

Abstract
The heterodimeric actin-capping protein (CP) regulates actin assembly and cell motility by binding tightly to the barbed end of the actin filament. Here we demonstrate that myotrophin/V-1 binds directly to CP in a 1:1 molar ratio with a Kd of 10–50 nM. V-1 binding inhibited the ability of CP to cap the barbed ends of actin filaments. The actin-binding COOH-terminal region, the “tentacle,” of the CP β subunit was important for binding V-1, with lesser contributions from the α subunit COOH-terminal region and the body of the protein. V-1 appears to be unable to bind to CP that is on the barbed end, based on the observations that V-1 had no activity in an uncapping assay and that the V-1·CP complex had no capping activity. Two loops of V-1, which extend out from the α-helical backbone of this ankyrin repeat protein, were necessary for V-1 to bind CP. Parallel computational studies determined a bound conformation of the β tentacle with V-1 that is consistent with these findings, and they offered insight into experimentally observed differences between the α1 and α2 isoforms as well as the mutant lacking the α tentacle. These results support and extend our “wobble” model for CP binding to the actin filament, in which the two COOH-terminal regions of CP bind independently to the actin filament, and bound CP is able to wobble when attached only via its mobile β-subunit tentacle. This model is also supported by molecular dynamics simulations of CP reported here. The existence of the wobble state may be important for actin dynamics in cells.
Myotrophin is a 12-kDa protein identified in hypertrophied rat hearts (1) and dilated cardiomyopathic human hearts (2). Myotrophin was named for its ability to stimulate hypertrophy in cardiac myocytes when added to cells in culture (1). Transgenic mice overexpressing myotrophin in cardiac myocytes develop cardiac hypertrophy and heart failure (3). Myotrophin was found to be identical to the protein V-1 (4), which had been identified based on increased expression in granule cell neurons during development of the rat cerebellum (5). V-1 expression correlates with morphogenetic changes of cerebellar differentiation. V-1 is expressed in many vertebrate cells and tissues (6). V-1 homologues are present and highly conserved across vertebrates, and similar protein sequences are predicted by the genomes of other animal species, but not by those of fungi or plants. V-1 is important for expression of catecholamine-synthesizing enzymes (7), differentiation and regeneration of skeletal muscle (8), folliculogenesis and corpus luteum formation in the ovary (9), and regulation of insulin secretion (10). We will refer to the myotrophin/V-1 protein as V-1 for simplicity.
Structurally, V-1 is a small ankyrin repeat protein. A solution structure of V-1 shows two full ankyrin repeat motifs in tandem, with additional incomplete repeats at the amino and carboxyl termini (11, 12). An ankyrin repeat consists of a β hairpin loop followed by a pair of anti-parallel α helices connected by a short turn sequence (13). Tandem repeats stack on each other, based on hydrophobic interactions between the helices. The loops protrude from this structural backbone, and their more variable sequences impart specificity to interactions with other proteins.
V-1 was found to bind and inhibit actin-capping protein, known as CapZ (14). Capping protein binds to the barbed ends of actin filaments with high affinity (Kd < 1 nM), which prevents the addition and loss of actin monomers at the end (15). In the cell, CP3 can cap the barbed ends of filaments created by nucleation. CP is a key component of the dendritic nucleation model (16), and CP is necessary for reconstitution of actin-based motility from pure proteins in vitro (17). In yeast, Drosophila, Dictyostelium, and mouse cells, loss of CP results in abnormalities of actin assembly, morphogenesis, and motility (15).
CP is an α/ β heterodimer (molecular mass of α ~36 kDa, β ~32 kDa). Vertebrates have two major isoforms of each subunit. In striated muscle cells, the β1 isoform is found at the Z-line, the β2 isoform is found elsewhere, and the two β isoforms cannot functionally substitute for one another (18). Non-muscle cells generally express only the β2 isoform (19). The COOH-terminal regions of the α and β subunits are on the surface of the protein (20), and both contribute to high affinity capping of the barbed end of actin (21). In previous work, CP was pulled down by TAP-tagged V-1 expressed in cultured 293T cells (14). The α subunit was the α1 isoform, and the β isoform was not determined. In those studies, purified V-1 was found to bind and inhibit recombinant CP α1β1 (14).
The mechanism and molecular details of the interaction of V-1 with CP and its functional implications in cells are not known. In this study, we investigate the interaction of V-1 with CP, including a structure-function analysis with mutational and computational approaches. The results have relevance to our understanding of how CP binds to the barbed end of the filament.
EXPERIMENTAL PROCEDURES
Reagents, General Procedures, Peptides, and Proteins
Restriction endonucleases and DNA-modifying enzymes were purchased from Invitrogen, Roche Applied Science, or New England Biolabs. Oligonucleotides were purchased from the Protein and Nucleic Acid Chemistry Laboratory, Washington University Medical School (St. Louis, MO). PCR was performed according to the manufacturer’s directions (PerkinElmer Life Sciences). Bacterial media preparation, restriction digests, and ligation of DNA were performed by standard protocols. Peptides corresponding to the COOH-terminal region of V-1 for generation and affinity purification of antibody (22) and corresponding to the COOH-terminal regions of theβ1 and β2isoformsofCP(21)were generated by an automated peptide synthesizer and purified by high pressure liquid chromatography (Biomolecules Midwest, St. Louis, MO). Actin, spectrin-F-actin seeds, CP, and CP mutants were purified as described (21).
To express GST-V-1 in bacteria, a 359-bp V-1 cDNA sequence was PCR-amplified with forward and reverse primers CCG GAA TTC TTA TGT GCG ACA AGG AGT TCA TG and CCC AAG CTT TCA CTG GAG AAG AGC TTT GAT TG from a human embryonic cDNA library and cloned into pGEX-KG (Amersham Biosciences) to produce pBJ 1526.
