

Abstract
A specialized vector backbone from the Protein Structure Initiative was used to express full-length human cytochrome b5 as a C-terminal fusion to His8-maltose binding protein in Escherichia coli. The fusion protein could be completely cleaved by tobacco etch virus protease, and a yield of ~18 mg of purified full-length human cytochrome b5 per liter of culture medium was obtained (2.3 mg per]of wet weight bacterial cells). In situ proteolysis of the fusion protein in the presence of chemically defined synthetic liposomes allowed facile spontaneous delivery of the functional peripheral membrane protein into a defined membrane environment without prior exposure to detergents or other lipids. The utility of this approach as a delivery method for production and incorporation of monotopic (peripheral) membrane proteins is discussed.
Keywords: Protein Structure Initiative, Homo sapiens, human, cytochrome b5, peripheral membrane protein, monotopic membrane protein, auto-induction
Introduction
Cytochrome b5 (cytb5) is present in bacteria, protozoans, yeasts, and mammals [1]. The protein consists of an N-terminal hydrophilic heme-binding domain of ~100 residues, and a C-terminal hydrophobic membrane anchor domain of ~30 residues [2, 3].
In mammals, isoforms of cytb5 are present in the endoplasmic reticulum, mitochondria, and erythrocytes (Table 1, [3]). The endoplasmic and mitochondrial cytb5 are expressed from different genes, while the erythrocyte isoform is thought to originate from post-translational proteolysis of the endoplasmic protein [4, 5]. Since erythrocyte cytb5 lacks the C-terminal membrane anchor domain, it functions as a soluble protein in blood cells [6]. Erythrocyte cytb5 is responsible for the reduction of non-functional ferric met-hemoglobin to the O2-binding ferrous form [7].
Table 1.
Physiological roles of cytochrome b5.
| Localization | Active form | Function | Reference |
|---|---|---|---|
| Mitochondrion | Membrane bound | Androgenesis | [9] |
| Endoplasmic reticulum | Membrane Bound | Biosynthesis of unsaturated fatty acids (Δ5, Δ6, Δ9, Δ12); plasmalogens; cholesterol | [3] |
| Endoplasmic reticulum | Membrane bound | Reduction of cytochrome P450s | [8] |
| Erythrocytes | Soluble | Reduction of hemoglobin | [7] |
In the endoplasmic reticulum, cytb5 is attached to the cellular membrane through the C-terminal membrane anchor domain, where it participates in the electron transfer steps of many essential physiological reactions including fatty acid desaturation, biosynthesis of plasmalogen and cholesterol, and reduction of cytochrome P450 [3, 8]. Moreover, membrane-bound mitochondrial cytb5 has been shown to be involved in androgenesis in rat Leydig cells [9].
Initial functional [10] and structural studies [11] of cytb5 were performed on samples purified from rabbit, rat, and cow liver due to the high concentration present in this tissue. Strittmatter and co-workers provided biochemical and biophysical characterization of the detergent solubilized full-length microsomal cytb5 [11–14] and the heme-binding domain liberated by proteolysis [15]. Notably, these workers showed that full-length cytb5 (fl-cytb5) would spontaneously associate with synthetic lipids [16], and that fl-cytb5 was required for stearoyl-CoA desaturase activity [17].
More recently, recombinant forms truncated to only the heme-binding cytb5 soluble domain (cytb5-sd) have been extensively used for biophysical analysis and structural determinations [2, 18]. The codon-optimized rat cytb5-sd [19] and the tobacco cytb5 [20] have been expressed in E. coli. Approaches for detergent-mediated isolation of the full-length cytb5 (fl-cytb5) from membranes [21], incorporation of heme into the solubilized rabbit protein [21], preparation of the bovine protein as a fusion to glutathione-S-transferase and recovery of the fusion protein from membranes by lipase treatment [22], and purification of the expressed mouse protein from E. coli membranes [23] have also been developed. Although successful, these latter preparation approaches are labor intensive and time consuming.
In this work, we explored whether fusion protein vectors developed for high-throughput protein expression as part of the Protein Structure Initiative might have use in the expression of human cytb5. By fusion to His8-maltose binding protein (His8-MBP), the fl-cytb5 could be expressed as a fully soluble entity. Furthermore, the fl-cytb5 could be liberated from the fusion by site-specific proteolysis, which permitted controlled, spontaneous incorporation into membrane vesicles. The utility of this approach as an in situ delivery method for production and incorporation of monotopic integral membrane proteins is discussed.
Materials and Methods
Materials
Unless otherwise stated, bacterial growth reagents, antibiotics, routine laboratory chemicals, and disposable labware were from Sigma-Aldrich (St. Louis, MO), Fisher (Pittsburgh PA), or other major distributors. Chloroprotoporphyrin IX was from Sigma-Aldrich. DNA sequencing was performed in the University of Wisconsin Biotechnology Center.
Expression Vectors
The expression vectors pVP55A and pVP56K were created from pQE80 (Qiagen, Valencia CA) by removal of a non-functional chloramphenicol acetyltransferase coding region and specifically in pVP56K by replacement of the beta-lactamase coding region with an aminoglycoside 3′-phosphotransferase coding region conferring kanamycin resistance. The vectors use the viral T5 promoter under control of an engineered double lac operator for recombinant gene expression [24]. The plasmid backbones also provide the strong lacIq promoter for elevated expression of the lac repressor, LacI. Elevated expression of LacI provides strong attenuation of basal expression. Other design features of the expression plasmids and examples of their use are reported elsewhere [24].
Vector pVP55A is used to add an N-terminal tag of His8 to the target protein; vector pVP56K is used to add an N-terminal tag of His8-maltose binding protein. The linker region between the tag and the target protein contains the recognition sequence for site-specific tobacco etch virus (TEV) protease [25].
Cloning
The gene for fl-cytb5 from human (accession number: BC015182; originally cloned from the Mammalian Gene Collection, [26]) was obtained from Open Biosystems (Hunstville AL, http://www.openbiosystems.com/). The gene was amplified using the forward primer 5′-TTCGGCGATCGCCGAAATGGCAGAACAAAGCGAC-3′ containing an SgfI restriction site (underlined) and the reverse primer 5′-AGCAGTTTAAACTTAGTCCTCTGCCATGTATAGGCG-3′ containing a PmeI restriction site (underlined).
The SgfI and PmeI restriction sites were used for Flexi Vector cloning (Promega, Madison, WI) as previously described [24]. Transfer of the amplified fl-cytb5 gene into pVP55A gave a vector for expression of His8-fl-cytb5. Transfer of the same amplified gene into pVP56K gave a vector for expression of the fusion protein His8-MBP-fl-cytb5. The linear map for the pVP56K vector is shown in Fig. 1A and the amino acid sequence of His8-MBP-fl-cytb5 is shown in Fig. 1B.
Fig. 1.

A, Linear map of the pVP56K-fl-cytb5 expression vector. B, amino acid sequence of the His8-MBP-fl-cytb5 fusion protein, where fl indicates the full-length protein. The His8 tag, TEV protease cleavage site, and the first five residues the fl-cytb5 liberated by TEV proteolysis are shown in bold letters. The C-terminal membrane anchor domain is underlined. C, schematic of the fusion proteins studied here.
The cytb5-sd was obtained by removal of the 37-residue C-terminal membrane anchor sequence using PCR amplification. The forward primer was the same as that used for cloning the full-length protein, while the reverse primer was 5′-AGCAGTTTAAACTTACGGTTCCGGCGGTTTGTTCAG-3′ (PmeI site underlined). The amplified gene encoded the 97 amino acids from the N-terminal soluble domain that contains the heme-binding region [11, 13, 27]. This amplified gene was transferred into pVP55A so that His8-cytb5-sd could be generated.
The human cytochrome b5 reductase gene (accession number: BC004821) was obtained from Open Biosystems and amplified using the forward primer 5′-TTCGGCGATCGCCATGAAGCTGTTCCAGCGCTCCACG-3′ and the reserve primer 5′-TCGTGTTTAAACTCAGAAGACGAAGCAGCGCTC-3′ (5′ SgfI and 3′ PmeI sites underlined). The amplified gene encodes a 23-residue deletion corresponding to the N-terminal membrane-binding residues [13]. Transfer of the amplified gene into pVP55A gave a vector for expression of the soluble form of human cytb5 reductase as a fusion to an N-terminal His8 tag (His8-cytb5R).
Schematic representations of each of the fusion protein constructs studied here are shown in Fig. 1C and results from their use are summarized in Table 2.
Table 2.
Vectors and expressed proteins.
| Vector | Expressed proteina | Expression medium | Cell yieldb g/L | Purified protein yieldc mg/g | Cofactor incorporationd |
|---|---|---|---|---|---|
| pVP55A | His8-cytb5-sd | Auto-induction | 25e | 0.6 | Yes, high |
| pVP55A | His8-fl-cytb5 | Auto-induction | 12 | 0 | No, insoluble |
| pVP56K | His8-MBP-fl-cytb5 | Auto-induction | 12 | 0.42 | Lost in purification |
| pVP56K | His8-MBP-fl-cytb5 | TB-IPTG | ~4 | 2.3f | Lost in purification |
| pVP55K | His8-cytb5R | Auto-induction | 25e | 0.6 | Yes, high |
Schematic representations of the expressed proteins are show in Fig. 1C. sd, soluble domain. fl, full-length.
