

Abstract
Alterations in the serotonin and norepinephrine neuronal functions have been observed in patients with major depression. Several antidepressants bind to both serotonin transporters (SERT) and norepinephrine transporters (NET). The ability to image NET in the human brain would be a useful step toward understanding how alterations in NET relate to disease. In this study, we report the synthesis and characterization of a new series of derivatives of iodo-nisoxetine (INXT), a known radioiodinated probe. The most promising, (R)-N-methyl-3-(3-iodopyridin-2-yloxy)-3-phenylpropylamine (PYINXT) 9, displayed a high and saturable binding to NET with a Kd value of 0.53 ± 0.03 nM. Biodistribution studies of [125I]PYINXT in rats showed moderate initial brain uptake (0.54 %dose/organ at 2 min) with a relatively fast washout from the brain (0.16 %dose/organ at 2 hr) as compared to [125I]INXT, 7. The hypothalamus (a NET rich region) to striatum (a region devoid of NET) ratio was found to be 2.14 at 4 hr post i.v. injection. The preliminary results suggest that this improved iodinated ligand, when labeled with 123I, may be useful for mapping NET binding sites with SPECT in the living human brain.
1. Introduction
The norepinephrine transporters (NET) are transmembrane proteins located presynaptically to the noradrenergic neurons. These transporters play a critical role in noradrenergic neurotransmission by regulating the amount of norepinephrine in the synaptic cleft via reuptake mechanism [1, 2]. A reduction in the level of NE in the synapse appears to down regulate the level of NET [3]. Recent research has clearly demonstrated a close association between the changes in the distribution of NET in the human brain and various diseases including CNS disorders like depression [4] attention deficit hyperactivity disorder (ADHD) [5, 6] and Alzheimer’s disease [7]. Non-invasive imaging with highly selective radiolabeled probes would allow us to monitor and quantify the effect of treatment (drug occupancy) or the lack thereof on NET.
It has been difficult to image NET with single photon emission computed tomography (SPECT) or positron emission tomography (PET) because of the lack of selective radiotracers that display good in vivo kinetics. An ideal radiopharmaceutical for mapping NET binding sites in the brain should meet most of the following criteria: a) high binding affinity to NET: Ki < 1 nM; b) high binding selectivity: Ki for other sites > 100 nM; c) easily labeled for imaging; d) good initial brain uptake (> 0.5% dose/organ at 2 min post-iv injection) and high target to non-target ratio (> 2); e) in vivo stability; f) distinctive NET-specific labeling illustrated by ex vivo autoradiography of rat brain sections [8].
Up to this point, the most promising NET specific radiotracer has been a methyl derivative of reboxetine, (S,S)-[11C]-(α-(2-methoxyphenoxy)benzyl)morpholine (MeNER) 2 (Fig.1), which is a PET imaging agent [9-11]. (S,S)-[11C]MeNER 2 showed the highest hypothalamus to striatum (target to non-target) ratio of 2.5 at 60 min post-iv injection in rats. In vitro autoradiography of rat brain slices indicated that its localization correlated well with the known distribution of NET [9]. Imaging studies in nonhuman primates using (S,S)-[11C]MeNER 2 exhibited a reasonable midbrain to striatum ratio of 1.4-1.6 [10]. One limitation was the longer time (60 min) required to obtain maximal specific binding due to slower clearance from the non-specific binding sites. The slower in vivo kinetics did not match the short half-life of carbon-11 (20 min) and diminished the clinical utility of this ligand. In comparison to (S,S)-[11C]MeNER 2, the corresponding (R)-[O-11CH3] -nisoxetine derivative 6 (Fig.1) exhibited very high non-specific binding in vivo [12, 13]. None of the other 11C labeled ligands for NET were as suitable as (S,S)-[11C]MeNER 2 [14, 15].
Figure 1.
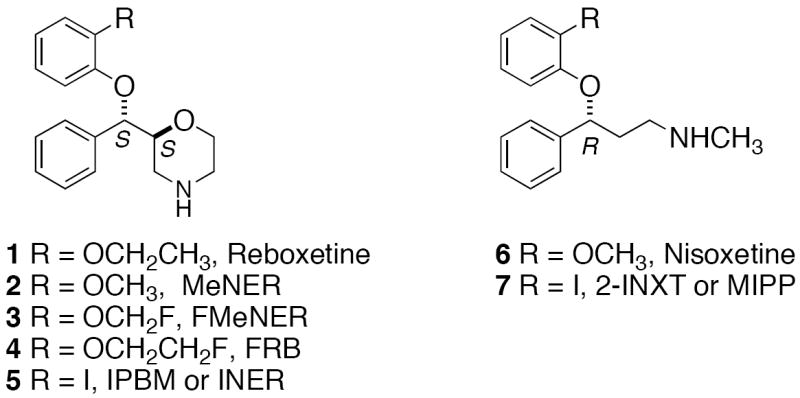
Structures of previously reported ligands targeting NET.
Due to the disadvantages of the 11C labeled imaging agents described above, there is strong impetus to develop 18F labeled probes for NET. 18F-labelled fluoromethyl [(S,S)-[18F]FMeNER] 3 (Fig.1) [16] as well as fluoroethyl [(S,S)[18F]FRB] 4 (Fig.1) [17, 18] derivatives of reboxetine have been reported. These ligands showed poor in vivo stability due to defluorination. Interestingly, deuterium substitution on the fluoroalkyl side-chain helped to reduce defluorination [16, 17].
Extensive research has also been directed towards developing radiotracers for brain imaging of NET using SPECT [8, 19, 20]. In one of the initial efforts, we have found that 125I-2-INXT 7, (Fig.1) an iodinated nisoxetine derivative, displayed saturable binding with a high affinity (Kd = 0.06 nM) in the homogenates prepared from rat cortical tissues as well as homogenates prepared from cells over-expressing NET [8, 23]. Biodistribution studies in rats showed a moderate target to non-target ratio (hypothalamus to striatum) of 1.5 at 3 hours post-iv. This was attributed to the higher non-specific binding of this ligand. Saji et al. evaluated the same compound (referred to as (R)-[125I]MIPP) as a potential radiopharmaceutical for imaging cardiac [19] and brain NET [20] with SPECT and reported similar results.
Recently, a novel reboxetine derivative iodinated at position 2 of the phenoxy ring, (S,S)-IPBM 5 (also named as [123I]INER) (Fig.1) has been reported [21, 22]. (S,S)-IPBM 5 displayed high affinity and selectivity for NET in binding assay experiments. The (S,S) diastereomer (Ki = 4.22 nM) was found to be more potent than the racemic (Ki = 10.4 nM) and the (R,R) (Ki = 80.4 nM) diastereomer [21]. In biodistribution experiments, (S,S)-125I-IPBM 5 provided a good initial uptake with maximum accumulation in the rat-brain at 30 minutes post–iv injection (0.54 % ID/gram at 30 min). The washout afterwards was relatively gradual. The regional cerebral distribution was consistent with the density of NET and a higher midbrain to striatum ratio was found at 180 minutes post injection [21]. A similar brain distribution pattern was observed in dynamic SPECT imaging of baboon using 5 [22]. Although the longer physical half-life of 123I (13.3 h) is compatible with the slower kinetics of this ligand, it is still far from ideal as a specific brain imaging agent for NET. None of the above ligands fully meet the criteria for a good NET imaging agent.
