

Summary
Huntington's Disease (HD) is a dominantly inherited neurodegenerative disorder caused by expansion of a translated CAG repeat in the N-terminus of the huntingtin protein. Here we describe the generation and characterization of a novel full-length HD Drosophila model to reveal a previously unknown disease mechanism that occurs early in the course of pathogenesis, before expanded huntingtin is cleaved and imported into the nucleus in detectable amounts. We find that expanded full-length huntingtin (128QhttFL) leads to behavioral, neurodegenerative, and electrophysiological phenotypes. These phenotypes are caused by a Ca2+-dependent increase in neurotransmitter release efficiency in 128QhttFL animals. Partial loss of function in synaptic transmission (Syntaxin, Snap, Rop) and voltage-gated Ca2+ channel genes suppresses both the electrophysiological and the neurodegenerative phenotypes. Thus, our data indicate that increased neurotransmission is at the root of neuronal degeneration caused by expanded full-length huntingtin during early stages of pathogenesis.
Keywords: Huntington's disease, neurodegeneration, neurotransmitter release, Drosophila, Syntaxin, Rop, Snap, Vo ATPase, calcium channel
Introduction
Huntington's Disease (HD) is one of at least nine disorders caused by expansion of a triplet repeat encoding glutamine. In the case of huntingtin (htt), the protein responsible for HD, expansion of the glutamine tract beyond 39 repeats invariably leads to psychiatric, motor, and cognitive disturbances. Most HD patients become symptomatic in the fourth or fifth decade of life, but large expansions (i.e. above 70 repeats) lead to juvenile forms of the disease demonstrating an inverse relationship between repeat size and disease onset (The Huntington's Disease Collaborative Research Group, 1993).
In HD, glutamine-expanded huntingtin forms ubiquitin-positive aggregates in the nuclei and neurites of brain neurons (DiFiglia et al., 1997; Rubinsztein, 2002). Wild-type huntingtin is a large (350kDa) soluble protein that is conserved between Drosophila and mammals and is detected in neurons and many other cell types. Although it has a broad intracellular distribution, huntingtin is most abundant in the cytoplasm where it associates with the Golgi complex, endoplasmic reticulum, and synaptic vesicles (Cattaneo et al., 2005). Huntingtin is essential for murine embryogenesis (Nasir et al., 1995), but its normal functions are still poorly understood. Protein interaction analyses implicate huntingtin in diverse processes including intracellular trafficking, axonal transport, transcriptional regulation, cytoskeletal organization and prevention of apoptosis (Cattaneo et al., 2005; Goehler et al., 2004; Harjes and Wanker, 2003; Li and Li, 2004b). Huntingtin has also been linked to neurotransmission, (reviewed in (Harjes and Wanker, 2003; Li et al., 2003; Smith et al., 2005). For example, huntingtin associates with clathrin-coated pits and vesicles at synaptic terminals (DiFiglia et al., 1995; Velier et al., 1998). Furthermore, increased neuronal input resistance, lower stimulus intensity to evoke action potentials (Klapstein et al., 2001), impaired long-term potentiation (LTP) (Hodgson et al., 1999; Klapstein et al., 2001; Murphy et al., 2000; Usdin et al., 1999), and abnormal responses to NMDA stimulation (Cepeda et al., 2001; Laforet et al., 2001) in HD neurons suggest that synaptic dysfunction may contribute to pathogenesis. Studies in the R6/2 N-terminal mouse model have also shown alterations in the corticostriatal pathway and altered levels of post-synaptic markers (Cepeda et al., 2003). However, it is not known whether alterations in synaptic function are early events or secondary to neuronal dysfunction during pathogenesis.
Numerous mouse models for HD have been generated. These include transgenic animals expressing either truncations of huntingtin or the entire protein, as well as “knock-ins” expressing the endogenous murine protein with an expanded polyglutamine tract (reviewed in (Menalled, 2005; Menalled and Chesselet, 2002; Rubinsztein, 2002). Most studies have been carried out using the first generated models that only express a small N-terminal portion of the protein (exon 1) containing the polyglutamine expansion. In general, mice expressing short truncations of the expanded protein have earlier and more severe phenotypes than mice expressing the entire protein. They also show formation of nuclear aggregates early in life. In contrast, full-length or longer N-terminal models exhibit cytoplasmic accumulation of huntingtin, and the nuclear localization or aggregation occurs only later in life (Hickey and Chesselet, 2003; Menalled, 2005; Menalled et al., 2002; Rubinsztein, 2002; Van Raamsdonk et al., 2005). In addition, N-terminal mouse models fail to reproduce the specificity of neuronal degeneration observed in HD patients, where neuronal loss occurs mainly in the striatum and cortex (Li and Li, 2004a). This selective neurodegeneration is best reproduced in models that express the full-length product (Van Raamsdonk et al., 2005).
HD has also been modeled in Drosophila, a system that allows for genetic screens and rapid experimentation using large numbers of animals. These models were used to demonstrate that nuclear (i.e. transcriptional dysregulation (Steffan et al., 2001)), and non-nuclear (i.e. fast axonal transport, (Lee et al., 2004)) mechanisms of pathogenesis, as well as posttranslational modifications of huntingtin are important for pathogenesis (Steffan et al., 2004). HD Drosophila models were also used to identify chemical compounds that may ameliorate huntingtin-induced toxicity (Bilen and Bonini, 2005; Marsh and Thompson, 2004; Sang and Jackson, 2005). However, no full-length model of HD has been reported in Drosophila.
Studies using N-terminal models have provided many important insights into HD, however it is now clear that protein context is important for pathogenesis (Yu et al., 2003). This is best illustrated by a transgenic mouse expressing 120 CAG repeats in the context of exons 1 and 2 of the huntingtin protein, which shows no neuronal dysfunction or degeneration in spite of abundant neuronal inclusions (Slow et al., 2005). The importance of appropriate protein context has also been documented in the case of other polyglutamine diseases. For example, in addition to polyglutamine expansion, neurodegeneration in spino-cerebellar ataxia type1 also requires phosphorylation of ataxin-1 (Emamian et al., 2003).
Here we report the development of a novel full-length Drosophila HD model to investigate the mechanisms by which expanded full-length huntingtin impairs synaptic transmission. We show that expression of expanded full-length huntingtin leads to an increased neurotransmitter release efficiency. This phenotype can be suppressed genetically by removing a single copy of genes encoding proteins required for proper neurotransmitter release. We also find that resting intracellular Ca2+ levels are increased in these flies when compared to controls. This suggests a defect in Ca2+ homeostasis in agreement with observations in mammalian systems, (Bezprozvanny and Hayden, 2004; Cepeda et al., 2001; Hodgson et al., 1999; Tang et al., 2005). Importantly, these abnormalities occur before we can detect the cleavage and nuclear translocation of the huntingtin protein, and we also show that mutations in certain voltage-gated Ca2+ channels restore the elevated Ca2+ levels and improve neurotransmitter release efficiency and neurodegenerative phenotypes.
Results
Full-length human huntingtin accumulates in the cytoplasm of Drosophila neurons and does not form visible aggregates
To develop a full-length model of HD in Drosophila, we generated transgenes to express the entire 3144aa protein with either 16 (16QhttFL, wild type) or 128 (128QhttFL, pathogenic) glutamines under the control of the UAS-GAL4 inducible system (Brand and Perrimon, 1993). Western analyses using the MAB2166 huntingtin-specific antibody (Lee et al., 2004) reveals a band corresponding to the ∼350KDa full-length protein in heads of Drosophila expressing either the 16QhttFL or the 128QhttFL transgenes with the GMR-GAL4 eye driver. In contrast, no such protein is detected in GMR-GAL4 controls (Figure 1A). The 16QhttFL protein runs slightly faster than the 128QhttFL protein, as may be expected from the different number of glutamine repeats. To further verify the difference in glutamine repeats between 16QhttFL and 128QhttFL transgenic lines, the same membrane was stripped and re-probed with the MAB1574 polyQ-specific antibody revealing that only the lanes containing expanded huntingtin produced a reactive band (data not shown).
Figure 1. Human full-length huntingtin accumulates in the cytoplasm of Drosophila neurons and does not form visible axonal aggregates.
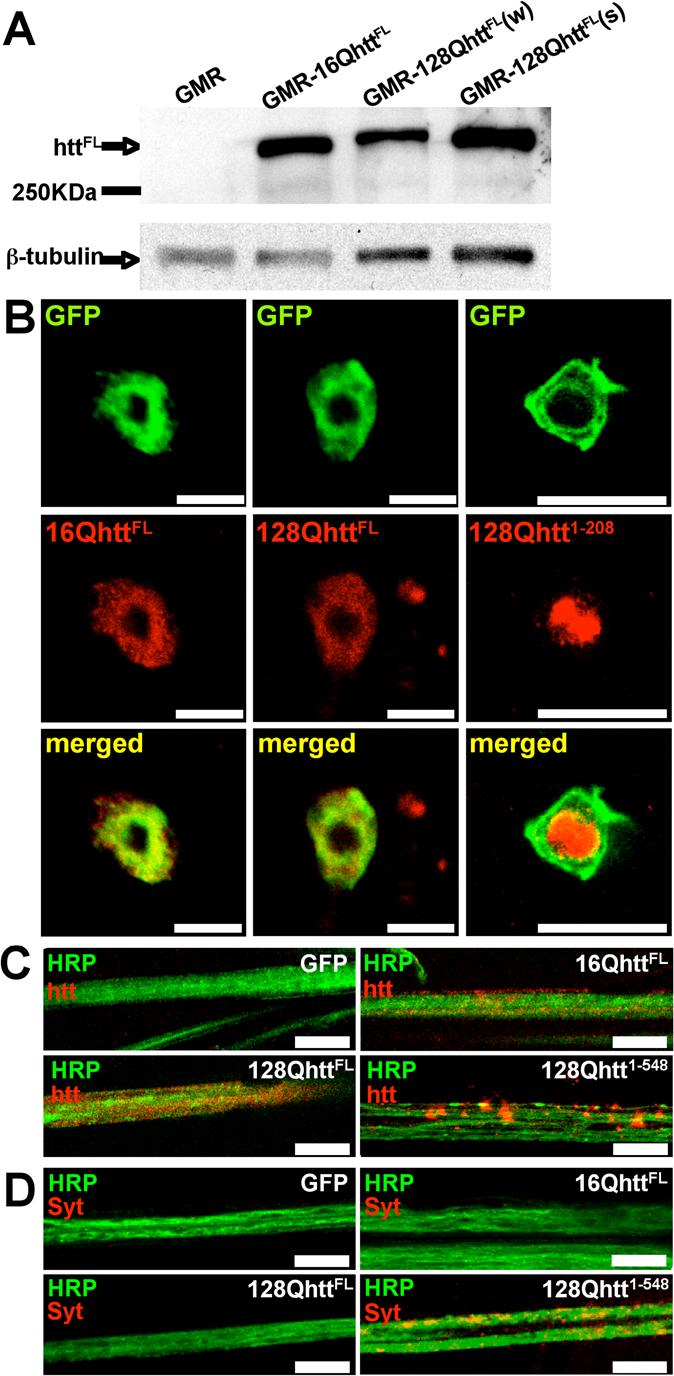
(A) Western blot analysis of transgenic Drosophila heads using MAB2166 huntingtin-specific antibody reveals a (∼350KDa) band corresponding to full-length (unexpanded and expanded) human huntingtin. Based on densitometry analysis, the 16Q line expresses higher/similar protein levels than the 128Q(w) and 128Q(s) lines (∼2-fold and ∼1-fold, respectively).
Genotypes: GMR (GMR-GAL4/UAS-GFP), GMR-16QhttFL (GMR-GAL4/UAS-16QhttFL[M28]), GMR-128QhttFL(w) (GMR-GAL4/+; UAS-128Qhtt(w)[F7]/+), GMR-128QhttFL(s) (GMR-GAL4/UAS-128Qhtt(s)[M36E2])
(B) Immunofluorescence confocal images of Drosophila apterous ventral nerve cord interneurons from 20-day-old flies expressing wild-type (left, 16QhttFL) or expanded (middle, 128QhttFL) full-length huntingtin. Right panels show similar images from animals expressing an expanded N-terminal huntingtin truncation (amino acids 1−208 excluding the polyglutamine tract, 128Qhtt1−208). Note co-localization of full-length huntingtin with the CD8-GFP cytoplasmic marker. The same results were obtained with 10-day-old and 30-day-old flies (data not shown). In contrast, the truncated version of huntingtin accumulates in the nucleus as early as the third-instar larval stage. MAB5374 was used at 1:100 for all stainings, which were done simultaneously. Flies raised at 27°C. Scale bar= 5μm.
