

Abstract
Pancreatic bi-hormones insulin and glucagon are the Yin and Yang in the regulation of glucose metabolism and homoeostasis. Insulin is synthesized primarily by pancreatic β-cells and is released in response to an increase in blood glucose levels (hyperglycaemia). By contrast, glucagon is synthesized by pancreatic α-cells and is released in response to a decrease in blood glucose (hypoglycaemia). The principal role of glucagon is to counter the actions of insulin on blood glucose homoeostasis, but it also has diverse non-hyperglycaemic actions. Although Type 1 diabetes is caused by insulin deficiency (insulin-dependent) and can be corrected by insulin replacement, Type 2 diabetes is a multifactorial disease and its treatment is not dependent on insulin therapy alone. Type 2 diabetes in humans is characterized by increased insulin resistance, increased fasting blood glucose, impaired glucose tolerance and the development of glomerular hyperfiltration and microalbuminuria, ultimately leading to diabetic nephropathy and end-stage renal disease. Clinical studies have suggested that an inappropriate increase in hyperglycaemic glucagon (hyperglucagonaemia) over hypoglycaemic insulin (not insulin deficiency until advanced stages) plays an important role in the pathogenesis of Type 2 diabetes. However, for decades, research efforts and resources have been devoted overwhelmingly to studying the role of insulin and insulin-replacement therapy. By contrast, the implication of glucagon and its receptor signalling in the development of Type 2 diabetic metabolic syndromes and end-organ injury has received little attention. The aim of this review is to examine the evidence as to whether glucagon and its receptor signalling play any role(s) in the pathogenesis of Type 2 diabetic renal injury, and to explore whether targeting glucagon receptor signalling remains only a theoretical antidiabetic strategy in Type 2 diabetes or may realize its promise in the future.
Keywords: diabetic nephropathy, glucagon, hyperglycaemia, insulin, metabolic syndrome, Type 2 diabetes
INTRODUCTION
Although numerous factors may influence blood glucose and body energy supply and consumption in animals and humans, pancreatic bi-hormones, namely insulin and glucagon, are undoubtedly the Yin and Yang in the regulation of glucose metabolism and homoeostasis [1–4]. Although insulin acts primarily to lower blood glucose levels by stimulating tissue and cellular glucose uptake and hepatic glycogen synthesis (glucose consumption), glucagon counters these regulatory actions of insulin by stimulating hepatic glycogenolysis and gluconeogenesis (glucose supply) [1–3]. Glucagon is a 29-amino-acid hormone that is synthesized mainly in pancreatic α-cells at the periphery of the islets of Langerhans, and is released in response to a decrease in serum glucose induced by insulin [3,5,6]. cDNAs encoding glucagon, GLPs (glucagon-like peptides) and glucagon receptors have been cloned [1,7–9]. Both glucagon and other GLPs are derived from a single proglucagon gene, whose translation is tightly regulated by different preglucagon convertases (PC2) in a cell-specific manner [3,10–12]. The rat and mouse glucagon receptors are 485-amino-acid seven-transmembrane-domain proteins, whereas the human glucagon receptors have 477 amino acids [1,8,9]. Both human and rodent glucagon receptors contain a common sequence motif (RLAR) in the third intracellular domain, which is clearly required for activation of Gs-proteins (stimulatory G-proteins) and downstream intracellular signalling [8,9].
Glucagon receptors are predominantly expressed in hepatocytes and renal cells, but also to a lesser extent in cardiovascular tissues and gastrointestinal cells [1,13,14]. All known hyperglycaemic effects of glucagon are mediated by glucagon receptors [1,2,8,9]. Previous studies in rodents and humans using specific glucagon receptor antagonists support this notion [1,15–17]. More recently, glucagon-receptor-knockout mice have been developed for glucose and metabolism research [18–20]. These mice have lower basal blood glucose levels and increased glucose tolerance than their wild-type controls, suggesting that glucagon is essential for maintaining normal glycaemia [18–20]. In vitro and in vivo, glucagon binds to its membrane-bound receptors through GTP-binding heterotrimeric Gs-proteins to activate adenylate cyclase, leading to increased intracellular cAMP levels [1,21,22]. cAMP is not only the major second messenger responsible for the hyperglycaemic actions of glucagon, but it also exerts a stimulatory effect on glucagon secretion from pancreatic α-cells via PKA (cAMP-dependent protein kinase)-dependent and PKA-independent pathways (Figure 1) [1,3]. The PKA-independent pathway involves activation of a protein named Epac (exchange protein directly activated by cAMP), which is expressed in pancreatic α-cells and plays an important role in cAMP-regulated glucagon exocytosis [3,23]. The second major intracellular signalling pathway activated by glucagon is the PLC (phospholipase C)/IP3 (inositol 1,4,5-trisphosphate)/Ca2+ pathway (Figure 1). Activation of glucagon receptors has been shown to increase [Ca2+]i (intracellular Ca2+) in pancreatic α-cells and hepatocytes [1,3,24]. Both the PLC/IP3/Ca2+ and PKA signalling pathways also play an important role in non-hyperglycaemic and/or growth-promoting effects of glucagon on pancreatic islets, as well as non-pancreatic cells. However, glucagon receptors and receptor-activated signalling have been studied primarily in hepatocytes and gastrointestinal cells [1]. The potential role(s) of glucagon and its receptor signalling in mediating the growth, hypertrophy and proliferation of glomerular mesangial cells, glucagon-induced glomerular hyperfiltration and in the development of Type 2 diabetic glomerular injury have not been explored. The aim of this review is to examine the evidence and explore the potential role(s) of glucagon and its receptor signalling in the development of Type 2 diabetic renal injury. We focus on the kidney in this review, because diabetic nephropathy is one of the most important complications in Type 2 diabetes. The general biology of glucagon synthesis and processing, its cellular distribution, the regulation of its secretion and glucagon receptors and its receptor signalling in other tissues have been comprehensively reviewed elsewhere [1–3], and further discussion on these areas is therefore beyond the scope of the present review.
Figure 1. Two classical GCGR-mediated intracellular signalling pathways in glucagon-targeted cells.
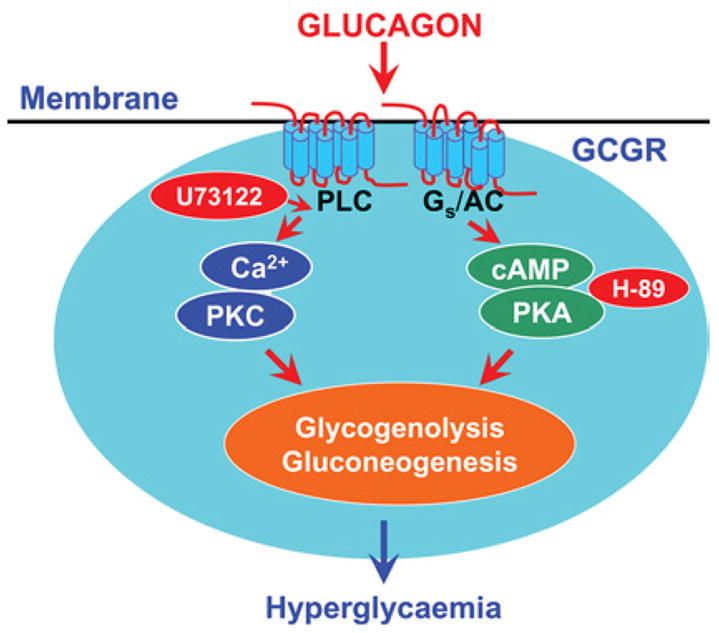
To induce an effect, glucagon binds to cell-surface Gs-protein-coupled receptors and activates the PLC/IP3/Ca2+ and PKA signalling pathways. Both signaling pathways are closely involved in mediating glucagon-induced glycogenolysis and gluconeogenesis, leading to hyperglycaemia. Pharmacological agents known to block these two signalling pathways (H-89, a selective PKA inhibitor, and U-73122, a selective PLC inhibitor) also inhibit glucagon-induced hyperglycaemic and growth effects in target cells. AC, adenylate cyclase; PKC, protein kinase C.
HYPERGLUCAGONAEMIA AND HUMAN TYPE 2 DIABETES
Clinical studies have shown that inappropriately elevated serum glucagon levels (also termed hyperglucagonaemia) are often associated with onset and progression of Type 2 diabetes [25–27], but the role of hyperglucagonaemia in the development of Type 2 diabetes and its cardiovascular and renal complications has remained controversial for several decades since its conception [27–29]. The classical view in diabetes research is that insulin deficiency is the most important cause of Type 1 (absolute) and Type 2 (relative) diabetes in humans and that glucagon plays little, if any, role in the pathogenesis of this disease. Indeed, it is not surprising that a random Medline or PubMed search on Type 2 diabetes easily yielded almost 20 times more citations on insulin than glucagon.