In our attempts to develop optimal radiotracers for imaging NET, we have focused on the modification of the nisoxetine core structure. For example, we replaced one of the benzene rings of 2-INXT 7, with a pyridine ring. This was expected to reduce the lipophilicity of [125I]7 without altering the binding affinity and selectivity for NET. In this paper, we are reporting the syntheses and initial screening of a novel series of pyridyl substituted nisoxetine derivatives, 8-11. For the most promising compound, 9 (PYINXT), (R)-N-methyl-3-(3-iodopyridin-2-yloxy)-3-phenylpropylamine, we synthesized the corresponding radiolabeled [125I]PYINXT. The results of biological evaluations have been encouraging, indicating that [125I]PYINXT may be a useful candidate for mapping NET binding sites in the brain.
2. Results and Discussion
2.1 Chemistry
The synthesis of Compounds 8-11 is depicted in Scheme 1. The starting material (S)-(3-methylamino)-1-phenylpropan-1-ol 12, was prepared according to a reported method [24]. Selective Boc-protection of the secondary amine led to the corresponding (S)-tert-butyl carbamate 13 in good yields. Compound 13 was used as a common intermediate in the synthesis of nisoxetine derivatives 8 and 10. The coupling product 15 was obtained by the Mitsunobu reaction [25] of commercially available 3-bromo-2-hydroxypyridine 14 with 13. Initial attempts to remove the Boc protecting group of 15 using TFA resulted in the cleavage of the pyridyl ether. Eventually, the deprotection was successfully carried out using trimethylsilyl trifluoromethanesulfonate (TMSOTf) and 2,6-lutidine in DCM at -40 °C [26] to provide Compound 8. A very similar protocol was employed for the preparation of 4-iodo-3-hydroxy-pyridyl nisoxetine derivative 10 from the known 4-iodo-3-hydroxypyridine 17 [27].
Scheme 1.
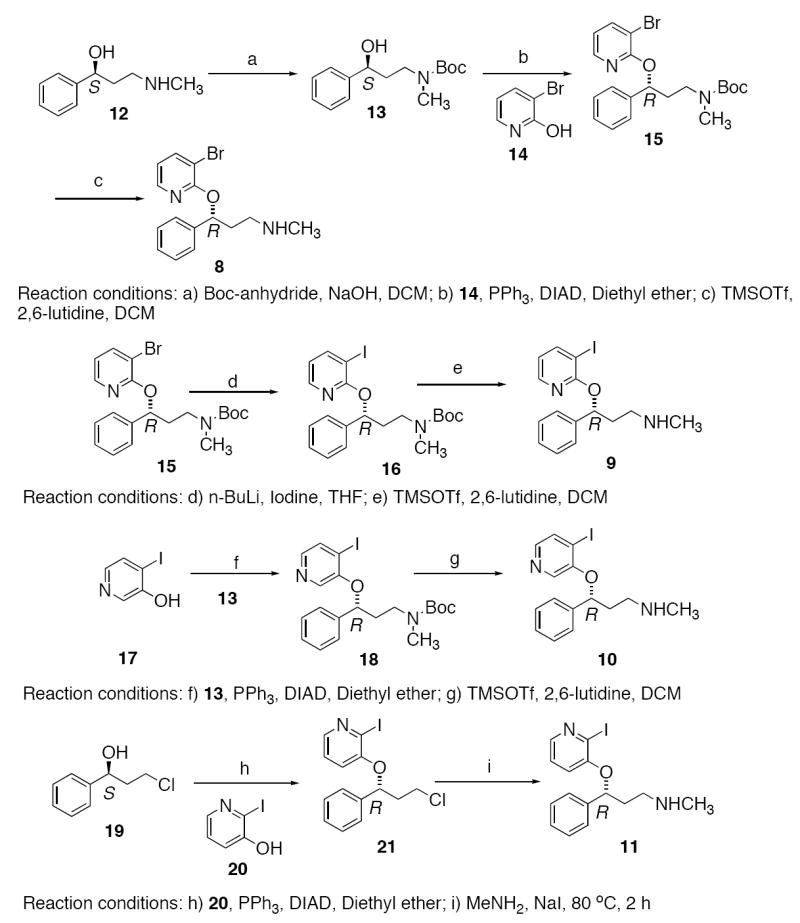
Synthesis of Compounds 8, 9, 10 and 11.
Lithium-halogen exchange of the bromo-compound 15 with n-BuLi-iodine followed by deprotection using TMSOTf furnished the ‘cold’ iodinated derivative 9 (PYINXT). The corresponding debrominated side-product was also observed in the halogen exchange reaction. In addition to routine NMR analysis, the purity of Compound 9 was confirmed by HPLC (Phenonemex Gemini column, 4.6 × 25 cm; isocratic solvent: CH3CN/ ammonium formate (1mM) = 9/1; flow rate: 0.5 ml/min, retention time 5.6 min).
A different approach was applied to the synthesis of Compound 11. The key step involved the Mitsunobu reaction of commercially available (S)-3-chloro-1-phenylpropan-1-ol 19 with 2-iodo-3-hydroxypyridine 20 to derive Compound 21. Substitution of chloride using aqueous methylamine afforded the expected product 11. The hydrochloride salt was obtained by treating the free amine with 2N HCl in ethyl acetate.
Palladium catalyzed stannylation of Compound 8 using hexabutylditin and Pd(PPh3)4 furnished Compound 22 which was used as the radioiodination precursor for the synthesis of [125I]PYINXT (Scheme 2). The radioiodinated ligand, [125I]PYINXT was compared with the standard cold compound 9 showing an identical retention time of 5.6-5.8 min on HPLC (Phenonemex Gemini column, 4.6 × 25 cm; isocratic solvent: CH3CN/1 mM ammonium formate = 9/1; flow rate: 0.5 ml/min). [125I]PYINXT has been shown to be stable in 100% ethanol for up to two months after radioiodination (> 95% radiochemical purity determined by HPLC).
Scheme 2.
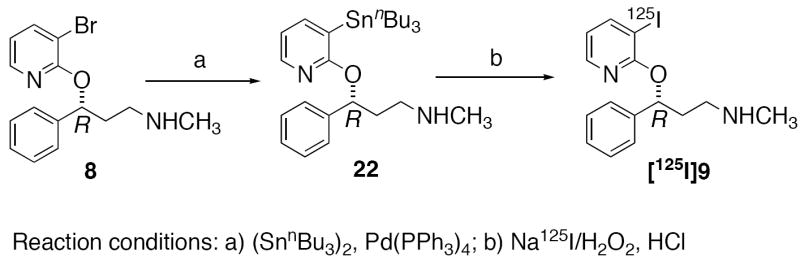
Synthesis of 22 and [125I]PYINXT.
2.2 Biological evaluations
The cloned cell-line, LLC-PK1, which over-expresses monoamine transporters, has been reported previously [28]. These cells are grown and maintained in our laboratory providing a convenient source of specific DAT, SERT or NET protein for in vitro binding assays. Because it is easy, readily available and provides comparable data for in vitro binding assays, our lab has used the cells expressing the specific transporter instead of membrane homogenates of rat frontal cortex or striatum to screen monoamine transporter inhibitors [29]. The compounds 8-11 synthesized and reported in this paper were subjected to binding evaluations for the monoamine transporters NET, SERT and DAT. As expected, these nisoxetine derivatives showed very high selectivity for NET with more than 100 to 1000 fold less affinity for the other two monoamine transporters SERT and DAT (Table 1). Nisoxetine, a known selective ligand for NET, competes effectively with [125I]2-INXT binding showing a Ki value of 4.8 ± 1.7 nM. Under similar assay conditions, the bromo derivative 8 and the iodo derivative PYINXT showed similar potency as nisoxetine with Ki values of 5.3 ± 0.3 and 4.5 ± 0.7 nM, respectively (Table 1). On the other hand, derivatives 10 and 11 had lower binding affinities with higher Ki values of 32.5 ± 5.5 and 13.5 ± 2.5 nM, respectively (Table 1). It is interesting to note that varying the position of nitrogen on the pyridine ring had a significant impact on binding affinity to NET. 2-Hydroxypyridine derivatives 8 and 9 (PYINXT) performed better than the 3-hydroxypyridine derivatives 10 and 11. Based on the initial screening results, the radioiodinated derivative [125I]PYINXT was prepared and subjected to further biological evaluation.