Wet cell weight obtained per liter of culture medium after preparative centrifugation as described in Materials and Methods.
Amount of purified protein obtained per gram of wet cells.
Protoporphyrin IX in cytb5 variants; FAD in His8-cytb5R.
From 10 liter fermentation.
Also detailed in Table 3; yield of cytb5 portion of the total mass of fusion protein purified.
Expression and Purification of fl-cytb5
Vectors pVP55A-fl-cytb5 and pVP56K-fl-cytb5 (Table 2) were individually transformed into E. coli BL21. For scale-up, a 10 mL culture was inoculated with a single colony from freshly transformed cells and incubated overnight in the non-inducing version of auto-induction medium [28]. The overnight scale-up culture was added to 1 L of auto-induction medium containing either 200 μg/mL of ampicillin (pVP55A) or 50 μg/mL of kanamycin (pVP56K). The growth and expression were continued for ~24 h at 25 °C. Cells were harvested by centrifugation at 5,000g for 30 min. The pVP55A-fl-cytb5 cells had no distinct color, while the pVP56K-fl-cytb5 cells were bright red in color. The yield of wet cell paste from 1 L of auto-induction medium was typically ~12 g (Table 2).
Cells transformed with pVP56K-fl-cytb5 were also grown at 37 °C in Terrific Broth medium until the OD600 reached ~0.6, isopropyl-βhiogalactopyranoside (IPTG) was added to a final concentration of 0.5 mM, and the temperature was dropped to 25 °C. After 4 h, the cells were harvested by centrifugation at 3000g and stored at −80 °C. These cells had no distinct color. The yield of wet cell paste from 1 L of IPTG induction medium and short time expression was typically ~4 g.
The His8-MBP-fl-cytb5 was purified by Ni immobilized metal affinity chromatography (Ni IMAC). The cell pellet (~12 g) was re-suspended in 50 mL of 25 mM HEPES pH 7.5, 20 mM imidazole, 500 mM NaCl, and 25 mg each of lysozyme, DNase, and RNase. The cell suspension was stirred for 20 min at 4 °C, followed by sonication for a total of 2 min with 10 s pulses. Insoluble proteins and cell debris were pelleted by centrifugation at 39,000g for 1 h. The supernatant was loaded onto a 5-mL HisTrap fast flow column (GE Healthcare, Piscataway NJ) equilibrated with 25 mM HEPES, pH 7.5, 20 mM imidazole, and 500 mM NaCl and washed with the same buffer until the OD280 reached baseline levels. The bound protein was eluted with a gradient of 20 to 300 mM imidazole. Peaks containing the fusion protein were detected at ~100 mM imidazole and ~180 mM imidazole.
The pooled fractions were concentrated by ultrafiltration using a YM30 membrane (Millipore, Bedford MA). Imidazole was removed from the sample by dialysis against 25 mM HEPES, pH 7.5, 100 mM NaCl. For short-term storage (several weeks), the dialysis buffer was amended to contain 10% (v/v) glycerol and the protein was stored at 4 °C. For long-term, the glycerol-amended His8-MBP-fl-cytb5 could also be drop frozen in liquid N2 and stored at −80 °C.
Incorporation of Heme into His8-MBP-fl-cytb5
The preparation of hemin chloride solutions and the incorporation of heme into His8-MBP-fl-cytb5 were adapted from elsewhere [21]. Briefly, a 1-mL sample was prepared by 40-fold dilution of the purified, concentrated, and dialyzed His-MBP-fl-cytb5 into 20 mM Tris, pH 8.0, containing 1 mM EDTA. The absorbance spectrum of the sample was recorded from 250 to 600 nm on an Agilent 8453 UV-spectrophotometer (Santa Clara, CA). Portions (1 μL) of 0.85 mM hemin chloride were titrated into the sample and the absorbance spectrum was monitored. Incorporation was judged to be complete when an increase in absorbance at 385 nm and a shift in the Soret peak from 412 to 410 nm were observed. The amount of heme required to reconstitute the remainder of the purified sample was calculated based on the small-scale titration result. To verify that the observed spectral changes were not due to adventitious binding of heme to MBP, hemin chloride was also titrated into free MBP as previously reported [29].
Expression and Purification of cytb5-sd
The cytb5-sd was expressed using a 10 L fermenter [30]. Vector pVP55A-cytb5-sd was transformed into E. coli BL21. A single colony was used to inoculate a 10-mL culture that was incubated overnight at 37 °C. The entire culture was used to inoculate 1 L of Terrific Broth medium containing 200 mg of ampicillin. After ~12 h at 37 °C, this culture was used to inoculate 9 L of auto-induction medium equilibrated at 25 °C in the fermenter vessel. After ~24 h, the cells were harvested by centrifugation at 5,000g for 30 min using an Avanti J-HC centrifuge (Beckman Coulter, Fullerton CA) with a JS-42 rotor. In the fermenter, the yield of wet cell paste from 1 L of auto-induction medium was typically ~20 g. The pVP55A-cytb5-sd cells had a distinct red color. The cell paste was stored at −80 °C.
The cell paste (~20 g) was re-suspended in 50 mL of 25 mM HEPES pH 7.5, 20 mM imidazole, 500 mM NaCl, and 25 mg each of lysozyme, DNase, and RNase, sonicated, centrifuged and separated by Ni IMAC as described above for fl-cytb5. A gradient of 20 to 300 mM imidazole was applied, and the bound cytb5-sd eluted from the Ni column at ~180 mM imidazole was concentrated and loaded into an Sephacryl S-100 column (5 cm id × 80 cm, GE Healthcare) equilibrated with 25 mM HEPES, pH 7.5, containing 100 mM NaCl. The fractions containing purified protein were identified by optical absorbance and denaturing gel electrophoresis.
Expression and Purification of Soluble His8-cytb5R
The soluble form of His8-cytb5R was expressed using a 10 L fermenter [30]. Vector pVP55A-cytb5R was transformed into E. coli BL21(DE3). A single colony was used to inoculate a 10-mL culture and incubated overnight at 37 °C. The entire culture was used to inoculate 1 L of Luria Bertani medium containing 50 mg of kanamycin. After ~12 h at 37 °C, this culture was used to inoculate 9 L of auto-induction medium equilibrated at 25 °C in the fermenter vessel. After ~24 h, the cells were harvested by centrifugation at 5,000g. The cell paste was stored at −80 °C.
The cell paste (~20 g) was re-suspended, sonicated, centrifuged, and separated by Ni IMAC as described above for cytb5-sd. His8-cytb5R was >95% pure after this single step. The pure protein was pooled, concentrated using ultrafiltration (YM10, Millipore, Bedford MD), and the buffer was exchanged by dialysis against 25 mM HEPES, pH 7.5, containing 100 mM NaCl. The purified His8-cytb5R was stored at −80 °C.
Enzyme Activity Measurements
The reduction of the different cytb5 preparations by His8-cytb5R was measured at 25 °C by following the change in absorbance at 413 nm in an Agilent 8453 UV-visible spectrophotometer. The concentration of cytb5 was varied from 0.3 μM to 15 μM in 1 mL of 25 mM HEPES, pH 7.5, containing 1.3 nM of His8-cytb5R. The reaction was initiated by the addition of NADH to a final concentration of 100 μM. Initial rate data were analyzed by non-linear least squares fitting using Kaleidagraph (Synergy Software, Reading, PA) and the Michaelis Menten equation v = kcat[S]/(KM + [S]). The concentrations of the cytb5 variants and cytb5R were determined by optical spectrometry using the extinction coefficients 117 mM−1 cm−1 at 412 nm and 11.3 mM−1 cm−1 at 466 nm, respectively [31, 32].
Liposome Preparation
Liposomes were prepared from 7.5 μmol of a lipid mix (85% 1-palmitoyl, 2-oleoyl phosphatidylcholine; 15% 1,2-dioleoyl phosphatidylserine, Avanti Polar Lipids, Alabaster, AL) labeled with 2 μCi of [3H]-1,2-dipalmitoyl phosphatidylcholine (NEN, Cambridge, UK). The lipid mix was dissolved in chloroform [33], the bulk of the organic solvent was removed by evaporation under a stream of N2 gas, and the remaining trace level of chloroform was removed by incubation for 30 min under vacuum. The dried lipid film was re-hydrated with 20 mM HEPES, pH 7.4, containing 100 mM KCl for 30 min, vortexed for 5 min, and subjected to 5 freeze-thaw cycles. Liposomes were formed by extrusion from an Avanti mini-extruder using 11 passes through 100 nm track-etch polycarbonate membranes (Nucleopore, Pleasanton, CA).
In situ Delivery of fl-cytb5 to Liposomes
TEV protease was expressed as an MBP fusion protein, liberated by auto-cleavage within the expression host, and purified as previously described [25]. TEV protease reactions were conducted in 20 mM sodium phosphate, pH 7.0, containing 100 mM NaCl, 5 mM DTT, and 1 mM EDTA. For initial testing, aliquots of 10 μM His8-MBP-fl-cytb5 were digested with TEV protease at ratios of fusion protein to protease ranging from 200:1 to 10:1. At a ratio of 10:1, the proteolysis was complete in less than 30 min. Thus all the preparative protease reactions were done at this ratio for 30 min.