However, patients with Type 2 diabetes do not normally have absolute insulin deficiency due to pancreatic β-cell dysfunction until the disease progresses to more advanced stages [4]. In fact, serum insulin levels are often subnormal, normal or even elevated (hyperinsulinaemia) in these patients and, therefore, increased insulin resistance remains the hallmark of Type 2 diabetes [4]. For instance, Reaven et al. [25] compared plasma glucose, insulin and glucagon levels over an 8-h period between normal subjects and patients with Type 2 diabetes. They found that day-long plasma glucose and insulin concentrations were significantly higher than normal in patients with Type 2 diabetes whether they were obese or non-obese. The elevated plasma glucose and insulin levels were accompanied by higher plasma glucagon concentrations in both non-obese and obese patients with Type 2 diabetes. Although these data do not directly implicate hyperglucagonaemia in the development of Type 2 diabetes, they suggest that increased plasma glucagon levels in patients with Type 2 diabetes may, in part, contribute to their persistent hyperglycaemia, despite the fact that plasma insulin levels are equal to or higher than normal [25].
Similar clinical observations have been reported for elevated proglucagon peptides in patients with Type 2 diabetes mellitus [26]. Orskov et al. [26] reported that, in the fasting state, proglucagon-like immunoreactivity was significantly elevated in the group with Type 2 diabetes compared with the control group. After an oral glucose load, insulin concentrations in plasma were lower, whereas the concentrations of all proglucagon products were elevated in the group with Type 2 diabetes compared with the control group [26]. Moreover, although serum glucagon levels are generally down-regulated by glucose and food intake in normal subjects, they are unable to decrease appropriately in response to glucose or food ingestion in patients with Type 2 diabetes [2,3,25,26,30]. This lack of suppression of glucagon secretion by hyperglycaemia is consistent with findings in isolated mouse pancreatic islets and clonal hamster In-R1-G9 glucagon-releasing cells [31]. In the latter study, low glucose (approx. 7 mmol/l) inhibited glucagon secretion, whereas higher glucose concentrations stimulated glucagon release at 25–30 mmol/l. These paradoxical findings may explain why patients with diabetes with pronounced hyperglycaemia still display hyperglucagonaemia [31]. Thus increased serum glucagon levels relative to insulin may play an important role in elevating or maintaining high serum and tissue glucose levels and, consequently, contributing to increased insulin resistance in patients with Type 2 diabetes [2,4,25,26,30].
HYPER-HAEMODYNAMIC EFFECTS OF GLUCAGON RELEVANT TO DIABETIC GLOMERULAR INJURY
Most previous studies on glucagon have focused primarily on the physiological regulation of blood glucose and body energy metabolism and its clinical usefulness in treating hypoglycaemia due to an overdose of insulin therapy in Type 1 diabetes [2,3]. In the kidney, studies on glucagon and its receptors have been limited to its acute effects on GFR (glomerular filtration rate) and urinary excretion of electrolytes. The potential role(s) of glucagon and its receptor signalling in the development of diabetic nephropathy have not been well studied.
Although the consequence of long-term hyperglucagonaemia on renal function and renal pathophysiology has not been studied, acute systemic or intrarenal infusion of glucagon has been shown to induce marked glomerular hyperfiltration, a hallmark of early Type 2 diabetic glomerular injury [32–35]. We [32] and others [33–35] have shown that infusion of glucagon at levels reported after a high-protein diet or in Type 2 diabetes increases GFR by approx. 50% (Figure 2), whereas an antibody that specifically neutralizes glucagon decreased GFR or glucose metabolism abnormalities in rats with streptozotocin-induced diabetes [36,37]. How glucagon increases GFR remains poorly understood, but increased levels of cAMP, prostaglandins or NO have been suggested to mediate glucagon-induced glomerular hyperfiltration [34,38]. Glucagon-induced glomerular hyperfiltration may also be due to increased blood pressure and cardiac performance [39,40] or increased glomerular filtration pressure (glomerular hypertension) due to glucagon-induced renal vasodilatation (Figure 2) [32–35]. Persistent glomerular hyperfiltration induced by glucagon may play an important role in the development of glomerular injury through stress- and pressure-mediated mesangial cell hypertrophy and/or proliferation, with subsequent mesangial expansion and ECM (extracellular matrix) deposition [40,41]. Further integrative studies in rodent models of experimental Type 2 diabetes are required to determine the long-term effects of hyperglucagonaemia on glomerular injury and renal pathophysiology.
Figure 2. Glucagon induces renal glomerular hyperfiltration in anaesthetized rats.
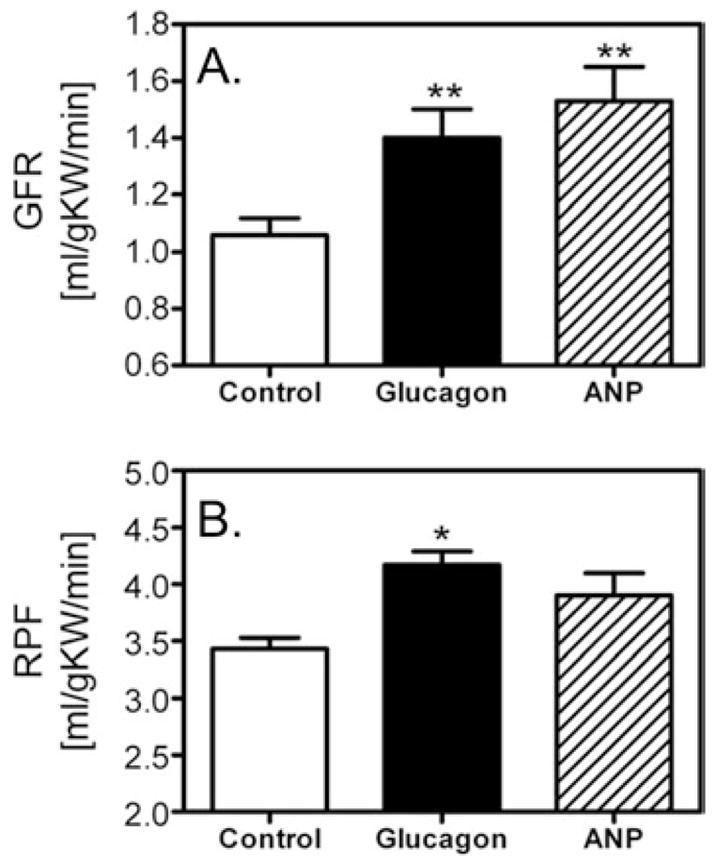
Intravenous glucagon infusion (50 ng · kg−1 of body weight · min−1) increased GFR by more than 30 % (A) as well as renal plasma flow (RPF) (B) over control (*P < 0.05 and **P < 0.01 compared with control). The extent of glucagon-induced hyperfiltration was similar to that induced by ANP (atrial natriuretic peptide), a well characterized renal vasodilator [32]. Data taken from [32].
NON-HYPERGLYCAEMIC AND GROWTH-PROMOTING EFFECTS OF GLUCAGON
Whether glucagon, acting on its receptors, has any direct, hyperglycaemic- and/or haemodynamic-independent effects on cardiovascular and renal tissues remains unknown. The prevailing view in current diabetes research is that the development and progression of Type 2 diabetes are the consequence of long-term hyperglycaemia due to insulin deficiency or increased insulin resistance [2–4,42]. Glucagon does not play an important role in Type 2 diabetes and, if it does, it is primarily due to its hyperglycaemic effects. Indeed, this view is supported by the difficulty, or lack of approaches, in discriminating the effects of hyperglycaemia secondary to glucagon stimulation from those caused directly by glucagon itself especially in animal models or human Type 2 diabetes [2,3]. It also gains considerable support from animal and human studies, in which acute glucagon administration induces hyperglycaemia, and from in vitro cell culture studies, in which high glucose concentrations (20–30 mmol/l) were used. For example, hyperglycaemia has been shown to induce proliferation of cultured vascular smooth muscle cells via activation of the JAK (Janus-activated kinase)/STAT (signal transducer and activator of transcription) pathway [43]. In the kidney, hyperglycaemia induced the phosphorylation of the MAPK (mitogen-activated protein kinase) ERK1/2 (extracellular-signal-regulated kinase 1/2) in primary cultured proximal tubule cells [44] and stimulated protein and ECM synthesis in mesangial cells [45]. It should be emphasized that most of these effects of hyperglycaemia have been demonstrated primarily in acute cell culture experiments in vitro. The direct effects of hyperglycaemia via long-term glucose infusion for weeks or even months in the presence of controlled plasma insulin and glucagon concentrations have not been well characterized in animals or humans.