V-1 mutants were generated via PCR-based mutagenesis using this plasmid as the template, as described (23). The forward and reverse primers, respectively, for the mutagenesis were as follows: V-1 Δ1–20, TAT GTG GCC AAG GGA GAA GAT GTC AAC, AAG TAT ACC ACC ACC ACC ACC GGA AAC, V-1 Δ1–94, TTC TCA TCG TGA CTG ACG ATC TGC CTC GCG CTG TTC, TTC TTT CAC CTC ATC CAA GTC TCC GTT TTT CAG GGC; V-1 Mut Loop1, GCA GCA ATT ACT CCT CTT CTG TCT GCT GTC TATGAG, AGC AGC TGG AGC ATT AAT ATC TGC TCC TTT CAG; V-1 Mut Loop2, GCA GCA AGG AAA CCT CTT CAT TAT GCA GCA GAT TGA AA, AGC AGC TGG AGC ATT AAT ATC TGC TCC TTT CAG CAG CAG TTC CAG. The numbers of the resulting plasmids were as follows: V-1 Δ1–20, pBJ 1683; V-1 Δ1–94, pBJ 1684; V-1 Mut Loop1, pBJ 1532; V-1 Mut Loop2, pBJ 1537. DNA sequencing and restriction digests confirmed the identity of the construct and the lack of mutations.
Expression was induced with 100 mM isopropyl-1-thio-D-galactopyranoside, and GST-V-1 was purified as described (24) with minor changes. The concentration of GST-V-1 was determined by absorbance at 280 with a calculated extinction coefficient of 50,570 M−1 cm−1. GST-V-1 on glutathione-agarose beads was cleaved with thrombin (Amersham Biosciences), according to the manufacturer’s instructions. The V-1 protein was tested for purity by SDS-PAGE, and the concentration was calculated by absorbance at 280 nm with a calculated extinction coefficient of 9520 M−1 cm−1. Purified GST-V-1 and thrombin-cleaved V-1 inhibited CP in a similar manner in functional assays. Cleaved V-1 was used in all of the functional and physical assays described here, except the uncapping assays, which were done with GST-V-1 protein. V-1 mutant proteins were purified similarly, and their folding was tested by circular dichroism spectroscopy as described (25) with a JASCO J-600 spectropolarimeter.
Chicken CP α1β1; CP truncation mutants CPα1(ΔC28) β1, CPα1β1(ΔC34), and CPα1(ΔC28) β1(ΔC34); and mouse CP α1β2 were expressed and purified as described (26) from pBJ 994, 1533, 1534, 1536, and 1219, respectively. A mouse CP α2β2 expression plasmid, pBJ 1603, was constructed using a similar strategy (26) in pET-24a (Novagen) by Dr. Ilgu Kang. The α2 and β2 sequences were PCR-amplified from cDNA-containing plasmids, pBJ 601 and 622, respectively (19, 27). Primers included restriction enzyme sites for NdeI/HindIII and NdeI/PvuI. The PCR-amplified α2 sequence was inserted between the NdeI and the HindIII sites of the vector, and β2 was inserted between the NdeI and PvuI sites. Each subunit had a separate ribosomal binding site to maximize translational efficiency. PCR-induced single base errors were corrected by site-directed mutagenesis. Purified CP was stored at −20°C in 10 mM Tris-HCl, pH 8.0, 40 mM KCl, 0.5 mM dithiothre-itol, and 50% glycerol. The concentration of CP was determined by absorbance at 280 with extinction coefficients of 76,300 M−1 cm−1 for α1β2, 78,450 M−1 cm−1 for α1β1, and 79,300 M−1 cm−1 for α2β2, calculated with ProtParam at www.ExPASy.org, which uses values from Pace et al. (28).
Actin Polymerization Assays
Actin was purified and labeled with pyrenyliodoacetamide (Molecular Probes, Inc., Eugene, OR), and actin polymerization and capping assays were performed as described (29). The uncapping assay was performed as described (30), using PIP2 as a positive control.
Fitting to Determine Binding Constants
Binding constants for capping of actin filaments by CP were determined by kinetic modeling of actin polymerization assays, using Berkeley Madonna as described (21). The kinetic mechanism used is shown in Reactions 1–3, where A represents actin monomer, Nb is free barbed ends, CP is capping protein, and V is V-1. The complex generated by CP and V-1 is represented as CPV. The on- and off-rate constants were determined from least-squares fitting for the full time course of the reaction.
| Reactions 1–3 |
The initial concentration of Nb was set equal to the con-centration of free barbed ends created by the spectrin-F-actin seeds alone in control polymerization reactions. The barbed end on-rate and off-rate constants for actin elongation were 11.6 μM−1 s−1 and 1.4 s−1, respectively (31). The rate constants k+cap and k−cap in Reaction 2 were fit with results from experiments with CP added, and those values were consistent with previous studies (21). When V-1 was added, the results were used to determine k+V and k−V in reaction 3. In this model, the CPV complex has no capping activity. CP mutants with single α or β COOH-terminal truncations can cap actin, so the concentration of the CP mutant was substituted for that of WT CP in Reactions 2 and 3. The CP mutant lacking both COOH-terminal regions, referred to as CPΔΔ, does not cap actin, so we performed a competition experiment with WT CP and added Reaction 4 to the kinetic mechanism.
| Reaction 4 |
For the steady-state assay, the reaction mechanisms were the same. The Kd value for CP binding to the barbed end was determined first, from a CP titration experiment. Next, with a constant CP concentration, a V-1 titration was performed. Those results were used to determine the Kd for V-1 binding to CP. Least-squares fitting by nonlinear regression was performed using Prism 4.0 (Graph-Pad Software, San Diego, CA). In this analysis, the number concentration of actin filament barbed ends is assumed to be much less than the concentration of CP, so that free [CP] is set equal to total [CP].
Tryptophan Fluorescence of CP Binding to V-1
Intrinsic tryptophan fluorescence emission spectra for WT and mutant CP proteins were collected as described (32). A continuous variation assay was done to determine the stoichiometry of CP α1β2 binding to V-1. The ratios of the two proteins were varied while maintaining the total protein concentration at 10 μM, which was well above the experimentally determined Kd of ~40 nM.