Genotypes: 16QhttFL (UAS-16QhttFL[M28]/UAS-CD8-GFP; apVNC-GAL4/+), 128QhttFL (UAS-128QhttFL(s)[M36E2]/UAS-CD8-GFP; apVNC-GAL4/+), 128Qhtt1−208 (UAS-CD8-GFP/+; UAS-128Qhtt1−208[M64]/apVNC-GAL4).
(C-D) Immunolabeling of huntingtin (htt) (C) or endogenous synaptotagmin I (Syt) (D) proteins in motor neuron axon bundles from third instar larvae of the genotype indicated in each panel. Note diffuse pattern and absence of aggregates in wild-type (16QhttFL) and expanded full-length huntingtin (128QhttFL) axons. In contrast, a long N-terminal huntingtin truncation (amino acids 1−548, 128Qhtt1−548) causes large huntingtin and synaptotagmin I axonal aggregates. MAB5374 1:100. Flies were at 29°C. Scale bar= 5μm.
Genotypes: GFP (Elav-GAL4/+; UAS-GFP/+), 16QhttFL (Elav-GAL4/+; UAS-16QhttFL[M28]/+), 128QhttFL (Elav-GAL4/+; UAS-128QhttFL(s)[M36E2]/+), 128Qhtt1−548 (Elav-GAL4/+; UAS-128QHtt1−548/+).
To determine the intracellular localization of full-length human huntingtin in Drosophila, we used a GAL4 driver directing expression to a specific subset of large central nervous system (CNS) neurons, the apVNC interneurons (Fernandez-Funez et al., 2000). Labeling using MAB5374 shows cytoplasmic accumulation of both 16QhttFL and 128QhttFL in apVNC interneurons of 20-day-old flies (Figure 1B, left and central panels). Similar results were observed in 10-day-old and 30-day-old flies (data not shown). The absence of detectable nuclear huntingtin staining is not a consequence of poor ability of this antibody to detect huntingtin in the nucleus because flies expressing an N-terminal truncated form of the protein (amino acids 1−208) with the same glutamine expansion show robust nuclear staining using the same antibody concentration and experimental conditions (Figure 1B right panels; see also (Jackson et al., 1998; Steffan et al., 2001) for nuclear staining of similar size huntingtin fragments in Drosophila). Thus, the cytoplasmic accumulation of 128QhttFL is reminiscent of data from HD murine models where full-length huntingtin localizes to the cytoplasm first, and nuclear staining is only evident months after birth (Menalled, 2005; Rubinsztein, 2002; Slow et al., 2003), a time well beyond the lifespan of flies.
We also investigated whether full-length huntingtin forms aggregates in Drosophila neurons. Large axonal aggregates blocking axonal transport have been reported in flies expressing an expanded N-terminal huntingtin fragment of 548 amino acids (Lee et al., 2004). In contrast to the 548 amino acid fragment (Figure 1C, lower right panel), 128QhttFL does not form obvious axonal aggregates (Figure 1C, lower left panel). To investigate whether axonal transport is impaired in 128QhttFL flies, we monitored the localization of synaptotagmin I, a synaptic vesicle associated protein that is transported along axons (Lee et al., 2004). We found that in animals expressing the htt 548 amino acid truncation, synaptotagmin I, like huntingtin, accumulates in the axons (Figure 1D, lower right panel). In contrast, larvae expressing the control protein GFP (Figure 1D, upper left panel), or the full-length huntingtin protein (expanded or unexpanded) (Figure 1D, lower left and upper right panels, respectively), do not show accumulation of synaptotagmin I in axons. Thus, although it is difficult to exclude the possibility of decreased axonal transport, we found no evidence of axonal blockages disrupting transport in 128QhttFL expressing animals.
Expression of 128QhttFL, but not 16QhttFL, in the Drosophila eye and central nervous system leads to progressive neurodegenerative phenotypes
The Drosophila eye provides an excellent system for performing genetic interaction screens (Karim et al., 1996), and has been utilized to successfully identify genetic modifiers of neurodegeneration (Auluck et al., 2002; Fernandez-Funez et al., 2000; Kazemi-Esfarjani and Benzer, 2000; Shulman and Feany, 2003; Warrick et al., 1999). To examine the consequences of expressing full-length huntingtin in the visual system, the 16QhttFL and 128QhttFL transgenes were expressed using GMR-GAL4 drivers. Examination of the external eye morphology, using light microscopy and scanning electron microscopy (SEM) reveals depigmentation and disorganization of ommatidia following expression of 128QhttFL(s) but not 16QhttFL from GMR-GAL4(s), a relatively strong driver (Figure 2A-D, left and center panels).
Figure 2. Progressive neurodegenerative eye phenotype produced by overexpression of expanded (128Q), but not unexpanded (16Q) full-length huntingtin.
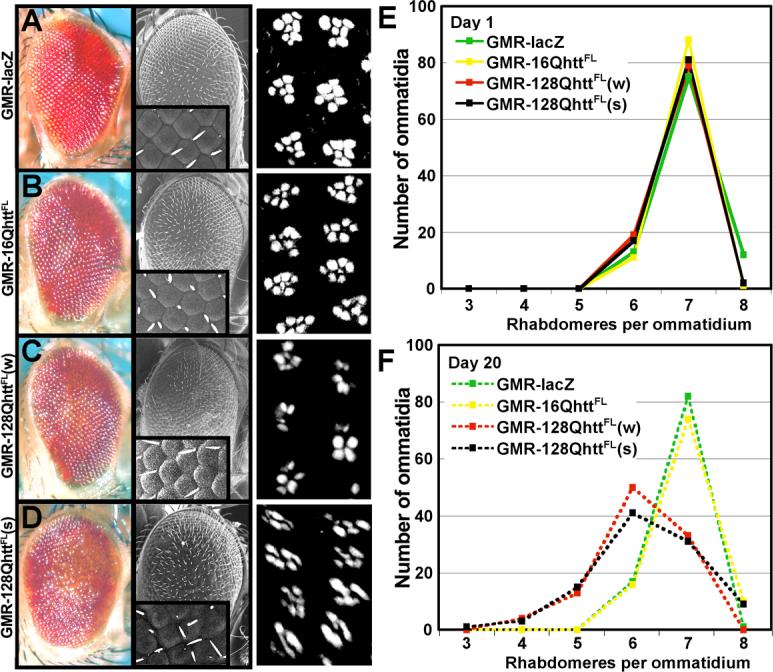
(A-D) Light microscope (left), and SEM (center) images of transgenic flies expressing (A) non-toxic LacZ control protein, (B) wild-type huntingtin, (C) low levels of expanded huntingtin, and (D) high levels of expanded huntingtin. Insets in SEM images show magnification (600×) of the ommatidia field. Note disorganization of ommatidia in D. Right panels show phalloidin staining of the corresponding eyes from 20-day-old flies showing arrangement of rhabdomeres. Flies for light microscope and SEM images were raised at 25°C. Flies for phalloidin staining were raised at 27°C.
Genotypes for light microscope and SEM: GMR-lacZ (GMR-GAL4(s)/UAS-lacZ), GMR-16QhttFL (GMR-GAL4(s)/UAS-16QhttFL[M28]), GMR-128QhttFL(w) (GMR-GAL4(s)/+; UAS-128QhttFL(w)[F7]/+), GMR-128QhttFL (GMR-GAL4(s)/UAS-128QhttFL(s)[M36E2]).
Genotypes for phalloidin staining: GMR-lacZ (GMR-GAL4/UAS-lacZ), GMR-16QhttFL (GMR-GAL4/UAS-16QhttFL[M28]), GMR-128QhttFL (GMR-GAL4/+; UAS-128QhttFL(w)[F7]), GMR-128QhttFL (GMR-GAL4/UAS-128QhttFL[M36E2]).
(E-F) Quantification of the number of rhabdomeres per ommatidium in 1-day-old (E) or 20-day-old (F) flies expressing the following proteins. Green: non-toxic LacZ control, yellow: wild-type huntingtin, red: expanded huntingtin at relatively low levels, and black: expanded huntingtin at relatively high levels. Flies raised at 27°C. n=100 ommatidia per genotype. The distribution of the rhabdomeres at day 20 for 128QhttFL(w) and 128QhttFL(s) is significantly different from LacZ and 16QhttFL controls (p<0.001, Mann-Whitney test).
Genotypes: GMR-lacZ (GMR-GAL4/UAS-lacZ), GMR-16QhttFL (GMR-GAL4/UAS-16QhttFL[M28]), GMR-128QhttFL(w) (GMR-GAL4/+; UAS-128QhttFL(w)[F7]), GMR-128QhttFL(s) (GMR-GAL4/UAS-128QhttFL(s)[M36E2]).
To investigate whether the eye phenotype is progressive, the same huntingtin transgenes were expressed using a weaker GMR-GAL4 eye driver to facilitate comparison of the internal retinal structure between animals of different genotypes. Phalloidin staining of dissected wild-type eyes reveals the organization of the Drosophila eye in ommatidia. Each ommatidium contains 8 rhabdomeres (the rod-shaped structures where the photopigment rhodopsin accumulates in photoreceptor neurons), 7 of which are typically visible in a single plane (Figure 2A, right panel). Retinal degeneration often results in fewer rhabdomeres (Cauchi and van den Heuvel, 2006; Marsh and Thompson, 2006; Sang and Jackson, 2005). We compared the number of rhabdomeres per ommatidium in 1-day-old and 20-day-old animals expressing 128QhttFL, 16QhttFL or a lacZ control transgene. In 1-day-old flies we observed no differences between the distributions of rhabdomeres in these genotypes, with 7 rhabdomeres visible in most ommatidia (Figure 2E). However, in flies aged for 20 days, we found significantly fewer rhabdomeres in 128QhttFL flies than in 16QhttFL and lacZ control flies (right panels of Figures 2A-D and Figure 2F, p<0.0001, Mann-Whitney test). Thus, the analyses of both external and internal eye phenotypes caused by full-length huntingtin using two GMR-GAL4 drivers of different strengths produced similar results. In addition, expression of 128QhttFL leads to progressive degeneration of photoreceptor cells.
Expression of 128QhttFL in the visual system shows that it has deleterious effects on eye photoreceptors. Hence, we also investigated the consequences of expressing 128QhttFL in CNS neurons. First, we compared the survival rates of 128QhttFL and GFP control animals following transgene expression in motor neurons using the C164-GAL4 driver (Pennetta et al., 2002). As shown in Figure 3A, flies expressing 128QhttFL show reduced survival in adult life when compared to controls expressing GFP (p<0.05, 0.01 and 0.01 for days 25, 30 and 35 respectively, Mann-Whitney test).
Figure 3. Reduced survival, impaired motor performance, and neuronal degeneration in Drosophila expressing expanded full-length huntingtin in the CNS.
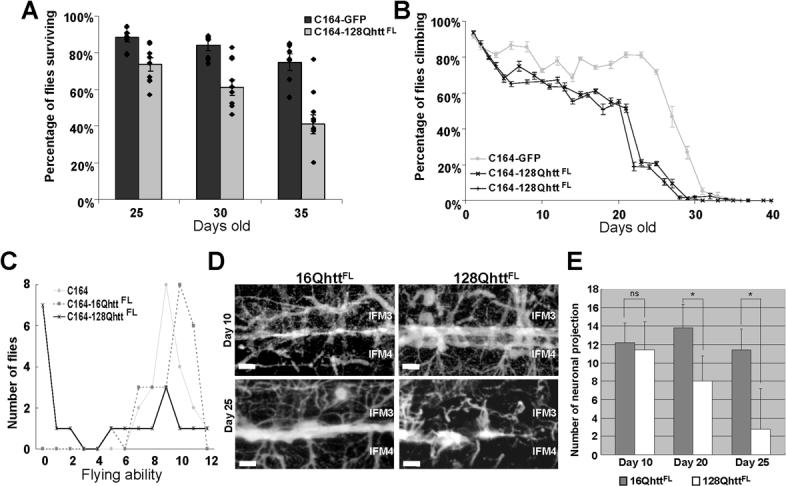
(A) Average survival of flies expressing GFP (dark bars) or 128QhttFL (light bars) in the CNS (C164-GAL4) at days 25, 30 and 35. The survival rate of flies expressing 128QhttFL is significantly lower than the survival rate of control flies expressing a non-toxic GFP protein (p<0.05, 0.01 and 0.01, respectively, Mann-Whitney test). Dots denote single data points for individual populations. Error bars= SEM. n= 7 and 10 populations for GFP and 128QhttFL respectively.