Table 1.
Inhibition constants (Ki) of NET ligands binding to homogenates prepared from LLC cells respectively expressing norepinephrine, serotonin and dopamine transporters
| Compound | LLC-NET (nM) | LLC-SERT (nM) | LLC-DAT (nM) |
|---|---|---|---|
| 8 | 5.3 ± 0.3 | >5,000 | >10,000 |
| 9 | 4.5 ± 0.7 | >1,500 | >10,000 |
| 10 | 32.5 ± 5.0 | >2,000 | >10,000 |
| 11 | 13.5 ± 2.5 | >1,000 | >30,000 |
| nisoxetine | 4.8 ± 1.7 | >2000 | >15,000 |
Studies were carried out with 0.08-0.08 nM [125I]INXT for NET, 0.1-0.2 nM [125I]IDAM for SERT and 0.1-0.2 nM [125I]IPT for DAT. Values are the mean ± SEM of three determinations performed in duplicate.
Specific binding of [125I]PYINXT to NET was further examined using membrane homogenates prepared from LLC-NET cells. Scatchard transformation of the saturation binding data gave a linear plot indicating one-site binding (data not shown). The mean values from 3 experiments gave a Kd value of 0.53± 0.05 nM, which is higher than the value obtained with the previous ligand [125I]2-INXT (Kd = 0.06 ± 0.01 nM) [8]. It is important to note that the Ki and the Kd values for 9 are different. The ten-fold difference between Ki and Kd is most likely due to the very high binding affinity of [125I]2-INXT (Kd = 0.06 nM in LLC-NET) used in the assay for Ki estimation.
The biodistribution of [125I]PYINXT in different organs and brain regions after an i.v. injection is shown in Table 2. The initial brain uptake at 2 minutes post-injection was moderate (0.54 %dose/organ) indicating a less efficient passage of the tracer through the intact blood-brain barrier. Radioactivity levels in all brain regions were the highest at 2 minutes and then gradually decreased over the 4 hour period. The washout rate was faster in striatum, where no NET binding sites are located, as compared to other brain regions (i.e. cerebellum, hippocampus, cortex and hypothalamus where different levels of NET are concentrated). At 240 minutes postinjection, regions containing norepinephrine innervation showed higher concentrations of radioactivity than the striatum (0.04, 0.04, 0.03, 0.06 and 0.03 % dose/gram for cerebellum, hippocampus, cortex, hypothalamus and striatum respectively). The ratio of hypothalamus to striatum, representing the target to non-target ratio, was about 2.1 (Table 2). This value is slightly higher than the ratio reported previously for [125I]INXT 7 [8] and it is comparable to the ratio reported for 11C-MeNER, 2, a reboxetine derivative [9]. Despite the lower in vitro binding affinity of [125I]PYINXT for NET (Kd = 0.53 nM in LLC-NET) as compared to [125I]INXT 7 (Kd = 0.06 nM in LLC-NET), it displayed more favorable in vivo kinetic properties (relative fast washout thus resulting in a slightly higher target (HY) vs. non-target (ST) ratio) (Table 2). As expected, the lipophilicity of [125I]PYINXT, because of the substitution of the phenyl ring with the pyridine ring, is lower than that of [125I]INXT 7 (log P = 1.45 for [125I]PYINXT and logP = 1.96 for [125I]INXT 7). This combination of lower lipophilicity together with the good selectivity and specific binding affinity of [125I]PYINXT for NET appears to account for the in vivo target vs non-target signal. The new NET probe [125I]PYINXT, reported here meets a majority of the criteria for an ideal NET imaging agent, i.e. a high binding affinity; a high selectivity (>100 fold); easily labeled and a ratio of target vs. non-target >2. The moderate initial brain uptake of [125I]PYINXT (0.54 %dose/organ at 2 min) is consistent with the values reported for other NET imaging agents in rats ([125I]INXT 7 = 0.56 at 2 min) [8]; ((S,S)-[11C]MeNER 2 = 0.53 at 5 min) [9]; ((S,S)-125I-IPBM 5 = 0.89 at 5 min) [21]. This general phenomenon, which is independent of the lipophilicity of the ligands, might be an inherent property of the common backbone structure for diffusion across the intact blood-brain barrier. This may be a limitation for the development of ideal imaging agents based on nisoxetine or reboxetine structures. Also noteworthy is the striking difference between the brain uptake in rats and mice. In mice, [125I]PYINXT showed a higher initial uptake (1.23 % dose/organ at 2 min post injection; data not shown) compared to the uptake in rats (0.54 % dose/organ at 2 min post injection). Saji et al. reported a very similar observation in the case of [125I]INXT 7 (0.97 % dose/organ at 2 min post injection in mice and 0.45 % dose/organ at 5 min post injection in rats) [20]. We have no simple explanation for this observation. However, it seems that in developing probes for NET, species differences may require careful evaluation.
Table 2.
Biodistribution in rats after an IV injection of [125I]PYINXT (% dose/organ, average of 3 rats ± SD)
| 2min | 30min | 60min | 120 min | 240 min | |
|---|---|---|---|---|---|
| Blood | 3.38±0.16 | 2.87±0.16 | 2.96±0.35 | 3.12±0.14 | 3.22±0.70 |
| Heart | 2.28±0.37 | 0.53±0.12 | 0.29±0.03 | 0.16±0.03 | 0.12±0.03 |
| Muscle | 24.14±13.37 | 13.48±2.10 | 15.39±1.19 | 9.40±0.90 | 5.98±0.82 |
| Lung | 10.21±1.85 | 3.06±0.77 | 1.43±0.20 | 0.75±0.09 | 0.37±0.03 |
| Kidney | 7.51±0.44 | 2.00±0.24 | 1.46±0.24 | 0.93±0.12 | 0.59±0.15 |
| Spleen | 1.04±0.51 | 0.76±0.21 | 0.56±0.03 | 0.26±0.03 | 0.12±0.02 |
| Liver | 7.37±2.01 | 3.48±0.08 | 2.91±0.28 | 2.43±0.03 | 2.08±0.15 |
| Skin | 7.09±3.01 | 9.13±0.69 | 9.55±0.74 | 8.33±0.81 | 7.12±1.76 |
| Brain | 0.54±0.08 | 0.46±0.00 | 0.33±0.03 | 0.16±0.01 | 0.07±0.01 |
| Thyroid | 0.05±0.01 | 0.05±0.02 | 0.05±0.05 | 0.02±0.01 | 0.32±0.19 |
| Regional Brain Distribution (% dose/g ± SD) | |||||
| Cerebellum | 0.41±0.02 | 0.28±0.00 | 0.18±0.03 | 0.09±0.01 | 0.04±0.00 |
| Striatum | 0.29±0.05 | 0.22±0.02 | 0.16±0.01 | 0.07±0.01 | 0.03±0.01 |
| Hippocampus | 0.28±0.06 | 0.26±0.02 | 0.20±0.01 | 0.11±0.01 | 0.04±0.01 |
| Cortex | 0.33±0.07 | 0.28±0.01 | 0.20±0.02 | 0.10±0.01 | 0.03±0.00 |
| Remainder | 0.30±0.05 | 0.26±0.01 | 0.18±0.02 | 0.09±0.01 | 0.04±0.00 |
| Hypothalamus | 0.31±0.04 | 0.33±0.09 | 0.23±0.04 | 0.12±0.01 | 0.06±0.01 |
| Ratio vs. ST (± SD) | |||||
| Cerebellum | 1.44±0.27 | 1.26±0.09 | 1.11±0.18 | 1.24±0.17 | 1.53±0.40 |
| Striatum | 1.00±0.26 | 1.00±0.09 | 1.00±0.11 | 1.00±0.10 | 1.00±0.33 |
| Hippocampus | 0.97±0.27 | 1.17±0.12 | 1.25±0.13 | 1.58±0.23 | 1.71±0.52 |
| Cortex | 1.14±0.32 | 1.23±0.10 | 1.20±0.16 | 1.40±0.16 | 1.21±0.34 |
| Remainder | 1.04±0.26 | 1.15±0.09 | 1.12±0.16 | 1.32±0.18 | 1.36±0.37 |
| Hypothalamus | 1.08±0.24 | 1.49±0.49 | 1.41±0.25 | 1.70±0.17 | 2.14±0.62 |
At 120 min post-injection of [125I]PYINXT, autoradiograms of rat brain sections showed an intense labeling in several regions [30], i.e. locus coerulus, thalamic and hypothalamic nuclei and bed nucleus of the strai terminalis (Fig.2, top row), areas known to have high densities of NET sites [31]. A medium level of labeling was observed in the median raphe, and dentate gyrus. Lower, but detectable labeling was also found in the frontal cortex. The caudate putamen showed the lowest labeling. The regional distribution observed with [125I]PYINXT is consistent with that reported for other NET ligands [32-34] indicating that the in vivo binding of [125I]PYINXT in rat brain strongly correlates with known NET density distribution in different brain regions. Nisoxetine pretreatment completely abolished the labeling for NET (shown in Fig.2, bottom row) indicating that [125I]PYINXT and nisoxetine compete for the same binding site (NET) in vivo.