For the proteolysis reaction, His8-MBP-fl-cytb5 (10 μM), TEV protease (1 μM) and 45 μL of the liposome preparation described above were incubated with agitation at 37 °C in a total volume of 75 μL of 20 mM HEPES, pH 7.4, containing 100 mM KCl, 5 mM dithiothreitol, and 1 mM EDTA. After 30 min, the reactions were mixed 1:1 with 80% (w/v) Accudenz (Accurate Chemical and Scientific, Westbury NY) made up in the same buffer used to perform the proteolysis. The mixture was transferred to Ultra-Clear centrifuge tubes (5 mm × 41 mm, Beckman Coulter, Fullerton CA) and sequentially overlaid with 350 μL of 30% Accudenz followed by 100 μL of the reaction buffer. The mixture was centrifuged for 4 h at 45,000 rpm (189,000 x g) and 4 °C in an L-60 ultracentrifuge (Beckman Coulter) using an SW-50.1 rotor with adaptors. After centrifugation, 60 μL fractions were collected from the top to the bottom of the gradient. Liposomes were detected by counting 5 μL of each fraction by liquid scintillation using an LS6500 counter (Beckman Coulter). An aliquot of 20 μL of 4X SDS-PAGE sample buffer was added to each fraction and 25 μL of the sample was loaded onto a Criterion SDS-PAGE polyacrylamide gel (4–20% gradient Tris-HCl, 1.0 mm, 26-well, BioRad, Richmond CA) and analyzed as previously reported [34].
Mass spectral analysis
TEV protease-treated samples of His8-MBP-cytb5 were analyzed by mass spectrometry at the University of Madison-Wisconsin Biotechnology Center. Purified protein samples were concentrated and desalted by acetone precipitation before addition (1:1) to an α-cyano-4-hydroxycinnamic acid matrix. MALDI-TOF analysis was performed with a 4800 MALDI TOF/TOF Analyzer from Applied Biosystems (Foster City, CA) calibrated with bovine serum albumin and deuterated bovine serum albumin.
Results
Vector
By use of the Flexi Vector system, vectors pVP55A and pVP56K allow transfer of the same sequence-verified cloned gene into multiple contexts for expression testing. Here we have investigated either His8- or His8-MBP-tagged proteins. This combination was used to investigate the possibility that His8-MBP might act as a carrier for a monotopic (peripheral) membrane protein for in situ delivery to a membrane environment. This is a different use for MBP than originally envisaged [35], and to our knowledge, one that has not been intentionally tested.
Protein Expression
His8-cytb5-sd was highly expressed from pVP55A in both shaken flask culture and in a 10 L fermenter. This over-expression required neither codon optimization of the gene nor codon adaptation of the expression host. By using auto-induction medium in the 10 L fermenter, a typical yield of ~250 g of cell paste was obtained, and the cell paste had an intense red color. Fig. 2, lane 1, shows a denaturing electrophoresis analysis of the purified His8-cytb5-sd, which could be obtained in >98% purity by the two-step purification procedure.
Fig. 2.
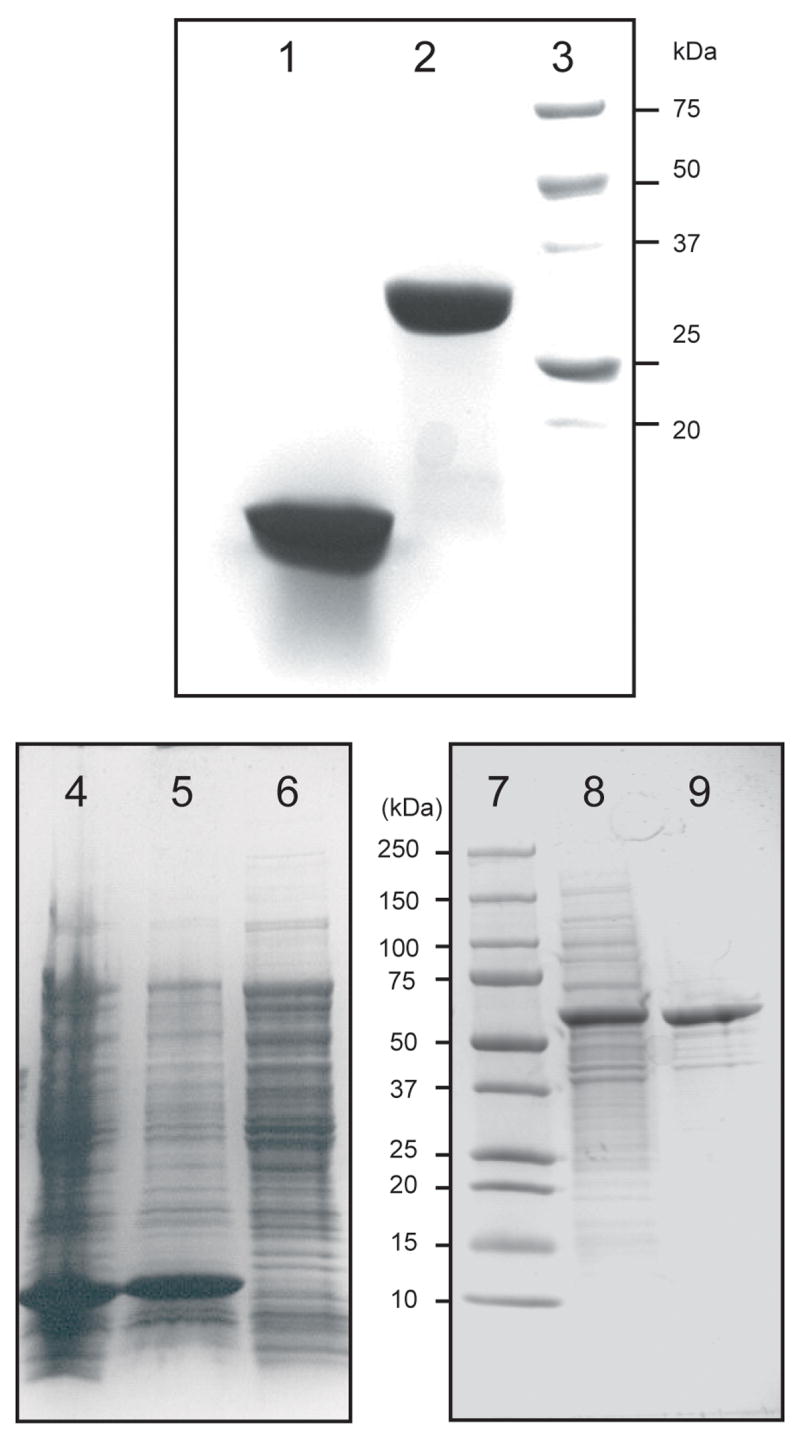
Coomassie-stained denaturing gel electrophoresis. 1, 100 μg of purified cytb5-sd, where sd indicates the soluble domain. 2, 100 μg of purified His8-cytb5R, where R indicates the reductase protein. 3, molecular weight markers. 4, total cell lysate from E. coli cells expressing His8-fl-cytb5, where fl indicates the full-length protein. 5, insoluble fraction. 6, soluble fraction. 7, molecular weight markers. 8, Soluble fraction from E. coli cells expressing His8-MBP-fl-cytb5. 9, His8-MBP-fl-cytb5 purified Ni IMAC chromatography.
His8-fl-cytb5 was also highly expressed from pVP55A (Fig. 2, lane 4), but accumulated entirely as an insoluble protein (compare lanes 5 and 6), and these cells had no distinct color. Since His8-fl-cytb5 was an insoluble apo-protein, it was not studied further.
His8-MBP-fl-cytb5 was also highly expressed from pVP56K in E. coli, but in this case, the fusion protein was entirely soluble (Fig. 2, lane 8), and the cells harvested from the auto-induction protocol were bright red. Interestingly, expression of His8-MBP-fl-cytb5 in Terrific Broth using a short time period induction with IPTG also gave strong expression of a soluble fusion protein (not shown), but these cells had no distinct color.
Protein Purification
The yields of purified His8-cytb5-sd and His8-MBP-fl-cytb5 are summarized in Table 2, and a summary of the purification of His8-MBP-fl-cytb5 is given in Table 3. All His8-tagged variants were purified using standard Ni IMAC to ~90% or greater purity as judged by denaturing gel electrophoresis.
Table 3.
Purification of His8-MBP-fl-cytb5.
| Step | Volume (mL) | Total protein (mg)a | Yield of fusion protein (%) |
|---|---|---|---|
| Sonicated cellsb | 50 | 789 | 100 |
| Cell-free lysate | 41 | 571 | 72 |
| Ni IMAC | 8.5 | 73c | 9.3 |
Determined by Bradford assay [58].
Prepared from 7.8 g of E. coli cells obtained from 2 L of IPTG-induced culture medium.
Yield of His8-MBP-fl-cytb5 fusion protein. A yield of 18 mg of fl-cytb5 (25% of total protein yield) was present in the heme-reconstituted fusion protein as determined by optical spectroscopy. This measurement was consistent with the 26% contribution of fl-cytb5 to the mass of the fusion protein.