Molecular Simulations
The starting point for our computational work was the crystal structure of CP (Protein Data Bank code 1IZN) and the NMR structure of V-1 (Protein Data Bank code 1MYO). We first selected the COOH-terminal portion of the β1 subunit of CP, which corresponds to the peptide used in the experiments, and simulated the molecular dynamics of the peptide structure using Gromacs 3.2 (33). Following minimization and heating to 300 K, we performed 10 ns of NVT simulations with the OPLS/AA force field, SPC waters, Particle Mesh Ewald, Berendsen temperature coupling (NVT ensemble), and 2-fs time steps. Structures were written out every 10 ps, resulting in an ensemble of 1000 structures. Each of these peptide structures was then used in docking studies using AutoDock 3.0 (34). Docking grid maps were created for 17 NMR V-1 structures in the Protein Data Bank file, and all 1000 tentacle structures were docked to each map, resulting in a total of 17,000 trajectories. The docking results were ranked by energy and clustered based on a root mean square deviation of 4 Å. The cluster analysis resulted in a single top-ranked structure, which was used in all subsequent analysis. In order to determine the interaction of V-1 with full-length CP α1β1, we used the β1 tentacle as a reference point and fit this top structure onto a CP α1β1 structure taken from a molecular dynamics simulation. The CP molecular dynamics simulation was performed using NAMD (35). Here we used the CHARMM27 force field, TIP3P waters, Particle Mesh Ewald, Berendsen temperature and pressure coupling (NPT ensemble), and 2-fs time steps. The heating was again performed in 50 K steps with α carbon constraints, equilibrated at 300K without constraints, followed by a 20-ns production trajectory. The simulation of the α tentacle-deleted protein (missing the 28 COOH-terminal residues of the α subunit, CPαΔC28) was carried out using exactly the same protocol.
RESULTS
Molecular Dynamics Simulation of CP
We used molecular dynamics to investigate the range of structures that CP might occupy in solution. Beginning with the x-ray crystal structure, we performed a 20-ns molecular dynamics simulation (see “Experimental Procedures” for details). For the full-length wild type protein, the position of the COOH-terminal α-helical region of the β subunit fluctuated to the highest degree, as illustrated in Fig. 1. The COOH-terminal α-helical region of the α subunit moved much less, remaining in position on the surface of the protein, primarily through the interactions of Trp279. The loop proximal to the helix and the COOH-terminal extension distal to the helix did show some movement, but the α-helix itself moved minimally. These results are further illustrated with plots of root mean square fluctuations in Fig. 2, C and D.
FIGURE 1. Molecular dynamics analysis of CP.

Range of dynamics of the α and β COOH termini (tentacles) based on a 20-ns molecular dynamics simulation.
FIGURE 2. Molecular dynamics analysis of the effect of truncation of the CP αCOOH terminus.

A, regions on the α subunit that show a significant difference in dynamics upon deletion of the αtentacle are shown in blue, orange, and purple, and those colors correspond to the bars in C. The translucent pink region represents the surface where V-1 is predicted to contact the α subunit. B, residues that differ between the α1 and α2 isoforms are labeled and shown in space-filling mode. They overlap extensively with the pink contact surface and with the colored increased fluctuation residues of A. C and D, root mean square fluctuations of the α and β subunit residues during a 20-ns MD simulation. Wild-type CP is compared with a CP mutant lacking the α subunit COOH-terminal region. C, the α subunit shows three regions of significant change, indicated by colors that match the corresponding regions in B. D, the β subunit shows only one small difference, in an α-helical segment that underlies the α-subunit COOH-terminal region, colored green on the graph and in the inset.
We then asked how truncating the actin-binding COOH-terminal regions of the subunits affected the structure of the rest of the protein. These mutants have been used in the past and are used here, below, to analyze how CP binds to the barbed end of the actin filament. Upon removal of the α subunit COOH-terminal region, fluctuations were significantly decreased in a region of the α subunit on the other side of the protein, near the COOH-terminal region of the β subunit, whereas the β subunit showed little change apart from the helix that contacts the α subunit COOH terminus. These results are illustrated in Fig. 2. This analysis is important for several reasons. The region of the α subunit exhibiting the change in dynamics is where most of the differences between the α1 and α2 isoforms are located, and this region is part of the contact site for V-1 predicted by our docking analysis. This correspondence is in line with experimental observations, described below, that the α1 and α2 isoforms have slightly different affinities and that the truncation of the α COOH terminus has a minor effect on the affinity for V-1. The mobility of the β subunit COOH terminus in the molecular dynamics simulation strongly suggested that its movements were independent of the rest of the protein. These points are addressed further under “Discussion.”
V-1 Inhibits the Actin-capping Activity of CP
To investigate whether V-1 influences the capping activity of CP α1β2, which includes the major β isoform expressed in nearly all vertebrate cells and tissues, we performed seeded actin polymerization assays. CP α1β2 inhibited barbed end polymerization in a dose-dependent manner (Fig. 3A). The equilibrium dissociation constant for CP binding to barbed ends (Kcap) was 0.1 nM, consistent with previous results (21). The addition of increasing concentrations of V-1 inhibited the capping activity of 2–4 nM CP, with complete inhibition at 500 nM V-1 (Fig. 3B). V-1 alone, without CP, had no effect. Kinetic modeling with a simple model of V-1 binding CP and completely inhibiting the ability of CP to cap the barbed end gave good fits (Fig. 3B, red lines), with a Kd of 40 ± 9 nM for the V-1·CP interaction (Table 1).
FIGURE 3. Inhibition of CP by V-1 in actin polymerization seeded growth assays.