There is evidence that, in addition to causing glomerular hyperfiltration, glucagon may induce glomerular injury through its potential role as a ‘growth factor’ or ‘proliferative cytokine’ [43,44,46–49]. At least in cell culture models, glucagon has been shown to induce cellular growth, hypertrophy and proliferation or activation of growth-related signalling proteins in pancreatic and renal cells in the absence of high glucose conditions. For instance, glucagon directly activates downstream signalling cascades involving the MAPKs ERK1/2, p38 MAPK and JNK (c-Jun N-terminal kinase)/SAPK (stress-activated protein kinase) in pancreatic islet cells [46,47,50,51]. These cytoplasmic serine/threonine-containing MAPKs are closely associated with cell differentiation, proliferation and transformation [46,47]. To minimize the confounding hyperglycaemic effects secondary to glucagon administration, as occurred in animal or human studies in vivo, we have recently used rat glomerular mesangial cells as an in vitro cell model to study whether glucagon has growth effects independent of hyperglycaemia or hyper-haemodynamics [48,49]. We found that rat glomerular mesangial cells have high-affinity GCGR (Gs-protein-coupled glucagon receptor) binding (Figure 3) and express GCGR mRNA, and that glucagon induced mesangial cell growth by increasing DNA and protein synthesis in the absence of high glucose concentrations, effects that were completely blocked by a specific glucagon receptor antagonist (Figure 4) [48,49]. We found further that these growth effects are mediated by glucagon-receptor-activated phosphorylation of ERK1/2 via PKA-, PLC/IP3/Ca2+-or PI3K (phosphoinositide 3-kinase)-mediated signalling [48,49] (Figure 4). Because these effects were induced by glucagon in the absence of hyperglycaemia and can be blocked by a glucagon receptor antagonist, our data suggest that glucagon may directly stimulate mesangial cell growth, at least in cell culture settings. However, whether these glucagon-induced responses in the cell culture studies have any role in Type 2 diabetic glomerular injury still awaits confirmation in animal and human studies.
Figure 3. Expression of high-affinity GCGRs, and glucagon-induced activation of PLC/Ca2+ and cAMP signalling in cultured rat glomerular mesangial cells.
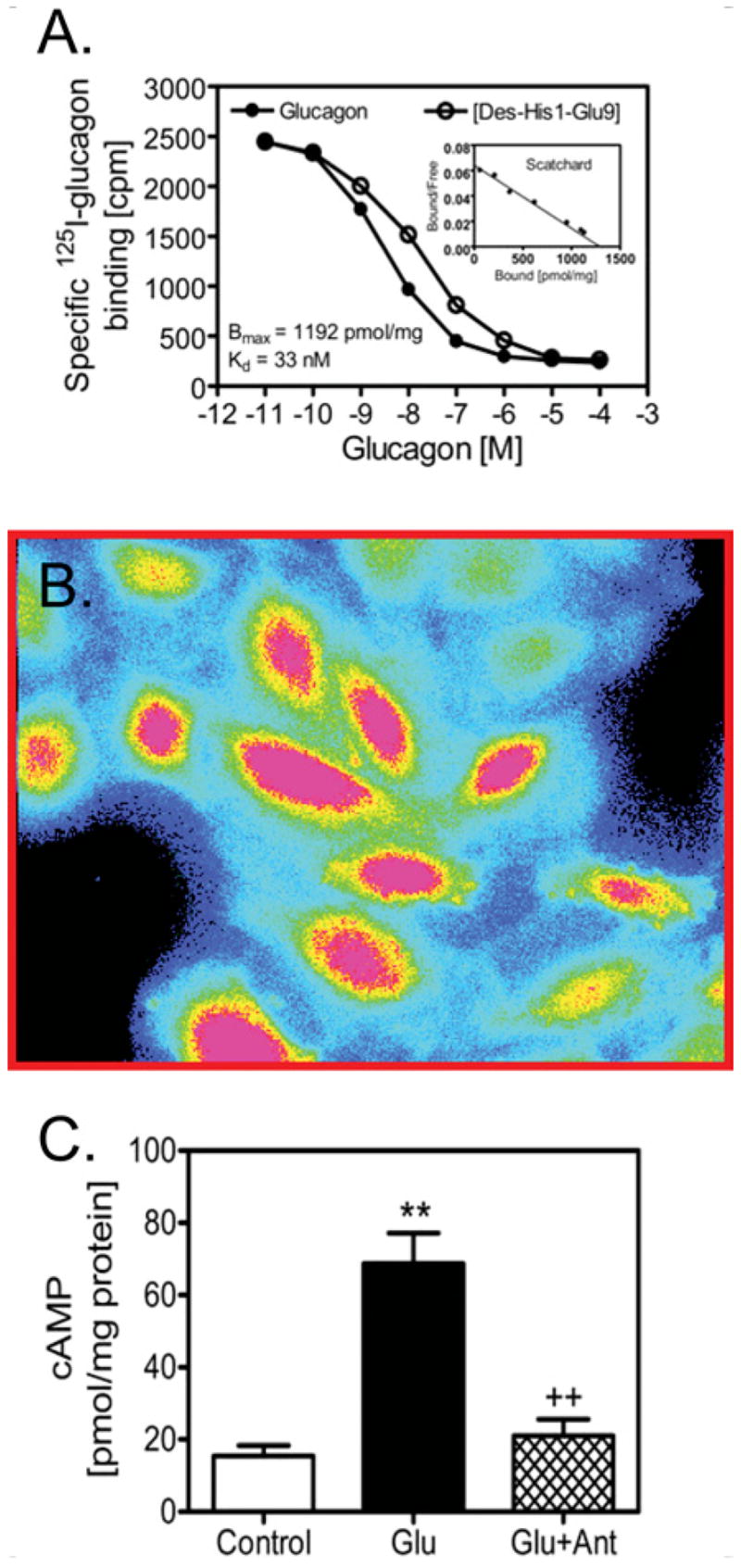
(A) Maximal binding sites (Bmax) and binding dissociation constant (Kd) of glucagon receptors in mesangial cells. (B) Fura 2 (340 nm/380 nm) ratiometric image showing glucagon-increased [Ca2+]i signalling, with the highest level of [Ca2+]i in red to background in black. (C) Glucagon-induced increases in intracellular cAMP is blocked by a glucagon receptor antagonist (1 μmol/l des-His1-[Glu9]glucagon amide; Ant). **P < 0.01 compared with control; and ++P < 0.01 compared with glucagon. Figures 3(A) and 3(C) are reproduced from [48] with permission. © (2006) Lippincott Williams & Wilkins (http://lww.com). Figure 3(B) is reprinted from Biochem. Pharmacol., 71, X.C. Li, O.A. Carretero and J.L. Zhuo, Cross-talk between angiotensin II and glucagon receptor signaling mediates phosphorylation of mitogen-activated protein kinases ERK 1/2 in rat glomerular mesangial cells, pp. 1711–1719, Copyright (2006), with permission from Elsevier.
Figure 4. Glucagon-induced growth of cultured rat glomerular mesangial cells in the absence of high glucose via the receptor-mediated activation of ERK1/2.
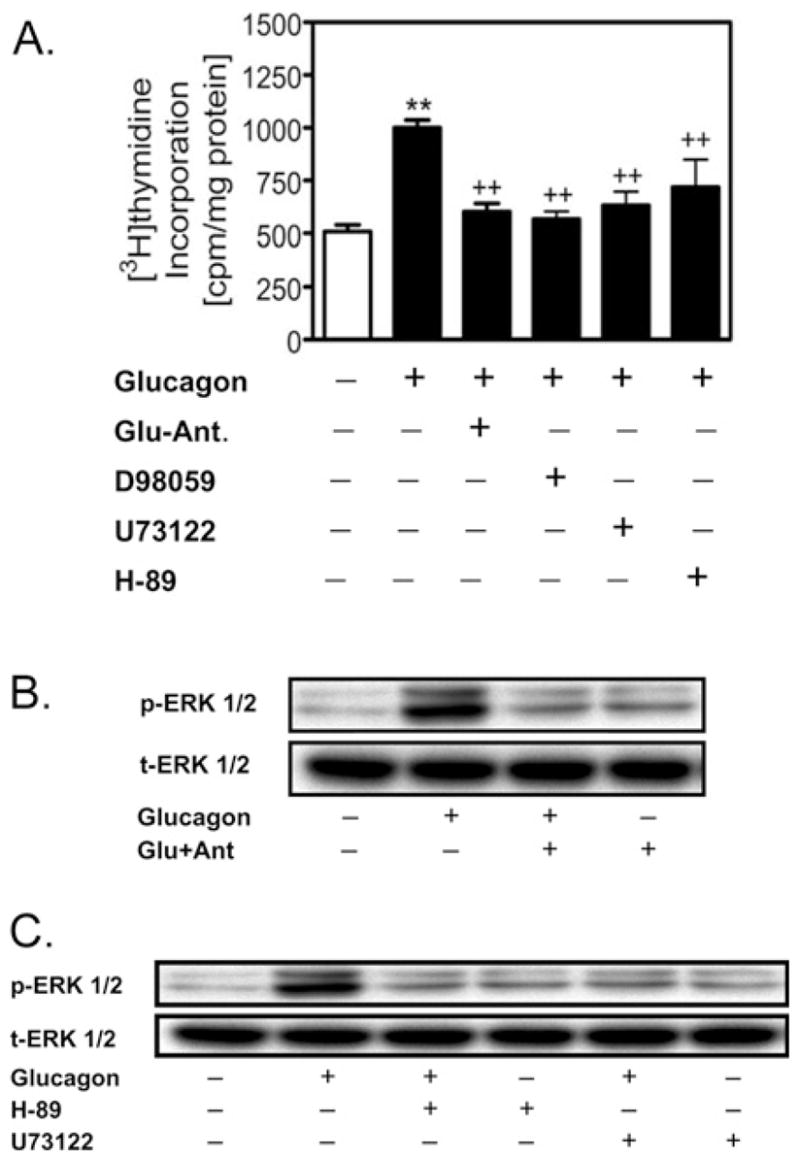
(A) Glucagon (1 nmol/l) significantly increased [3H]thymidine incorporation, an index of DNA synthesis, compared with control, and the response was blocked by des-His1-[Glu9]glucagon amide (Glu-Ant.; 1 μmol/l), the MAPK inhibitor D-98059 (1μmol/l), the PKA inhibitor H-89 (1 μmol/l) or the PLC inhibitor U-73122 (1 μmol/l) respectively. **P < 0.01 compared with control; ++P < 0.01 compared with glucagon. (B) Glucagon induced the activation of ERK1/2, which was blocked by des-His1-[Glu9]glucagon amide (Glu-Ant.; 1 μmol/l). (C) Inhibition of -dependent pathway by U-73122 (1 μmol/l) or the PKA pathway the PLC/IP3/Ca2+ by H-89 (1 μmol/l) blocked glucagon-induced activation of ERK1/2 signalling. p-ERK 1/2, phosphorylated ERK1/2; t-ERK 1/2, total ERK1/2. Reproduced from [48] with permission. © (2006) Lippincott Williams & Wilkins (http://lww.com).
