Scholar]
- 46.Kinnula VL, Crapo JD. Superoxide dismutases in the lung and human lung diseases. Am J Respir Crit Care Med. 2003;167:1600–1619. doi: 10.1164/rccm.200212-1479SO. [DOI] [PubMed] [Google Scholar]
- 47.Flohe L. Glutathione peroxidase. Basic Life Sci. 1988;49:663–668. doi: 10.1007/978-1-4684-5568-7_104. [DOI] [PubMed] [Google Scholar]
- 48.Beck MA, Esworthy RS, Ho YS, Chu FF. Glutathione peroxidase protects mice from viral-induced myocarditis. Faseb J. 1998;12:1143–1149. doi: 10.1096/fasebj.12.12.1143. [DOI] [PubMed] [Google Scholar]
- 49.Esworthy RS, Aranda R, Martin MG, Doroshow JH, Binder SW, Chu FF. Mice with combined disruption of Gpx1 and Gpx2 genes have colitis. Am J Physiol Gastrointest Liver Physiol. 2001;281:G848–855. doi: 10.1152/ajpgi.2001.281.3.G848. [DOI] [PubMed] [Google Scholar]
- 50.Cheng WH, Zheng X, Quimby FR, Roneker CA, Lei XG. Low levels of glutathione peroxidase 1 activity in selenium-deficient mouse liver affect c-Jun N-terminal kinase activation and p53 phosphorylation on Ser-15 in pro-oxidant-induced aponecrosis. Biochem J. 2003;370:927–934. doi: 10.1042/BJ20021870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Franek WR, Horowitz S, Stansberry L, Kazzaz JA, Koo HC, Li Y, Arita Y, Davis JM, Mantell AS, Scott W, et al. Hyperoxia inhibits oxidant-induced apoptosis in lung epithelial cells. J Biol Chem. 2001;276:569–575. doi: 10.1074/jbc.M004716200. [DOI] [PubMed] [Google Scholar]
- 52.Liu ZG, Hsu H, Goeddel DV, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kappaB activation prevents cell death. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- 53.Ho YS, Chen CH, Wang YJ, Pestell RG, Albanese C, Chen RJ, Chang MC, Jeng JH, Lin SY, Liang YC, et al. Tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) induces cell proliferation in normal human bronchial epithelial cells through NFkappaB activation and cyclin D1 up-regulation. Toxicol Appl Pharmacol. 2005;205:133–148. doi: 10.1016/j.taap.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 54.Mangelus M, Galron R, Naor Z, Sokolovsky M. Involvement of nuclear factor-kappaB in endothelin-A-receptor-induced proliferation and inhibition of apoptosis. Cell Mol Neurobiol. 2001;21:657–674. doi: 10.1023/A:1015195803445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.De Smaele E, Zazzeroni F, Papa S, Nguyen DU, Jin R, Jones J, Cong R, Franzoso G. Induction of gadd45beta by NF-kappaB downregulates pro-apoptotic JNK signalling. Nature. 2001;414:308–313. doi: 10.1038/35104560. [DOI] [PubMed] [Google Scholar]
- 56.Deng Y, Ren X, Yang L, Lin Y, Wu X. A JNK-dependent pathway is required for TNFalpha-induced apoptosis. Cell. 2003;115:61–70. doi: 10.1016/s0092-8674(03)00757-8. [DOI] [PubMed] [Google Scholar]
- 57.Gao M, Labuda T, Xia Y, Gallagher E, Fang D, Liu YC, Karin M. Jun turnover is controlled through JNK-dependent phosphorylation of the E3 ligase Itch. Science. 2004;306:271–275. doi: 10.1126/science.1099414. [DOI] [PubMed] [Google Scholar]
- 58.Chang L, Kamata H, Solinas G, Luo JL, Maeda S, Venuprasad K, Liu YC, Karin M. The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIP(L) turnover. Cell. 2006;124:601–613. doi: 10.1016/j.cell.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 59.Yamamoto K, Ichijo H, Korsmeyer SJ. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol Cell Biol. 1999;19:8469–8478. doi: 10.1128/mcb.19.12.8469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fan M, Goodwin M, Vu T, Brantley-Finley C, Gaarde WA, Chambers TC. Vinblastine-induced phosphorylation of Bcl-2 and Bcl-XL is mediated by JNK and occurs in parallel with inactivation of the Raf-1/MEK/ERK cascade. J Biol Chem. 2000;275:29980–29985. doi: 10.1074/jbc.M003776200. [DOI] [PubMed] [Google Scholar]