Fig.2.
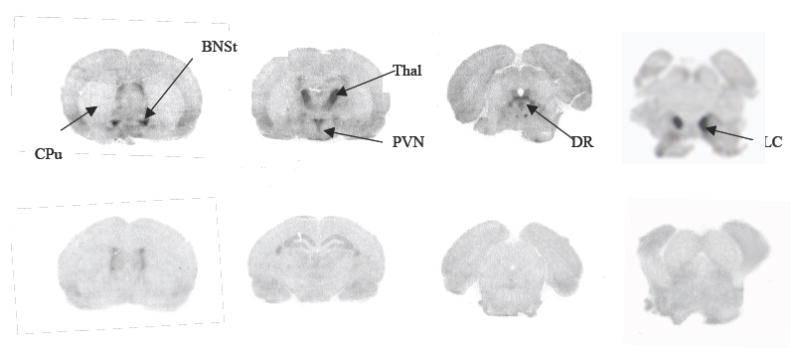
Ex vivo autoradiography of rat brains at 2 hours after an iv injection of [125I]PYINXT. Top row: saline treated (control one); bottom row: nisoxetine pretreatment (10 mg/kg, 10 min prior to radiotracer administration). Abbreviations: CPu: caudate putamen; BNSt: bed nucleus of the stria terminals; DR: dorsal raphe nucleus; LC: locus coeruleus; PVN: paraventricular nucleus of the hypothalamus; Thal: thalamic nucleus.
The development of an ideal imaging agent for NET is challenging due to the very low density of NET binding sites in the brain. Previous estimations in our laboratory have shown that the Bmax of NET is significantly lower than that of SERT and DAT (55 fmol/mg protein, 194 fmol/mg protein and 2000 fmol/mg protein for NET, SERT and DAT respectively) [8]. Hence, an important prerequisite for a useful NET tracer is that it should display very high binding affinity and selectivity. Despite the high binding affinity and selectivity in vitro, iodinated ligands reported so far such as [125I]INXT 7 and (S,S)-125I-IPBM 5, displayed rather slower in vivo kinetics. For [125I]INXT 7 higher non-specific binding was observed. Assuming that the non-specific binding could be minimized by adjusting the lipophilicity of the molecule, we synthesized a new pyridyl substituted nisoxetine derivative [125I]PYINXT. It was successfully characterized in vitro and in vivo as targeting NET. The binding affinity of [125I]PYINXT to NET was lower than that of [125I]INXT 7, but [125I]PYINXT exhibited comparable initial uptake, improved kinetics and specific binding in the rat brain. Furthermore, the in vivo stability of [125I]PYINXT to be qualified as a suitable platform structure remains to be determined. The new ligand, [125I]PYINXT is not yet ideal, but undoubtedly, it represents progress in the quest for better imaging agents for NET using SPECT. In addition, it may be interesting to evaluate the potential of [125I]PYINXT for imaging cardiac sympathetic innervation. Presently, we are also working on the extension of the same strategy to develop suitable pyridyl-reboxetine analogues targeting NET.
In conclusion, in a search for potential SPECT imaging agents for NET, a new series of nisoxetine derivatives was developed by replacing one of the phenyl rings with a pyridine ring. These novel nisoxetine derivatives displayed good binding affinities to NET (Ki values between 5 and 30 nM). The radioiodinated probe [125I]PYINXT displayed a high and selective binding to NET in the rat brain. Further evaluation of this ligand and its congeners may lead to useful imaging agents for mapping NET sites in the living human brain.
3. Materials and methods
3.1 General
All reagents used in the synthesis were commercial products and were used without purification, unless otherwise indicated. 1H and 13C nuclear magnetic resonance (NMR) spectra were obtained with a Bruker DPX 200 spectrometer for all compounds except 21 and 11 (DPX 400). Chemical shifts are reported as δ values with respect to residual CHCl3 proton in CDCl3 for 1H NMR and residual CHCl3 carbons in CDCl3 for 13C NMR, unless otherwise mentioned. Coupling constants are reported in Hertz. Multiplicities are defined as: s (singlet), d (doublet), dd (doublet of doublet), t (triplet), br (broad), m (multiplet). Preparative thin layer chromatography (PTLC) was performed with Analtech Uniplate (20 × 20; 2000 μm). Chromatography refers to flash chromatography on silica gel, unless otherwise indicated. Electrospray ionization mass spectra were recorded with LC MSD TOF, Agilent Technologies. High resolution mass spectra for compounds 21 and 11 were obtained with JMS-HX110/110 Tandem Mass Spectrometer, JEOL Ltd. Optical rotations were measured with a Perkin Elmer 243B polarimeter. [ 125I]NaI (0.1 N NaOH solution) was purchased from DuPont NEN Research (Boston, MA).
3.1.1 (S)-tert-butyl-3-hydroxy-3-phenylpropyl(methyl)carbamate (13)
A solution of (S)-3-(methylamino)-1-phenylpropan-1-ol (12) (1.27 g, 7.6 mmol) in DCM (20 mL) was stirred at room temperature. 4N NaOH (337 mg, 8.4 mmol) was added to this dropwise. After 5 minutes Boc-anhydride (1.67 g, 7.6 mmol) was added and the reaction mixture stirred for 4 h. Water (20 mL) was added and the mixture was extracted with DCM (3 × 50 mL). The combined organic layers were washed with brine, dried over anhydrous sodium sulfate and the solvent evaporated. The residue was purified by chromatography (75:25 hexane-ethyl acetate) to give Compound 13 (1.2 g, 59 %). (c = 0.94, CHCl3). 1H NMR (200 MHz, CDCl3): δ 7.35-7.23 (m, 5H), 4.59 (d, J = 7.7 Hz, 1H), 3.86 (br, 1H), 3.08 (br, 1H), 2.86 (s, 3H), 2.02-1.86 (m, 2H), 1.46 (s, 9H).