(B) Climbing performance as a function of age in control and 128QhttFL-expressing flies. Normal decline in climbing performance is observed after day 25 in flies expressing the non-toxic GFP protein (silver line). In contrast, flies expressing 128QhttFL (black lines) perform poorly after day 20. All flies raised at 27°C. Two independent experiments are shown for flies expressing 128QhttFL. Error bars= SEM of 10 trials per time point.
(C) Flying ability in 25-day-old control and huntingtin-expressing flies. Flying ability is represented in arbitrary units with 12 being a perfect ability and 0 being no ability (see Experimental Procedures). Note that most control flies that carry only the motor neuron driver (silver line) or express wild-type huntingtin (16QhttFL, grey dotted lines) perform well in this assay with no significant difference between them (p>0.1, Mann-Whitney test). Most flies expressing expanded huntingtin (128QhttFL, black lines), on the other hand, show impaired flying ability when compared to either control (p<0.05, Mann-Whitney test).
(D) Stacks of confocal images of neurons projecting into indirect flight muscles (IFM) 3 and 4 of 10-day (upper panels) and 25-day (lower panels) old flies expressing wild-type (left) or expanded (right) huntingtin. Note the loss of neuronal projections and NMJs in flies expressing expanded huntingtin. Scale bar=10μm.
(E) Quantification of the number of neuronal projections in a specified 100μm × 100μm area of IFM 3/ 4 shown in D. 20-day and 25-day, but not 10-day old flies expressing 128QhttFL have significantly fewer neuronal projections than control flies of the same age expressing wild-type 16QhttFL protein (ns: p>0,05; *: p<0.05, Student-t test; n=5).
All flies raised at 27°C.
Genotypes: C164 (C164-GAL4/+), C164-GFP (C164-GAL4/UAS-GFP), C164−16QhttFL (C164-GAL4/UAS-16QhttFL[M28]), C164−128QhttFL (C164-GAL4/UAS-128QhttFL(s)[M36E2]).
We also compared motor performance in 128QhttFL and control animals. First we measured motor performance in a climbing assay that takes advantage of the strong negative geotaxis behavior of Drosophila (Le Bourg and Lints, 1992). This assay measures the ability of flies to climb the wall of a vial as a function of age. As shown in Figure 3B, we found impaired motor performance in 128QhttFL flies as compared to controls expressing GFP. In addition, we used a flight assay to measure the flying ability of 128QhttFL flies. In this assay, flying ability is determined as a function of flight distance after flies are dropped from the top of a transparent cylinder (Pesah et al., 2004). We found that 25-day-old 128QhttFLflies exhibit a severe flight impairment phenotype, while GFP control and 16QhttFL flies of the same age behave normally (Figure 3C).
To more directly probe for neurodegeneration that would correlate with flight impairments in the 128QhttFL expressing flies, we investigated the neuronal projections into the Indirect Flight Muscles (IFM). Interestingly, labeling of the motor neurons innervating the IFM shows a neurodegenerative phenotype in aged 128QhttFL flies but not 16QhttFL control flies of the same age (Figures 3D-E, p<0.05, Mann-Whitney test). Together, these data indicate that expression of expanded full-length huntingtin causes progressive degenerative phenotypes in peripheral nervous system (PNS) and CNS neurons in Drosophila.
Huntingtin is unevenly distributed across synaptic boutons but does not affect the distribution of key synaptic proteins
We investigated the mechanism by which 128QhttFL triggers the eye and CNS phenotypes described above. Potentially important mechanisms of pathogenesis in HD are transcriptional dysregulation (Sugars and Rubinsztein, 2003), impaired axonal transport (Gunawardena and Goldstein, 2005; Li and Li, 2004a), synaptic dysfunction (Smith et al., 2005) and abnormal Ca2+ homeostasis (Bezprozvanny and Hayden, 2004). Since we did not detect 128QhttFL nuclear accumulation, or axonal aggregation of 128QhttFL or synaptotagmin, we focused on synaptic dysfunction and Ca2+ homeostasis.
First we investigated the distribution of full-length huntingtin in synaptic terminals. Interestingly, huntingtin, driven by the Elav-GAL4 paneural driver, shows a non-uniform distribution across boutons within a single third instar larval neuromuscular junction (NMJ) (Figure 4A). We also studied the distribution of proteins involved in synaptic transmission in 128QhttFL and control animals. Figure 4B shows that soluble NSF-attachment protein, Snap (Ordway et al., 1994), Ras opposite (Rop; (Schulze et al., 1994)) and Syntaxin 1A (Syx (Schulze et al., 1995)) are normally distributed in boutons expressing 128QhttFL. Furthermore, we did not observe noticeable abnormalities in bouton number, branching or morphology in 128QhttFL larvae (data not shown). In 128QhttFL flies, huntingtin is expressed in the pre-synaptic neurons and is not present in the post-synaptic muscles, therefore, we do not expect significant changes in post-synaptic glutamate receptor clusters. Nonetheless, to visualize neurotransmitter receptor clusters at the NMJ, we labeled 128QhttFL and 16QhttFL control flies with GluRIIA antibodies (Schuster et al., 1991). Receptor cluster abundance is not significantly different in 128QhttFL and 16QhttFL (Figure 4B, upper most row panels, p>0.5, Mann-Whitney test). Hence, post-synaptic neurotransmitter receptor clustering appears unaffected by neuronal expression of expanded full-length huntingtin. These observations suggest that abnormal bouton morphology and/or altered synaptic protein accumulation are not responsible for the eye and CNS phenotypes observed in 128QhttFL flies.
Figure 4. Uneven distribution of full-length huntingtin across boutons does not affect distribution of key synaptic proteins.
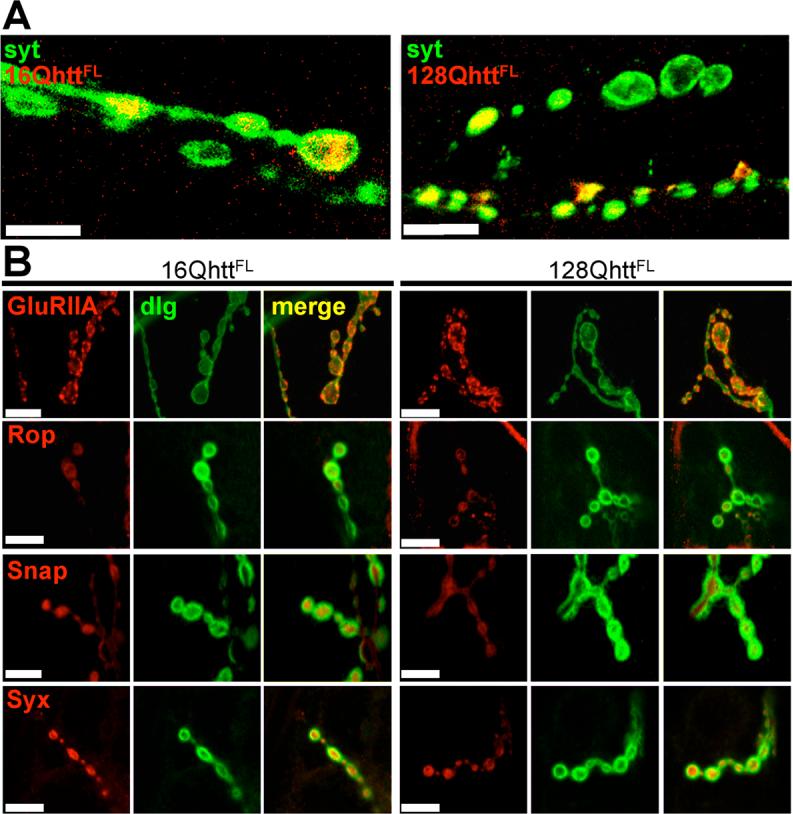
(A) Immunofluorescence confocal images of neuromuscular junctions from third-instar larvae expressing wild-type (left) or expanded (right) huntingtin reveal its uneven distribution from one bouton to another within individual axons. Boutons are visualized with anti-synaptotagmin antibody (green) and huntingtin is labeled in red. Scale bar= 10μm.
(B) Immunofluorescence confocal images of neuromuscular junctions from third-instar larvae expressing wild-type (left) or expanded (right) huntingtin show morphologically normal boutons with no abnormal distribution of proteins involved in neurotransmitter secretion. Boutons are visualized with anti-dlg (green) and stained for (from top to bottom) GluRIIA, Rop, Snap and Syx (red). No differences are detected in the patterns of accumulation of any of these proteins in boutons from larvae expressing expanded huntingtin or boutons from control larvae expressing wild-type huntingtin (p>0.5, 0.1, 0.1 and 0.1, respectively). Scale bar= 5μm.
All larvae raised at 29°C.
Genotypes: 16QhttFL (Elav-GAL4/+; UAS-16QhttFL(s)[M28]/+), 128QhttFL (Elav-GAL4/+; UAS-128QhttFL(s)[M36E2]/+).
Increased neurotransmitter release probability upon expression of 128QhttFL
Since the immunolabeling studies of the NMJ using confocal microscopy did not reveal major differences in abundance or distribution of synaptic proteins between 128QhttFL and control animals, we investigated possible electrophysiological defects. To determine the properties of neurotransmitter release in neurons expressing 128QhttFL presynaptically, we recorded excitatory junctional potentials (EJPs) at the third-instar larval NMJ. Although EJPs recorded from animals expressing either 128QhttFL or GFP in the nervous system (Elav-128QhttFL or Elav-GFP) are similar at 1.2mM extracellular Ca2+ (Elav-GFP: 40.5±2.0 mV and Elav-128QhttFL 44.2 ± 0.6 mV, t-test: p>0.1, data not shown), EJPs recorded at lower extracellular Ca2+ are clearly different. As shown in Figure 5A-C, EJPs recorded from Elav-128QhttFL animals in 0.25 mM Ca2+ are increased about 5-fold compared to those of Elav-GFP controls (Elav-GFP: 2.9 ± 0.3 mV and Elav-128QhttFL: 16.0 ± 2.7 mV, t-test: p<0.001), whereas EJPs recorded in 0.6 mM Ca2+ are increased by ∼30% (Elav-GFP: 27.7 ± 3.0 mV and Elav-128QhttFL: 36.3 ± 2.4 mV, t-test: p<0.05). The increased neurotransmission is specific to expression of 128QhttFL since expression of wild-type 16QhttFL does not produce this effect (Figure 5B-C; Elav-16QhttFL in 0.25mM Ca2+: 3.6 ± 0.8 mV, t-test: p>0.7). Furthermore, this phenotype is not simply an effect of the 128QhttFL transgene insertion site since flies carrying the transgene in the absence of the Elav driver do not differ from GFP controls (128QhttFL: 3.08 ± 0.63mV and Elav-GFP: 2.9 ± 0.3mV). Thus, expression of expanded huntingtin protein in neurons leads to increased synaptic transmission at the third instar larval Drosophila NMJ.
Figure 5. Neurotransmitter release probability is increased upon expression of full-length expanded huntingtin.

(A-B) Quantification of EJP amplitudes recorded at (A) 1Hz in 0.6mM Ca2+ or (B) 0.25mM Ca2+ in Drosophila larvae expressing a non-toxic control protein (GFP), wild-type or expanded full-length huntingtin or controls carrying the huntingtin transgene without a GAL4 driver. ns: p>0.05; *p<0.05; **p<0.01 throughout the figure.
(C) Sample EJP traces recorded from Drosophila larvae expressing GFP control protein and expanded full-length huntingtin in HL3 buffer at 0.6mM Ca2+ (top) or 0.25mM Ca2+ (bottom). For 0.25mM Ca2+, sample traces are also shown for transgenic larvae expressing wild-type huntingtin and for controls carrying the expanded huntingtin transgene without a GAL4 driver. Arrows indicate blanked-out stimulus artifact.
(D-E) Average (D) frequency and (E) amplitude of mEJPs recorded from abdominal muscle 6 or 7 in GFP control and larvae expressing expanded huntingtin. Error bars: SEM. Number inside bars is the number of animals studied.