- 61.Alikhani M, Alikhani Z, Raptis M, Graves DT. TNF-alpha in vivo stimulates apoptosis in fibroblasts through caspase-8 activation and modulates the expression of pro-apoptotic genes. J Cell Physiol. 2004;201:341–348. doi: 10.1002/jcp.20067. [DOI] [PubMed] [Google Scholar]
- 62.Aggarwal S, Gollapudi S, Gupta S. Increased TNF-alpha-induced apoptosis in lymphocytes from aged humans: changes in TNF-alpha receptor expression and activation of caspases. J Immunol. 1999;162:2154–2161. [PubMed] [Google Scholar]
- 63.Iocca HA, Isom HC. Tumor necrosis factor-alpha acts as a complete mitogen for primary rat hepatocytes. Am J Pathol. 2003;163:465–476. doi: 10.1016/s0002-9440(10)63676-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pastorino JG, Hoek JB. Ethanol potentiates tumor necrosis factor-alpha cytotoxicity in hepatoma cells and primary rat hepatocytes by promoting induction of the mitochondrial permeability transition. Hepatology. 2000;31:1141–1152. doi: 10.1053/he.2000.7013. [DOI] [PubMed] [Google Scholar]
- 65.Segura-Valdez L, Pardo A, Gaxiola M, Uhal BD, Becerril C, Selman M. Upregulation of gelatinases A and B, collagenases 1 and 2, and increased parenchymal cell death in COPD. Chest. 2000;117:684–694. doi: 10.1378/chest.117.3.684. [DOI] [PubMed] [Google Scholar]
- 66.Yokohori N, Aoshiba K, Nagai A. Increased levels of cell death and proliferation in alveolar wall cells in patients with pulmonary emphysema. Chest. 2004;125:626–632. doi: 10.1378/chest.125.2.626. [DOI] [PubMed] [Google Scholar]
- 67.Calabrese F, Giacometti C, Beghe B, Rea F, Loy M, Zuin R, Marulli G, Baraldo S, Saetta M, Valente M. Marked alveolar apoptosis/proliferation imbalance in end-stage emphysema. Respir Res. 2005;6:14. doi: 10.1186/1465-9921-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rangasamy T, Cho CY, Thimmulappa RK, Zhen L, Srisuma SS, Kensler TW, Yamamoto M, Petrache I, Tuder RM, Biswal S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest. 2004;114:1248–1259. doi: 10.1172/JCI21146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schwartz J, Weiss ST. Dietary factors and their relation to respiratory symptoms. The Second National Health and Nutrition Examination Survey. Am J Epidemiol. 1990;132:67–76. doi: 10.1093/oxfordjournals.aje.a115644. [DOI] [PubMed] [Google Scholar]
- 70.Hu G, Zhang X, Chen J, Peto R, Campbell TC, Cassano PA. Dietary vitamin C intake and lung function in rural China. Am J Epidemiol. 1998;148:594–599. doi: 10.1093/oxfordjournals.aje.a009685. [DOI] [PubMed] [Google Scholar]
- 71.Hu G, Cassano PA. Antioxidant nutrients and pulmonary function: the Third National Health and Nutrition Examination Survey (NHANES III). Am J Epidemiol. 2000;151:975–981. doi: 10.1093/oxfordjournals.aje.a010141. [DOI] [PubMed] [Google Scholar]
- 72.Schunemann HJ, Grant BJ, Freudenheim JL, Muti P, Browne RW, Drake JA, Klocke RA, Trevisan M. The relation of serum levels of antioxidant vitamins C and E, retinol and carotenoids with pulmonary function in the general population. Am J Respir Crit Care Med. 2001;163:1246–1255. doi: 10.1164/ajrccm.163.5.2007135. [DOI] [PubMed] [Google Scholar]
- 73.Hertog MG, Hollman PC, Katan MB, Kromhout D. Intake of potentially anticarcinogenic flavonoids and their determinants in adults in The Netherlands. Nutr Cancer. 1993;20:21–29. doi: 10.1080/01635589309514267. [DOI] [PubMed] [Google Scholar]