3.1.2 (R)-tert-butyl-3-(3-bromopyridin-2-yloxy)-3-phenylpropyl(methyl)carbamate (15)
Under inert atmosphere, 3-Bromo-2-hydroxypyridine (14) (489 mg, 2.81 mmol) was added to a solution of Compound 13 (572 mg, 2.16 mmol) and Pph3 (848 mg, 3.24 mmol) in diethylether (15 mL). The resulting suspension was stirred at 0 °C for 10 minutes followed by the dropwise addition of DIAD (0.63 mL, 3.24 mmol) over 5 minutes. The reaction mixture was warmed to room temperature and stirred for 3 h. The solvent was evaporated and the residue was adsorbed on silica gel. Purification by chromatography (75:25 hexane-ethyl acetate) afforded the product 15 (535 mg, 58 %). (c = 1.3, CHCl3). 1H NMR (200 MHz, CDCl3): δ 7.92 (d, J = 4.8 Hz, 1H), 7.71 (d, J = 7.6 Hz, 1H), 7.43-7.20 (m, 5H), 6.66-6.60 (m, 1H), 6.15 (dd, J = 4.8 Hz, 7.8 Hz, 1H), 3.50-3.36 (m, 2H), 2.82 (s, 3H), 2.34-2.04 (m, 2H), 1.37 (s, 9H).
3.1.3 (R)-N-methyl-3-(3-bromopyridin-2-yloxy)-3-phenylpropan-1-amine (8)
A solution of the Boc-protected compound 15 (263 mg, 0.62 mmol) and 2,6- lutidine (401 mg, 3.75 mmol) in DCM (7 mL) was stirred at -40 °C for 10 minutes under inert atmosphere. Trimethylsilyl trifluoromethanesulfonate (555 mg, 2.5 mmol) was carefully added to this dropwise. The reaction mixture was allowed to warm to room temperature over 2 h. After the addition of aqueous Na2CO3 (10 %, 10 mL), the reaction mixture was extracted with DCM (3 × 20 mL). The organic phase was washed with brine, dried over anhydrous Na2SO4 and the solvent removed under reduced pressure. The pure product 8 (165 mg) was isolated from the residue by gradient chromatography (8:2 hexane-ethylacetate to 9:1:1 DCM-MeOH-triethylamine) in 82 % yield. (c = 0.95, CHCl3). 1H NMR (200 MHz, CDCl3): δ 7.97 (dd, J = 1.6 Hz, 4.8 Hz, 1H), 7.76 (dd, J = 1.6 Hz, 7.6 Hz, 1H), 7.53-7.18 (m, 5H), 6.70 (dd, J = 4.8 Hz, 7.6 Hz, 1H), 6.28-6.22 (m, 1H), 5.28 (br, 1H), 2.84 (t, J = 7.1 Hz, 2H), 2.49 (s, 3H), 2.39-2.24 (m, 2H). 13C NMR (50 MHz, CDCl3): δ 158.57, 145.09, 141.41, 140.48, 128.05, 127.24, 125.69, 117.68, 107.10, 75.45, 46.99, 35.42, 34.74. HRMS (ESI) calcd for C15H18BrN2O [M + H]+ 321.0603, found 321.0605.
3.1.4 (R)-tert-butyl-3-(3-iodopyridin-2-yloxy)-3-phenylpropyl(methyl)carbamate (16)
To a solution of the bromo-compound 15 (142 mg, 0.33 mmol) and TMEDA (78 μL, 0.53 mmol) in anhydrous THF (3 mL) stirred at -78 °C under inert atmosphere, n-BuLi (1.6 M in hexanes, 281 μL, 0.44 mmol) was added dropwise. The reaction mixture was stirred at -78 °C for 1 h before the addition of iodine (133 mg, 0.53 mmol) in THF (2 mL). After stirring for another hour at -78 °C, the reaction mixture was warmed to room temperature and stirred for an additional 15 minutes. Saturated aqueous NH4Cl (5 mL) was added and the resulting mixture extracted with DCM (3 × 15 mL). The organic phase was washed with brine, dried over anhydrous Na2SO4 and the solvent removed under reduced pressure. The crude reaction mixture was purified by careful chromatography (9:1 hexane-ethyl acetate) to afford 16 as colorless oil (46 mg, 30 % yield). The debrominated side product was also isolated (25 mg, 22 %). (c = 1.36, CHCl3). 1H NMR (200 MHz, CDCl3): δ 8.00–7.96 (m, 2H), 7.44-7.18 (m, 5H), 6.56 (dd, J = 5.3 Hz, 7.0 Hz, 1H), 6.11 (dd, J = 4.8 Hz, 7.9 Hz, 1H), 3.50-3.36 (m, 2H), 2.83 (s, 3H), 2.22-2.07 (m, 2H), 1.37 (s, 9H).
3.1.5 (R)-N-methyl-3-(3-iodopyridin-2-yloxy)-3-phenylpropan-1-amine (9)
The procedure described before for the deprotection of Compound 15 was applied to Compound 16 (45 mg, 0.096 mmol). The reaction mixture was purified by PTLC (9:1:1 DCM-MeOH-triethylamine) to obtain the title Compound 9 (25 mg, 71 %) as colorless oil. (c = 0.72, CHCl3). 1H NMR (200 MHz, CDCl3): δ 8.02–7.99 (m, 2H), 7.44-7.20 (m, 5H), 6.64-6.58 (m, 1H), 6.27-6.21 (m, 1H), 3.06-2.99 (m, 2H), 2.57 (s, 3H), 2.47 (br, 2H). 13C NMR (50 MHz, CDCl3): δ 160.21, 147.97, 146.10, 139.61, 127.95, 127.09, 125.65, 117.94, 80.13, 75.30, 46.09, 33.68, 33.33 HRMS (ESI) calcd for C15H18IN2O [M + H]+: 369.0464, found 369.0421.
3.1.6 (R)-tert-butyl-3-(4-iodopyridin-3-yloxy)-3-phenylpropyl(methyl)carbamate (18)
Mitsunobu reaction of 4-iodo-3-hydroxypyridine 17 (183 mg, 1.05 mmol) with Compound 13 (215 mg, 0.81 mmol) was carried out as described for the preparation of Compound 15. The reaction mixture was purified by chromatography (7:3 hexane-ethyl acetate) to provide the product 18 (151 mg, 39 %). (c = 1.48, CHCl3). 1H NMR (200 MHz, CDCl3): δ 7.83 (s, 1H), 7.75 (d, J = 4.8 Hz, 1H), 7.67 (d, J = 4.8 Hz, 1H), 7.33-7.26 (m, 5H), 5.28 (dd, J = 4.1 Hz, 8.3 Hz, 1H), 3.53-3.45 (m, 2H), 2.86 (s, 3H), 2.35-2.03 (m, 2H), 1.37 (s, 9H).
3.1.7 (R)-N-methyl-3-(4-iodopyridin-3-yloxy)-3-phenylpropan-1-amine (10)
The procedure described before for the deprotection of Compound 15 was applied to Compound 18 (99 mg, 0.21 mmol). The reaction mixture was purified by PTLC (95:5:0.5 DCM-MeOH-NH4OH) to afford the title Compound 10 (35 mg, 45 %). (c = 1.35, CHCl3). 1H NMR (200 MHz, CDCl3): δ 7.87 (s, 1H), 7.74 (d, J = 4.9 Hz, 1H), 7.66 (d, J = 4.9 Hz, 1H), 7.34-7.25 (m, 5H), 5.45 (dd, J = 4.7 Hz, 8.0 Hz, 1H), 3.60 (br, 1H), 2.94-2.77 (m, 2H), 2.47 (s, 3H), 2.42-2.05 (m, 2H). 13C NMR (50 MHz, CDCl3): δ 152.78, 142.69, 138.59, 135.53, 133.81, 128.70, 128.15, 125.56, 97.81, 79.10, 46.60, 36.11, 34.07. HRMS (ESI) calcd for C15H18IN2O [M + H]+: 369.0464, found 369.0443.