INTERACTIONS BETWEEN GLUCAGON, HYPERGLYCAEMIA AND ANGII (ANGIOTENSIN II)/ANGII RECEPTOR SIGNALLING IN DIABETIC NEPHROPATHY
It is well recognized that activation of the RAS (renin–angiotensin system) plays an important role in the development and progression of cardiovascular and renal complications in Type 1 and Type 2 diabetes. The widespread use of ACE (angiotensin-converting enzyme) inhibitors and ARBs [AT1 (AngII type 1) receptor blockers] to treat proteinuria in patients with diabetic nephropathy strongly supports the involvement of the RAS in Type 1 and Type 2 diabetes [52–54]. Glomerular hyperfiltration (increased GFR) and microalbuminuria are two major complications of early Type 2 diabetic glomerular injury [38,42,55,56]. A recent prospective multicentre double-blind clinical study in patients with early Type 2 diabetic nephropathy showed that the ACE inhibitor enalapril and the ARB telmisartan are equally effective in preventing glomerular hyperfiltration and attenuating urinary albumin excretion [57].
We have studied the interactions between glucagon and AngII or AngII receptor signalling in cultured rat mesangial cells as well as in glucagon-infused mice [48,49,58,59]. The findings suggest that hyperglucagonaemia may cause mesangial cell or glomerular injury by interacting with AngII/AT1 receptor signaling [49,58,59]. In vitro, we have shown that glucagon moderately increased AngII production in cultured mesangial cells independent of hyperglycaemia [49]. Activation of the RAS has been reported in experimental diabetic rats in vivo [60–62] and in cultured mesangial cells exposed to hyperglycaemia [63,64]. ACE inhibitors and ARBs, which block the RAS, also inhibited glucagon-receptor-mediated activation of PKA and PLC-dependent [Ca2+]i mobilization, as well as Akt/PKB (protein kinase B)/PI3K signalling, in mesangial cells [58,59]. In vivo, we have demonstrated that the ACE inhibitor enalapril prevented glucagon-induced glomerular hyper-filtration in anaesthetized rats [58]. These results suggest that glucagon receptor signalling may interact with that of AngII/AT1 receptor signalling in the glomeruli or mesangial cells. More recently, we have infused glucagon for 4 weeks to induce hyperglucagonaemia in mice, which produced metabolic and renal phenotypes of early Type 2 diabetes, including increased fasting blood glucose, impaired glucose tolerance, development of microalbuminuria and mesangial expansion, and increased ECM deposition in the glomeruli [59]. The effects of hyper-glucagonaemia could be blocked by concurrent administration with a glucagon receptor antagonist, des-His1[Glu9]glucagon amide, or the ACE inhibitor captopril, which blocks AngII production [59]. AngII, a powerful growth factor and proliferative cytokine, may increase TGF-β1 (transforming growth factor-β1) or activate JAK and STAT proteins, which in turn mediate the synthesis of ECM in mesangial cells [65–67]. This information helps explain the underlying mechanisms by which ACE inhibitors and ARBs may be used to prevent and treat Type 2 diabetic renal and cardiovascular complications. Nevertheless, our studies do not provide direct evidence that hyperglucagonaemia alone induced Type 2 diabetic metabolic and renal phenotypes independent of its effect on hyperglycaemia in vivo [59]. More studies or new experimental approaches are needed in order to discriminate the direct effects of glucagon from those induced secondary to hyperglycaemia following glucagon administration in animals or humans.
GLUCAGON RECEPTORS AS A POTENTIAL TARGET IN TREATING TYPE 2 DIABETIC CARDIOVASCULAR AND RENAL COMPLICATIONS
Although Type 1 diabetes can be treated with insulin replacement, pancreatic β-cell transplantation and even adult or embryonic stem cells, such approaches are unlikely to be practical for patients with Type 2 diabetes, because approx. 95% of patients have Type 2, rather than Type 1, diabetes (based on the data provided by the National Institute of Diabetes, Digestive and Kidney Diseases). Increased insulin resistance, rather than insulin deficiency, plays a key role in the development of Type 2 diabetes [3], but the factors that contribute to increased insulin resistance are not well understood. Because glucagon plays a counter-regulatory role in glucose metabolism and there is an imbalance between the influences of insulin and glucagon in favouring the latter in Type 2 diabetes, pharmacological blockade of glucagon receptors or its receptor signalling has been pursued as a potential treatment for Type 2 diabetes.
In fact, the concept that glucagon or glucagon receptors may be targeted for treatment of Type 2 diabetes is not new [2,3]. On the basis of the concept that glucose metabolism and homoeostasis is dually controlled by the pancreatic bi-hormones insulin and glucagon, rather than insulin alone, the pharmaceutical industry has attempted to develop potent and selective glucagon receptor antagonists for over two decades. As early as in 1982, Johnson et al. [68] reported that a glucagon peptide analogue antagonist THG [(l-N α-trinitrophenylhistidine,12-homo-arginine)glucagon] significantly lowered blood glucose concentrations by 30–65% in rats made diabetic with streptozotocin. In vitro, THG is a potent antagonist of glucagon-activated hepatic adenylate cyclase. This study provided the first evidence that a glucagon receptor antagonist could substantially reduce blood glucose levels in diabetic animals without insulin replacement [68]. Another glucagon receptor antagonist, des-His1-[Glu9]glucagon amide, was developed a few years later, which also inhibited glucagon-induced hyperglycaemia or lowered blood glucose in streptozotocin-induced diabetic rats in vivo [15]. Although these antagonists are potent and selective in blocking the effect of glucagon on glucose metabolism, their usefulness for treatment of diabetes is restricted because, as a peptide, they cannot be taken orally.
There have been significant advances on several fronts in the development of non-peptidyl glucagon receptor antagonists or blockers for the treatment of Type 2 diabetes over the last few years [1–3,69–71]. Madsen et al. [72] described the first non-peptide competitive human glucagon receptor antagonist NNC 92-1687 [2-(benzimidazol-2-ylthio)-1-(3,4-dihydroxyphenyl)-1-ethan one]. Although this antagonist inhibited glucagon receptor binding in a competitive manner, its binding affinity or inhibitory constants were at micromolar ranges. Ling et al. [69] also identified and characterized alkylidene hydrazides as non-peptidyl antagonist(s) for the human glucagon receptor to block glucagon-receptor-mediated cAMP production and glucose metabolism. Furthermore, the efficacy and safety of the non-peptidyl glucagon receptor antagonist Bay 27-9955 (Bayer) has been the first to be reported in a double blind placebo-controlled crossover human study [17]. In this study, Bay 27-9955 at 200 mg significantly blunted hyperglucagonaemia-induced hyperglycaemia. However, long-term antidiabetic benefits, as well as side effects, of this compound in patients with Type 2 diabetes remain unknown, because no follow-up studies have been reported. Recently, the effects of antisense oligonucleotides targeting glucagon receptors have been evaluated in db/db diabetic fatty mice, a rodent model of human Type 2 diabetes [73,74]. In both studies, small molecular antisense oligonucleotides were found to inhibit glucagon receptor expression in hepatocytes and ameliorate or reverse the diabetic metabolic syndrome in db/db mice [73,74]. Although it remains to be determined how glucagon receptor antisense oligonucleotides can be used in treating human Type 2 diabetes, it supports the concept that targeting glucagon receptor expression and, therefore, its signalling may have clinical benefits for patients with Type 2 diabetes in the future.