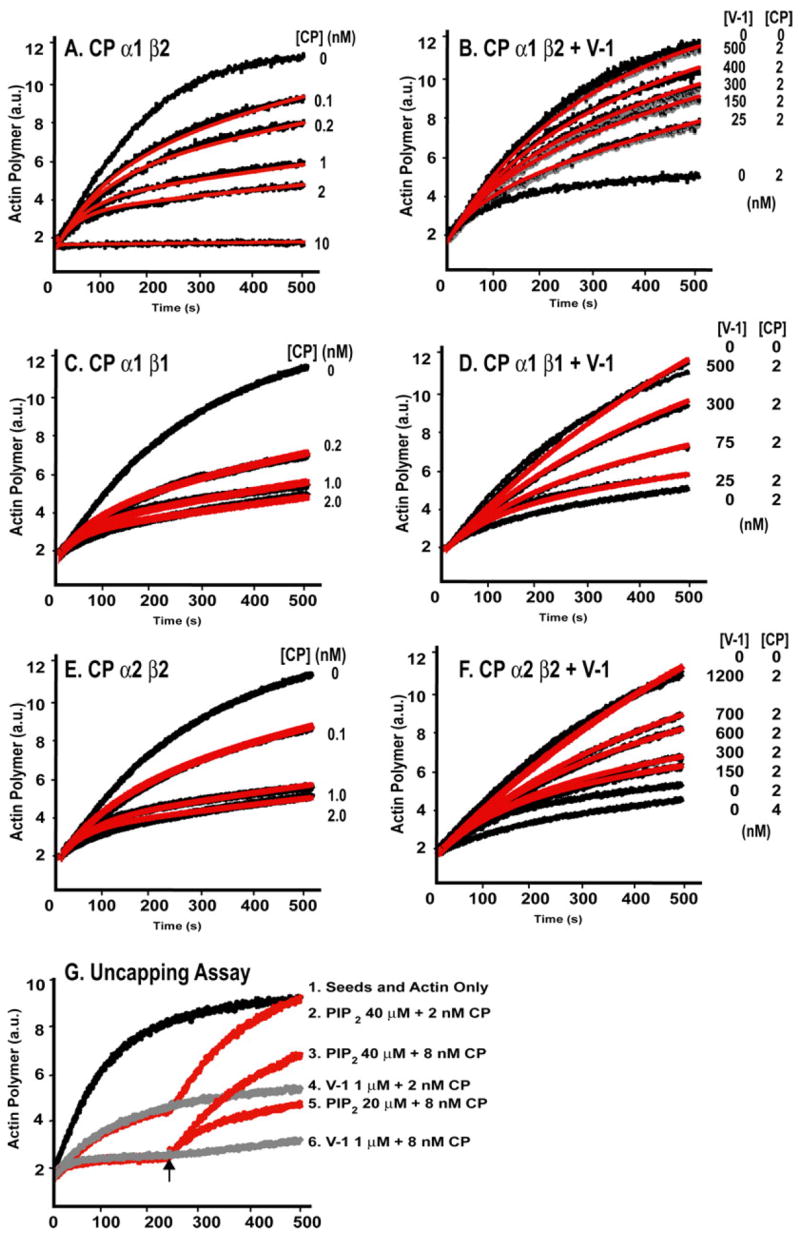
Actin polymer concentration based on fluorescence (arbitrary units; a.u.) of pyrene-labeled actin (2 μM; 4% pyrene) is plotted versus time. Polymerization was nucleated with spectrin-actin seeds. A, C, and E, CP α1β2, α1β1, and α2β2, respectively, cap the barbed end of actin at indicated concentrations (black lines). Best fit curves from kinetic modeling with Reactions 1 and 2(see “Experimental Procedures”) are shown in red. B, D, and F, V-1 inhibits capping by CP α1β2, α1β1, and α2β2, respectively. Actin polymerization is shown versus time with CP and V-1 concentrations as indicated. Best fit curves from kinetic modeling with Reactions1–3 (see “Experimental Procedures”)are shown in red. G,V-1does not uncap barbed ends capped with CP. Actin polymerization versus time in the presence of 2 or 8 nM CP. After 250 s, PIP2 or GST-V-1 was added (arrow). PIP2 causes rapid uncapping, and GST-V-1 has no effect.
TABLE 1. Equilibrium dissociation constants for binding of WT CP, mutant CP, and the CP β peptide with WT or mutant V-1, calculated from modeling the actin polymerization kinetics experiments.
Most Kd values are the mean of 3–4 independent experiments, and they are listed as such, with S.E. In cases with two independent experiments, the two values are listed.
| CP | Kd |
|---|---|
| nM | |
| CP and V-1 | |
| WT CP α1β2 3 V-1 | 40 ± 9 |
| WT CP α1β1 3 V-1 | 45 ± 3 |
| WT CP α2β2 3 V-1 | 153 ± 20 |
|
| |
| CP mutants and V-1 | |
| CPα1(ΔC28) β1 + V-1 | 65 ± 7 |
| CPα1β1(ΔC34) + V-1 | 270 ± 6 |
| CPα1(ΔC28) β1(ΔC34) + V-1 | 262 ± 35 |
| CP β1 28-aa peptide + V-1 | 70, 100 |
| CP β1 34-aa peptide + V-1 | 64, 70 |
|
| |
| CP and V-1 mutants | |
| WT CP + V-1 Mut Loop 1 | 4450, 4506 |
| WT CP + V-1 Mut Loop 2 | 1363, 1547 |
| WT CP + V-1 Δ1–94 | 6300, 6750 |
| WT CP + V-1 Δ1–20 | 59, 67 |
The β1 isoform of CP is specific for the Z-line of striated muscle. Assays with CP α1β1 gave similar results, with a Kd of 45 ± 3 nM (Fig.3, C and D, Table 1). The α1 and α2 isoforms of CP are widely expressed, in a ratio that varies among cell and tissue types (27). CP α2β2 capped actin with a Kcap of 0.2 nM (Fig. 3E). V-1 inhibited the capping activity of CP α2β2, and kinetic modeling gave a Kd of 153 ± 20 nM (Fig. 3F, Table 1).
In addition to actin polymerization growth assays, we tested the binding affinity of V-1 for CP α1β2 with a steady-state polymerization assay. Increasing concentrations of CP caused the steady-state level of F-actin to decrease and plateau, characteristic of barbed end capping, which increases the critical monomer concentration to that of the pointed end (Fig. 4A). Increasing concentrations of V-1 reversed the effect of CP (Fig.4B). The Kd for V-1 binding to CP was 10–30 nM in three experiments, based on fitting to the same simple reaction mechanism used for the kinetics assays.
FIGURE 4. V-1 inhibition of CP in a steady-state assay.
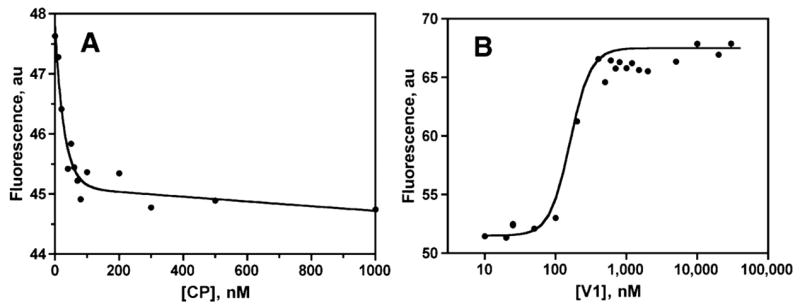
Pyrene-actin fluorescence, which is proportional to the concentration of filamentous actin, is plotted. A, increasing concentrations of CP α1β2, in the absence of V-1, causes the F-actin level to fall, because the critical concentration rises to that of the pointed end. B, increasing concentrations of V-1 added to 75 nM CP antagonize this inhibitory effect and promote actin polymerization.