3.1.8 (R)-3-(3-chloro-1-phenylpropoxy)-2-iodopyridine (21)
A solution of (S)-3-chloro-1-phenylpropan-1-ol 19 (340 mg, 2 mmol) and PPh3 (530 mg, 2.02 mmol) in anhydrous THF (2 mL) was cooled to 0 °C. 2-iodo-3-hydroxypyridine (442 mg, 2 mmol) was added to the reaction mixture and it was stirred at 0 °C for 5 min before the dropwise addition of DIAD (424 mg, 2.1 mmol) over another 5 min. After 30 minutes, the solvent was evaporated and the residue dissolved in DCM and purified by flash chromatography (4:1 hexane-ethyl acetate) to provide 21 (485 mg, 65 %) as a clear oil. 1H NMR (400 MHz, CDCl3): δ 7.91 (m, 1H), 7.42-7.34 (m, 5H), 7.28 (m, 2H), 5.72 (m, 1H), 3.83 (m, 1H), 3.64 (m, 1H), 2.62 (m, 1H), 2.38 (m, 1H).
3.1.9 (R)-N-methyl-3-(2-iodopyridin-3-yloxy)-3-phenylpropan-1-amine (11)
A solution of 21 (190 mg, 0.5 mmol) and methyl amine (2 mL, 40 % wt. in water) in ethanol (2 mL) was heated in a sealed flask at 80 °C for 2 h. The solvent was removed and the residue purified by flash chromatography (5:1 ethyl acetate-methanol) to provide 11 (126 mg, 67 %) as a clear oil. The free amine was dried under vacuum for 2 days and stored in the freezer. 1H NMR (400 MHz, CD3OD): δ 7.90 (d, J = 3.2 Hz, 1H), 7.38 (m, 5H), 6.99 (dd, J = 1.6 Hz, 3.2 Hz 1H), 6.86 (dd, J = 1.6 Hz, 3.2 Hz, 2H), 5.63 (m, 1H), 3.34 (m, 1H), 3.20 (m, 1H), 2.78 (m, 2H). 2.74 (s, 3H). HRMS (FAB+) calcd for C15H18IN2O [M + H]+: 369.0464, found 369.0453.
3.2.0 (R)-N-methyl-3-[3-(tributylstannyl)pyridin-2-yloxy]-3-phenylpropan-1-amine (22)
A suspension of bromo-compound 8 (89 mg, 0.28 mmol) and Pd(PPh3)4 (33 mg, 0.028 mmol) in hexabutylditin (0.75 mL, 1.4 mmol) was refluxed at 110 °C for 6 h. After cooling to room temperature, the reaction mixture was treated with aqueous KF solution (10 %, 10 mL) followed by aqueous Na2CO3 solution (10 %, 10 mL). It was extracted with DCM-MeOH (95:5) mixtures (5 × 15 mL). The organic phase was washed with brine, dried over anhydrous Na2SO4 and the solvent removed under reduced pressure. The crude residue was purified by PTLC (9:1 DCM-MeOH) to give the expected product 22 , albeit in low yield (19 mg, 13 %). 1H NMR (200 MHz, CDCl3): δ 8.01 (dd, J = 1.9 Hz, 4.9 Hz, 1H), 7.63 (dd, J = 2.0 Hz, 6.8 Hz, 1H), 7.42-7.23 (m, 5H), 6.81-6.79 (m, 1H), 6.19 (t, J = 6.8 Hz, 1H), 3.81 (br, 1H), 2.84-2.62 (m, 2H), 2.49 (s, 3H), 2.34-2.16 (m, 2H), 1.60-1.21 (m, 15H), 1.07-0.82 (m, 12H).
3.2 Preparation of [125I]PYINXT
Preparation of the radioiodinated tracer, [125I]PYINXT was carried out by an iododestannylation reaction from the corresponding tin precursor catalyzed by hydrogen peroxide as described previously [35]. The no-carrier-added product [125I]PYINXT with a high specific activity (2,200 Ci/mmol) was obtained via a simple C4 mini-column. In brief, after quenching with sodium bisulfite and neutralizing with sodium bicarbonate, the reaction mixture was loaded on a reversed phase C4 mini-column (Vydac BioSelect, CA, 1 × 6 cm). After washing the column with 3 ml of 10% EtOH, the desired product [125I]PYINXT was then eluted with 0.5 ml of ethanol. The radiochemical purity of the ligand was examined on HPLC using a reversed-phase column (Phenomenex Gemini C18 analytical column, 4.6 × 250 mm, 5 μm, CH3CN/Ammonium formate buffer (1 mM) 9/1; flow rate 0.5 mL/min).
3.3 In vitro binding assays
[125I]2-INXT [8], [125I]IDAM [36] and [125I]IPT [37] were prepared according to the procedures described previously. Nisoxetine, Citalopram and GBR12909 were obtained from Sigma Biochemical Inc. LLC-PK1 cells over-expressing three monoamine transporters, named respectively as norepinephrine transporters (LLC-NET), serotonin transporters (LLC-SERT) and dopamine transporters (LLC-DAT), were kindly provided by Dr. G. Rudnick of Yale University. Cells were plated and grown to confluence as a monolayer as described before [28]. The membrane homogenates were prepared and used for the binding assays with the known characterized radioiodinated ligands [125I]2-INXT for NET, [125I]IDAM for SERT and [125I]IPT for DAT. Binding assays were performed in glass tubes (12 × 75 mm) with a final volume of 0.5 ml. In saturation experiments, aliquots (100 μl corresponding to 40-60 μg of protein) of membrane suspensions were mixed with buffer (50 mM Tris-HCl, pH 7.4, 120 mM NaCl, 5 mM KCl) containing 0.05-2.0 nM [125I]PYINXT. Competition experiments were performed using 0.05-0.06 nM [125I]2-INXT for NET, 0.1 nM [125I]IDAM for SERT and 0.2 nM [125I]IPT for DAT and 8-10 concentrations (10-10-10-5M) of competing drugs. Inhibitors were serially diluted with the buffer (as above) containing 0.1% BSA to overcome the stickiness and loss due to dilution. Nonspecific binding was defined with 1 μM nisoxetine (for NET) or 1 μM citalopram (for SERT) or 1 μM GBR12909 (for DAT). Incubation was carried out for 60 min at room temperature and then terminated by separation of bound from free radioligand by filtration through glass fiber filters (Schleicher & Schuell No. 25, Keene, NH) presoaked with 1% polyethylenimine. The filters were then washed three times with 3 ml of ice-cold 20 mM Tris buffer and counted in a gamma counter (Cobra II, Perkin Elmer) with 70 % efficiency. The results of saturation and competition experiments were subjected to nonlinear regression analysis using EBDA [38] to obtain Kd and Ki values.