A unique class of new antidiabetic drugs targeting the serine protease DPP-IV (dipeptidyl peptidase IV) has recently been approved by the USA FDA (Food and Drug Administration) for treatment of Type 2 diabetes in humans. DPP-IV, or CD26, is a membrane-associated peptidase widely expressed in the digestive and non-digestive tissues [3,75,76]. It degrades both GLP-1 and glucagon [77,78]. GLP-1 is a major gut-secreted incretin hormone released in response to glucose or food intake [75–78]. Upon its release, GLP-1 is rapidly degraded by DPP-IV and eliminated by renal clearance, therefore having limited physiological or therapeutic effects [75–78]. However, GLP-1 has been shown to stimulate glucose-dependent insulin secretion from pancreatic β-cells in vivo and in vitro [75,79,80]. These findings led to the development of various DPP-IV inhibitors, which aim to raise circulating GLP-1 levels by inhibiting GLP-1 degradation by DPP-IV. Two DPP-IV inhibitors developed recently, sitagliptin and vildagliptin, may have particular potential for treatment of Type 2 diabetes. Phase III clinical trials have shown that JANUVIA™ (sitagliptin; developed by Merck and approved by the FDA) is well tolerated and can significantly improve glycaemic control by decreasing HbA1c (glycated haemoglobin) levels [80–82]. Its antidiabetic effect is mediated primarily through increases in GLP-1, which stimulates insulin release from pancreatic β-cells [81]. Whether sitagliptin suppresses glucagon secretion from pancreatic α-cells or not has not been well characterized. By contrast, the DPP-IV inhibitor vildagliptin (LAF237; developed by Novartis) both increases insulin secretion from β-cells and potently suppresses glucagon secretion from pancreatic α-cells [83,84]. The cellular mechanisms by which DPP-IV inhibitors decrease serum glucagon levels are not well understood. As glucagon is also a substrate for the enzyme DPP-IV [78], inhibition of this enzyme by sitagliptin or vildagliptin is expected to increase, rather than decrease, serum glucagon. Thus it is possible that these inhibitors may initially increase serum glucagon, together with GLP-1, which subsequently suppresses glucagon secretion from the pancreatic α-cells. The dual insulin-promoting and glucagon-suppressing effects of DPP-IV inhibitors is particularly relevant to clinical Type 2 diabetes, since these patients often have insulin resistance and hyperglucagonaemia [25,26,30].
Because GLP-1 is a major gut-secreted incretin hormone that induces insulin secretion from pancreatic β-cells and simultaneously inhibits glucagon secretion from α-cells, a number of incretin enhancers or GLP-1 agonists have been pursued for the treatment of Type 2 diabetes [3,75,76,80]. Exenatide and liraglutide are two major GLP-1 agonists that have been shown to have beneficial antidiabetic effects in both animal models and clinical trials [85–87]. Both agonists appear to activate GLP-1 receptors and have similar antidiabetic properties to the native GLP-1. However, as peptide agonists, exenatide and liraglutide are often administered by injection subcutaneously or systemically [88,89], therefore the clinical relevance of these compounds in treating Type 2 diabetes remains to be determined.
PERSPECTIVES
Diabetes is a major risk factor for the development of cardiovascular and renal organ damage, leading to hypertension, atherosclerosis, ischaemic heart disease and nephropathy. Type 1 and Type 2 diabetes affect over 20 million Americans and, together, rank as the sixth leading cause of disease-related death in the USA alone. Nearly 1 million new cases (most representing Type 2 diabetes) are diagnosed every year, and the magnitude of this public health problem as well as its economic impacts continues to escalate. Moreover, children with diabetes are usually assumed to have Type 1 diabetes, but the percentage of children diagnosed with Type 2 diabetes has increased dramatically over recent years (according to the data provided by the National Institute of Diabetes, Digestive and Kidney Diseases). It is therefore imperative that the factors contributing to the development and progression of Type 2 diabetes should be identified and new therapeutic targets beyond insulin replacement explored. Since glucagon plays a critical role in the regulation of glucose metabolism and homoeostasis in humans by promoting glycogenolysis and gluconeogenesis, glucagon and its receptors may be potential targets for the treatment of Type 2 diabetes (Figure 5). Furthermore, clinical studies have shown that patients with Type 2 diabetes often have hyperglucagonaemia, which is not decreased proportionally in response to glucose or food intake. By contrast, serum insulin may be normal or subnormal depending on the progression of the disease. We propose that imbalance in the actions of insulin and glucagon in favouring the latter may contribute to increased insulin resistance, impaired glucose tolerance, persistent hyperglycaemia and target organ injury in Type 2 diabetes (Figure 5). Thus in view of the diverse hyperglycaemic, pro-growth and hyperhaemodynamic effects of glucagon, pharmacological blockade of glucagon receptors or its receptor signalling with orally active and highly selective antagonists remains an attractive antidiabetic strategy to improve insulin sensitivity and treat Type 2 diabetic cardiovascular and renal complications in humans. However, more innovative approaches are required to study whether long-term hyperglucagonaemia plays any role in the development of target organ injury in animal models of Type 2 diabetes or human Type 2 diabetes. New challenges also remain with respect to designing and/or discovering more selective, potent and orally active compounds with few side effects. One likely side effect of glucagon receptor blockade may be hypoglycaemia or to slow the recovery from hypoglycaemia due to an insulin overdose [3]. Interestingly, studies in mice have shown that glucagon-receptor knockout only moderately lowers basal blood glucose levels [18–20]. This suggests that, with proper dosage titrations, glucagon receptor antagonism is unlikely to lead to hypoglycaemia, as seen during insulin overdose.
Figure 5. Potential role of hyperglucagonaemia in the development of metabolic and renal glomerular phenotypes in Type 2 diabetes.
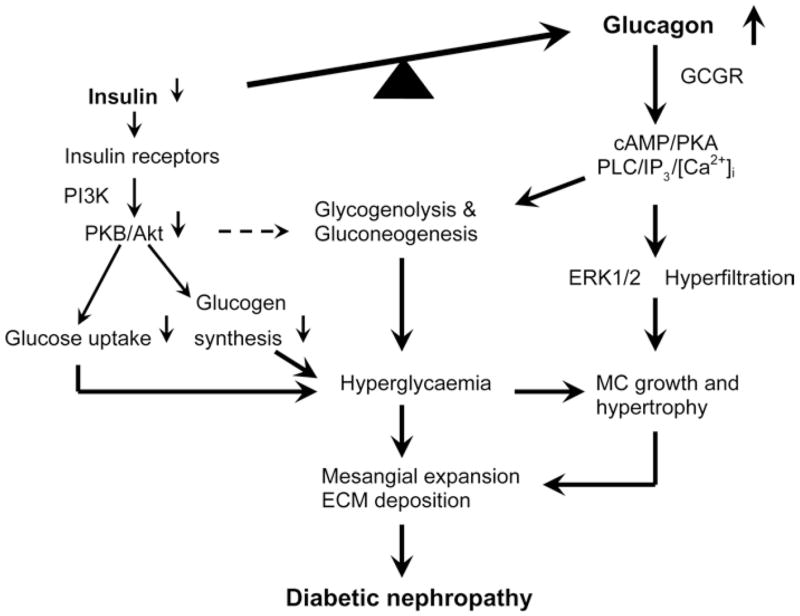
In human Type 2 diabetes, there is an inappropriate increase in the hyperglycaemic hormone glucagon over the hypoglycaemic hormone insulin (also termed hyperglucagonaemia). Imbalance in the actions of insulin and glucagon in favouring the latter leads to increases in glycogenolysis and gluconeogenesis in the liver and other tissues, resulting in hyperglycaemia, increased insulin resistance and impaired glucose metabolism. Moreover, glucagon, via direct activation of GCGR-mediated signalling pathways (PKA and ERK1/2), stimulates mesangial cell (MC) growth and hypertrophy and induces glomerular mesangial expansion and ECM deposition. Glucagon receptor antagonists are expected to block the metabolic and growth effects of hyperglucagonaemia in Type 2 diabetes.
Acknowledgments
This work was supported in part by a National Institute of Diabetes, Digestive and Kidney Diseases Grant (5RO1DK067299 to J.L.Z.), a Henry Ford Health System Institutional Grant (A10217 to J.L.Z.), and a National Kidney Foundation of Michigan Grant (to X.C.L). We apologize to those investigators whose excellent work is not cited due to space restrictions.