(F) Percent failures measured at 1Hz in 0.25mM Ca2+ following stimulation at 2−3× threshold of motor neurons in larvae expressing a non-toxic GFP protein, wild-type huntingtin, expanded huntingtin or controls carrying the huntingtin transgene without a GAL4 driver.
All experiments done at 29°C.
Genotypes: Elav-GFP (Elav-GAL4/+; UAS-GFP/+), Elav-16QhttFL (Elav-GAL4/+; UAS-16QhttFL[M28]/+), 128QhttFL (UAS-128QhttFL(s)[M36E2]/+), Elav-128QhttFL (Elav-GAL4/+; UAS-128QhttFL(s)[M36E2]/+).
Since our data argue against abnormal glutamate receptor clustering (see above), the increased synaptic transmission in 128QhttFL animals could be caused by increased neurotransmitter release from individual synaptic vesicles or increased release efficiency. To test whether neurotransmitter release from single vesicles is altered in Elav-128QhttFL animals, we recorded spontaneous fusion events in the absence of stimulation (mEJPs or minis; 0.1mM Ca2+ and 5M TTX). As is shown in Figure 5D-E, mEJP frequency and amplitude are similar in Elav-128QhttFL and Elav-GFP animals, indicating that transmitter loading as well as post-synaptic glutamate receptor clustering are unaffected.
Finally, to determine release probability, we recorded the number of failures (non-events) when motor neurons were stimulated 2−3 times above threshold in low (0.25mM) Ca2+. Interestingly, whereas controls (Elav-GFP, Elav-16QhttFL and 128QhttFL with no driver) fail to release about 20−25% of the time (Figure 5F), animals that express 128QhttFL reliably release neurotransmitter upon stimulation with very low failure rates (0.3 ± 0.3% failures, t-test: p<0.001). Hence, the increased synaptic transmission observed in animals that express 128QhttFL is, at least in part, caused by an increase in neurotransmitter release probability.
Partial loss of function of genes required for proper synaptic transmission suppresses neurotransmitter release abnormalities caused by 128QhttFL
Since expression of 128QhttFL causes increased synaptic transmission and release probability, an attractive possibility is that reducing the activity of components of the neurotransmitter release machinery may ameliorate these phenotypes, providing a potential therapeutic strategy.
To test this hypothesis, we recorded EJP amplitudes in 0.25mM Ca2+ and determined the release probability in 128QhttFL flies that lack one functional copy of either Soluble NSF attachment protein (Snap), Syntaxin1A (Syx), or proteins that are known to bind Syx, like Ras Opposite (Rop) (also named Unc18, or Munc18) (Schulze et al., 1994) and Vha100−1 (Hiesinger et al., 2005). Syx is involved in synaptic transmission as well as non-neuronal secretion and is a plasma membrane associated member of the SNARE complex that mediates fusion between the vesicular and synaptic membranes (Schulze et al., 1995). Snap associates with the SNARE complex and assists NSF (N-ethylmaleimide Sensitive Fusion Protein) in the dissociation of SNARE (Soluble NSF attachment Receptor) complexes (Ordway et al., 1994). Rop, the Drosophila Sec1 homologue, interacts with syntaxin in vivo, and has both positive and inhibitory functions on neurotransmission (Wu et al., 1998). Interestingly, whereas animals that overexpress 128QhttFL in the nervous system show increased EJP amplitude and decreased failure rate (Figure 5B and F), removing one copy of Snap (Elav-GAL4/+; UAS-128QhttFL(s)[M36E2]/+; SnapM4/+) suppresses these phenotypes and restores the EJP amplitude and failure rates to control levels (Figure 6A, B, I and L). This suppression effect is not a result of reduced transmission in animals that have lost one copy of Snap, as the EJP amplitude and failure rates in Elav-GAL4/+; SnapM4/+ larvae are not different from controls (Elav-GAL4/+; UAS-GFP/+) (Figure 6A, B, J and K). Similarly, although removing one copy of Rop (Elav-GAL4/+; RopG27/+) (Figure 6C, D, M and N) or one copy of Syx (Elav-GAL4/+; Syx229/+) (Figure 6E and F) does not affect EJP amplitudes or failure rates compared to controls at 0.25mM Ca2+, it efficiently suppresses the increased EJP amplitude and decreased failure rates observed in Elav-128QhttFL animals.
Figure 6. Heterozygous mutations in genes affecting secretion suppress the increased release probability caused by full-length expanded huntingtin.

(A-H) Quantification of EJP amplitudes (A, C, E and G) and percent failures (B, D, F and H) in larvae expressing a non-toxic control protein GFP, heterozygous mutant for (A,B) Snap, (C,D) Rop, (E,F) Syntaxin 1A, or (G, H) Vha100kDa, expressing expanded huntingtin or expressing expanded huntingtin as well as heterozygous mutant for either (A,B) Snap, (C,D) Rop, (E,F) Syntaxin 1A, or (G,H) Vha100−1 at 29°C. Error bars: SEM. Number inside bars is the number of animals studied. ns, p>0.05; *, p<0.05; **, p<0.01. Experiments done at 29°C.
(I-N) Sample EJP traces (5 consecutive traces recorded at 1Hz interval) recorded from (I) larvae expressing expanded huntingtin, (J) the control protein, GFP, (K) heterozygous mutant for Snap, (L) expressing expanded huntingtin and heterozygous mutant for Snap, (M) heterozygous mutant for Rop and (N) expressing expanded huntingtin and heterozygous mutant for Rop. Arrows indicate blanked-out stimulus artifact.
All experiments done at 29°C. All recordings at 1Hz in HL3 buffer at 0.25mM Ca2+.
Genotypes: GFP (Elav-GAL4/+; UAS-GFP/+), 128QhttFL (Elav-GAL4/+; UAS-128QhttFL(s)[M36E2]/+), SnapM4/+ (Elav-GAL4/+; SnapM4/+), 128QhttFL/SnapM4 (Elav-GAL4/+; UAS-128QhttFL(s)[M36E2]/+; SnapM4/+), Rop27/+ (Elav-GAL4/+; RopG27/+), 128QhttFL/RopG27 (Elav-Gal4/+; UAS-128QhttFL(s)[M36E2]/+; RopG27/+), Syx229/+ (Elav-GAL4/+; Syx229/+), 128QhttFL/Syx229 (Elav-Gal4/+; UAS-128QhttFL(s)[M36E2]/+; Syx229/+), vha1/+ (Elav-GAL4/+; vha1/+), 128QhttFL/vha1 (Elav-GAL4/+; UAS-128QhttFL(s)[M36E2]/+; vha1/+).
However, not all components of the release machinery are able to suppress the increased release in Elav-128QhttFL larvae to the same extent. The Vha100−1 subunit is an important component of the synaptic vesicle release machinery, recently shown to act downstream of the SNARE complex in neurons (Hiesinger et al., 2005; Strompen et al., 2005). Removing one copy of Vha100−1 in 128QhttFL-expressing animals (Elav-GAL4/+; UAS-128QhttFL(s)[M36E2]/+; vha1/+), does not fully suppress the increased EJP amplitude seen in Elav-128QhttFL larvae, and fails to suppress the increased release probability (Figure 6G-H). Taken together, these data suggest that neuronal expression of expanded huntingtin facilitates neurotransmitter release by acting upon a general process that promotes vesicle release.
Synaptic transmission mutations also suppress the eye neurodegeneration and motor performance phenotypes caused by 128QhttFL
If increased neurotransmitter release is an important mechanism by which expanded full-length huntingtin exerts its toxicity on 128QhttFL flies, one may expect that suppressors of the electrophysiological phenotype also suppress other defects observed in these animals. To test this hypothesis we assessed the ability of neurotransmitter release mutations to suppress the degenerative phenotype observed in eye photoreceptors of 128QhttFL flies. At 20 days, flies expressing 128QhttFL (GMR-GAL4/+; UAS-128QhttFL(w)[F7]/+) show fewer rhabdomeres per ommatidium than controls (GMR-GAL4/UAS-GFP) (Figures 2F and 7A-B). However, animals expressing 128QhttFL in the context of partial loss of function of Snap (GMR-GAL4/+; UAS-128QhttFL(w)[F7]/SnapM4) show a reversal of this neurodegenerative phenotype (Figures 7A and C, p<0.001, Mann-Whitney test). Similar results were also obtained with animals having only one functional copy of the Syntaxin 1A gene (GMR-GAL4/+; UAS-128QhttFL(w)[F7]/Syx229, Figures 7A and D, p<0.001, Mann-Whitney test), or the Rop gene (GMR-GAL4/+; UAS-128QhttFL(w)[F7]/RopG27, Figure 7A and E, p<0.001, Mann-Whitney test). Partial loss of function of Vha100−1, on the other hand, did not improve the eye degeneration (GMR-GAL4/+; UAS-128QhttFL(w)[F7]/vha 1, Figure 7A and G, p>0.5, Mann-Whitney test). Moreover, we observe suppression of the 128QhttFL external eye phenotype with reduced activity of the neurotransmitter release gene Syx (Figures 7J-L). These data indicate a link between increased neurotransmission and neuronal degeneration in 128QhttFL animals.
Figure 7. Heterozygous mutations in genes affecting neurotransmitter secretion also suppress neurodegeneration and motor impairments in animals expressing expanded full-length huntingtin.

(A) Number of rhabdomeres per ommatidium in 20-day-old flies of the indicated genotypes. The distribution of the rhabdomeres is significantly different (p<0.001, Mann-Whitney test) between 128QhttFL and 128QhttFL animals also carrying one mutant copy of SNAP, Syx, Rop, or Dmca1D. Flies grown at 27°C.
Genotypes: 128QhttFL (GMR-GAL4/+; UAS-128QhttFL(w)[F7]), 128QhttFL/SnapM4 (GMR-GAL4/+; UAS-128QhttFL(w)[F7]/SnapM4), 128QhttFL/Syx229 (GMR-GAL4/+; UAS-128QhttFL(w)[F7]/Syx229), 128QhttFL/RopG27 (GMR-GAL4/+; UAS-128QhttFL(w)[F7]/RopG27), 128QhttFL/Dmca1DX10 (GMR-GAL4/Dmca1DX10; UAS-128QhttFL(w)[F7]/+), 128QhttFL/vha1 (GMR-GAL4/+; UAS-128QhttFL(w)[F7]/vha1), GFP (GMR-GAL4/ UAS-GFP).
(B-H) Phalloidin staining of dissected retinas from 20-day-old flies of the genotypes indicated in each panel. Scale bar= 5μM. Flies grown at 27°C
Genotypes in order: (B)128QhttFL (GMR-GAL4/+; UAS-128QhttFL(w)[F7]/+), (C)128QhttFL/SnapM4 (GMR-GAL4/+; UAS-128QhttFL(w)[F7]/SnapM4), (D) 128QhttFL/Syx229 (GMR-GAL4/+; UAS-128QhttFL(w)[F7]/Syx229), (E) 128QhttFL/RopG27 (GMR-GAL4/+; UAS-128QhttFL(w)[F7]/RopG27), (F) 128QhttFL/Dmca1DX10 (GMR-GAL4/Dmca1DX10; UAS-128QhttFL(w)[F7]/+),.(G) 128QhttFL/Vha1 (GMR-GAL4/+; UAS-128QhttFL(w)[F7]/Vha1), (H) GFP (GMR-GAL4/UAS-GFP).
(I) Suppression of climbing performance phenotype in flies expressing 128QhttFL that also carry a heterozygous loss-of-function mutation in Syx. Flies expressing the non-toxic GFP protein or 16QhttFL show normal decline in climbing performance with age. Climbing performance impairment occurs prematurely in flies expressing 128QhttFL, but it is restored to almost normal levels in a background heterozygous mutant for Syx. Error bars= SEM of 10 trials per time point. Flies grown at 25°C. Genotypes: GFP (C164-GAL4/UAS-GFP), 16QhttFL (C164-GAL4/UAS-16QhttFL[M28]), 128QhttFL (C164-GAL4/UAS-128QhttFL(s)[M36E2]), 128QhttFL/Syx229 (C164-GAL4/UAS-128QhttFL(s)[M36E2]; Syx229/+).
(J-L) SEM eye images of flies expressing (J) the non-toxic control protein GFP, (K) 128QhttFL (L) 128QhttFL and heterozygous mutant for Syx. Note partial suppression of the disorganized ommatidia phenotype in Syx heterozygous mutant animals. Genotypes: lacZ (GMR-GAL4(s)/UAS-GFP), 128QhttFL (GMR-GAL4(s)/UAS-128QhttFL(s)[M36E2]), 128QhttFL(w)/Syx229 (GMR-GAL4(s)/UAS-128QhttFL(s)[M36E2]/+; Syx229/+). Flies grown at 25°C.