- 74.Tabak C, Arts IC, Smit HA, Heederik D, Kromhout D. Chronic obstructive pulmonary disease and intake of catechins, flavonols, and flavones: the MORGEN Study. Am J Respir Crit Care Med. 2001;164:61–64. doi: 10.1164/ajrccm.164.1.2010025. [DOI] [PubMed] [Google Scholar]
- 75.Kasielski M, Nowak D. Long-term administration of N-acetylcysteine decreases hydrogen peroxide exhalation in subjects with chronic obstructive pulmonary disease. Respir Med. 2001;95:448–456. doi: 10.1053/rmed.2001.1066. [DOI] [PubMed] [Google Scholar]
- 76.Sadowska AM, van Overveld FJ, Gorecka D, Zdral A, Filewska M, Demkow UA, Luyten C, Saenen E, Zielinski J, De Backer WA. The interrelationship between markers of inflammation and oxidative stress in chronic obstructive pulmonary disease: modulation by inhaled steroids and antioxidant. Respir Med. 2005;99:241–249. doi: 10.1016/j.rmed.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 77.Olsson B, Johansson M, Gabrielsson J, Bolme P. Pharmacokinetics and bioavailability of reduced and oxidized N-acetylcysteine. Eur J Clin Pharmacol. 1988;34:77–82. doi: 10.1007/BF01061422. [DOI] [PubMed] [Google Scholar]
- 78.Pendyala L, Creaven PJ. Pharmacokinetic and pharmacodynamic studies of N-acetylcysteine, a potential chemopreventive agent during a phase I trial. Cancer Epidemiol Biomarkers Prev. 1995;4:245–251. [PubMed] [Google Scholar]
- 79.Pela R, Calcagni AM, Subiaco S, Isidori P, Tubaldi A, Sanguinetti CM. N-acetylcysteine reduces the exacerbation rate in patients with moderate to severe COPD. Respiration. 1999;66:495–500. doi: 10.1159/000029447. [DOI] [PubMed] [Google Scholar]
- 80.Boman G, Backer U, Larsson S, Melander B, Wahlander L. Oral acetylcysteine reduces exacerbation rate in chronic bronchitis: report of a trial organized by the Swedish Society for Pulmonary Diseases. Eur J Respir Dis. 1983;64:405–415. [PubMed] [Google Scholar]
- 81.Black PN, Morgan-Day A, McMillan TE, Poole PJ, Young RP. Randomised, controlled trial of N-acetylcysteine for treatment of acute exacerbations of chronic obstructive pulmonary disease [ISRCTN21676344]. BMC Pulm Med. 2004;4:13. doi: 10.1186/1471-2466-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Decramer M, Rutten-van Molken M, Dekhuijzen PN, Troosters T, van Herwaarden C, Pellegrino R, van Schayck CP, Olivieri D, Del Donno M, De Backer W, et al. Effects of N-acetylcysteine on outcomes in chronic obstructive pulmonary disease (Bronchitis Randomized on NAC Cost-Utility Study, BRONCUS): a randomised placebo-controlled trial. Lancet. 2005;365:1552–1560. doi: 10.1016/S0140-6736(05)66456-2. [DOI] [PubMed] [Google Scholar]
- 83.Jovanavic S, Steenken S, Simic M. Reduction potentials of flavanoid and model phenoxyl radicals. J Chem Soc Perkins Trans. 1996;2:2497–2503. [Google Scholar]
- 84.Jovanavic S, Hara Y, Steenken S, Simic M. Antioxidant potential of theaflavins. A pulse radiolysis study. J Am Chem Soc. 1997;119:5337–5343. [Google Scholar]
- 85.Cantin AM, North SL, Hubbard RC, Crystal RG. Normal alveolar epithelial lining fluid contains high levels of glutathione. J Appl Physiol. 1987;63:152–157. doi: 10.1152/jappl.1987.63.1.152. [DOI] [PubMed] [Google Scholar]
- 86.van der Vliet A, O'Neill CA, Cross CE, Koostra JM, Volz WG, Halliwell B, Louie S. Determination of low-molecular-mass antioxidant concentrations in human respiratory tract lining fluids. Am J Physiol. 1999;276:L289–296. doi: 10.1152/ajplung.1999.276.2.L289. [DOI] [PubMed] [Google Scholar]