Abbreviations
- ACE
angiotensin-converting enzyme
- AngII
angiotensin II
- AT1
AngII type 1
- ARB
AT1 receptor blocker
- [Ca2+]i
intracellular Ca2+
- DPP-IV
dipeptidyl peptidase IV
- ECM
extracellular matrix
- ERK1/2
extracellular-signal-regulated kinase 1/2
- FDA
Food and Drug Administration
- GFR
glomerular filtration rate
- GLP
glucagon-like peptide
- Gs-protein
stimulatory G-protein
- GCGR
Gs-protein-coupled glucagon receptor
- IP3
inositol 1,4,5-trisphosphate
- MAPK
mitogen-activated protein kinase
- PI3K
phosphoinositide 3-kinase
- PKA
cAMP-dependent protein kinase
- PLC
phospholipase C
- RAS
renin–angiotensin system
- THG
(l-N α-trinitrophenylhistidine,12-homoarginine)glucagon
References
- 1.Mayo KE, Miller LJ, Bataille D, et al. International Union of Pharmacology. XXXV The glucagon receptor family. Pharmacol Rev. 2003;55:167–194. doi: 10.1124/pr.55.1.6. [DOI] [PubMed] [Google Scholar]
- 2.Jiang G, Zhang BB. Glucagon and regulation of glucose metabolism. Am J Physiol Endocrinol Metab. 2003;284:E671–E678. doi: 10.1152/ajpendo.00492.2002. [DOI] [PubMed] [Google Scholar]
- 3.Gromada J, Franklin I, Wollheim CB. α cells of the endocrine pancreas: 35 years of research but the enigma remains. Endocr Rev. 2007;28:84–116. doi: 10.1210/er.2006-0007. [DOI] [PubMed] [Google Scholar]
- 4.Matthaei S, Stumvoll M, Kellerer M, Haring HU. Pathophysiology and Pharmacological treatment of insulin resistance. Endocr Rev. 2000;21:585–618. doi: 10.1210/edrv.21.6.0413. [DOI] [PubMed] [Google Scholar]
- 5.Bromer WW, Sinn LG, Staub A, Behrens OK. The amino acid sequence of glucagon. J Am Chem Soc. 1956;78:3858–3859. doi: 10.2337/diab.6.3.234. [DOI] [PubMed] [Google Scholar]
- 6.Baum J, Simon BF, Unger RH, Madison LL. Localization of glucagon in the α cells in the pancreatic islet by immunofluorescent techniques. Diabetes. 1962;11:371–374. [PubMed] [Google Scholar]
- 7.Lund PK, Goodman RH, Dee PC, Habener JF. Pancreatic preproglucagon cDNA contains two glucagon-related coding sequences arranged in tandem. Proc Natl Acad Sci USA. 1982;79:345–349. doi: 10.1073/pnas.79.2.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jelinek LJ, Lok S, Rosenberg GB, et al. Expression cloning and signaling properties of the rat glucagon receptor. Science. 1993;259:1614–1616. doi: 10.1126/science.8384375. [DOI] [PubMed] [Google Scholar]
- 9.Lok S, Kuijper JL, Jelinek LJ, et al. The human glucagon receptor encoding gene: structure, cDNA sequence and chromosomal localization. Gene. 1994;140:203–209. doi: 10.1016/0378-1119(94)90545-2. [DOI] [PubMed] [Google Scholar]
- 10.Furuta M, Zhou A, Webb G, et al. Severe defect in proglucagon processing in islet A-cells of prohormone convertase 2 null mice. J Biol Chem. 2001;20:27197–27202. doi: 10.1074/jbc.M103362200. [DOI] [PubMed] [Google Scholar]
- 11.Rouille Y, Westermark G, Martin SK, Steiner DF. Proglucagon is processed to glucagon by prohormone convertase PC2 in αTC1-TC6 cells. Proc Natl Acad Sci USA. 1994;91:3242–3246. doi: 10.1073/pnas.91.8.3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Webb GC, Akbar MS, Zhao C, Swift HH, Steiner DF. Glucagon replacement via micro-osmotic minipump corrects hypoglycemia and α-cell hyperplasia in prohormone convertase 2 knockout mice. Diabetes. 2002;51:398–405. doi: 10.2337/diabetes.51.2.398. [DOI] [PubMed] [Google Scholar]
- 13.Butlen D, Morel F. Glucagon receptors along the nephron: [125I]glucagon binding in rat tubules. Pflugers Arch. 1985;404:348–353. doi: 10.1007/BF00585347. [DOI] [PubMed] [Google Scholar]
- 14.Marks J, Debnam ES, Dashwood MR, Srai SK, Unwin RJ. Detection of glucagon receptor mRNA in the rat proximal tubule: potential role for glucagon in the control of renal glucose transport. Clin Sci. 2003;104:253–258. doi: 10.1042/CS20020336. [DOI] [PubMed] [Google Scholar]
- 15.Unson CG, Gurzenda EM, Merrifield RB. Biological activities of des-His1[Glu9]glucagon amide, a glucagon antagonist. Peptides. 1989;10:1171–1177. doi: 10.1016/0196-9781(89)90010-7. [DOI] [PubMed] [Google Scholar]
- 16.Buggy JJ, Heurich RO, MacDougall M, et al. Role of the glucagon receptor COOH-terminal domain in glucagon-mediated signaling and receptor internalization. Diabetes. 1997;46:1400–1405. doi: 10.2337/diab.46.9.1400. [DOI] [PubMed] [Google Scholar]
- 17.Petersen KF, Sullivan JT. Effects of a novel glucagon receptor antagonist (Bay 27–9955) on glucagon-stimulated glucose production in humans. Diabetologia. 2001;44:2018–2024. doi: 10.1007/s001250100006. [DOI] [PubMed] [Google Scholar]
- 18.Gelling RW, Du XQ, Dichmann DS, et al. Lower blood glucose, hyperglucagonemia, and pancreatic α cell hyperplasia in glucagon receptor knockout mice. Proc Natl Acad Sci USA. 2003;100:1438–1443. doi: 10.1073/pnas.0237106100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vuguin PM, Kedees MH, Cui L, et al. Ablation of the glucagon receptor gene increases fetal lethality and produces alterations in islet development and maturation. Endocrinology. 2006;147:3995–4006. doi: 10.1210/en.2005-1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parker JC, Andrews KM, Allen MR, Stock JL, McNeish JD. Glycemic control in mice with targeted disruption of the glucagon receptor gene. Biochem Biophys Res Commun. 2002;290:839–843. doi: 10.1006/bbrc.2001.6265. [DOI] [PubMed] [Google Scholar]
- 21.Rodbell M, Birnbaumer L, Pohl SL, Krans HM. The glucagon-sensitive adenylyl cyclase system in plasma membranes of rat liver. V an obligatory role of guanylnucleotides in glucagon action. J Biol Chem. 1971;246:1877–1882. [PubMed] [Google Scholar]
- 22.Wakelam MJ, Murphy GJ, Hruby VJ, Houslay MD. Activation of two signal-transduction systems in hepatocytes by glucagon. Nature. 1986;323:68–71. doi: 10.1038/323068a0. [DOI] [PubMed] [Google Scholar]
- 23.Seino S, Shibasaki T. PKA-dependent and PKA-independent pathways for cAMP-regulated exocytosis. Physiol Rev. 2005;85:1303–1342. doi: 10.1152/physrev.00001.2005. [DOI] [PubMed] [Google Scholar]
- 24.Hansen LH, Gromada J, Bouchelouche P, Whitmore T, Jelinek LJ, Kingsvogel W, Nishimura E. Glucagon-mediated Ca2+ signaling in BHK cells expressing cloned human glucagon receptors. Am J Physiol. 1998;274:C1552–C1562. doi: 10.1152/ajpcell.1998.274.6.C1552. [DOI] [PubMed] [Google Scholar]
- 25.Reaven GM, Chen YD, Golay A, Swislocki AL, Jaspan JB. Documentation of hyperglucagonemia through the day in nonobese and obese patients with noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab. 1987;64:106–110. doi: 10.1210/jcem-64-1-106. [DOI] [PubMed] [Google Scholar]
- 26.Orskov C, Jeppesen J, Madsbad S, Holst JJ. Proglucagon products in plasma of noninsulin-dependent diabetics and nondiabetic controls in the fasting state and after oral glucose and intravenous arginine. J Clin Invest. 1991;87:415–423. doi: 10.1172/JCI115012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barazzoni R, Zanetti M, Davanzo G, et al. Increased fibrinogen production in type 2 diabetic patients without detectable vascular complications: correlation with plasma glucagon concentrations. J Clin Endocrinol Metab. 2000;85:3121–3125. doi: 10.1210/jcem.85.9.6779. [DOI] [PubMed] [Google Scholar]
- 28.Unger RH. Role of glucagon in the pathogenesis of diabetes: the status of the controversy. Metab Clin Exp. 1978;27:1691–1709. doi: 10.1016/0026-0495(78)90291-3. [DOI] [PubMed] [Google Scholar]
- 29.Unger RH. Glucagon physiology and pathophysiology in the light of new advances. Diabetologia. 1985;28:574–578. doi: 10.1007/BF00281991. [DOI] [PubMed] [Google Scholar]