3.4 Biodistribution in rats
Male Sprague-Dawley rats weighing 225-275 g were used in the studies. Three rats per group were used for biodistribution study. While under isoflurane anesthesia, 0.2 ml of a saline solution containing 10 μCi of radioactive tracer was injected into the femoral vein. The rats were sacrificed at the time indicated by cardiac excision while under anesthesia. Organs of interest were removed and weighed, and the radioactivity was counted. The percent dose per organ was calculated by a comparison of the tissue counts to counts of 1% of the initial dose (100 times diluted aliquots of the injected material) measured at the same time. Regional brain distribution in rats was measured after an i.v. injection of the radioactive tracer. Samples from different brain regions [cortex (CX), striatum (ST), hippocampus (HP), cerebellum (CB) and hypothalamus (HY)] were dissected, weighed and counted. The percentage dose/g of each sample was calculated by comparing sample counts with the counts of the diluted initial dose described above. The striatum region containing no NET was used as a background region and ratios of target to background region were obtained.
3.5 Ex vivo autoradiography of [125I]PYINXT in rat brain
Rats under isoflurane anesthesia were injected intravenously with 0.3 ml of a saline solution containing 300 μCi of [125I]PYINXT. At 120 min post i.v. injection, the animals were sacrificed by cardiac excision while under isoflurane anesthesia. The brains were rapidly removed, placed in embedding medium (Tissue Tek, Miles Laboratory, Elkhart, IN) and frozen with powdered dry ice. After reaching equilibrium at -20 °C, consecutive 20 μm coronal sections were cut on a cryostat microtome (Hacker Instruments, Fairfield, NJ), thaw-mounted on microscope slides, and air-dried at room temperature. The slides containing the brain sections were exposed to Kodak Biomax MR film along with 20 μm thick 125I standards (Amersham Corp., Arlington, Ill) for three days in autoradiographic cassettes. A second group of rats was pretreated with nisoxetine (10 mg/kg, i.v. 10 min prior to [125I]PYINXT) and then injected with [125I]PYINXT and given the same treatment described above. The ex vivo autoradiograms of nisoxetine-treated brains were compared to the untreated control brains for NET labeling.
3.6 Partition coefficient
Partition coefficients were measured by mixing the [125I]PYINXT with 3g each of 1-octanol and buffer (0.1 M phosphate, pH 7.4) in a test tube. The test tube was vortexed for 3 min at room temperature, followed by centrifugation for 5 min. Two weighed samples (0.5 g each) from the 1-octanol and buffer layers were counted in a well counter. The partition coefficient was determined by calculating the ratio of cpm/g of 1-octanol to that of buffer. Samples from the 1-octanol layer were re-partitioned until consistent partitions of coefficient values were obtained. The measurement was done in triplicate and repeated three times.
Acknowledgments
The work was supported by the grants awarded from the National Institute of Health (NS5049438 to H.F. Kung) and (R21 MH074008-01A1 to R. Waterhouse).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Amara SG, Kuhar MJ. Neurotransmitter transporters: recent progress. Annu Rev Neurosci. 1993;16:73–93. doi: 10.1146/annurev.ne.16.030193.000445. [DOI] [PubMed] [Google Scholar]
- 2.Blakely RD, Bauman AL. Biogenic amine transporters: regulation in flux. Current Opinion in Neurobiology. 2000;10:328–336. doi: 10.1016/s0959-4388(00)00088-x. [DOI] [PubMed] [Google Scholar]
- 3.Zhu M-Y, Ordway GA. Down-regulation of norepinephrine transporters on PC12 cells by transporter inhibitors. J Neurochem. 1997;68:134–41. doi: 10.1046/j.1471-4159.1997.68010134.x. [DOI] [PubMed] [Google Scholar]
- 4.Klimek V, Stockmeier C, Overholser J, Meltzer HY, Kalka S, Dilley G, Ordway GA. Reduced Levels of Norepinephrine Transporters in the Locus Coeruleus in Major Depression. J Neurosci. 1997;17:8451–58. doi: 10.1523/JNEUROSCI.17-21-08451.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Biederman J, Spencer T. Attention-deficit/hyperactivity disorder (ADHD) as a noradrenergic disorder. Biol Psychiatry FIELD Full Journal Title:Biological psychiatry. 1999;46:1234–42. doi: 10.1016/s0006-3223(99)00192-4. [DOI] [PubMed] [Google Scholar]
- 6.Viggiano D, Ruocco LA, Arcieri S, Sadile AG. Involvement of norepinephrine in the control of activity and attentive processes in animal models of Attention Deficit Hyperactivity Disorder. Neural Plas. 2004;11:133–49. doi: 10.1155/NP.2004.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tejani-Butt SM, Yang J, Zaffar H. Norepinephrine transporter sites are decreased in the locus coeruleus in Alzheimer’s disease. Brain Res FIELD Full Journal Title:Brain research. 1993;631:147–50. doi: 10.1016/0006-8993(93)91201-3. [DOI] [PubMed] [Google Scholar]
- 8.Kung MP, Choi SR, Hou C, Zhuang ZP, Foulon C, Kung HF. Selective binding of 2-[125I]iodo-nisoxetine to norepinephrine transporters in the brain. Nucl Med Biol. 2004;31:533–41. doi: 10.1016/j.nucmedbio.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 9.Wilson AA, Patrick Johnson D, Mozley D, Hussey D, Ginovart N, Nobrega J, Garcia A, Meyer J, Houle S. Synthesis and in vivo evaluation of novel radiotracers for the in vivo imaging of the norepinephrine transporter. Nucl Med Biol. 2003;30:85–92. doi: 10.1016/s0969-8051(02)00420-1. [DOI] [PubMed] [Google Scholar]
- 10.Schou M, Halldin C, Sovago J, Pike VW, Gulyas B, Mozley PD, Johnson DP, Hall H, Innis RB, Farde L. Specific in vivo binding to the norepinephrine transporter demonstrated with the PET radioligand, (S,S)-[(11)C]MeNER. Nucl Med Biol. 2003;30:707–14. doi: 10.1016/s0969-8051(03)00079-9. [DOI] [PubMed] [Google Scholar]
- 11.Ding YS, Lin KS, Garza V, Carter P, Alexoff D, Logan J, Shea C, Xu Y, King P. Evaluation of a new norepinephrine transporter PET ligand in baboons, both in brain and peripheral organs. Synapse. 2003;50:345–52. doi: 10.1002/syn.10281. [DOI] [PubMed] [Google Scholar]
- 12.Haka M, Kilbourn M. Synthesis and regional mouse brain distribution of [11C]-nisoxetine, a norepinephrine uptake inhibitor. Nucl Med Biol. 1989;16:771–74. doi: 10.1016/0883-2897(89)90160-8. [DOI] [PubMed] [Google Scholar]
- 13.Ding Y-S, Lin K-S, Logan J, Benveniste H, Carter P. Comparative evaluation of positron emission tomography radiotracers for imaging the norepinephrine transporter: (S,S) and (R,R) enantiomers of reboxetine analogs ([11C]methylreboxetine, 3-Cl-[11C]methylreboxetine and [18F]fluororeboxetine), (R)-[11C]nisoxetine, [11C]oxaprotiline and [11C]lortalamine. J Neurochem. 2005;94:337–51. doi: 10.1111/j.1471-4159.2005.03202.x. [DOI] [PubMed] [Google Scholar]