Some inhibitors of CP, such as CARMIL or PIP2, can rapidly reverse the effect of CP on actin, implying that they can dissociate CP from the capped barbed ends in addition to preventing CP from binding to the barbed end (30, 36). We tested V-1 in an uncapping assay (Fig. 3G). The addition of GST-V-1 at concentrations up to 1 μM, which was sufficient to completely inhibit the capping activity of CP in Fig. 3B, had no effect in the uncapping assay, for two CP concentrations (Fig. 3G, curves 4 and 6). As a positive control, PIP2 produced a rapid increase in the polymerization rate, approaching the maximal rate of actin polymerization in the absence of capping protein (Fig. 3G, curves 2, 3, and 5), which is consistent with complete uncapping.
Stoichiometry of CP Binding V-1
These analyses assume a 1:1 stoichiometry for CP binding to V-1, which is supported by previous results with native PAGE (14). To measure the stoichiometry, we used intrinsic tryptophan fluorescence and the continuous variation method (37). The two proteins were mixed in various ratios, with a constant total protein concentration of 10 μM, well above the Kd. CP has higher intrinsic fluorescence, due to its 9 tryptophan residues, than does V-1, with a single tryptophan residue. The presence of V-1 increased the intrinsic fluorescence of CP. The maximum fluorescence intensity was seen at concentrations of 5 μM for each of CP and V-1 (Fig. 5), indicating a 1:1 molar ratio for the complex.
FIGURE 5. Continuous variation analysis of the stoichiometry of V-1 interaction with CP, assayed by intrinsic tryptophan fluorescence.

Fluorescence in arbitrary units (a.u.) is plotted against the concentrations of the proteins. Values from two independent experiments are shown.
V-1 Interaction with CP Mutants Lacking the COOH-terminal Actin-binding Regions
We asked which regions of CP bound to V-1. We tested truncation mutants of CP α1β1 lacking one or both COOH-terminal actin-binding sites with functional assays of actin capping. CPα1(ΔC28) β1, which lacks 28 amino acid residues at the COOH terminus of the α1 subunit, capped actin with a Kcap of 640 ± 240 nM, consistent with previous studies (21). V-1 inhibited the capping activity of CPα1(ΔC28) β1 (Fig. 6A, black lines), and kinetic modeling gave good fits (red lines) with a Kd of 65 ± 7 nM (Table 1), slightly weaker than for full-length CP α1β1.
FIGURE 6. V-1 inhibition of CP COOH-terminal truncation mutants in actin polymerization growth assays.

Actin polymer concentration based on arbitrary units of pyrene fluorescence is plotted versus time. A, CPα1(Δ28) β1; B, CPα1β1(ΔC34). Conditions were as in Fig. 1, with the indicated concentrations of V-1 and mutant CP. Best fits are shown in red from modeling with Reactions 1–3 (see “Experimental Procedures”). C, V-1 interaction with the CPα1(Δ28) β1(Δ34) mutant. This CP mutant does not cap actin, but it competes with WT CP for binding to V-1. Concentrations of the proteins (in nM) were as indicated on the right. Best fits from kinetic modeling, shown in red, were determined with Reactions 1– 4 (see “Experimental Procedures”).
CPα1β (ΔC34), lacking the COOH-terminal 34 amino acids of the β subunit, capped actin with a Kcap value of 41.3 ± 2.2 nM, consistent with previous findings (21). V-1 also inhibited the capping activity of this mutant (Fig. 6B, black lines), and modeling gave a good fit with a Kd of 270 ±6 nM (Table 1), ~6-fold weaker than that of full-length WT CP α1β1.
A CP mutant with truncation of both COOH-terminal regions CPα1(ΔC28) β1(Δ34) had no capping activity, as in previous studies (21). To measure binding of V-1 to this CP mutant, we performed actin polymerization assays in the presence of WT CP, asking if increasing concentrations of the mutant CP would compete with WT CP for binding to V-1. The addition of the CPα1(ΔC28) β1(ΔC34) mutant neutralized the inhibitory effect of V-1 and restored the activity of WT CP to a nearly normal level. We modeled the reaction assuming that V-1 can bind the mutant or WT CP. The affinity of V-1 for WT CPα1β1 was set to the value determined above. For V-1 binding to the CPα1(ΔC28) β1(Δ34) mutant, the Kd value was 262 ± 35 nM (Table 1), ~6-fold higher than that of WT CP and similar to that of the CP mutant lacking only the β tentacle. Thus, the results of the actin-capping assays with the CP truncation mutants indicate that the β subunit COOH-terminal region is important for binding V-1 and that the α subunit COOH-terminal region and the body of CP make lesser but significant contributions.
CP β COOH-terminal Peptides Bind V-1
The above results show that the CPβ COOH terminus is important for CP to bind V-1. We asked whether peptides corresponding to the β subunit tentacle in CP α1β1 would be sufficient to bind V-1, using functional actin-capping assays. Two peptides, of 28 and 34 aa, were tested. Each peptide was able to cap barbed ends, with Kcap values of 500 ± 9 nM, similar to previous findings (21). In the presence of a 5 μM concentration of the 28-aa β1 peptide (Fig. 7C), V-1 concentrations up to 1.5 μM were able to partially inhibit the capping activity. For the 34-aa peptide, we added 0.5 and 0.8 μM V-1 to 1.5 μM of the peptide and saw partial inhibition (Fig. 7D). Modeling gave a Kd of ~65 nM for the 34-aa peptide and ~85 nM for the 28-aa peptide (Table 1), which are similar to the Kd of 65 nM for V-1 binding to the CPα COOH terminus truncation mutant and greater than the Kd of 40 nM for WT CP. Thus, the CP β1 COOH-terminal tentacle is sufficient as well as necessary for binding to V-1.
FIGURE 7. V-1 interaction with peptides corresponding to the COOH-terminal region of the β subunit, both β1 and β2 isoforms, assayed with actin polymerization growth assays (2 μM actin, 4% pyrene-labeled).

A, high concentrations of the 34-aa β2 peptide are able to cap. B, the addition of V-1 at concentrations up to 6 μM inhibits the capping activity, almost completely. C, the 28-aa β1 peptide caps actin at 5 μM, with partial inhibition by V-1 at concentrations up to 1.5 μM. D, the 34-aa β1 peptide partially caps actin at 1.5 μM, with partial inhibition by V-1 at concentrations up to 0.8 μM.
We also tested a 34-aa peptide corresponding to the COOH terminus of the β2 isoform (21). Much higher concentrations of this peptide were needed to cap actin (Fig. 7A), presumably because the peptide folds poorly, so we were not able to analyze the data with kinetic modeling. We did observe that V-1 was able to inhibit the capping activity of the β2 peptide (Fig. 7B). V-1 binding to both the β1 and β2 peptides of CP corroborates our finding that V-1 is able to bind to both the β isoforms of CP in the context of full-length heterodimeric protein.