- 30.Shah P, Vella A, Basu A, Basu R, Schwenk WF, Rizza RA. Lack of suppression of glucagon contributes to postprandial hyperglycemia in subjects with type 2 diabetes mellitus. J Clin Endocrinol Metab. 2000;85:4053–4059. doi: 10.1210/jcem.85.11.6993. [DOI] [PubMed] [Google Scholar]
- 31.Salehi A, Vieira E, Gylfe E. Paradoxical stimulation of glucagon secretion by high glucose concentrations. Diabetes. 2006;55:2318–2323. doi: 10.2337/db06-0080. [DOI] [PubMed] [Google Scholar]
- 32.Harris PJ, Skinner SL, Zhuo JL. The effects of atrial natriuretic peptide and glucagon on proximal glomerulotubular balance in anesthetized rats. J Physiol. 1988;402:29–42. doi: 10.1113/jphysiol.1988.sp017192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Premen AJ, Hall JE, Smith LJ. Postprandial regulation of renal hemodynamics: role of pancreatic glucagon. Am J Physiol. 1985;248:F656–F662. doi: 10.1152/ajprenal.1985.248.5.F656. [DOI] [PubMed] [Google Scholar]
- 34.Tolins JP. Mechanisms of glucagon-induced renal vasodilation: role of prostaglandins and endothelium-derived relaxing factor. J Lab Clin Med. 2004;120:941–948. [PubMed] [Google Scholar]
- 35.Ahloulay M, Decaux M, Laborde K, Bankir L. Influence of glucagon on GFR and on urea and electrolyte excretion: direct and indirect effects. Am J Physiol. 1995;38:F225–F235. doi: 10.1152/ajprenal.1995.269.2.F225. [DOI] [PubMed] [Google Scholar]
- 36.Ahloulay M, Bankir L. Glucagon antibody infusion reduces GFR in diabetic rats. J Am Soc Nephrol. 1996;7:1868a. [Google Scholar]
- 37.Sorensen H, Brand CL, Neschen S, et al. Immunoneutralization of endogenous glucagon reduces hepatic glucose output and improves long-term glycemic control in diabetic ob/ob mice. Diabetes. 2006;55:2843–2848. doi: 10.2337/db06-0222. [DOI] [PubMed] [Google Scholar]
- 38.Ahloulay M, Dechaux M, Hassler C, Bouby N, Bankir L. Cyclic AMP is a hepatorenal link influencing natriuresis and contributing to glucagon-induced hyperfiltration in rats. J Clin Invest. 1996;98:2251–2258. doi: 10.1172/JCI119035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nikolaidis LA, Elahi D, Hentosz T, et al. Recombinant glucagon-like peptide-1 increases myocardial glucose uptake and improves left ventricular performance in conscious dogs with pacing-induced dilated cardiomyopathy. Circulation. 2004;110:955–961. doi: 10.1161/01.CIR.0000139339.85840.DD. [DOI] [PubMed] [Google Scholar]
- 40.Gartner K. Glomerular hyperfiltration during the onset of diabetes mellitus in two strains of diabetic mice (c57bl/6j db/db and c57bl/ksj db/db) Diabetologia. 1978;15:59–63. doi: 10.1007/BF01219330. [DOI] [PubMed] [Google Scholar]
- 41.Levine DZ, Iacovitti M, Robertson SJ, Mokhtar GA. Modulation of single-nephron GFR in the db/db mouse model of type 2 diabetes mellitus. Am J Physiol Regul Integr Comp Physiol. 2006;290:R975–R981. doi: 10.1152/ajpregu.00693.2005. [DOI] [PubMed] [Google Scholar]
- 42.Breyer MD, Bottinger E, Brosius FC, III, et al. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2005;16:27–45. doi: 10.1681/ASN.2004080648. [DOI] [PubMed] [Google Scholar]
- 43.Amiri F, Venema VJ, Wang X, Ju H, Venema RC, Marrero MB. Hyperglycemia enhances angiotensin II-induced janus-activated kinase/STAT signaling in vascular smooth muscle cells. J Biol Chem. 1999;274:32382–32386. doi: 10.1074/jbc.274.45.32382. [DOI] [PubMed] [Google Scholar]
- 44.Park SH, Woo CH, Kim JH, et al. High glucose down-regulates angiotensin II binding via the PKC-MAPK-cPLA2 signal cascade in renal proximal tubule cells. Kidney Int. 2002;61:913–925. doi: 10.1046/j.1523-1755.2002.00204.x. [DOI] [PubMed] [Google Scholar]
- 45.Singh R, Alavi N, Singh AK, Leehey DJ. Role of angiotensin II in glucose-induced inhibition of mesangial matrix degradation. Diabetes. 1999;48:2066–2073. doi: 10.2337/diabetes.48.10.2066. [DOI] [PubMed] [Google Scholar]
- 46.Jiang Y, Cypess AM, Muse ED, et al. Glucagon receptor activates extracellular signal-regulated protein kinase 1/2 via cAMP-dependent protein kinase. Proc Natl Acad Sci USA. 2001;98:10102–10107. doi: 10.1073/pnas.131200398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dalle S, Longuet C, Costes S, et al. Glucagon promotes cAMP-response element-binding protein phosphorylation via activation of ERK 1/2 in MIN6 cell line and isolated islets of Langerhans. J Biol Chem. 2004;279:20345–20355. doi: 10.1074/jbc.M312483200. [DOI] [PubMed] [Google Scholar]
- 48.Li XC, Carretero OA, Shao Y, Zhuo JL. Glucagon receptor-mediated ERK 1/2 phosphorylation in rat mesangial cells: role of protein kinase A and phospholipase C. Hypertension. 2006;47:580–585. doi: 10.1161/01.HYP.0000197946.81754.0a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li XC, Carretero OA, Zhuo JL. Cross-talk between angiotensin II and glucagon receptor signaling mediates phosphorylation of mitogen-activated protein kinases ERK 1/2 in rat glomerular mesangial cells. Biochem Pharmacol. 2006;71:1711–1719. doi: 10.1016/j.bcp.2006.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gomez E, Pritchard C, Herbert TP. cAMP-dependent protein kinase and Ca2+ influx through L-type voltage-gated calcium channels mediated raf-independent activation of extracellular regulated kinase in response to glucagon-like peptide-1 in pancreatic β cells. J Biol Chem. 2002;277:48146–48151. doi: 10.1074/jbc.M209165200. [DOI] [PubMed] [Google Scholar]
- 51.Kemp DM, Habener JF. Insulinotrophic hormone glucagon-like peptide 1 (GLP-1) activation of insulin gene promoter inhibited by p38 mitogen-activated protein kinase. Endocrinology. 2001;142:1179–1187. doi: 10.1210/endo.142.3.8026. [DOI] [PubMed] [Google Scholar]
- 52.Brenner BM, Cooper ME, de Zeeuw D, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001;345:861–869. doi: 10.1056/NEJMoa011161. [DOI] [PubMed] [Google Scholar]
- 53.Ravid M, Savin H, Jutrin I, Bental T, Katz B, Lishner M. Long-term stabilizing effect of angiotensin-converting enzyme inhibition on plasma creatinine and on proteinuria in normotensive type 2 diabetic patients. Ann Intern Med. 1993;118:577–581. doi: 10.7326/0003-4819-118-8-199304150-00001. [DOI] [PubMed] [Google Scholar]
- 54.Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting enzyme inhibition on diabetic nephropathy. N Engl J Med. 1993;329:1456–1462. doi: 10.1056/NEJM199311113292004. [DOI] [PubMed] [Google Scholar]
- 55.Hostetter TH. Hyperfiltration and glomerulosclerosis. Semin Nephrol. 2003;23:194–199. doi: 10.1053/anep.2003.50017. [DOI] [PubMed] [Google Scholar]
- 56.Sochett EB, Cherney DZ, Curtis JR, Dekker MG, Scholey JW, Miller JA. Impact of renin angiotensin system modulation on the hyperfiltration state in type 1 diabetes. J Am Soc Nephrol. 2006;17:1703–1709. doi: 10.1681/ASN.2005080872. [DOI] [PubMed] [Google Scholar]
- 57.Barnett AH, Bain SC, Bouter P, et al. Angiotensin-receptor blockade versus converting-enzyme inhibition in type 2 diabetes and nephropathy. N Engl J Med. 2004;351:1952–1961. doi: 10.1056/NEJMoa042274. [DOI] [PubMed] [Google Scholar]
- 58.Zhuo JL, Harris PJ, Skinner SL. ACE inhibition prevents glucagon-induced glomerular hyperfiltration in anesthetized rats. FASEB J. 2003;17:A923. [Google Scholar]
- 59.Li XC, Shao Y, D’Ambrosio M, Zhuo JL. Long-term hyperglucagonemia induces metabolic and renal phenotypes of early type 2 diabetes in mice: effects of glucagon receptors and angiotensin II. Hypertension. 2006;48:e75. [Google Scholar]
- 60.Wilkes BM, Mento PF, Vernace MA. Angiotensin responsiveness in hyperfiltering and nonhyperfiltering diabetic rats. J Am Soc Nephrol. 1993;4:1346–1353. doi: 10.1681/ASN.V461346. [DOI] [PubMed] [Google Scholar]