- 87.Comhair SA, Lewis MJ, Bhathena PR, Hammel JP, Erzurum SC. Increased glutathione and glutathione peroxidase in lungs of individuals with chronic beryllium disease. Am J Respir Crit Care Med. 1999;159:1824–1829. doi: 10.1164/ajrccm.159.6.9810044. [DOI] [PubMed] [Google Scholar]
- 88.Moodie FM, Marwick JA, Anderson CS, Szulakowski P, Biswas SK, Bauter MR, Kilty I, Rahman I. Oxidative stress and cigarette smoke alter chromatin remodeling but differentially regulate NF-kappaB activation and proinflammatory cytokine release in alveolar epithelial cells. FASEB J. 2004;18:1897–1899. doi: 10.1096/fj.04-1506fje. [DOI] [PubMed] [Google Scholar]
- 89.Ito K, Ito M, Elliott WM, Cosio B, Caramori G, Kon OM, Barczyk A, Hayashi S, Adcock IM, Hogg JC, et al. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N Engl J Med. 2005;352:1967–1976. doi: 10.1056/NEJMoa041892. [DOI] [PubMed] [Google Scholar]
- 90.Bridgeman MM, Marsden M, Selby C, Morrison D, MacNee W. Effect of N-acetyl cysteine on the concentrations of thiols in plasma, bronchoalveolar lavage fluid, and lung tissue. Thorax. 1994;49:670–675. doi: 10.1136/thx.49.7.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cogdell R. The functions of pigments in chloroplasts: 1988 Academic Press. [Google Scholar]
- 92.Shi J, Le Maguer M. Lycopene in tomatoes: chemical and physical properties affected by food processing. Crit Rev Biotechnol. 2000;20:293–334. doi: 10.1080/07388550091144212. [DOI] [PubMed] [Google Scholar]
- 93.Prince MR, Frisoli JK. Beta-carotene accumulation in serum and skin. Am J Clin Nutr. 1993;57:175–181. doi: 10.1093/ajcn/57.2.175. [DOI] [PubMed] [Google Scholar]
- 94.Stahl W, Schwarz W, von Laar J, Sies H. All-trans beta-carotene preferentially accumulates in human chylomicrons and very low density lipoproteins compared with the 9-cis geometrical isomer. J Nutr. 1995;125:2128–2133. doi: 10.1093/jn/125.8.2128. [DOI] [PubMed] [Google Scholar]
- 95.Donnelly LE, Newton R, Kennedy GE, Fenwick PS, Leung RH, Ito K, Russell RE, Barnes PJ. Anti-inflammatory effects of resveratrol in lung epithelial cells: molecular mechanisms. Am J Physiol Lung Cell Mol Physiol. 2004;287:L774–783. doi: 10.1152/ajplung.00110.2004. [DOI] [PubMed] [Google Scholar]
- 96.Wheeler DS, Catravas JD, Odoms K, Denenberg A, Malhotra V, Wong HR. Epigallocatechin-3-gallate, a green tea-derived polyphenol, inhibits IL-1 beta-dependent proinflammatory signal transduction in cultured respiratory epithelial cells. J Nutr. 2004;134:1039–1044. doi: 10.1093/jn/134.5.1039. [DOI] [PubMed] [Google Scholar]
- 97.Walle T, Hsieh F, DeLegge MH, Oatis JE, Jr., Walle UK. High absorption but very low bioavailability of oral resveratrol in humans. Drug Metab Dispos. 2004;32:1377–1382. doi: 10.1124/dmd.104.000885. [DOI] [PubMed] [Google Scholar]
- 98.Lu H, Meng X, Li C, Sang S, Patten C, Sheng S, Hong J, Bai N, Winnik B, Ho CT, et al. Glucuronides of tea catechins: enzymology of biosynthesis and biological activities. Drug Metab Dispos. 2003;31:452–461. doi: 10.1124/dmd.31.4.452. [DOI] [PubMed] [Google Scholar]
- 99.Lambert JD, Lee MJ, Lu H, Meng X, Hong JJ, Seril DN, Sturgill MG, Yang CS. Epigallocatechin-3-gallate is absorbed but extensively glucuronidated following oral administration to mice. J Nutr. 2003;133:4172–4177. doi: 10.1093/jn/133.12.4172. [DOI] [PubMed] [Google Scholar]
- 100.Chen L, Lee MJ, Li H, Yang CS. Absorption, distribution, elimination of tea polyphenols in rats. Drug Metab Dispos. 1997;25:1045–1050. [PubMed] [Google Scholar]