The Loops of V-1 Are Needed to Interact with CP
We asked which regions of V-1 are necessary for its interaction with CP. Ankyrin repeat proteins often interact with other proteins by their loop regions (13), and V-1 has two such loops, which we refer to as ANK1 and ANK2 (Fig. 8A). Loop regions from different ankyrin repeat protein families generally show little sequence similarity (13), but the sequences of the loops of V-1 are nearly identical across vertebrates (Fig. 8B).
FIGURE 8. Effect of V-1 mutants on capping activity of CP.

A, NMR solution structure of V-1 adapted from Yang et al. (11). The α helices are labeled as α 1–α8. The loops of the complete ankyrin repeats are labeled ANK1 and ANK2. Residues EGGR (arrow; ANK1) and KHHI (arrow; ANK2) were mutated to alanine. Arrows mark the start of the truncation mutants V-1 Δ1–20 (deletion of the first 20 amino acids) and V-1 Δ1–94 (deletion of the first 94 amino acids). B, alignment of V-1 sequences from various organisms, with some features from A labeled. A period indicates a residue identical to the one in the top line, mouse V-1. The mutated loop regions, boxed, have very similar sequences among vertebrates. The alignment was performed with ClustalW. For C–F, actin polymerization is plotted versus time for a seeded growth assay. 2 μM actin, 4% pyrene-labeled, was used with indicated concentrations of V-1 mutants and mouse CP α1β2. The best fit from kinetic modeling of Reactions 1–3 (see “Experimental Procedures”) is shown in red. C, effect of the V-1 ANK1 mutant, with a magnified view of the early time course of the reactions. D, effect of the V-1 ANK2 mutant, again with a magnified view of early time. E, effect of the V-1 Δ1–94 mutant. F, effect of the V-1 Δ1–20 mutant.
We assayed the ability of V-1 mutants to inhibit CP in seeded actin polymerization growth assays. To investigate the importance of the ANK1 loop, we mutated four residues at the tip of the loop, Glu33–Arg36, to alanine. This V-1 mutant showed normal α-helical content by circular dichroism, as did all of the V-1 mutants except as noted. The mutant had very little effect on CP activity, even with a 10 μM concentration of the V-1 mutant and 4 nM CP (Fig. 8C, compare with Fig. 3A). Kinetic modeling gave fits (Fig. 8C, redlines, insetis a zoomed image of initial time) with aKd of ~4500 nM (Table 1), which is ~100-fold weaker than the Kd of WT V-1 for CP. We tested the ANK2 loop in a similar way by changing the four tip residues, Lys66–Ile69, to alanine (Fig. 8A). The mutant also had only a weak effect on the capping activity of WT CP (black lines). Kinetic modeling gave good fits (Fig. 8D, red lines; inset shows zoomed fit of initial time) with a Kd of ~1450 nM, ~30 times weaker than the WT V-1·CP interaction (Table 1).
To test the role of the amino terminus of V-1, we generated a truncation mutant lacking the first 20 residues of the NH2 terminus, V-1 Δ1–20. The mutant completely inhibited CP (Fig. 8F). Kinetic modeling gave good fits (Fig. 8F, red lines) with a Kd of ~60 nM (Table 1). Finally, we tested a V-1 Δ1–94 mutant, lacking the two complete ankyrin repeats. This mutant had no α-helical structure by circular dichroism, as expected. The mutant had very little effecton CP activity (Fig. 8E, Table1), and the mutant alone had no effect on actin polymerization. Thus, the loops of the V-1 ankyrin repeats are necessary for its interaction with CP.
Molecular Docking of CP and V-1
In order to gain more structural insight into the binding of V-1 and CP, we performed molecular dynamics on the β1 tentacle and used these structures in molecular docking studies with the NMR solution structure of V-1 (see “Experimental Procedures”). Analysis of these results provided us with a clear prediction for the V-1· β1 tentacle complex. The tentacle binds between the ankyrin repeats on V-1, making significant interaction with both loop regions (Fig. 9). Therefore, this structure agrees well with the in vitro mutagenesis results. Taking this structure for the peptide·V-1 complex and fitting it back onto a full-length CP structure, generated from a molecular dynamics simulation, showed that V-1 appears to bind between the β tentacle and the main body of CP (Fig. 9).
FIGURE 9. Model illustrating the interaction of V-1 and CP based on computational docking.

A, the COOH-terminal tentacle of CP β1 (green) associates with V-1 (pink). B, magnified view of several residues important in the interaction, including the two loops of V-1.
DISCUSSION
In the present study, we characterized the binding and inhibition of the nonmuscle and sarcomeric isoforms of CP by V-1, and we investigated the regions of CP and V-1 involved in this interaction. In addition, we used computational simulation and docking approaches to construct a model for the interaction of CP with V-1. Predictions from that model agree with the results from the mutagenesis studies.
Based on physical binding assays, we found that V-1 binds to CP in a 1:1 complex. Actin polymerization assays of the capping activity of CP gave a Kd value of ~40 nM, using a simple model in which the CP·V-1 complex was 1:1 and had no ability to cap the barbed end. These results and conclusions are similar to those of a previous study (14).
This simple model fit all of the data well, and more complex models in which CP·V-1 complex had weak capping activity did not improve the fit. At high concentrations of V-1, the level of inhibition saturated at zero capping activity for CP. The protein CARMIL also binds and inhibits CP, but the CARMIL ·CP complex has detectable, albeit ~50-fold decreased, capping activity (36). The proteins CKIP-1 and CD2AP also bind and inhibit CP (36, 38, 39), as does PIP2 and other polyphosphoinositides (30).
PIP2 and CARMIL are efficient “uncappers”; they can rapidly remove CP from the barbed ends to which it is bound (30, 36). Here, we found that V-1 had no detectable uncapping activity. Thus, our results provide no evidence for the existence of a ternary V-1·CP·F-actin complex. This conclusion is in contrast to CAR-MIL, for which one must posit the existence of a ternary CARMIL·CP·F-actin state in order to explain the uncapping activity of CARMIL and the capping activity of the CARMIL·CP complex (36).