In addition to the eye assay, we tested the ability of one mutant copy of Syx to suppress the motor performance defects caused by expression of 128QhttFL in the CNS. Flies expressing 128QhttFL (C164-GAL4/+; UAS-128QhttFL(s)[M36E2]/+) perform poorly in the climbing assay relative to controls, but this phenotype is suppressed in 128QhttFL flies that are heterozygous mutant for Syx (C164-GAL4/+; UAS-128QhttFL(s)[M36E2]/+; Syx229/+); see Figure 7I. These results validate that loss-of-function mutations in components of the neurotransmitter release machinery act as suppressors in different assays, and they underscore the importance of synaptic transmission dysfunction in 128QhttFL-induced neurodegeneration.
Increased Ca2+ levels in synaptic terminals of Drosophila expressing 128QhttFL are restored by mutations in components of the neurotransmitter release machinery
Intracellular Ca2+ levels have been found to be elevated in lymphoblasts of HD mouse models, and expanded huntingtin has been shown to potentiate NMDAR (NR1/NR2B)-induced apoptosis and Ca2+ transients (Cepeda et al., 2001; Hodgson et al., 1999; Tang et al., 2005). Hence, we investigated whether elevated presynaptic Ca2+ levels may account for increased vesicle release at 128QhttFL synapses by measuring presynaptic Ca2+ levels at the NMJ of 128QhttFL third instar larvae. To assess resting intracellular Ca2+ levels at presynaptic terminals, we forward-filled motor axons of control and 128QhttFL larvae with the ratiometric dye, Fura-2 Dextran. Upon Ca2+ binding, the maximal excitation of Fura-2 shifts from ∼380nm to ∼340nm. Therefore, the fluorescence ratio, F340/F380, provides a relative measure of Ca2+ levels. We found that resting synaptic Ca2+ levels are elevated on average ∼2-fold in 128QhttFL larvae compared to GFP controls. A similar effect was also found in16QhttFL larvae (Figure 8A). In addition, Ca2+ levels are more variable at 128QhttFL synapses compared to GFP controls (f-test, p < 0.001) with a bimodal distribution of fluorescence ratios indicative of bouton populations with both normal and aberrant intracellular Ca2+ levels (Figure 8A, open diamonds denote individual data points). This observation is in agreement with the uneven distribution of 128QhttFL in synaptic boutons described above (Figure 4A).
Figure 8. Increased Ca2+ levels caused by expanded full-length huntingtin can be suppressed genetically.

(A) Fluorescence ratios (F340/F380) measured from fura dextran-filled larval NMJs. Neuronal expression of 128QhttFL and 16QhttFL leads to increased fluorescence ratios compared to controls (GFP), indicating elevated resting Ca2+ levels at these synapses (* denotes p< 0.05, t-test). These elevated fluorescence ratios are restored to control levels in 128QhttFL animals that are also heterozygous for either Syntaxin or Dmca1D mutations (* denotes p< 0.05, t-test). Genotypes: Elav-Gal4/+; UAS-128QhttFL(s)[M36E2]/+; Syx229/+ and Elav-GAL4/+; UAS-128QhttFL(s)[M36E2]/Dmca1DX10. Note that animals carrying Syx or Dmca1D heterozygous mutations but no huntingtin transgene show no effect on resting Ca2+ levels compared to GFP controls. Genotypes: Elav-GAL4/+; Syx229/+ and Elav-GAL4/+; Dmca1DX10/+. Boutons were measured from 2−3 synapses in 6 larvae per genotype. Dots denote single data points from individual boutons.
(B) Fluorescence from boutons forward-filled with fura dextran and excited at both 340nm and 380nm. Ca2+ -free dye absorbs optimally at 380nm while Ca2+-bound dye is excited primarily at 340nm. NMJ synapses loaded with fura dextran were clearly visualized (top panel) and individual boutons could be spatially resolved (dashed box). In contrast to controls (Elav-GAL4/+; UAS-GFP/+), NMJ boutons from larvae expressing expanded huntingtin (Elav-GAL4/+; UAS-128QhttFL(s)[M36E2]/+) emit more intensely when excited at 340nm compared to 380nm. However, Syx heterozygosity (Elav-Gal4/+; UAS-128QhttFL(s)[M36E2]/+; Syx229/+) alleviates this phenotype.
(C-D) Quantification of EJP amplitudes and percent failures recorded at 1Hz in 0.25mM Ca2+ in larvae expressing a non-toxic GFP control protein, expressing 128QhttFL, heterozygous mutant for Dmca1D (C), Dmca1A (D), or expressing 128QhttFL as well as heterozygous mutant for either Dmca1D (C) or Dmca1A (D). All experiments at done at 29°C. Error bars: SEM. The number of recordings from at least 3 animals is indicated inside the bars. ns, p>0.05; *, p<0.05; **, p<0.01 throughout the figure. Genotypes: GFP (Elav-GAL4/+; UAS-GFP/+), 16QhttFL (Elav-GAL4/+; UAS-16QhttFL(s)[M28]/+), 128QhttFL (Elav-GAL4/+; UAS-128QhttFL(s)[M36E2]/+), Syx229/+ (Elav-GAL4/+; Syx229/+), 128QhttFL/Syx229 (Elav-Gal4/+; UAS-128QhttFL(s)[M36E2]/+; Syx229/+), Dmca1DX10/+ (Elav-GAL4/+; Dmca1DX10/+), 128QhttFL/Dmca1DX10 (Elav-GAL4/+; UAS-128QhttFL(s)[M36E2]/Dmca1DX10), Dmca1AHC129/+ (Elav-GAL4/Dmca1AHC129), 128QhttFL/Dmca1AHC129 (Elav-GAL4/Dmca1AHC129; UAS-128QhttFL(s)[M36E2]/+).
In order to mediate efficient release, vesicles are positioned closely to sites of Ca2+ influx via interactions between presynaptic release proteins, including Syntaxin, SNAP-25, and the synprint region of voltage-gated Ca2+ channels (Katz and Miledi, 1965; Swayne et al., 2005). Interestingly, huntingtin interacts with subunits of voltage-gated Ca2+ channels present at the synapse and may functionally alter Ca2+ channel function (Swayne et al., 2005). These data suggest that while interactions of expanded huntingtin may interfere with components of the release machinery, they may also disturb Ca2+ homeostasis at the synapses, resulting in increased neurotransmitter release.
To investigate whether the elevated presynaptic Ca2+ levels stem from interactions between expanded huntingtin and the synaptic release machinery, we determined resting Ca2+ levels in larvae overexpressing 128QhttFL but lacking one functional copy of Syx. Compared to animals expressing 128QhttFL alone, which have elevated synaptic Ca2+ levels (Figure 8A-B, Elav-GAL4/+; UAS-128QhttFL(s)[M36E2]/+: F340/F380=0.83 ± 0.42), 128QhttFL animals with only one functional copy of Syx exhibit a reduction in Ca2+ (Figure 8A-B, Elav-GAL4/+; UAS-128QhttFL(s)[M36E2]/+; Syx229/+: F340/F380=0.39 ± 0.07), and show levels similar to controls (Figure 8A-B, Elav-GAL4/+; UAS-GFP/+: F340/F380=0.28 ± 0.04). Since voltage-gated Ca2+ channels are critical purveyors of presynaptic calcium entry and synaptic release, we also investigated whether reduced calcium channel activity also corrects the 128httFL-induced elevation in calcium levels. Two Drosophila voltage-gated Ca2+ channels pore subunits are expressed in neurons (Kawasaki et al., 2004; Zheng et al, 1995). Dmca1D is an L-type voltage-gated Ca2+ channel, while Dmca1A, also known as Cacophony, is a P/Q-type voltage-gated channel, both mediating Ca2+ influx at the presynaptic terminal (Kawasaki et al., 2000; Zheng et al., 1995). Interestingly, when we remove one copy of Dmca1D in 128QhttFL expressing animals, the increased Ca2+ levels return to levels measured in controls (Elav-GAL4/+; UAS-128QhttFL(s)[M36E2]/Dmca1DX10: F340/F380=0.29 ± 0.04). Importantly, this reduction is not due to the loss of either Syx or Dmca1D alone since Elav-GAL4/+; Syx229 /+ and Elav-GAL4/+; Dmca1Dx10/+ have similar resting Ca2+ levels as controls (Figure 8A, Elav-GAL4/+; Syx229/+: F340/F380=0.30 ± 0.05, Elav-GAL4/+; Dmca1DX10/+: F340/F380=0.24 ± 0.05). Since Ca2+ is required upstream of vesicle fusion, this suggests that alterations in neurotransmission at synapses that accumulate 128QhttFL may stem from an elevation in synaptic Ca2+ levels. In addition, interactions between 128QhttFL and the vesicle release machinery may serve to modulate synaptic efficacy by regulating synaptic Ca2+ levels.
Increased transmission, decreased failures and photoreceptor degeneration in 128QhttFL animals are restored by reducing the activity of voltage-gated Ca2+ channels
To test if Ca2+ levels are a cause of increased transmission and decreased failures in 128QhttFL animals, we recorded EJPs at 0.25mM Ca2+ in Elav-128QhttFL flies that lack one copy of Dmca1D or Dmca1A (Figure 8C-D). As shown in Figure 8C-D, although neither Elav-GAL4; Dmca1Dx10/+ or Elav-GAL4, Dmca1AHC129 affect EJP amplitude or failure rate compared to controls (Elav-GFP), both the increased EJP amplitude and the decreased failures observed in Elav-128QhttFL animals are significantly suppressed by removing one copy of either Dmca1D or Dmca1A. Similarly, the photoreceptor degeneration observed in 128QhttFL-expressing flies is suppressed by removing one copy of Dmca1D (Figures 7A, F; p<0.001, Mann-Whitney test). These data further support the hypothesis that altered intracellular Ca2+ levels in neurons that express expanded huntingtin, lead to increased release probability and neuronal degeneration.
Discussion
Here we develop a novel Drosophila model of HD to investigate the mechanisms by which expanded full-length human huntingtin causes neuronal dysfunction and degeneration. We used the GAL4/UAS system to target 128QhttFL expression to specific neuronal subpopulations. Using this system we found that 128QhttFL causes neuronal degeneration both in eye photoreceptors and in the CNS, motor performance impairments, reduced life span and increased neurotransmitter release. Importantly, these abnormalities occur in the absence of detectable huntingtin nuclear import or axonal blockages and are specific to 128QhttFL, as expression of similar levels of wild-type 16QhttFL does not cause abnormal phenotypes.
Expression of 128QhttFL in the eye using GMR-GAL4 leads to progressive photoreceptor neuron degeneration. Histological examination of the internal eye structure in flies of different ages reveals that the number and arrangement of rhabdomeres in photoreceptor cells is relatively normal in 1-day-old flies (Figure 2E), but degeneration is evident at day 20 (Figure 2F). We also found that targeted expression of 128QhttFL to motor neurons leads to motor impairment phenotypes. The 128QhttFL animals perform as controls in a climbing assay when they are young, but their motor performance declines prematurely as they age. Moreover, flying ability is impaired in aged 128QhttFL flies, and they also show progressive loss of NMJs at the IFM. In addition, these flies show a reduced survival rate when compared to controls (Figures 3A-E).
These neurodegenerative phenotypes are not likely a consequence of transcriptional dysregulation as they occur in the absence of obvious nuclear huntingtin even in aged flies. In full-length mouse models, expanded huntingtin is first diffuse in the cytoplasm, but these animals gradually develop nuclear staining later in life (Menalled, 2005). The accumulation of huntingtin in the nucleus requires proteolytic cleavage of the protein, a process in which capase-6 plays a key role (Graham et al., 2006). In agreement with the absence of nuclear staining, we do not detect cleavage in 128QhttFL flies. Therefore, Drosophila might not be able to process human huntingtin, providing a rationale for the lack of detectable nuclear huntingtin accumulation. However, this possibility is unlikely because the Drosophila genome contains orthologues of caspase-6 as well as other proteins implicated in huntingtin cleavage (http://flybase.bio.indiana.edu/genes/). We therefore favor the alternative explanation that cleavage and nuclear import of huntingtin requires more time than the relatively short life span of Drosophila.