- 14.Schou M, Pike VW, Varrone A, Gulyas B, Farde L, Halldin C. Synthesis and PET evaluation of (R)-[S-methyl-11C]thionisoxetine, a candidate radioligand for imaging brain norepinephrine transporters. J Lab Compds Radiopharm. 2006;49:1007–19. [Google Scholar]
- 15.Schou M, Pike VW, Sovago J, Gulyas B, Gallagher PT, Dobson DR, Walter MW, Rudyk H, Farde L, Halldin C. Synthesis of 11C-labelled (R)-OHDMI and CFMME and their evaluation as candidate radioligands for imaging central norepinephrine transporters with PET. Bioorg Med Chem. 2007;15:616–25. doi: 10.1016/j.bmc.2006.10.065. [DOI] [PubMed] [Google Scholar]
- 16.Schou M, Halldin C, Sovago J, Pike VW, Hall H, Gulyas B, Mozley PD, Dobson D, Shchukin E, Innis RB, Farde L. PET evaluation of novel radiofluorinated reboxetine analogs as norepinephrine transporter probes in the monkey brain. Synapse. 2004;53:57–67. doi: 10.1002/syn.20031. [DOI] [PubMed] [Google Scholar]
- 17.Lin KS, Ding YS, Kim SW, Kil KE. Synthesis, enantiomeric resolution, F-18 labeling and biodistribution of reboxetine analogs: promising radioligands for imaging the norepinephrine transporter with positron emission tomography. Nucl Med Biol. 2005;32:415–22. doi: 10.1016/j.nucmedbio.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 18.Ding Y-S, Lin K-S, Logan J. PET imaging of norepinephrine transporters. Curr Pharm Des. 2006;12:3831–45. doi: 10.2174/138161206778559687. [DOI] [PubMed] [Google Scholar]
- 19.Kiyono Y, Kanegawa N, Kawashima H, Fujiwara H, Iida Y, Nishimura H, Saji H. A new norepinephrine transporter imaging agent for cardiac sympathetic nervous function imaging: radioiodinated (R)-N-methyl-3-(2-iodophenoxy)-3-phenylpropanamine. Nucl Med Biol. 2003;30:697–706. doi: 10.1016/s0969-8051(03)00085-4. [DOI] [PubMed] [Google Scholar]
- 20.Kiyono Y, Kanegawa N, Kawashima H, Kitamura Y, Iida Y, Saji H. Evaluation of radioiodinated (R)-N-methyl-3-(2-iodophenoxy)-3-phenylpropanamine as a ligand for brain norepinephrine transporter imaging. Nucl Med Biol. 2004;31:147–53. doi: 10.1016/j.nucmedbio.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 21.Kanegawa N, Kiyono Y, Kimura H, Sugita T, Kajiyama S, Kawashima H, Ueda M, Kuge Y, Saji H. Synthesis and evaluation of radioiodinated (S,S)-2-(alpha-(2-iodophenoxy)benzyl)morpholine for imaging brain norepinephrine transporter. Eur J Nucl Med Mol Imaging. 2006;33:639–47. doi: 10.1007/s00259-005-0017-y. [DOI] [PubMed] [Google Scholar]
- 22.Tamagnan GD, Brenner E, Alagille D, Staley JK, Haile C, Koren A, Early M, Baldwin RM, Tarazi FI, Baldessarini RJ, Jarkas N, Goodman MM, Seibyl JP. Development of SPECT imaging agents for the norepinephrine transporters: [123I]INER. Bioorg Med Chem Lett. 2007;17:533–37. doi: 10.1016/j.bmcl.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chumpradit S, Kung M-P, Panyachotipun C, Prapansiri V, Foulon C, Brooks BP, Szabo SA, Tejani-Butt S, Frazer A, Kung HF. Iodinated tomoxetine derivatives as selective ligands for serotonin and norepinephrine uptake sites. J Med Chem. 1992;35:4492–97. doi: 10.1021/jm00101a029. [DOI] [PubMed] [Google Scholar]
- 24.Robertson DW, Krushinski JH, Fuller RW, Leander JD. Absolute configurations and pharmacological activities of the optical isomers of fluoxetine, a selective setotonin-uptake inhibitor. J Med Chem. 1988:1412–17. doi: 10.1021/jm00402a027. [DOI] [PubMed] [Google Scholar]
- 25.Mitsunobu O. The use of diethyl azodicarboxylate and triphenylphosphine in synthesis and transformation of natural products. Synthesis. 1981:1–25. [Google Scholar]
- 26.Bastiaans HMM, van der Baan JL, Ottenheijm HCJ. Flexible and Convergent Total Synthesis of Cyclotheonamide B. J Org Chem. 1997;62:3880–89. [Google Scholar]
- 27.Lindstroem S, Ripa L, Hallberg A. Synthesis of Two Conformationally Constrained Analogues of the Minor Tobacco Alkaloid Anabasine. Org Lett. 2000;2:2291–93. doi: 10.1021/ol006056f. [DOI] [PubMed] [Google Scholar]
- 28.Gu H, Wall SC, Rudnick G. Stable expression of biogenic amine transporters reveals differences in inhibitor sensitivity, kinetics, and ion dependence. J Biol Chem. 1994;269:7124–30. [PubMed] [Google Scholar]
- 29.Oya S, Choi SR, Kung MP, Kung HF. 5-Chloro-2-(2’-((dimethylamino)methyl)-4’-iodophenylthio)benzenamine: a new serotonin transporter ligand. Nucl Med Biol. 2007;34:129–39. doi: 10.1016/j.nucmedbio.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paxinos G, Watson C. The Rat Brain In Stereotaxic Coordinates. New York: Academic Press; 1986. [Google Scholar]
- 31.Cortes R, Soriano E, Pazos A, Probst A, Palacios JM. Autoradiography of antidepressant binding sites in the human brain: localization using [3H]paroxetine. Nueroscience. 1988;27:473–96. doi: 10.1016/0306-4522(88)90282-5. [DOI] [PubMed] [Google Scholar]
- 32.De Souza EB, Kuyatt BL. Autoradiographic localization of 3H-paroxetine-labeled serotonin uptake sites in rat brain. Synapse. 1987;1:488–96. doi: 10.1002/syn.890010513. [DOI] [PubMed] [Google Scholar]
- 33.Kovachich GB, Aronson CE, Brunswick DJ, Frazer A. Quantitative autoradiography of serotonin uptake sites in rat brain using [3H]cyanoimipramine. Brain Res. 1988;454:78–88. doi: 10.1016/0006-8993(88)90805-0. [DOI] [PubMed] [Google Scholar]
- 34.Biegon A, Mathis CA, Hanrahan SM, Jagust WJ. [125I]5-Iodo-6-nitroquipazine: a potent and selective ligand for the 5-hydroxytryptamine uptake complex II in vivo studies in rats. Brain Res. 1993;619:236–46. doi: 10.1016/0006-8993(93)91617-2. [DOI] [PubMed] [Google Scholar]
- 35.Kung MP, Hou C, Zhuang Z-P, Zhang B, Skovronsky DM, Gur T, Lee VM-Y, Trojanowski JQ, Kung HF. IMPY:An improved thioflavin-T derivative for in vivo Labeling of b-amyloid plaques. Brain Res. 2002;956:202–10. doi: 10.1016/s0006-8993(02)03436-4. [DOI] [PubMed] [Google Scholar]
- 36.Kung MP, Hou C, Oya S, Mu M, Acton PD, Kung HF. Characterization of [123I]IDAM as a novel single-photon emission tomography tracer for serotonin transporters. Eur J Nucl Med. 1999;26:844–53. doi: 10.1007/s002590050458. [DOI] [PubMed] [Google Scholar]
- 37.Kung M-P, Essman WD, Frederick D, Meegalla S, Goodman M, Mu M, Lucki I, Kung HF. IPT: a novel iodinated ligand for the CNS dopamine transporter. Synapse. 1995;20:316–24. doi: 10.1002/syn.890200405. [DOI] [PubMed] [Google Scholar]
- 38.Munson PJ, Rodbard D. LIGAND: a versatile computerized approach for characterization of ligand-binding systems. Anal Biochem. 1980;107:220–39. doi: 10.1016/0003-2697(80)90515-1. [DOI] [PubMed] [Google Scholar]