During Ni IMAC purification of His8-MBP-fl-cytb5, two peaks that contained the fusion protein were detected from the gradient elution. Table 4 summarizes results of mass spectral analysis of these two peaks. The first peak from the IMAC column was more red colored than the second peak, and mass spectral analysis showed that the protein from this peak had a mass of 57648 Da, representing a loss of ~1950 Da from the mass of 59594 Da calculated for the complete fusion protein (assuming that the N-terminal Met is efficiently removed; note the close match in masses calculated and observed for His8-MBP). In contrast, the second peak from the IMAC column had a mass of 59626 Da, which matched that calculated for the fusion protein within 0.05%. Thus it appears that the first peak contained a truncated form of cytb5 while the second peak contained the intact fusion protein, His8-MBP-fl-cytb. Fig. 3 shows that two distinct forms of cytb5 were also obtained after TEV proteolysis of the fusion protein.
Table 4.
Mass spectral analysis.
| Protein | Calculated mass | Observed mass (% difference from calculated mass) | Assignment |
|---|---|---|---|
| His8-MBP-fl-cytb5a | 59594 | ||
| Peak 1 | 57648 (−3.3%) | C-terminal truncationb | |
| Peak 2 | 59626 (−0.05%) | His8-MBP-fl-cytb5 | |
| His8-MBP-fl-cytb5c | 59756 | 59626 (−0.2%) | His8-MBP-fl-cytb5 |
| His8-MBPd | 43897 | 43898 (0.00%) | His8-MBP |
| fl-cytb5e | 15715 | 15729 ( + 0.09%) | fl-cytb5 |
From the auto-induction protocol. Peaks 1 and 2 were separated by gradient elution from Ni IMAC. Calculated masses for the His8-MBP portion up to the Gln residue of the tobacco etch virus protease site in the linker include removal of the N-terminal Met residue.
Proposed to arise from deletion of the sequence ISAVAVALMYRLYMAED from the C-terminus of the fusion protein during auto-induction. The truncated fusion protein has a calculated mass of 57695, which is within −0.08% of the mass observed for peak 1.
From the IPTG induction protocol.
Proteins separated by treatment of His8-MBP-fl-cytb5 obtained from the IPTG induction protocol with TEV protease.
Fig. 3.
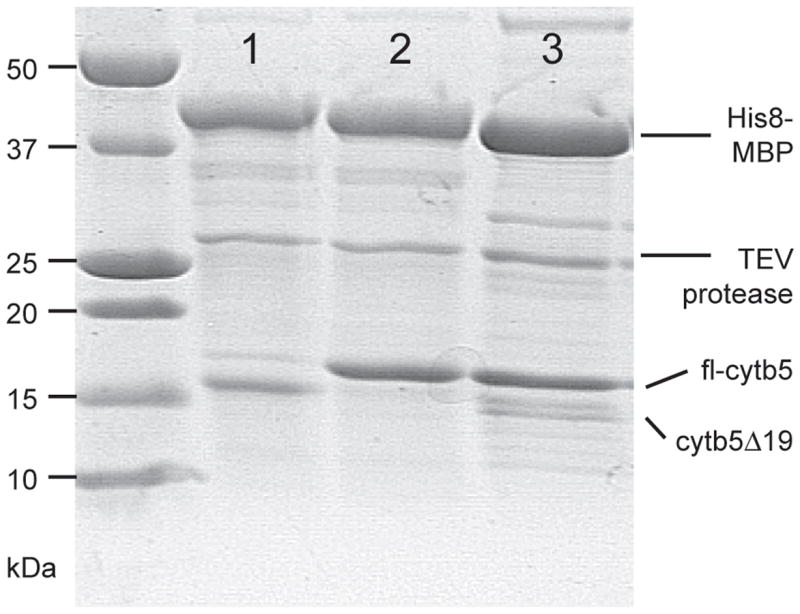
Difference in post-translational processing caused by long-term auto-induction or short-term IPTG induction. TEV proteolysis emphasizes the difference in size of the cytb5 variants. Lane 1, peak 1 from Ni IMAC purification of His8-MBP-fl-cytb5 expressed by auto-induction; cytb5 19 designates a truncated form lacking 19 residues from the C-terminus. Lane 2, peak 2 from Ni IMAC purification of His8-MBP-fl-cytb5 expressed by auto-induction. Lane 3, peak from Ni IMAC purification of His8-MBP-fl-cytb5 expressed by IPTG induction.
Since both peaks were purified using Ni IMAC and an imidazole gradient, it was unlikely that the N-terminus of the His8-MBP portion of the fusion protein was truncated. Instead, the mass of the smaller protein matched within 0.08% of the mass of 57695 Da calculated for removal of 17 residues from the C-terminus of cytb5 (corresponding to the sequence ISAVAVALMYRLYMAED, see Fig. 1B and Table 4).
During Ni IMAC purification of His8-MBP-fl-cytb5 obtained from IPTG induction, a single peak that contained the fusion protein was detected from the gradient elution. Table 4 summarizes results of mass spectral analysis of this peak, and Fig. 3 shows that the predominant form after TEV protease treatment was fl-cytb5.
Spectral Properties of His8-MBP-fl-cytb5
Purified His8-MBP-fl-cytb5 has a Soret peak at 413 nm arising from the bis-imidazolate ligation of the ferric center in oxidized cytb5 (Fig. 4). Even though the cell paste obtained from auto-induction containing His8-MBP-fl-cytb5 was bright red, the purified protein obtained from Ni IMAC chromatography contained less than 10% of the heme content expected. Apparently, the high imidazole concentration required for elution of both bound forms of the fusion protein caused dissociation of the heme prosthetic group. In this case, the imidazole was removed from the protein preparation by dialysis, and the heme content was simply restored by titration with free heme [21] as detected by a characteristic increase in Soret band absorbance (413 nm, 117 mM−1 cm−1, as compared to the 385 nm absorption maximum observed from free heme, 56 mM−1 cm−1). As previously reported [29], control experiments showed that titration of heme into preparations of His8-MBP did not give a shift in the heme optical spectrum. The heme-incorporated His8-MBP-fl-cytb5 preparation could be stored at 4 °C in buffer amended with 10% v/v glycerol for up to 2 months with no apparent deleterious effects as judged by catalytic properties, optical spectrum, and denaturing electrophoresis. Samples drop frozen in the amended buffer and stored at −80 °C for extended periods gave no change in these properties when thawed.
Fig. 4.

Spectra changes in His8-MBP-fl-cytb5 during a time-course reduction with soluble His8-cytb5R in the presence of NADH. As assembled, the reaction went to completion in ~30 s. A blank was obtained with buffer solution containing NADH, and the spectral contribution of His8-cytb5R (1.3 nM) was negligible.
Interestingly, Ni IMAC was also used to purify His8-cytb5-sd, but in this case, the heme was retained in the purified protein to a high level upon the basis of the quantification of heme and protein and upon the lack of a diagnostic spectral shift when incubated with additional heme.
Purification and Characterization of His8-cytb5R
For these studies, cytb5R was purified as a soluble N-terminal His8 fusion by using Ni IMAC. The protein obtained from the single step purification was >95% pure as judged by denaturing gel electrophoresis (Fig. 2, lane 2). The purified protein had an optical spectrum consistent with stoichiometric incorporation of FAD and quantitative HPLC analysis [36] also revealed high percentage incorporation of the cofactor in the purified protein.
His8-MBP-fl-cytb5 is a Substrate for His8-cytb5R
His8-MBP-fl-cytb5 and His8-cytb5R were mixed in the presence of NADH in order to determine whether the fusion protein was recognized as a substrate by cytb5R. Fig. 4 shows that His8-MBP-fl-cytb5 was used by His8-cytb5R as an electron acceptor. Over an ~2 min period, the Soret peak was shifted from 413 nm to 423 nm and the peak at 555 nm increased in intensity. The resulting spectrum matched that of the reduced state of detergent-solubilized fl-cytb5 [15, 37]. In control experiments, free heme was not reduced by His8-cytb5R in the time-scale and NADH concentration used for the cytb5 reduction.
Steady-state kinetic parameters for reduction of both His8-MBP-fl-cytb5 and His8-cytb5-sd were determined in order to investigate the influence of the His8-MBP fusion on the interactions between cytb5 and His8-cytb5R. Fig. 5 shows that both forms of cytb5 acted as saturable substrates for the electron transfer reaction, as the experimental data were well fit by the simple Michaelis-Menten equation (Table 5). The apparent KM values for both forms of cyt5 were the same. Interestingly, however, the His8-MBP-fl-cytb5 had an apparent Vmax that was ~3-fold higher than that of His8-cytb5-sd.
Fig. 5.