- 101.Hong J, Lambert JD, Lee SH, Sinko PJ, Yang CS. Involvement of multidrug resistance-associated proteins in regulating cellular levels of (−)-epigallocatechin-3-gallate and its methyl metabolites. Biochem Biophys Res Commun. 2003;310:222–227. doi: 10.1016/j.bbrc.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 102.Burton GW, Traber MG, Acuff RV, Walters DN, Kayden H, Hughes L, Ingold KU. Human plasma and tissue alpha-tocopherol concentrations in response to supplementation with deuterated natural and synthetic vitamin E. Am J Clin Nutr. 1998;67:669–684. doi: 10.1093/ajcn/67.4.669. [DOI] [PubMed] [Google Scholar]
- 103.Rahman I, MacNee W. Lung glutathione and oxidative stress: implications in cigarette smoke-induced airway disease. Am J Physiol Lung Cell Mol Physiol. 1999;277:L1067–L1088. doi: 10.1152/ajplung.1999.277.6.L1067. [DOI] [PubMed] [Google Scholar]
- 104.Tabak C, Smit HA, Rasanen L, Fidanza F, Menotti A, Nissinen A, Feskens EJ, Heederik D, Kromhout D. Dietary factors and pulmonary function: a cross sectional study in middle aged men from three European countries. Thorax. 1999;54:1021–1026. doi: 10.1136/thx.54.11.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Foronjy RF, Mirochnitchenko O, Propokenko O, Lemaitre V, Jia Y, Inouye M, Okada Y, D'Armiento JM. Superoxide dismutase expression attenuates cigarette smoke- or elastase-generated emphysema in mice. Am J Respir Crit Care Med. 2006;173:623–631. doi: 10.1164/rccm.200506-850OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cuzzocrea S, McDonald MC, Mazzon E, Filipe HM, Centorrino T, Lepore V, Terranova ML, Ciccolo A, Caputi AP, Thiemermann C. Beneficial effects of tempol, a membrane-permeable radical scavenger, on the multiple organ failure induced by zymosan in the rat. Crit Care Med. 2001;29:102–111. doi: 10.1097/00003246-200101000-00022. [DOI] [PubMed] [Google Scholar]
- 107.Zhu M, Chen Y, Li RC. Oral absorption and bioavailability of tea catechins. Planta Med. 2000;66:444–447. doi: 10.1055/s-2000-8599. [DOI] [PubMed] [Google Scholar]
- 108.Summers QA, Fleming JS, Dai Y, Perring S, Honeywell R, Gough KJ, Renwick AG, Clark AR, Nassim MA, Holgate ST. The pulmonary deposition of two aerosol preparations of nedocromil sodium delivered by MDI assessed by single photon emission computed tomography. J Aerosol Med. 1996;9(Suppl 1):S93–109. doi: 10.1089/jam.1996.9.suppl_1.s-93. [DOI] [PubMed] [Google Scholar]
- 109.Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, Cherniack RM, Rogers RM, Sciurba FC, Coxson HO, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350:2645–2653. doi: 10.1056/NEJMoa032158. [DOI] [PubMed] [Google Scholar]
- 110.Chan HK, Daviskas E, Eberl S, Robinson M, Bautovich G, Young I. Deposition of aqueous aerosol of technetium-99m diethylene triamine pentaacetic acid generated and delivered by a novel system (AERx) in healthy subjects. Eur J Nucl Med. 1999;26:320–327. doi: 10.1007/s002590050393. [DOI] [PubMed] [Google Scholar]
- 111.Jaimes EA, DeMaster EG, Tian RX, Raij L. Stable compounds of cigarette smoke induce endothelial superoxide anion production via NADPH oxidase activation. Arterioscler Thromb Vasc Biol. 2004;24:1031–1036. doi: 10.1161/01.ATV.0000127083.88549.58. [DOI] [PubMed] [Google Scholar]
- 112.Deliconstantinos G, Villiotou V. Gas phase oxidants of cigarette smoke increase nitric oxide synthase and xanthine oxidase activities of rabbit brain synaptosomes. Neurochem Res. 2000;25:769–774. doi: 10.1023/a:1007505221453. [DOI] [PubMed] [Google Scholar]