We also investigated the possibility that axonal blockages trigger the phenotypes observed in 128QhttFL flies. Axonal blockages and impaired fast axonal transport have been reported following expression of polyglutamine tracts alone or in the context of other polypeptides (Gunawardena et al., 2003) including expanded huntingtin (Lee et al., 2004; Li et al., 2001; Szebenyi et al., 2003; Gauthier et al., 2004). However, we did not detect huntingtin or synaptotagmin accumulation in the axons of 128QhttFL flies even though we could reproduce the observation of axonal blockages reported with an expanded huntingtin fragment (Figure 1C-D). These data suggest that, like nuclear import, the formation of axonal aggregates requires huntingtin cleavage. Despite the absence of huntingtin and synaptotagmin visible aggregates, the possibility that intracellular transport is decreased cannot be excluded (Szebenyi et al., 2003; Gauthier et.al., 2004). However, we did not observe mislocalization or aberrant distribution of known synaptic markers that rely on vesicular transport for their proper synaptic localization (Figure 4B).
All together, these data suggest that the presynaptic accumulation of 128QhttFL impairs the function of factors involved in neurotransmitter release. This hypothesis is in agreement with abundant data describing protein interactions between huntingtin and components of the synaptic machinery (Smith et al., 2005), and with findings in R6/1 and R6/2 mouse models that suggested a role for altered neurotransmitter release as a potential mechanism of HD pathogenesis. In R6/2 mice, synapsin phosphorylation is partially defective (Lievens et al., 2002), and in R6/1 mice glutamate levels are reduced and aspartate and GABA are increased (Nicniocaill et al., 2001). Moreover, increased NMDA receptor activity has been reported in full-length HD mice (Cepeda et al., 2001; Zeron et al., 2002) leading to a postsynaptic increase in Ca2+ influx and abnormal synaptic transmission. In addition, Ca2+ levels were found to be increased by almost two-fold in CA1 pyramidal neurons in full-length HD mice (Hodgson et al., 1999). However, no defects were observed in paired pulse facilitation, questioning the biological relevance of this finding. In addition, mutant huntingtin has been implicated in aberrant mitochondrial Ca2+ buffering (Panov et al., 2005), and it also increases the sensitivity of the inositol 1,4,5-triphosphate (IP3) receptor to IP3, causing enhanced Ca2+ release following mGluR1/5 activation (Tang et al., 2003). These data suggest that cytosolic Ca2+ levels play a role in HD pathogenesis (Bezprozvanny and Hayden, 2004).
To test the hypothesis that expanded huntingtin impairs the normal function of proteins involved in synaptic transmission we used a genetic approach using the 128QhttFL animals. We found that partial loss of function of Snap, Syntaxin or Rop restore the increased EJP amplitude observed in 128QhttFL larvae to near-normal levels (Figure 6A, C and E). Moreover, the lack of neurotransmitter release failures is also suppressed by these mutations (Figure 6B, D and F). These observations suggest that neurodegeneration in 128QhttFL flies is caused by increased synaptic transmission. In agreement with this hypothesis we found a progressive neurodegenerative phenotype in the NMJ of adult 128QhttFL animals (Figure 3 D and E). Most importantly, further support for this hypothesis comes from the observation that the same synaptic transmission mutants that restore the EJP amplitude and release failure abnormalities,also suppress motor impairment and/ or photoreceptor degeneration in 128QhttFL adult animals (Figure 7 and Supplemental Figure).
Interestingly, Ca2+ levels have a bimodal distribution in 128QhttFL flies with some boutons showing high Ca2+ levels and other boutons within the same neuromuscular junction showing levels in the normal range (Figure 8A, note also that 16QhttFL flies show a similar effect). This distribution can be correlated with the accumulation pattern of huntingtin, which is present in some boutons and absent in others within a given neuromuscular junction (Figure 4A). We tested the hypothesis that Ca2+ levels are relevant for the increased transmission and decreased failures observed in 128QhttFL animals using mutations in voltage-gated Ca2+ channels. We found that Ca2+ levels are restored within normal range in 128QhttFL flies carrying heterozygous mutations in either Syx or the Dmca1D Ca2+ channel (Figure 8A, B). Furthermore, heterozygous mutants for either the Dmca1A or Dmca1D Ca2+ channels also show suppression of the increased transmission and decreased failure phenotypes (Figure 8 C, D). We also tested Dmca1D in the context of the eye assay, and found that its partial loss of function suppresses photoreceptor degeneration (Figures 7A and F). These data support the hypothesis that increased Ca2+ levels play an important role in the observed increased transmission in neurons of 128QhttFL animals. Interestingly, mutations in K+ channels cause neurodegeneration in flies (Fergestad et al., 2006), and in humans (Waters et al., 2006) further supporting the idea that the increased release is responsible, at least in part, for neuronal degeneration caused by expanded huntingtin.
The findings described in this report unveil a mechanism of pathogenesis for expanded huntingtin that does not require its cleavage and nuclear accumulation in detectable amounts. The increased synaptic transmission phenotype exerted by full-length huntingtin likely represents a mechanism of pathogenesis taking place at early stages of disease progression. In later stages, cleavage of huntingtin would compound the toxic effects of the full-length protein with fast axonal transport impairments and transcriptional dysregulation caused by N-terminal fragments. These findings point to increased synaptic transmission as a therapeutic target with the potential of delaying HD onset, and thus likely impacting disease progression. The genetic data showing suppression of the synaptic transmission and neurodegenerative phenotypes further define specific therapeutic targets and support the idea that Ca2+ channel antagonists, and perhaps other inhibitors of neurotransmission, offer an attractive therapeutic option because of their specificity and wide usage.
Experimental Procedures
Drosophila Genetics and Generation of Huntingtin Constructs
Drosophila were maintained on standard medium at 25°C unless otherwise specified for a particular experiment. A full-length 16QhttFL cDNA was generously donated by Dr. Tagle (National Human Genome Research Institute, NIH), and 128QhttFL was generated by replacing an N-terminal fragment of 16QhttFL with the corresponding 128Q N-terminal construct previously generated in the lab (Kaltenbach et al., 2007). The constructs were subsequently cloned into the pUAST transformation vector (Brand and Perrimon, 1993) and injected in a y1w118 strain by standard procedures. Four 128QhttFL and thre16QhttFL lines were generated.
GMR and Elav GAL4 drivers were obtained from the Bloomington Stock Center, GMR(s)-GAL4 from Dr. P. Jin (Emory Univ. School Med.), C164-GAL4 from Dr. V. Budnik, (Univ. Massachusetts, Amherst), and apVNC–GAL4 was generated in our lab (Fernandez-Funez et al., 2000). Snap mutants were obtained from Dr. L.J Pallanck (Univ. Washington, Seattle), and Rop, Syx, vha mutants were generated in the Bellen lab. Dmca1DX10 mutants were obtained from Dr. L. Hall (Univ. California, Davis) and Dmca1AHC129 mutants were obtained from Dr. R. Ordway (Penn State Univ.)
Western Blot Analysis
To test the expression level of huntingtin in transgenic flies, pUAST-htt lines were crossed to flies carrying the GMR-Gal4 driver at 27°C. 8 females of each individual genotype were aged 1 day before isolating heads and homogenizing in sample buffer containing 8M urea followed by boiling. The proteins were separated on 4−20% gradient SDS/PAGE gels (BIORAD). After electrophoresis, gels were electroblotted overnight onto nitrocellulose membranes (Laemmli, 1970). The membranes were immunoblotted with mouse monoclonal anti-htt antibody (MAB2166, Chemicon) at 1:2000, and immunoreactive bands were visualized using ECLTM Western blotting detection reagents from Amersham. To discriminate between expanded and unexpanded htt, the same procedure was used using mouse anti-polyQ MAB1574 (Chemicon). Densitometry analysis was done using a Molecular Dynamics Personal Densitometer SI and analyzed with Image Quant 5.2 software.
Immunohistochemistry and histology
Immunohistochemistry on third-instar larva NMJs and brains, or adult brains was performed as described (Fernandez-Funez et al., 2000; Verstreken et al., 2003) with the following modifications. Larvae and adult neuronal systems were fixed in 4% formaldehyde for 1 hr and washed in 1XPBS with 0.1% Triton X-100. For GluRIIA labeling, larvae were fixed in 50% picric acid, 2.5% formaldehyde in 1XPBS for 20 min and washed in 1XPBS with 0.1% Triton X-100. After blocking with 2% BSA, samples were incubated with primary antibodies overnight. Primary antibodies were used at the following concentrations: htt MAB5374 (1:200, Chemicon), HRP (1:500), GFP (1:200), Syt (1:200), Dlg (1:200), GluIIRA (1:100), Rop (1:100), SNAP (1:100) and Syntaxin (1:200). After washing steps, secondary antibodies conjugated to Cy3 or Alexa 488 (Jackson Immunolabs, Molecular Probes) were used at 1:200.
Phalloidin staining to monitor rhabdomere degeneration in adult fly visual system was performed as described (Sang and Ready, 2002). Adult eye retinas were removed from the head capsule in 4% formaldehyde and fixed for 1 hour. Fixed, dissected eyes were washed in PBS, 0.1% Triton X-100 and incubated in phalloidin (1: 200, Molecular Probes) and washed again.
For visualizing the neuronal projections in to IFMs, fixed adult thoraxes from 25-day-old adult flies were dissected along the midline and neurons were labeled with anti-HRP antibody. The number of neuronal projections were counted in confocal stacks of a specified 100μm × 100μm area of IFMs 3 and 4 (located 100μm away from the cuticle, shown in Figure 3D) using the whole mount preparation of each hemithorax.
All immunostained samples were visualized using a Zeiss 510 confocal microscope and ImageJ Software was used to obtain a 2D image by stacking the optically sectioned images. These stacked images were used for further analysis. Image panels were processed using Photoshop (Adobe).
For light microscopy images, whole flies were frozen at −20°C for at least 24 hours and pictures were taken with a Leica MZ16 stereomicroscope using a MagnaFire™ SP Digital Camera and images were collected with Image-Pro Plus 4.5 Analytical Imaging Software. For SEM images, whole flies were dehydrated in ethanol, critical-point dried and analyzed with a JEOL JSM 6100 election microscope.
Adult Behavioral Assays
Climbing assays were performed on adult virgin female flies incubated as described (Ganetzky and Flanagan, 1978). ∼30 flies were placed in a plastic vial and gently tapped to the bottom of the vial. The number of flies above the 5cm line after 18 seconds were counted and recorded. Ten trials were performed at each time point. The results shown represent the performance of one batch of flies tested over 30 days.
Fliying tests were performed on 25-day-old female virgins incubated. Flying ability was estimated by measuring flying distances after they were dropped from the top of a transparent plastic cylinder as described (Pesah et al., 2004).
Larval Electrophysiological Assays
Third instar physiology was performed as described (Verstreken et al., 2003). L3 NMJ recordings were performed in modified HL3 (in mM): NaCl (110), KCl (5), NaHCO3 (10), HEPES (5), sucrose (30), trehalose (5), CaCl2 (5), and MgCl2 (20) (pH 7.2). mEJPs were recorded in modified HL-3 with 5μM TTX and 0.5mM Ca2+. Current clamp recordings were made from muscles 6 or 7 using high resistance electrodes (100 MW) filled with 2 M KAc and 0.2 M KCl. EJPs were evoked at 2−3× threshold using a suction electrode. Data was digitized and stored on a PC with pClamp and analyzed with Clampfit (Axon) or Mini analysis (Synaptosoft). EJP amplitudes in low Ca2+ were determined using only the successful events.
Larval Ca2+ Imaging
After dissecting larvae in Schneider's medium (Gibco), a drop (∼20μL) of 5mM Fura-2 Dextran (Invitrogen) was placed on the preparation and a small amount of dye as well as a cut motor axon taken into a suction electrode. Excess dye was removed and the preparation washed and incubated in Schneider's medium for ∼40min. Following dye loading, Schneider's medium was replaced with modified HL3 (NaCl (110mM), KCl (5mM), NaHCO3 (10mM), HEPES (5mM), sucrose (30mM), trehalose (5mM), and MgCl2 (20mM), pH 7.2) with 1mM Ca2+. Images were obtained after allowing the preparation to equilibrate with HL3 for ∼15min. Excitation at 340nm and 380nm was aided by a filter wheel (Sutter Lambda 10−2) and fluorescence images collected using a 40× water immersion objective (Zeiss Achroplan) and Zeiss AxioCam MRm CCD camera. Images were processed with Amira 2.2.