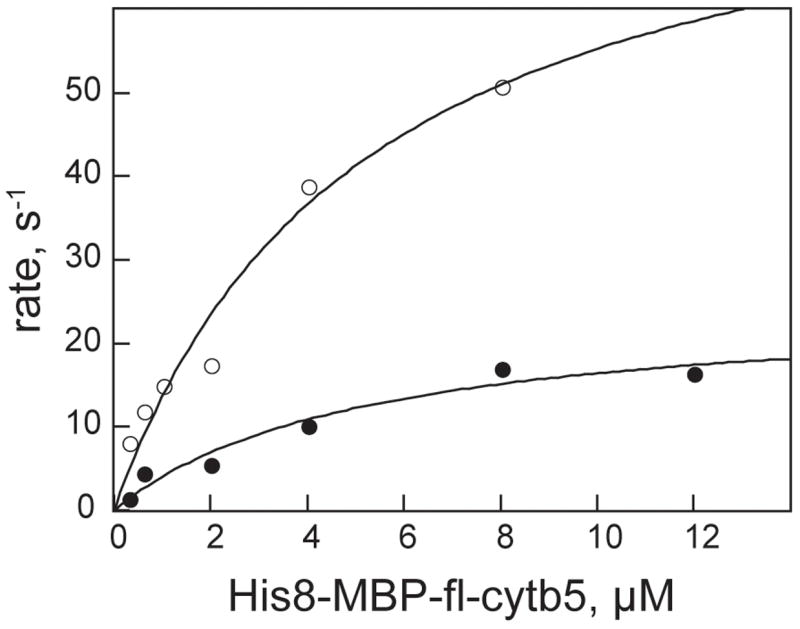
Steady-state reduction of His8-MBP-fl-cytb5 (○) and His8-cytb5-sd (●) by His8-cytb5R. The lines are least square fits to the simple Michaelis-Menten assumption.
Table 5.
Kinetic parameters for the reduction of cytochrome b5 variants by His8-cytochrome b5 reductase. a
TEV Protease Reaction
Vectors pVP55A and pVP56K allow TEV protease-dependent cleavage of a fusion protein to release the target protein from either His8- or His8-MBP, respectively. Fig. 6 shows that fl-cytb5 could be stoichiometrically released from His8-MBP-fl-cytb5 by treatment with TEV protease. For these studies, a satisfactory reaction was obtained at a ratio of His8-MBP-fl-cytb5 to TEV protease of 10:1 (reaction completed in 30 min). Lower amounts of TEV protease also catalyzed the proteolysis reaction, albeit with a longer, less practical amount of time required to reach completion.
Fig. 6.
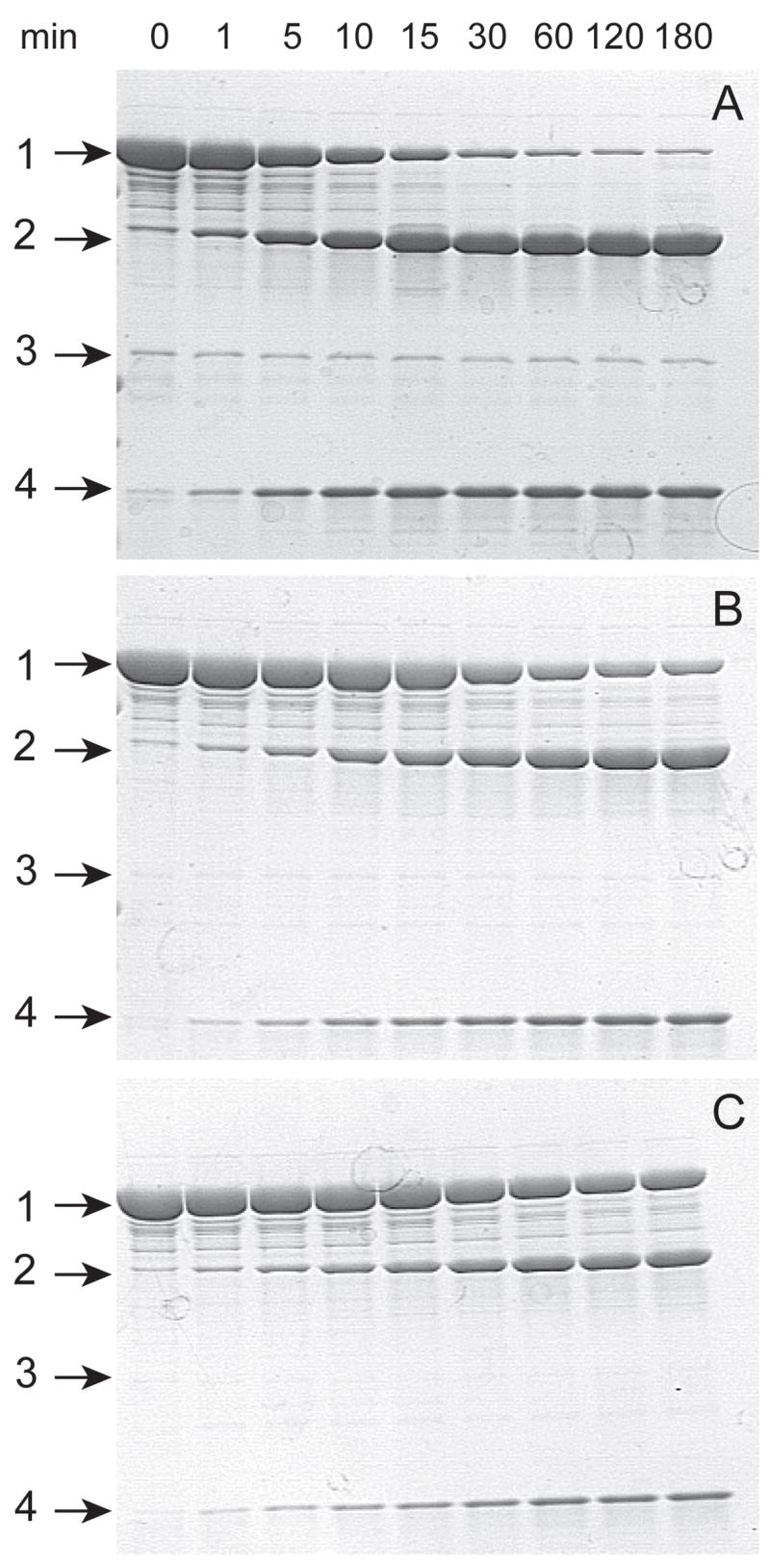
Coomassie-stained denaturing gel electrophoresis showing the time and concentration dependence of proteolysis of His8-MBP-fl-cytb5 in the presence of TEV protease to release fl-cytb5. A, the TEV protease was present at ratio of 1:10 relative to His8-MBP-fl-cytb5. (1) is His8-MBP-fl-cytb5, (2) is His8-MBP, (3) is TEV protease, and (4) is fl-cytb5. B, TEV protease to His8-MBP-fl-cytb5 ratio of 1:50. C, TEV protease to His8-MBP-fl-cytb5 ratio of 1:100.
Incorporation of cytb5 into Liposomes
We investigated whether the highly soluble and easily handled His8-MBP-fl-cytb5 could also be used for the in situ delivery of fl-cytb5 to a membrane environment. Fig. 7A shows a schematic representation of this approach. For the experiment, the TEV protease reaction was performed in the presence of radiolabeled liposomes. The liposome fraction was then fractionated by density gradient ultracentrifugation. The liposomes and bound protein float owing to their low density. In contrast, proteins not associated with the liposomes will have a higher density and will thus be separated by the ultracentrifugation. Samples were taken from different points in the gradient and analyzed for lipid content by scintillation counting and for protein content by denaturing gel electrophoresis.
Fig. 7.