CP is a central element of the dendritic nucleation model for Arp2/3 complex-mediated actin assembly, which may account for much of actin-based motility in cells (16). In vitro, CP is constitutively active, binding barbed ends with high affinity. Certain results suggest that inhibitors of CP may exist within the cell. For example, the apparent affinity of CP for barbed ends in vivo is ~100-fold weaker than the affinity observed with pure proteins in vitro (40). V-1 is a candidate to play this role, along with the other protein and lipid inhibitors mentioned above. The cytoplasmic concentrations of V-1 and CP are estimated to be higher than the Kd values, and endogenous cellular V-1 and CP can be co-immunoprecipitated (14), confirming the potential for physiological relevance.
The Role of the V-1 Loops in Binding to CP
We asked which regions of V-1 were responsible for binding CP. V-1 is an ankyrin repeat protein, with two loops protruding from the structural backbone of α-helices. Among ankyrin repeat proteins, the loops generally mediate protein interactions, and the loop sequences are not conserved (13). The sequences of the two V-1 loops are highly conserved among vertebrates, and both loops of V-1 are important for binding to CP. Mutations in either loop decreased the binding affinity by a very large amount. Truncation of 20 residues from the NH2 terminus of V-1, which should not affect the ankyrin repeats, had no effect on CP binding.
Role of the COOH-terminal Regions of CP in Binding V-1
Since V-1 inhibits actin capping by CP, and the COOH-terminal regions (“tentacles”) of the CP α and β subunits are responsible for binding actin (21), we reasoned that V-1 might bind to the COOH-terminal regions. We found that a CP mutant lacking the COOH-terminal region of CP β showed a substantial loss of binding affinity for V-1. In addition, a peptide corresponding to the CP β COOH-terminal region was able to bind V-1, indicating that this region is sufficient to bind V-1. Other regions of CP may also contribute to V-1 binding, as evidenced by the ability of CP mutants lacking the CP β COOH-terminal region to bind V-1. Truncation of the α subunit tentacle decreased the CP·V-1 affinity by a small amount, as did a change of the α subunit isoform from α1 to α2, which involves only residues on the body, not the tentacle. Based on the physical dimensions of CP, it does not seem possible that V-1 could interact with both tentacles simultaneously; however, molecular simulations were able to lend insight into these phenomena. Molecular dynamics simulations of CP with a truncated α subunit tentacle showed a distinct change in dynamics in the body of the α subunit. Interestingly, the region where this changeis observed corresponds to the same vicinity of where V-1 would be predicted to interact with the α subunit, and this region is likewise an area where the α1 and α2 isoforms differ in sequence.
The β tentacle is the only region of difference between the β1 and β2 isoforms of CP, because they are produced by alternative splicing from a single gene (19). The sequences are quite different, but both are predicted to form amphipathic α-helices and appear as such in the crystal structure (20). Remarkably, we find that the β1 and β2 isoforms bind V-1 equally well, and they are known to bind the actin filament equally well (30). The sequence conservation between the β1and β2 tentacles is greater on the hydrophobic side of the helix, so this surface may be the one that contacts both actin and V-1, which would be consistent with these results, along with the observed competitive inhibition.
Relevance to the Mechanism of Capping
These results support and refine the model for how CP binds to the barbed end of the actin filament. In the current version of the “wobble” hypothesis, the α and β COOH-terminal regions of CP bind actin independently, each with a dissociation rate constant of ~0.2/s (21). When CP is bound to the barbed end, and the α COOH-terminal region dissociates, then the CP can wobble about, because the attachment of the β COOH-terminal region to the body of the protein is mobile, like a tentacle. In this wobble state, CARMIL can bind to CP, because CARMIL does not require either COOH-terminal region (36), and thus prevent rebinding by steric inhibition. The CARMIL·CP complex should then dissociate from the barbed end with the dissociation rate constant of the β tentacle, ~0.2/s. In contrast, V-1 requires the β tentacle, at least for maximal binding, so V-1 is predicted to not bind the wobble state and thus not uncap. Indeed, we found that V-1 did not uncap and that V-1 could not bind in a ternary complex with CP and the barbed end, as predicted. Additional structural and mutational studies will be important to test this model. The model has potential significance for cells, where inhibition of capping and its reversal (i.e. uncapping) may be important for actin assembly and movement.
Acknowledgments
We thank Dr. Hanspeter Niederstrasser for help with V-1·CP structural analyses, Dr. Mark Crankshaw (Protein and Nucleic Acid Chemistry Laboratory, Washington University) for circular dichroism studies, Dr. Ilgu Kang for helping to construct the CP α2β2 expression plasmid, and Dr. Martin Wear for invaluable advice and assistance with experiments, data analysis, and the manuscript.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants GM38542 (to J. A. C.) and GM067246 (to D. S.).
The abbreviations used are: CP, capping protein; PIP2, phosphatidylinositol 4,5-bisphosphate; GST, glutathione S-transferase; WT, wild type; aa, amino acid(s).