Acknowledgements
Thanks to R. Atkinson, the Baylor MRDDRC core (HD24062) and J. Barish for assistance with the laser confocal- and scanning electron- microscopes, A. Singh and the Joiner lab for help with the plastic resin sections and H. Y. Zoghbi for comments to the manuscript. We are grateful to the Developmental Studies Hybridoma Bank for antibodies, the Bloomington Drosophila Stock Center, P. Jin, V. Budnik, Leo Pallanck, Linda Hall, and Richard Ordway for mutant strains and to D. Tagle for human huntingtin cDNA. We thank Amir Fayyazuddin for sharing expertise pertaining to the adult NMJ. This work was supported by NINDS grant NS42179 to JB. CVL is supported by an NRSA fellowship from the NINDS (F30NS056520). G-HC was supported by an HDSA fellowship (GCha-05/06-F1) and a postdoctoral fellowship from KOSEF. PV was supported by a R.L. Kirchstein NRS award, and HJB is a HHMI investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
Supplemental Figure. Rescue of flying impairments in 128QhttFL flies by one mutant copy of Syntaxin 1A
Flying ability of 25-day-old flies was measured as in Figure 3C. Note that flying ability is significantly different in 128QhttFL flies and 128QhttFL flies also carrying a heterozygous loss of function mutation in Syx (p<0.05, Mann-Whitney test).
Genotypes: C164 (C164-GAL4/+), C164-GFP (C164-GAL4/UAS-GFP), C164−16QhttFL (C164-GAL4/UAS-16QhttFL[M28]), C164−128QhttFL (C164-GAL4/UAS-128QhttFL(s)[M36E2]), C164−128QhttFL/Syx229 (C164-GAL4/UAS-128QhttFL(s)[M36E2]; Syx229/+).
References
- The Huntington's Disease Collaborative Research Group A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM. Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson's disease. Science. 2002;295:865–868. doi: 10.1126/science.1067389. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Hayden MR. Deranged neuronal calcium signaling and Huntington disease. Biochem Biophys Res Commun. 2004;322:1310–1317. doi: 10.1016/j.bbrc.2004.08.035. [DOI] [PubMed] [Google Scholar]
- Bilen J, Bonini NM. Drosophila as a model for human neurodegenerative disease. Annu Rev Genet. 2005;39:153–171. doi: 10.1146/annurev.genet.39.110304.095804. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Cattaneo E, Zuccato C, Tartari M. Normal huntingtin function: an alternative approach to Huntington's disease. Nat Rev Neurosci. 2005;6:919–930. doi: 10.1038/nrn1806. [DOI] [PubMed] [Google Scholar]
- Cauchi RJ, van den Heuvel M. The fly as a model for neurodegenerative diseases: is it worth the jump? Neurodegen Dis. 2006;3:338–356. doi: 10.1159/000097303. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Ariano MA, Calvert CR, Flores-Hernandez J, Chandler SH, Leavitt BR, Hayden MR, Levine MS. NMDA receptor function in mouse models of Huntington disease. J Neurosci Res. 2001;66:525–539. doi: 10.1002/jnr.1244. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Hurst RS, Calvert CR, Hernandez-Echeagaray E, Nguyen OK, Jocoy E, Christian LJ, Ariano MA, Levine MS. Transient and progressive electrophysiological alterations in the corticostriatal pathway in a mouse model of Huntington's disease. J Neurosci. 2003;23:961–969. doi: 10.1523/JNEUROSCI.23-03-00961.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase K, Schwarz C, Meloni A, Young C, Martin E, Vonsattel JP, Carraway R, Reeves SA, et al. Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron. 1995;14:1075–1081. doi: 10.1016/0896-6273(95)90346-1. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- Emamian ES, Kaytor MD, Duvick LA, Zu T, Tousey SK, Zoghbi HY, Clark HB, Orr HT. Serine 776 of ataxin-1 is critical for polyglutamine-induced disease in SCA1 transgenic mice. Neuron. 2003;38:375–387. doi: 10.1016/s0896-6273(03)00258-7. [DOI] [PubMed] [Google Scholar]
- Fergestad T, Ganetzky B, Palladino MJ. Neuropathology in Drosophila membrane excitability mutants. Genetics. 2006;172:1031–1042. doi: 10.1534/genetics.105.050625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Funez P, Nino-Rosales ML, de Gouyon B, She WC, Luchak JM, Martinez P, Turiegano E, Benito J, Capovilla M, Skinner PJ, et al. Identification of genes that modify ataxin-1-induced neurodegeneration. Nature. 2000;408:101–106. doi: 10.1038/35040584. [DOI] [PubMed] [Google Scholar]
- Ganetzky B, Flanagan JR. On the relationship between senescence and age-related changes in two wild-type strains of Drosophila melanogaster. Exp Gerontol. 1978;13:189–196. doi: 10.1016/0531-5565(78)90012-8. [DOI] [PubMed] [Google Scholar]
- Gauthier LR, Charrin BC, Borrell-Pagès M, Dompierre JP, Rangone H, Cordelières FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, Saudou F. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004 Jul 9;118(1):127–38. doi: 10.1016/j.cell.2004.06.018. 2004. [DOI] [PubMed] [Google Scholar]
- Goehler H, Lalowski M, Stelzl U, Waelter S, Stroedicke M, Worm U, Droege A, Lindenberg KS, Knoblich M, Haenig C, et al. A protein interaction network links GIT1, an enhancer of huntingtin aggregation, to Huntington's disease. Mol Cell. 2004;15:853–865. doi: 10.1016/j.molcel.2004.09.016. [DOI] [PubMed] [Google Scholar]
- Graham RK, Deng Y, Slow EJ, Haigh B, Bissada N, Lu G, Pearson J, Shehadeh J, Bertram L, Murphy Z, et al. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell. 2006;125:1179–1191. doi: 10.1016/j.cell.2006.04.026. [DOI] [PubMed] [Google Scholar]
- Gunawardena S, Goldstein LS. Polyglutamine diseases and transport problems: deadly traffic jams on neuronal highways. Arch Neurol. 2005;62:46–51. doi: 10.1001/archneur.62.1.46. [DOI] [PubMed] [Google Scholar]
- Gunawardena S, Her LS, Brusch RG, Laymon RA, Niesman IR, Gordesky-Gold B, Sintasath L, Bonini NM, Goldstein LS. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron. 2003;40:25–40. doi: 10.1016/s0896-6273(03)00594-4. [DOI] [PubMed] [Google Scholar]
- Harjes P, Wanker EE. The hunt for huntingtin function: interaction partners tell many different stories. Trends Biochem Sci. 2003;28:425–433. doi: 10.1016/S0968-0004(03)00168-3. [DOI] [PubMed] [Google Scholar]
- Hebbar S, Fernandes JJ. Pruning of motor neuron branches establishes the DLM innervation pattern in Drosophila. J Neurobiol. 2004;60:499–516. doi: 10.1002/neu.20031. [DOI] [PubMed] [Google Scholar]
- Hickey MA, Chesselet MF. The use of transgenic and knock-in mice to study Huntington's disease. Cytogenet Genome Res. 2003;100:276–286. doi: 10.1159/000072863. [DOI] [PubMed] [Google Scholar]
- Hiesinger PR, Fayyazuddin A, Mehta SQ, Rosenmund T, Schulze KL, Zhai RG, Verstreken P, Cao Y, Zhou Y, Kunz J, Bellen HJ. The v-ATPase V0 subunit a1 is required for a late step in synaptic vesicle exocytosis in Drosophila. Cell. 2005;121:607–620. doi: 10.1016/j.cell.2005.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgson JG, Agopyan N, Gutekunst CA, Leavitt BR, LePiane F, Singaraja R, Smith DJ, Bissada N, McCutcheon K, Nasir J, et al. A YAC mouse model for Huntington's disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. 1999;23:181–192. doi: 10.1016/s0896-6273(00)80764-3. [DOI] [PubMed] [Google Scholar]
- Jackson GR, Salecker I, Dong X, Yao X, Arnheim N, Faber PW, MacDonald ME, Zipursky SL. Polyglutamine-expanded human huntingtin transgenes induce degeneration of Drosophila photoreceptor neurons. Neuron. 1998;21:633–642. doi: 10.1016/s0896-6273(00)80573-5. [DOI] [PubMed] [Google Scholar]
- Kaltenbach LS, Romero E, Becklin RR, Chettier R, Bell R, Phansalkar A, Strand A, Torcassi C, Savage J, Hurlburt A, et al. Huntingtin Interacting Proteins Are Genetic Modifiers of Neurodegeneration. PLoS Genet. 2007;3:e82. doi: 10.1371/journal.pgen.0030082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim FD, Chang HC, Therrien M, Wassarman DA, Laverty T, Rubin GM. A screen for genes that function downstream of Ras1 during Drosophila eye development. Genetics. 1996;143:315–329. doi: 10.1093/genetics/143.1.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz B, Miledi R. The Effect of Calcium on Acetylcholine Release from Motor Nerve Terminals. Proc R Soc Lond B Biol Sci. 1965;161:496–503. doi: 10.1098/rspb.1965.0017. [DOI] [PubMed] [Google Scholar]
- Kawasaki F, Felling R, Ordway RW. A temperature-sensitive paralytic mutant defines a primary synaptic calcium channel in Drosophila. J Neurosci. 2000;20:4885–4889. doi: 10.1523/JNEUROSCI.20-13-04885.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki F, Zou B, Xu X, Ordway RW. Active zone localization of presynaptic calcium channels encoded by the cacophony locus of Drosophila. J Neurosci. 2004;24:282–285. doi: 10.1523/JNEUROSCI.3553-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazemi-Esfarjani P, Benzer S. Genetic suppression of polyglutamine toxicity in Drosophila. Science. 2000;287:1837–1840. doi: 10.1126/science.287.5459.1837. [DOI] [PubMed] [Google Scholar]
- Klapstein GJ, Fisher RS, Zanjani H, Cepeda C, Jokel ES, Chesselet MF, Levine MS. Electrophysiological and morphological changes in striatal spiny neurons in R6/2 Huntington's disease transgenic mice. J Neurophysiol. 2001;86:2667–2677. doi: 10.1152/jn.2001.86.6.2667. [DOI] [PubMed] [Google Scholar]
- Koh TW, Verstreken P, Bellen HJ. Dap160/intersectin acts as a stabilizing scaffold required for synaptic development and vesicle endocytosis. Neuron. 2004;43:193–205. doi: 10.1016/j.neuron.2004.06.029. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Laforet GA, Sapp E, Chase K, McIntyre C, Boyce FM, Campbell M, Cadigan BA, Warzecki L, Tagle DA, Reddy PH, et al. Changes in cortical and striatal neurons predict behavioral and electrophysiological abnormalities in a transgenic murine model of Huntington's disease. J Neurosci. 2001;21:9112–9123. doi: 10.1523/JNEUROSCI.21-23-09112.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bourg E, Lints FA. Hypergravity and aging in Drosophila melanogaster. 4. Climbing activity. Gerontology. 1992;38:59–64. doi: 10.1159/000213307. [DOI] [PubMed] [Google Scholar]
- Lee WC, Yoshihara M, Littleton JT. Cytoplasmic aggregates trap polyglutamine-containing proteins and block axonal transport in a Drosophila model of Huntington's disease. Proc Natl Acad Sci U S A. 2004;101:3224–3229. doi: 10.1073/pnas.0400243101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Li SH, Yu ZX, Shelbourne P, Li XJ. Huntingtin aggregate-associated axonal degeneration is an early pathological event in Huntington's disease mice. J Neurosci. 2001;21:8473–8481. doi: 10.1523/JNEUROSCI.21-21-08473.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JY, Plomann M, Brundin P. Huntington's disease: a synaptopathy? Trends Mol Med. 2003;9:414–420. doi: 10.1016/j.molmed.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Li SH, Li XJ. Huntingtin and its role in neuronal degeneration. Neuroscientist. 2004a;10:467–475. doi: 10.1177/1073858404266777. [DOI] [PubMed] [Google Scholar]