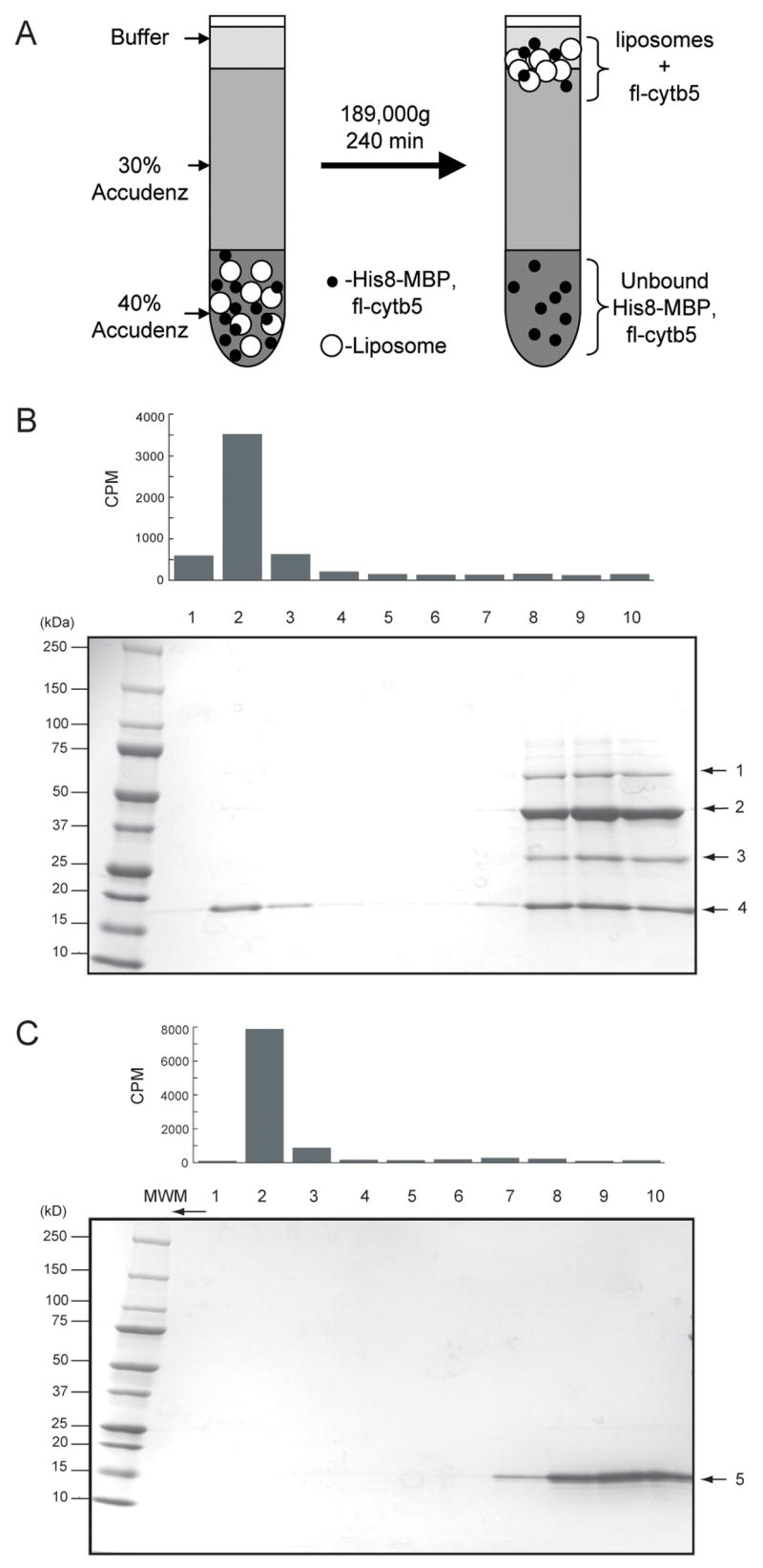
Incorporation of fl-cytb5 into phospholipids vesicles. A, a schematic representation of the liposome floating experiment. The open circles represent the liposomes and the solid black circles represent proteins, including His8-MBP-fl-cytb5 and fl-cytb5 and TEV protease. Protein bound to the liposome will migrate to the interface between buffer and 30% Accudenz, while unbound proteins will remain in the 40% Accudenz layer. B, Alignment of scintillation counting results (detects [3H]-1,2-dipalmitoyl phosphatidylcholine added as a tracer to the liposome preparation) of the fractions collected from top to bottom from the liposome floating experiment with a Coomassie-stained denaturing gel. Lanes 1–3 contain the liposomes and fl-cytb5 (4). His8-MBP and TEV protease did not bind to liposomes. In control experiments (not shown), His8-MBP-fl-cytb5 was also bound to liposomes in the absence of TEV protease. Lanes 8–10 contain residual His8-MBP-fl-cytb5 (1), His8-MBP (2), TEV protease (3), and unbound fl-cytb5 (4). C, Alignment of scintillation counting results ([3H] lipid) with Coomassie-stained denaturing gel for exposure of cytb5-sd to liposomes. Cytb5-sd (5) did not bind to liposomes.
Fig. 7B shows the results of the liposome incorporation experiment performed with His8-MBP-fl-cytb5. The bar graph above the gel shows the position of 3H-labeled phosphatidylcholine detected in the samples taken from the ultracentrifugation gradient, while the denaturing electrophoresis gel shows the proteins present in the different fractions from the gradient. Near quantitative proteolysis of fl-cytb5 was obtained. Moreover, the lipid containing fractions contained a significant fraction (~25–50%) of the total released fl-cyt5. His8-MBP did not associate with the liposomes.
Fig. 7C shows a comparable liposome incorporation experiment performed with His8-cytb5-sd, which lacks the requisite membrane anchor. In this case, no incorporation of cytb5-sd into the vesicle fraction was observed.
Discussion
Vector Platform
Vectors pVP55A and pVP56K are part of a platform developed in the Protein Structure Initiative to support high-throughput structural biology studies [24]. The modular design allows systematic variation of promoters, selectable markers, fusion tags, and patterns of either in vivo or in vitro proteolysis of the fusion proteins. The vectors also allow high-fidelity transfer of cloned and sequence-verified genes between different expression contexts for bacterial and wheat germ and insect cell-free translation. In this case, we investigated His8- and His8-MBP as vehicles for expression and purification of the monotopic membrane protein human cytochrome b5 retaining the functionally significant membrane anchor domain.
Auto-induction
F.W. Studier introduced auto-induction to high-throughput structural biology [38]. This powerful approach is based on bacterial diauxic growth in the presence of glucose and lactose. Some principles uncovered on the use of auto-induction with the vector platform described here are reported elsewhere [28]. In our previous work on the use of lactose as an inducer of recombinant protein expression, we found that this approach can lead to high-level incorporation of iron into iron-containing proteins [39]. We postulated that this favorable result arose from the slow onset of recombinant protein expression and continued cellular metabolism allowed by lactose-derived induction. This would contrast with the strong disruption of cellular growth and metabolism enforced by batch addition of IPTG.
When auto-induction was used to express the soluble forms of cytb5, whether as a domain alone or as a fusion to MBP, an increased accumulation of cellular heme was revealed by the intense red color of the harvested cells. We assumed that this would also correspond to a unitary stoichiometry of heme in the expressed cytb5, and indeed, this was true for cytb5-sd, which could be purified by Ni IMAC with a high level of heme incorporation. In contrast, expression of the insoluble His8-fl-cytb5 or a short time induction of soluble His8-MBP-fl-cytb5 with IPTG did not stimulate heme incorporation.
These finding are both consistent with the concept that auto-induction allows a smooth transition of the expression host from an un-induced to induced state under control of natural metabolic processes [28, 38, 39]. In this case, the need for increased heme biosynthesis was apparently signaled by accumulation of a soluble heme binding protein, but not by expression of an insoluble variant. With soluble expression, the auto-induction process permitted the transcription and translation of the natural biosynthetic genes needed to increase heme content. This is remarkably different from the diversion of many important cellular functions initiated by the batch addition of IPTG, and correspondingly, a lowered prospect for obtaining heme incorporation.
Given the favorable aspect of heme incorporation given by auto-induction, it was vexing to observe that the desired membrane anchor was not present in a substantial fraction of the protein purified. Since intact fl-cytb5 was obtained from the short time IPTG induction and TEV protease treatment of His8-MBP-fl-cytb5, we have concluded that no cryptic TEV protease recognition site is present in His8-MBP-fl-cytb5. Therefore, the observed heterogeneity must have arisen from in vivo events associated with the continuation of many cellular processes during the longer time required by auto-induction. At this point it is not possible to conclude whether the truncation arises from partial degradation of an mRNA or from proteolysis of the fusion protein. Nevertheless, this discovery signals a caveat in the use of auto-induction for high-throughput studies, and an obvious approach to address this variability would be to continue the practice of evaluating multiple approaches for bacterial expression screening prior to deciding on the best method for scale-up [25, 40–43].
Purification of fl-cytb5
Several procedures for the expression, solubilization, and incorporation of heme into fl-cytb5 expressed in E. coli have been developed. These require solubilization of the precipitated protein from bacterial membranes by addition of chaotropes and detergents, refolding of the solubilized proteins, heme incorporation, and further purification steps [21, 44].
In this work, we show how expression of His8-MBP-fl-cytb5 for a short time with IPTG induction gives rise to a fully soluble fusion protein that can be purified to homogeneity without the use of detergents and without the requirement for refolding the protein. A part of this work, we tested whether amylose affinity chromatography could be used to purify His8-MBP-fl-cytb5 and possibly avoid loss of heme. However, this method gave a considerably less pure protein preparation after a single step with significant protein loss and furthermore did not address the low heme incorporation in fl-cytb5 obtained from IPTG induction. Due to these experimental deficiencies and also due to the higher cost of the amylose resin, Ni IMAC and in vitro heme reconstitution emerged as the best option for purification of the His8-MBP-fl-cytb5 obtained from IPTG induction.
His8-MBP-fl-cytb5 was found to be a suitable substrate for soluble cytb5R and the V/K values for the fusion protein and cytb5-sd alone differed by only ~3-fold (Table 5). The mechanism of electron transfer between cytb5 and cytb5R is not clearly understood, but recent reports suggest that the interaction and electron transfer between cytb5 and its partners occur via a dynamic docking mechanism in which a large ensemble of weakly bound protein-protein configurations contribute to binding, but only a few are productive in electron transfer [45–47]. The minor differences in Vmax observed for His8-MBP-fl-cytb5 and cytb5-sd may represent differences in this docking process leading to the productive electron transfer complex.
In situ Delivery of Monotopic Membrane Proteins to Liposomes
Several elegant experiments have shown that fl-cytb5 spontaneously associates with cellular membranes [48, 49]. This natural property was exploited in order to use His8-MBP-fl-cytb5 as a delivery vehicle for incorporation into biologically relevant membrane environments (in this case, a synthetic liposome). The results of Fig. 7 show that while fl-cytb5 liberated by proteolysis could be effectively transferred into the liposome, cytb5-sd lacking the membrane anchor could not. Optimization of the ratio of liposome and time for membrane insertion should allow higher level of capture of the released fl-cytb5. This in situ delivery method proceeds directly in the presence of the desired liposome, and is not complicated by the presence of additional detergents. Bulk proteolysis in a rapid manner is also facilitated by the availability of gram quantities of highly active TEV protease obtained from an alternative use of this vector platform and auto-induction [25].