- 61.Singh R, Singh AK, Leehey DJ. A novel mechanism for angiotensin II formation in streptozotocin-diabetic rat glomeruli. Am J Physiol Renal Physiol. 2005;288:F1183–F1190. doi: 10.1152/ajprenal.00159.2003. [DOI] [PubMed] [Google Scholar]
- 62.Carey RM, Siragy HM. The intrarenal renin-angiotensin system and diabetic nephropathy. Trends Endocrinol Metab. 2003;14:274–281. doi: 10.1016/s1043-2760(03)00111-5. [DOI] [PubMed] [Google Scholar]
- 63.Singh R, Singh AK, Alavi N, Leehey DJ. Mechanism of increased angiotensin II levels in glomerular mesangial cells cultured in high glucose. J Am Soc Nephrol. 2003;14:873–880. doi: 10.1097/01.asn.0000060804.40201.6e. [DOI] [PubMed] [Google Scholar]
- 64.Vidotti DB, Casarini DE, Cristovam PC, Leite CA, Schor N, Boim MA. High glucose concentration stimulates intracellular renin activity and angiotensin II generation in rat mesangial cells. Am J Physiol Renal Physiol. 2004;286:F1039–F1045. doi: 10.1152/ajprenal.00371.2003. [DOI] [PubMed] [Google Scholar]
- 65.Riser BL, Denichilo M, Cortes P, et al. Regulation of connective tissue growth factor activity in cultured rat mesangial cells and its expression in experimental diabetic glomerulosclerosis. J Am Soc Nephrol. 2000;11:25–38. doi: 10.1681/ASN.V11125. [DOI] [PubMed] [Google Scholar]
- 66.Isono M, Cruz MC, Chen S, Hong SW, Ziyadeh FN. Extracellular signal-regulated kinase mediates stimulation of TGF-β1 and matrix by high glucose in mesangial cells. J Am Soc Nephrol. 2000;11:2222–2230. doi: 10.1681/ASN.V11122222. [DOI] [PubMed] [Google Scholar]
- 67.Banes AK, Shaw S, Jenkins J, et al. Angiotensin II blockade prevents hyperglycemia-induced activation of JAK and STAT proteins in diabetic rat kidney glomeruli. Am J Physiol Renal Physiol. 2004;286:F653–F659. doi: 10.1152/ajprenal.00163.2003. [DOI] [PubMed] [Google Scholar]
- 68.Johnson DG, Goebel CU, Hruby VJ, Bregman MD, Trivedi D. Hyperglycemia of diabetic rats decreased by a glucagon receptor antagonist. Science. 1982;215:1115–1116. doi: 10.1126/science.6278587. [DOI] [PubMed] [Google Scholar]
- 69.Ling A, Hong Y, Gonzalez J, et al. Identification of alkylidene hydrazides as glucagon receptor antagonists. J Med Chem. 2001;44:3141–3149. doi: 10.1021/jm000547o. [DOI] [PubMed] [Google Scholar]
- 70.Madsen P, Ling A, Plewe M, et al. Optimization of alkylidene hydrazide based human glucagon receptor antagonists. Discovery of the highly potent and orally available 3-cyano-4-hydroxybenzoic acid [1-(2,3,5,6-tetramethylbenzyl)-1H-indol-4-ylmethylene]hydrazide. J Med Chem. 2002;45:5755–5775. doi: 10.1021/jm0208572. [DOI] [PubMed] [Google Scholar]
- 71.Qureshi SA, Rios CM, Xie D, et al. A novel glucagon receptor antagonist inhibits glucagon-mediated biological effects. Diabetes. 2004;53:3267–3273. doi: 10.2337/diabetes.53.12.3267. [DOI] [PubMed] [Google Scholar]
- 72.Madsen P, Knudsen LB, Wiberg FC, Carr RD. Discovery and structure-activity relationship of the first non-peptide competitive human glucagon receptor antagonists. J Med Chem. 1998;41:5150–5157. doi: 10.1021/jm9810304. [DOI] [PubMed] [Google Scholar]
- 73.Liang Y, Osborne MC, Monia BP, et al. Reduction in glucagon receptor expression by an antisense oligonucleotide ameliorates diabetic syndrome in db/db mice. Diabetes. 2004;53:410–417. doi: 10.2337/diabetes.53.2.410. [DOI] [PubMed] [Google Scholar]
- 74.Sloop KW, Cao JX, Siesky AM, et al. Hepatic and glucagon-like peptide-1-mediated reversal of diabetes by glucagon receptor antisense oligonucleotide inhibitors. J Clin Invest. 2004;113:1571–1581. doi: 10.1172/JCI20911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Drucker DJ. Biologic actions and therapeutic potential of the proglucagon-derived peptides. Nat Clin Pract Endocrinol Metab. 2005;1:22–31. doi: 10.1038/ncpendmet0017. [DOI] [PubMed] [Google Scholar]
- 76.Holst JJ. Glucagon-like peptide-1: from extract to agent. Diabetologia. 2006;49:253–260. doi: 10.1007/s00125-005-0107-1. [DOI] [PubMed] [Google Scholar]
- 77.Deacon CF, Nauck MA, Toft-Nielsen M, Pridal L, Willms B, Holst JJ. Both subcutaneously and intravenously administered glucagon-like peptide I are rapidly degraded from the NH2-terminus in type II diabetic patients and in healthy subjects. Diabetes. 1995;44:1126–1131. doi: 10.2337/diab.44.9.1126. [DOI] [PubMed] [Google Scholar]
- 78.Hinke SA, Pospisilik JA, Demuth HU, et al. Dipeptidyl peptidase IV (DPIV/CD26) degradation of glucagon. J Biol Chem. 2000;276:3827–3834. doi: 10.1074/jbc.275.6.3827. [DOI] [PubMed] [Google Scholar]
- 79.Fehmann HC, Habener JF. Insulinotropic hormone glucagon-like peptide-I(7–37) stimulation of proinsulin gene expression and proinsulin biosynthesis in insulinoma β TC-1 cells. Endocrinology. 1992;130:159–166. doi: 10.1210/endo.130.1.1309325. [DOI] [PubMed] [Google Scholar]
- 80.Gutniak M, Orskov C, Holst JJ, Ahren B, Efendic S. Antidiabetogenic effect of glucagon-like peptide-1 (7–36)amide in normal subjects and patients with diabetes mellitus. N Engl J Med. 1992;326:1316–1322. doi: 10.1056/NEJM199205143262003. [DOI] [PubMed] [Google Scholar]
- 81.Deacon CF. MK-431 (Merck) Curr Opin Invest Drugs. 2005;6:419–426. [PubMed] [Google Scholar]
- 82.Herman GA, Bergman A, Stevens C, et al. Effect of single oral doses of sitagliptin, a dipeptidyl peptidase-4 inhibitor, on incretin and plasma glucose levels after an oral glucose tolerance test in patients with type 2 diabetes. J Clin Endocrinol Metab. 2006;91:4612–4619. doi: 10.1210/jc.2006-1009. [DOI] [PubMed] [Google Scholar]
- 83.Mari A, Sallas WM, He YL, et al. Vildagliptin, a dipeptidyl peptidase-IV inhibitor, improves model-assessed β-cell function in patients with type 2 diabetes. J Clin Endocrinol Metab. 2005;90:4888–4894. doi: 10.1210/jc.2004-2460. [DOI] [PubMed] [Google Scholar]
- 84.Ahren B, Landin-Olsson M, Jansson PA, Svensson M, Holmes D, Schweizer A. Inhibition of dipeptidyl peptidase-4 reduces glycemia, sustains insulin levels and reduces glucagon levels in type 2 diabetes. J Clin Endocrinol Metab. 2004;89:2078–2084. doi: 10.1210/jc.2003-031907. [DOI] [PubMed] [Google Scholar]
- 85.Parkes DG, Pittner R, Jodka C, Smith P, Young A. Insulinotropic actions of exendin-4 and glucagon-like peptide-1 in vivo and in vitro. Metab Clin Exp. 2001;50:583–589. doi: 10.1053/meta.2001.22519. [DOI] [PubMed] [Google Scholar]
- 86.Buse JB, Henry RR, Han J, et al. Effects of exenatide (exendin-4) on glycemic control over 30 weeks in sulfonylurea-treated patients with type 2 diabetes. Diabetes Care. 2004;27:2628–2635. doi: 10.2337/diacare.27.11.2628. [DOI] [PubMed] [Google Scholar]
- 87.Madsbad S, Schmitz O, Ranstam J, Jakobsen G, Matthews DR. Improved glycemic control with no weight increase in patients with type 2 diabetes after once-daily treatment with the long-acting glucagon-like peptide 1 analog liraglutide (NN2211): a 12-week, double-blind, randomized, controlled trial. Diabetes Care. 2004;27:1335–13342. doi: 10.2337/diacare.27.6.1335. [DOI] [PubMed] [Google Scholar]
- 88.Egan JM, Clocquet AR, Elahi D. The insulinotropic effect of acute exendin-4 administered to humans: comparison of nondiabetic state to type 2 diabetes. J Clin Endocrinol Metab. 2002;87:1282–1290. doi: 10.1210/jcem.87.3.8337. [DOI] [PubMed] [Google Scholar]
- 89.Feinglos MN, Saad MF, Pi-Sunyer FX, An B, Santiago O. Effects of liraglutide (NN2211), a long-acting GLP-1 analogue, on glycemic control and body weight in subjects with Type 2 diabetes. Diabetic Med. 2005;22:1016–1023. doi: 10.1111/j.1464-5491.2005.01567.x. [DOI] [PubMed] [Google Scholar]