References
- 1.Sen S, Kundu G, Mekhail N, Castel J, Misono K, Healy B. J Biol Chem. 1990;27:16635–16643. [PubMed] [Google Scholar]
- 2.Sil P, Misono K, Sen S. Circ Res. 1993;1:98–108. doi: 10.1161/01.res.73.1.98. [DOI] [PubMed] [Google Scholar]
- 3.Sarkar S, Leaman DW, Gupta S, Sil P, Young D, Morehead A, Mukherjee D, Ratliff N, Sun Y, Rayborn M, Hollyfield J, Sen S. J Biol Chem. 2004;19:20422–20434. doi: 10.1074/jbc.M308488200. [DOI] [PubMed] [Google Scholar]
- 4.Pennica D, Shaw KJ, Luoh SM, Wood WI. Gene (Amst) 1995;158:305–306. doi: 10.1016/0378-1119(95)00131-o. [DOI] [PubMed] [Google Scholar]
- 5.Taoka M, Isobe T, Okuyama T, Watanabe M, Kondo H, Yamakawa Y, Ozawa F, Hishinuma F, Kubota M, Minegishi A. J Biol Chem. 1994;269:9946–9951. [PubMed] [Google Scholar]
- 6.Anderson KM, Berrebi-Bertrand I, Kirkpatrick RB, McQueney MS, Underwood DC, Rouanet S, Chabot-Fletcher M. J Mol Cell Cardiol. 1999;4:705–719. doi: 10.1006/jmcc.1998.0903. [DOI] [PubMed] [Google Scholar]
- 7.Yamakuni T, Yamamoto T, Hoshino M, Song SY, Yamamoto H, Kunikata-Sumitomo M, Minegishi A, Kubota M, Ito M, Konishi S. J Biol Chem. 1998;273:27051–27054. doi: 10.1074/jbc.273.42.27051. [DOI] [PubMed] [Google Scholar]
- 8.Furukawa Y, Hashimoto N, Yamakuni T, Ishida Y, Kato C, Ogashiwa M, Kobayashi M, Kobayashi T, Nonaka I, Mizusawa H, Song SY. Neuromuscul Disord. 2003;1:32–41. doi: 10.1016/s0960-8966(02)00185-2. [DOI] [PubMed] [Google Scholar]
- 9.Song SY, Asakai R, Kenmotsu N, Taoka M, Isobe T, Yamakuni T. Endocrinology. 1996;137:1423–1428. doi: 10.1210/endo.137.4.8625920. [DOI] [PubMed] [Google Scholar]
- 10.Poy MN, Eliasson L, Krutzfeldt J, Kuwajima S, Ma X, Macdonald PE, Pfeffer S, Tuschl T, Rajewsky N, Rorsman P, Stoffel M. Nature. 2004;432:226–230. doi: 10.1038/nature03076. [DOI] [PubMed] [Google Scholar]
- 11.Yang Y, Nanduri S, Sen S, Qin J. Structure. 1998;5:619–626. doi: 10.1016/s0969-2126(98)00063-x. [DOI] [PubMed] [Google Scholar]
- 12.Yang Y, Rao NS, Walker E, Sen S, Qin J. Protein Sci. 1997;6:1347–1351. doi: 10.1002/pro.5560060625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mosavi LK, Cammett TJ, Desrosiers DC, Peng ZY. Protein Sci. 2004;6:1435–1448. doi: 10.1110/ps.03554604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taoka M, Ichimura T, Wakamiya-Tsuruta A, Kubota Y, Araki T, Obinata T, Isobe T. J Biol Chem. 2003;278:5864–5870. doi: 10.1074/jbc.M211509200. [DOI] [PubMed] [Google Scholar]
- 15.Wear MA, Cooper JA. Trends Biochem Sci. 2004;8:418–428. doi: 10.1016/j.tibs.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 16.Pollard TD, Borisy GG. Cell. 2003;4:453–465. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- 17.Loisel TP, Boujemaa R, Pantaloni D, Carlier MF. Nature. 1999;6753:613–616. doi: 10.1038/44183. [DOI] [PubMed] [Google Scholar]
- 18.Hart MC, Cooper JA. J Cell Biol. 1999;6:1287–1298. doi: 10.1083/jcb.147.6.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schafer DA, Korshunova YO, Schroer TA, Cooper JA. J Cell Biol. 1994;2:453–465. doi: 10.1083/jcb.127.2.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamashita A, Maéda K, Maéda Y. EMBO J. 2003;7:1529–1538. doi: 10.1093/emboj/cdg167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wear MA, Yamashita A, Kim K, Maéda Y, Cooper JA. Curr Biol. 2003;13:1531–1537. doi: 10.1016/s0960-9822(03)00559-1. [DOI] [PubMed] [Google Scholar]
- 22.Sil P, Mukherjee D, Sen S. Circ Res. 1995;6:1020–1027. doi: 10.1161/01.res.76.6.1020. [DOI] [PubMed] [Google Scholar]
- 23.Ghosh J, Postle K. Mol Microbiol. 2004;51:203–213. doi: 10.1046/j.1365-2958.2003.03816.x. [DOI] [PubMed] [Google Scholar]
- 24.Braun P, Hu Y, Shen B, Halleck A, Koundinya M, Harlow E, LaBaer J. Proc Natl Acad Sci U S A. 2002;99:2654–2659. doi: 10.1073/pnas.042684199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mosavi LK, Minor DLJ, Peng ZY. Proc Natl Acad Sci U S A. 2002;99:16029–16034. doi: 10.1073/pnas.252537899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Palmgren S, Ojala PJ, Wear MA, Cooper JA, Lappalainen P. J Cell Biol. 2001;2:251–260. doi: 10.1083/jcb.200106157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hart MC, Korshunova YO, Cooper JA. Cell Motil Cytoskeleton. 1997;2:120–132. doi: 10.1002/(SICI)1097-0169(1997)38:2<120::AID-CM2>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 28.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carlsson AE, Wear MA, Cooper JA. Biophys J. 2004;2:1074–1081. doi: 10.1016/S0006-3495(04)74182-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schafer DA, Jennings PB, Cooper JA. J Cell Biol. 1996;1:169–179. doi: 10.1083/jcb.135.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pollard TD. J Cell Biol. 1986;103:2747–2754. doi: 10.1083/jcb.103.6.2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wear MA, Cooper JA. J Biol Chem. 2004;14:14382–14390. doi: 10.1074/jbc.M313412200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. J Comput Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 34.Rosenfeld RJ, Goodsell DS, Musah RA, Morris GM, Goodin DB, Olson AJ. J Comput Aided Mol Des. 2003;17:525–536. doi: 10.1023/b:jcam.0000004604.87558.02. [DOI] [PubMed] [Google Scholar]
- 35.Kale L, Skeel R, Bhandarkar M, Brunner R, Gursoy A, Krawetz N, Phillips J, Shinozaki A, Varadarajan K, Schulten K. J Comput Phys. 1999;151:283–312. [Google Scholar]
- 36.Yang C, Pring M, Wear MA, Huang M, Cooper JA, Svitkina TM, Zigmond SH. Dev Cell. 2005;2:209–221. doi: 10.1016/j.devcel.2005.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang CY. Methods Enzymol. 1982;87:509–525. doi: 10.1016/s0076-6879(82)87029-8. [DOI] [PubMed] [Google Scholar]
- 38.Canton DA, Olsten ME, Kim K, Doherty-Kirby A, Lajoie G, Cooper JA, Litchfield DW. Mol Cell Biol. 2005;9:3519–3534. doi: 10.1128/MCB.25.9.3519-3534.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Uruno T, Remmert K, Hammer JA. J Biol Chem. 2006 doi: 10.1074/jbc.M513186200. [DOI] [PubMed] [Google Scholar]
- 40.Hug C, Jay PY, Reddy I, McNally JG, Bridgman PC, Elson EL, Cooper JA. Cell. 1995;81:591–600. doi: 10.1016/0092-8674(95)90080-2. [DOI] [PubMed] [Google Scholar]