Other control experiments showed that His8-MBP-fl-cytb5 would also associate with the liposome fraction in the absence of TEV protease. Two conclusions can be made from this observation: 1) the cytb5 membrane anchor might be used as a C-terminal fusion to facilitate spontaneous association of other proteins with a membrane fraction, which has recently been shown in cell-free translation reactions [50]; and 2) proteolysis of His8-MBP-fl-cytb5 may occur either in solution or as the membrane-associated complex.
Utility
fl-cytb5 is used in a number of important enzymatic reactions as a specific electron donor (Table 1). The method described here allows controlled introduction of this protein into membrane environments possibly containing medically relevant enzymes such as stearoyl-CoA Δ9 desaturase or several cyt P450s involved in drug metabolism.
There are also numerous other monotopic integral membrane proteins known [51, 52], including examples such as full-length cytb5R, peptidoglycan glycosyltransferases (N-terminal membrane anchors, [16, 53]), cyclooxygenase-2, prostaglandin H2 synthase-1 (internal sequences and secondary structures serving as the membrane anchor, [54, 55]), monoamine oxidase B, fatty acid amide hydrolase (C-terminal membrane anchors, [56, 57]), and many others. For those proteins that have a C-terminal membrane anchor, the potential use of His8-MBP as a carrier for the full length, otherwise insoluble protein, and the potential for in situ transfer into a biological membrane environment for further studies is an attractive topic for more thorough investigation. The concept that an N-terminal membrane anchor may also serve as the 22 linker region between a solubilization tag and the desired monotopic membrane protein is also an intriguing possibility. In the vector format described here, proteolysis of a properly constructed fusion protein might release a hidden N-terminal membrane anchor to allow controlled association to proceed.
Acknowledgments
This work was funded by GM50853 to BGF. PS was American Heart Association postdoctoral fellow. P.G. Blommel and BGF developed the expression vectors and protocols for preparation of TEV protease with funding from Protein Structure Initiative grant NIH U54 GM074901 (J.L Markley, PI; G.N. Phillips, Jr. and B.G. Fox, Co-Investigators).
This work was supported by the National Institute of Health Grant GM-50853 (to B.G.F.). P.S. was supported by an American Heart Association Postdoctoral Fellowship.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mathews FS. The structure, function and evolution of cytochromes. Prog Biophys Mol Biol. 1985;45:1–56. doi: 10.1016/0079-6107(85)90004-5. [DOI] [PubMed] [Google Scholar]
- 2.Argos P, Mathews FS. The structure of ferrocytochrome b5 at 2.8 A resolution. J Biol Chem. 1975;250:747–751. [PubMed] [Google Scholar]
- 3.Vergeres G, Waskell L. Cytochrome b5, its functions, structure and membrane topology. Biochimie. 1995;77:604–620. doi: 10.1016/0300-9084(96)88176-4. [DOI] [PubMed] [Google Scholar]
- 4.Slaughter SR, Hultquist DE. Membrane-bound redox proteins of the murine Friend virus-induced erythroleukemia cell. J Cell Biol. 1979;83:231–239. doi: 10.1083/jcb.83.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Slaughter SR, Williams CH, Jr, Hultquist DE. Demonstration that bovine erythrocyte cytochrome b5 is the hydrophilic segment of liver microsomal cytochrome b5. Biochim Biophys Acta. 1982;705:228–237. doi: 10.1016/0167-4838(82)90182-0. [DOI] [PubMed] [Google Scholar]
- 6.Lloyd E, Ferrer JC, Funk WD, Mauk MR, Mauk AG. Recombinant human erythrocyte cytochrome b5. Biochemistry. 1994;33:11432–11437. doi: 10.1021/bi00204a005. [DOI] [PubMed] [Google Scholar]
- 7.Hegesh E, Hegesh J, Kaftory A. Congenital methemoglobinemia with a deficiency of cytochrome b5. N Engl J Med. 1986;314:757–761. doi: 10.1056/NEJM198603203141206. [DOI] [PubMed] [Google Scholar]
- 8.Porter TD. The roles of cytochrome b5 in cytochrome P450 reactions. J Biochem Mol Toxicol. 2002;16:311–316. doi: 10.1002/jbt.10052. [DOI] [PubMed] [Google Scholar]
- 9.Ogishima T, Kinoshita JY, Mitani F, Suematsu M, Ito A. Identification of outer mitochondrial membrane cytochrome b5 as a modulator for androgen synthesis in Leydig cells. J Biol Chem. 2003;278:21204–21211. doi: 10.1074/jbc.M301698200. [DOI] [PubMed] [Google Scholar]
- 10.Spatz L, Strittmatter P. A form of reduced nicotinamide adenine dinucleotide-cytochrome b 5 reductase containing both the catalytic site and an additional hydrophobic membrane-binding segment. J Biol Chem. 1973;248:793–799. [PubMed] [Google Scholar]
- 11.Fleming PJ, Dailey HA, Corcoran D, Strittmatter P. The primary structure of the nonpolar segment of bovine cytochrome b5. J Biol Chem. 1978;253:5369–5372. [PubMed] [Google Scholar]
- 12.Fleming PJ, Strittmatter P. The nonpolar peptide segment of cytochrome b5. Binding to phospholipid vesicles and identification of the fluorescent tryptophanyl residue. J Biol Chem. 1978;253:8198–8202. [PubMed] [Google Scholar]
- 13.Spatz L, Strittmatter P. A form of cytochrome b5 that contains an additional hydrophobic sequence of 40 amino acid residues. Proc Natl Acad Sci U S A. 1971;68:1042–1046. doi: 10.1073/pnas.68.5.1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dailey HA, Strittmatter P. Orientation of the carboxyl and NH2 termini of the membrane-binding segment of cytochrome b5 on the same side of phospholipid bilayers. J Biol Chem. 1981;256:3951–3955. [PubMed] [Google Scholar]
- 15.Strittmatter P, Fleming P, Connors M, Corcoran D. Purification of cytochrome b5. Methods Enzymol. 1978;52:97–101. doi: 10.1016/s0076-6879(78)52010-7. [DOI] [PubMed] [Google Scholar]
- 16.Enoch HG, Fleming PJ, Strittmatter P. Cytochrome b5 and cytochrome b5 reductase-phospholipid vesicles. Intervesicle protein transfer and orientation factors in protein-protein interactions. J Biol Chem. 1977;252:5656–5660. [PubMed] [Google Scholar]
- 17.Enoch HG, Catala A, Strittmatter P. Mechanism of rat liver microsomal stearyl-CoA desaturase. Studies of the substrate specificity, enzyme-substrate interactions, and the function of lipid. J Biol Chem. 1976;251:5095–5103. [PubMed] [Google Scholar]
- 18.Yao P, Wu J, Wang YH, Sun BY, Xia ZX, Huang ZX. X-ray crystallography. CD and kinetic studies revealed the essence of the abnormal behaviors of the cytochrome b5 Phe35–> Tyr mutant. Eur J Biochem. 2002;269:4287–4296. doi: 10.1046/j.1432-1033.2002.03120.x. [DOI] [PubMed] [Google Scholar]
- 19.Beck von Bodman S, Schuler MA, Jollie DR, Sligar SG. Synthesis, bacterial expression, and mutagenesis of the gene coding for mammalian cytochrome b5. Proc Natl Acad Sci U S A. 1986;83:9443–9447. doi: 10.1073/pnas.83.24.9443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith MA, Napier JA, Stymne S, Tatham AS, Shewry PR, Stobart AK. Expression of a biologically active plant cytochrome b5 in Escherichia coli. Biochem J. 1994;303( Pt 1):73–79. doi: 10.1042/bj3030073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mulrooney SB, Waskell L. High-level expression in Escherichia coli and purification of the membrane-bound form of cytochrome b(5) Protein Expr Purif. 2000;19:173–178. doi: 10.1006/prep.2000.1228. [DOI] [PubMed] [Google Scholar]
- 22.Lin YW, Zhao DX, Wang ZH, Yu WH, Huang ZX. Expression of lipase-solubilized bovine liver microsomal cytochrome b5 in Escherichia coli as a glutathione S-transferase fusion protein (GST-cyt b5) Protein Expr Purif. 2006;45:352–358. doi: 10.1016/j.pep.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 23.Sergeev GV, Gilep AA, Estabrook RW, Usanov SA. Expression of outer mitochondrial membrane cytochrome b5 in Escherichia coli. Purification of the recombinant protein and studies of its interaction with electron-transfer partners. Biochemistry (Mosc) 2006;71:790–799. doi: 10.1134/s0006297906070121. [DOI] [PubMed] [Google Scholar]
- 24.Blommel PG, Martin PA, Wrobel RL, Steffen E, Fox BG. High efficiency single step production of expression plasmids from cDNA clones using the Flexi Vector cloning system. Protein Expr Purif. 2006;47:562–570. doi: 10.1016/j.pep.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 25.Blommel PG, Fox BG. A combined approach to improving large-scale production of tobacco etch virus protease. Protein Expr Purif. 2007;55:55–68. doi: 10.1016/j.pep.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Strausberg RL, Feingold EA, Klausner RD, Collins FS. The mammalian gene collection. Science. 1999;286:455–457. doi: 10.1126/science.286.5439.455. [DOI] [PubMed] [Google Scholar]