- Li SH, Li XJ. Huntingtin-protein interactions and the pathogenesis of Huntington's disease. Trends Genet. 2004b;20:146–154. doi: 10.1016/j.tig.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Lievens JC, Woodman B, Mahal A, Bates GP. Abnormal phosphorylation of synapsin I predicts a neuronal transmission impairment in the R6/2 Huntington's disease transgenic mice. Mol Cell Neurosci. 2002;20:638–648. doi: 10.1006/mcne.2002.1152. [DOI] [PubMed] [Google Scholar]
- Marsh JL, Thompson LM. Can flies help humans treat neurodegenerative diseases? Bioessays. 2004;26:485–496. doi: 10.1002/bies.20029. [DOI] [PubMed] [Google Scholar]
- Marsh JL, Thompson LM. Drosophila in the study of neurodegenerative disease. Neuron. 2006;52:169–178. doi: 10.1016/j.neuron.2006.09.025. [DOI] [PubMed] [Google Scholar]
- Menalled LB. Knock-in mouse models of Huntington's disease. NeuroRx. 2005;2:465–470. doi: 10.1602/neurorx.2.3.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menalled LB, Chesselet MF. Mouse models of Huntington's disease. Trends Pharmacol Sci. 2002;23:32–39. doi: 10.1016/s0165-6147(00)01884-8. [DOI] [PubMed] [Google Scholar]
- Menalled LB, Sison JD, Wu Y, Olivieri M, Li XJ, Li H, Zeitlin S, Chesselet MF. Early motor dysfunction and striosomal distribution of huntingtin microaggregates in Huntington's disease knock-in mice. J Neurosci. 2002;22:8266–8276. doi: 10.1523/JNEUROSCI.22-18-08266.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy KP, Carter RJ, Lione LA, Mangiarini L, Mahal A, Bates GP, Dunnett SB, Morton AJ. Abnormal synaptic plasticity and impaired spatial cognition in mice transgenic for exon 1 of the human Huntington's disease mutation. J Neurosci. 2000;20:5115–5123. doi: 10.1523/JNEUROSCI.20-13-05115.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasir J, Floresco SB, O'Kusky JR, Diewert VM, Richman JM, Zeisler J, Borowski A, Marth JD, Phillips AG, Hayden MR. Targeted disruption of the Huntington's disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 1995;81:811–823. doi: 10.1016/0092-8674(95)90542-1. [DOI] [PubMed] [Google Scholar]
- Nicniocaill B, Haraldsson B, Hansson O, O'Connor WT, Brundin P. Altered striatal amino acid neurotransmitter release monitored using microdialysis in R6/1 Huntington transgenic mice. Eur J Neurosci. 2001;13:206–210. doi: 10.1046/j.0953-816x.2000.01379.x. [DOI] [PubMed] [Google Scholar]
- Ordway RW, Pallanck L, Ganetzky B. Neurally expressed Drosophila genes encoding homologs of the NSF and SNAP secretory proteins. Proc Natl Acad Sci U S A. 1994;91:5715–5719. doi: 10.1073/pnas.91.12.5715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panov AV, Lund S, Greenamyre JT. Ca2+-induced permeability transition in human lymphoblastoid cell mitochondria from normal and Huntington's disease individuals. Mol Cell Biochem. 2005;269:143–152. doi: 10.1007/s11010-005-3454-9. [DOI] [PubMed] [Google Scholar]
- Pennetta G, Hiesinger PR, Fabian-Fine R, Meinertzhagen IA, Bellen HJ. Drosophila VAP-33A directs bouton formation at neuromuscular junctions in a dosage-dependent manner. Neuron. 2002;35:291–306. doi: 10.1016/s0896-6273(02)00769-9. [DOI] [PubMed] [Google Scholar]
- Pesah Y, Pham T, Burgess H, Middlebrooks B, Verstreken P, Zhou Y, Harding M, Bellen H, Mardon G. Drosophila parkin mutants have decreased mass and cell size and increased sensitivity to oxygen radical stress. Development. 2004;131:2183–2194. doi: 10.1242/dev.01095. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC. Lessons from animal models of Huntington's disease. Trends Genet. 2002;18:202–209. doi: 10.1016/s0168-9525(01)02625-7. [DOI] [PubMed] [Google Scholar]
- Sang TK, Jackson GR. Drosophila models of neurodegenerative disease. NeuroRx. 2005;2:438–446. doi: 10.1602/neurorx.2.3.438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang TK, Ready DF. Eyes closed, a Drosophila p47 homolog, is essential for photoreceptor morphogenesis. Development. 2002;129:143–154. doi: 10.1242/dev.129.1.143. [DOI] [PubMed] [Google Scholar]
- Schulze KL, Broadie K, Perin MS, Bellen HJ. Genetic and electrophysiological studies of Drosophila syntaxin-1A demonstrate its role in nonneuronal secretion and neurotransmission. Cell. 1995;80:311–320. doi: 10.1016/0092-8674(95)90414-x. [DOI] [PubMed] [Google Scholar]
- Schulze KL, Littleton JT, Salzberg A, Halachmi N, Stern M, Lev Z, Bellen HJ. rop, a Drosophila homolog of yeast Sec1 and vertebrate n-Sec1/Munc-18 proteins, is a negative regulator of neurotransmitter release in vivo. Neuron. 1994;13:1099–1108. doi: 10.1016/0896-6273(94)90048-5. [DOI] [PubMed] [Google Scholar]
- Schuster CM, Ultsch A, Schloss P, Cox JA, Schmitt B, Betz H. Molecular cloning of an invertebrate glutamate receptor subunit expressed in Drosophila muscle. Science. 1991;254:112–114. doi: 10.1126/science.1681587. [DOI] [PubMed] [Google Scholar]
- Shulman JM, Feany MB. Genetic modifiers of tauopathy in Drosophila. Genetics. 2003;165:1233–1242. doi: 10.1093/genetics/165.3.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slow EJ, Graham RK, Osmand AP, Devon RS, Lu G, Deng Y, Pearson J, Vaid K, Bissada N, Wetzel R, et al. Absence of behavioral abnormalities and neurodegeneration in vivo despite widespread neuronal huntingtin inclusions. Proc Natl Acad Sci U S A. 2005;102:11402–11407. doi: 10.1073/pnas.0503634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slow EJ, van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y, Oh R, Bissada N, Hossain SM, Yang YZ, et al. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. 2003;12:1555–1567. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- Smith R, Brundin P, Li JY. Synaptic dysfunction in Huntington's disease: a new perspective. Cell Mol Life Sci. 2005;62:1901–1912. doi: 10.1007/s00018-005-5084-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan JS, Agrawal N, Pallos J, Rockabrand E, Trotman LC, Slepko N, Illes K, Lukacsovich T, Zhu YZ, Cattaneo E, et al. SUMO modification of Huntingtin and Huntington's disease pathology. Science. 2004;304:100–104. doi: 10.1126/science.1092194. [DOI] [PubMed] [Google Scholar]
- Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–743. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- Strompen G, Dettmer J, Stierhof YD, Schumacher K, Jurgens G, Mayer U. Arabidopsis vacuolar H-ATPase subunit E isoform 1 is required for Golgi organization and vacuole function in embryogenesis. Plant J. 2005;41:125–132. doi: 10.1111/j.1365-313X.2004.02283.x. [DOI] [PubMed] [Google Scholar]
- Sugars KL, Rubinsztein DC. Transcriptional abnormalities in Huntington disease. Trends Genet. 2003;19:233–238. doi: 10.1016/S0168-9525(03)00074-X. [DOI] [PubMed] [Google Scholar]
- Swayne LA, Chen L, Hameed S, Barr W, Charlesworth E, Colicos MA, Zamponi GW, Braun JE. Crosstalk between huntingtin and syntaxin 1A regulates N-type calcium channels. Mol Cell Neurosci. 2005;30:339–351. doi: 10.1016/j.mcn.2005.07.016. [DOI] [PubMed] [Google Scholar]
- Szebenyi G, Morfini GA, Babcock A, Gould M, Selkoe K, Stenoien DL, Young M, Faber PW, MacDonald ME, McPhaul MJ, Brady ST. Neuropathogenic forms of huntingtin and androgen receptor inhibit fast axonal transport. Neuron. 2003;40:41–52. doi: 10.1016/s0896-6273(03)00569-5. [DOI] [PubMed] [Google Scholar]
- Tang TS, Slow E, Lupu V, Stavrovskaya IG, Sugimori M, Llinas R, Kristal BS, Hayden MR, Bezprozvanny I. Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington's disease. Proc Natl Acad Sci U S A. 2005;102:2602–2607. doi: 10.1073/pnas.0409402102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang TS, Tu H, Chan EY, Maximov A, Wang Z, Wellington CL, Hayden MR, Bezprozvanny I. Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron. 2003;39:227–239. doi: 10.1016/s0896-6273(03)00366-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usdin MT, Shelbourne PF, Myers RM, Madison DV. Impaired synaptic plasticity in mice carrying the Huntington's disease mutation. Hum Mol Genet. 1999;8:839–846. doi: 10.1093/hmg/8.5.839. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk JM, Murphy Z, Slow EJ, Leavitt BR, Hayden MR. Selective degeneration and nuclear localization of mutant huntingtin in the YAC128 mouse model of Huntington disease. Hum Mol Genet. 2005;14:3823–3835. doi: 10.1093/hmg/ddi407. [DOI] [PubMed] [Google Scholar]
- Velier J, Kim M, Schwarz C, Kim TW, Sapp E, Chase K, Aronin N, DiFiglia M. Wild-type and mutant huntingtins function in vesicle trafficking in the secretory and endocytic pathways. Exp Neurol. 1998;152:34–40. doi: 10.1006/exnr.1998.6832. [DOI] [PubMed] [Google Scholar]
- Verstreken P, Koh TW, Schulze KL, Zhai RG, Hiesinger PR, Zhou Y, Mehta SQ, Cao Y, Roos J, Bellen HJ. Synaptojanin is recruited by endophilin to promote synaptic vesicle uncoating. Neuron. 2003;40:733–748. doi: 10.1016/s0896-6273(03)00644-5. [DOI] [PubMed] [Google Scholar]
- Warrick JM, Chan HY, Gray-Board GL, Chai Y, Paulson HL, Bonini NM. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat Genet. 1999;23:425–428. doi: 10.1038/70532. [DOI] [PubMed] [Google Scholar]
- Waters MF, Minassian NA, Stevanin G, Figueroa KP, Bannister JP, Nolte D, Mock AF, Evidente VG, Fee DB, Müller U, Dürr A, Brice A, Papazian DM, Pulst SM. Mutations in voltage-gated potassium channel KCNC3 cause degenerative and developmental central nervous system phenotypes. Nat Genet. 2006;38:447–451. doi: 10.1038/ng1758. [DOI] [PubMed] [Google Scholar]
- Wu MN, Littleton JT, Bhat MA, Prokop A, Bellen HJ. ROP, the Drosophila Sec1 homolog, interacts with syntaxin and regulates neurotransmitter release in a dosage-dependent manner. Embo J. 1998;17:127–139. doi: 10.1093/emboj/17.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu ZX, Li SH, Evans J, Pillarisetti A, Li H, Li XJ. Mutant huntingtin causes context-dependent neurodegeneration in mice with Huntington's disease. J Neurosci. 2003;23:2193–2202. doi: 10.1523/JNEUROSCI.23-06-02193.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeron MM, Hansson O, Chen N, Wellington CL, Leavitt BR, Brundin P, Hayden MR, Raymond LA. Increased sensitivity to N-methyl-D-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington's disease. Neuron. 2002;33:849–860. doi: 10.1016/s0896-6273(02)00615-3. [DOI] [PubMed] [Google Scholar]
- Zheng W, Feng G, Ren D, Eberl DF, Hannan F, Dubald M, Hall LM. Cloning and characterization of a calcium channel alpha 1 subunit from Drosophila melanogaster with similarity to the rat brain type D isoform. J Neurosci. 1995;15:1132–1143. doi: 10.1523/JNEUROSCI.15-02-01132.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure. Rescue of flying impairments in 128QhttFL flies by one mutant copy of Syntaxin 1A
Flying ability of 25-day-old flies was measured as in Figure 3C. Note that flying ability is significantly different in 128QhttFL flies and 128QhttFL flies also carrying a heterozygous loss of function mutation in Syx (p<0.05, Mann-Whitney test).
Genotypes: C164 (C164-GAL4/+), C164-GFP (C164-GAL4/UAS-GFP), C164−16QhttFL (C164-GAL4/UAS-16QhttFL[M28]), C164−128QhttFL (C164-GAL4/UAS-128QhttFL(s)[M36E2]), C164−128QhttFL/Syx229 (C164-GAL4/UAS-128QhttFL(s)[M36E2]; Syx229/+).