- 27.Dailey HA, Strittmatter P. Structural and functional properties of the membrane binding segment of cytochrome b5. J Biol Chem. 1978;253:8203–8209. [PubMed] [Google Scholar]
- 28.Blommel PG, Becker KJ, Duvnjak P, Fox BG. Enhanced bacterial protein expression during auto-induction obtained by alteration of lac repressor dosage and medium composition. Biotechnol Prog. 2007;23:585–598. doi: 10.1021/bp070011x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kroliczewski J, Szczepaniak A. In vitro reconstitution of the spinach chloroplast cytochrome b6 protein from a fusion protein expressed in Escherichia coli. Biochim Biophys Acta. 2002;1598:177–184. doi: 10.1016/s0167-4838(02)00369-2. [DOI] [PubMed] [Google Scholar]
- 30.Studts JM, Fox BG. Application of fed-batch fermentation to the preparation of isotopically labeled- or selenomethionine-labeled proteins. Protein Expr Purif. 1999;16:109–119. doi: 10.1006/prep.1999.1067. [DOI] [PubMed] [Google Scholar]
- 31.Estabrook RW, Werringloer J. The measurement of difference spectra: application to the cytochromes of microsomes. Methods Enzymol. 1978;52:212–220. doi: 10.1016/s0076-6879(78)52024-7. [DOI] [PubMed] [Google Scholar]
- 32.Mihara K, Sato R. Partial purification of NADH-cytochrome b5 reductase from rabbit liver microsomes with detergents and its properties. J Biochem (Tokyo) 1972;71:725–735. [PubMed] [Google Scholar]
- 33.Burgess SW, Massenburg D, Yates J, Lentz BR. Poly(ethylene glycol)-induced lipid mixing but not fusion between synthetic phosphatidylcholine large unilamellar vesicles. Biochemistry. 1991;30:4193–4200. doi: 10.1021/bi00231a013. [DOI] [PubMed] [Google Scholar]
- 34.Sreenath HK, Bingman CA, Buchan BW, Seder KD, Burns BT, Geetha HV, Jeon WB, Vojtik FC, Aceti DJ, Frederick RO, Phillips GN, Jr, Fox BG. Protocols for production of selenomethionine-labeled proteins in 2-L polyethylene terephthalate bottles using auto-induction medium. Protein Expr Purif. 2005;40:256–267. doi: 10.1016/j.pep.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 35.Kapust RB, Waugh DS. Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Science. 1999;8:1668–1674. doi: 10.1110/ps.8.8.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hausinger RP, Honek J, Walsh FC. Separation of flavins and flavin analogs by high-performance liquid chromatography. Methods Enzymol. 1986;122:199–209. doi: 10.1016/0076-6879(86)22171-0. [DOI] [PubMed] [Google Scholar]
- 37.Yoshida Y, Tamura-Higashimaki Y, Sato R. Purification and characterization of intact cytochrome b5 from yeast microsomes. Arch Biochem Biophys. 1983;220:467–476. doi: 10.1016/0003-9861(83)90437-x. [DOI] [PubMed] [Google Scholar]
- 38.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 39.Hoffman BJ, Broadwater JA, Johnson P, Harper J, Fox BG, Kenealy WR. Lactose fed-batch overexpression of recombinant metalloproteins in Escherichia coli BL21(DE3): Process control yielding high levels of metal incorporated, soluble protein. Protein Expr Purif. 1995;6:646–654. doi: 10.1006/prep.1995.1085. [DOI] [PubMed] [Google Scholar]
- 40.Alzari PM, Berglund H, Berrow NS, Blagova E, Busso D, Cambillau C, Campanacci V, Christodoulou E, Eiler S, Fogg MJ, Folkers G, Geerlof A, Hart D, Haouz A, Herman MD, Macieira S, Nordlund P, Perrakis A, Quevillon-Cheruel S, Tarandeau F, van Tilbeurgh H, Unger T, Luna-Vargas MP, Velarde M, Willmanns M, Owens RJ. Implementation of semi-automated cloning and prokaryotic expression screening: the impact of SPINE. Acta Crystallogr D Biol Crystallogr. 2006;62:1103–1113. doi: 10.1107/S0907444906029775. [DOI] [PubMed] [Google Scholar]
- 41.Doyle SA. Screening for the expression of soluble recombinant protein in Escherichia coli. Meth Mol Biol. 2005;310:115–121. doi: 10.1007/978-1-59259-948-6_8. [DOI] [PubMed] [Google Scholar]
- 42.Knaust RK, Nordlund P. Screening for soluble expression of recombinant proteins in a 96-well format. Anal Biochem. 2001;297:79–85. doi: 10.1006/abio.2001.5331. [DOI] [PubMed] [Google Scholar]
- 43.Nguyen H, Martinez B, Oganesyan N, Kim R. An automated small-scale protein expression and purification screening provides beneficial information for protein production. J Struct Funct Genomics. 2004;5:23–27. doi: 10.1023/B:JSFG.0000029195.73810.86. [DOI] [PubMed] [Google Scholar]
- 44.Holmans PL, Shet MS, Martin-Wixtrom CA, Fisher CW, Estabrook RW. The high-level expression in Escherichia coli of the membrane-bound form of human and rat cytochrome b5 and studies on their mechanism of function. Arch Biochem Biophys. 1994;312:554–565. doi: 10.1006/abbi.1994.1345. [DOI] [PubMed] [Google Scholar]
- 45.Liang ZX, Jiang M, Ning Q, Hoffman BM. Dynamic docking and electron transfer between myoglobin and cytochrome b(5) J Biol Inorg Chem. 2002;7:580–588. doi: 10.1007/s00775-001-0332-0. [DOI] [PubMed] [Google Scholar]
- 46.Liang ZX, Nocek JM, Huang K, Hayes RT, Kurnikov IV, Beratan DN, Hoffman BM. Dynamic docking and electron transfer between Zn-myoglobin and cytochrome b(5) J Am Chem Soc. 2002;124:6849–6859. doi: 10.1021/ja0127032. [DOI] [PubMed] [Google Scholar]
- 47.Wheeler KE, Nocek JM, Cull DA, Yatsunyk LA, Rosenzweig AC, Hoffman BM. Dynamic docking of cytochrome b5 with myoglobin and alpha-hemoglobin: heme-neutralization “squares” and the binding of electron-transfer-reactive configurations. J Am Chem Soc. 2007;129:3906–3917. doi: 10.1021/ja067598g. [DOI] [PubMed] [Google Scholar]
- 48.Yabal M, Brambillasca S, Soffientini P, Pedrazzini E, Borgese N, Makarow M. Translocation of the C terminus of a tail-anchored protein across the endoplasmic reticulum membrane in yeast mutants defective in signal peptide-driven translocation. J Biol Chem. 2003;278:3489–3496. doi: 10.1074/jbc.M210253200. [DOI] [PubMed] [Google Scholar]
- 49.Stefanovic S, Hegde RS. Identification of a targeting factor for posttranslational membrane protein insertion into the ER. Cell. 2007;128:1147–1159. doi: 10.1016/j.cell.2007.01.036. [DOI] [PubMed] [Google Scholar]
- 50.Nomura SI, Kondoh S, Asayama W, Asada A, Nishikawa S, Akiyoshi K. Direct preparation of giant proteo-liposomes by in vitro membrane protein synthesis. J Biotechnol. 2007 doi: 10.1016/j.jbiotec.2007.08.023. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 51.Lomize MA, Lomize AL, Pogozheva ID, Mosberg HI. OPM: orientations of proteins in membranes database. Bioinformatics. 2006;22:623–625. doi: 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- 52.Jaysinghe S, Hristova K, Wimley W, Snider C, White SH. University of California; Irvine, Irvine, CA: 2007. http://blanco.biomol.uci.edu/index.html. [Google Scholar]
- 53.Lovering AL, de Castro LH, Lim D, Strynadka NC. Structural insight into the transglycosylation step of bacterial cell-wall biosynthesis. Science. 2007;315:1402–1405. doi: 10.1126/science.1136611. [DOI] [PubMed] [Google Scholar]
- 54.MirAfzali Z, Leipprandt JR, McCracken JL, DeWitt DL. Topography of the prostaglandin endoperoxide H2 synthase-2 in membranes. J Biol Chem. 2006;281:28354–28364. doi: 10.1074/jbc.M605206200. [DOI] [PubMed] [Google Scholar]
- 55.Picot D, Loll PJ, Garavito RM. The X-ray crystal structure of the membrane protein prostaglandin H2 synthase-1. Nature. 1994;367:243–249. doi: 10.1038/367243a0. [DOI] [PubMed] [Google Scholar]
- 56.Binda C, Hubalek F, Li M, Edmondson DE, Mattevi A. Crystal structure of human monoamine oxidase B, a drug target enzyme monotopically inserted into the mitochondrial outer membrane. FEBS Lett. 2004;564:225–228. doi: 10.1016/S0014-5793(04)00209-1. [DOI] [PubMed] [Google Scholar]
- 57.Bracey MH, Hanson MA, Masuda KR, Stevens RC, Cravatt BF. Structural adaptations in a membrane enzyme that terminates endocannabinoid signaling. Science. 2002;298:1793–1796. doi: 10.1126/science.1076535. [DOI] [PubMed] [Google Scholar]
- 58.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–252. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]