

Abstract
Heat stress (HS) is known to induce delayed preconditioning against myocardial infarction 24 h later, but the exact signaling pathway of this response remains to be elucidated. In previous studies, we have shown an implication of protein kinase C (PKC) and mitogen activated protein kinase (p38 MAPK) in the HS-induced reduction in infarct size. Furthermore, in their phosphorylated state, small heat shock proteins (Hsp27) seem to confer cytoskeletal protection. In the present study, we sought to determine the influence of HS on the subcellular distribution of PKC isoforms and on Hsp27 phosphorylation.
Rats were subjected to either HS (42°C for 15 min, HS group) or sham anaesthesia (Sham group) before their hearts were excised. Myocardial tissue extracts obtained 20 min or 24 h after HS were processed for Western blot analysis.
In the HS group, PKC-ε translocated from the cytosolic to the particulate fraction (4426±128 vs 6258±316 arbitrary units, P=0.002). Chelerythrine (5 mg kg−1, ip), a PKC inhibitor, abolished this translocation. Western blot analysis of Hsp27, 24 h after HS, showed a marked increase in protein expression and phosphorylation in the particulate fraction.
In the present study, we have shown that HS induces the translocation of PKCε from the cytosolic to the particulate fraction. Along with our previous observation that PKC is a trigger of the HS-induced myocardial preconditioning, the results of the present study suggest an important role of the epsilon isoform of PKC in this cardioprotective mechanism. Furthermore, we have also demonstrated that the cytoprotective protein Hsp27 is phosphorylated following HS. We therefore can conclude that PKC and MAPK/Hsp27 are involved in the signaling pathway of the HS-induced cardioprotection.
Keywords: Animals; Blotting, Western; Heat-Shock Proteins; metabolism; Hyperthermia, Induced; Ischemic Preconditioning, Myocardial; Isoenzymes; metabolism; Male; Phosphorylation; Protein Kinase C; metabolism; Protein Kinase C-alpha; Protein Kinase C-delta; Protein Kinase C-epsilon; Protein Transport; Rats; Rats, Wistar; Signal Transduction
Introduction
Prior heat stress (HS) is known to induce delayed cardiac preconditioning, improving post-ischaemic mechanical function in both rat [1] and rabbit [2] myocardium. This cardioprotection is initiated by the liberation of different triggers following HS, such as catecholamines [3], reactive oxygen species [4] and nitric oxide (NO) [5], leading to the activation of different protein kinases. We have previously shown that the early activation of one or more PKC subtypes triggers HS-induced cardioprotection [6]. Similar results were obtained in vivo in the rat [7]. Furthermore, specific PKC isoforms are translocated from soluble to particulate fractions in response to multiple stresses. Indeed, PKC alpha, delta and epsilon are activated in rat myocardium after ischaemic preconditioning [8, 9] and numerous studies have shown the obligatory role of the epsilon isoform in various late cardioprotective mechanisms, such as ischaemic [10, 11], NO-induced [12] and hypoxic [13] preconditionings. On the other hand, it seems that mitogen-activated protein kinase (MAPK) cascade, and in particular the p38 MAPK [14], is also involved in the signaling pathway of the HS preconditioning. Moreover, MAPKAP-kinase 2, one of the substrates of p38 MAPK, can in turn phosphorylate small heat stress proteins (Hsp) Hsp27 [15], which is one potential mediator of the HS-induced cardioprotection.
The aim of the present study was two-fold. First, we thought to investigate whether PKC alpha, delta and epsilon isoform are involved in the HS-induced cardioprotective phenomenon, by measuring their translocations after HS. Secondly, we tested the hypothesis that HS also induces Hsp27 expression and phosphorylation, in the rat heart.
Methods
Experimental treatment groups
In accordance with French law and local ethical committee guidelines for animal research, male Wistar rats (280–300 g IFFA CREDO, Lyon, France) were housed in climate controlled conditions and provided with standard rat chow.
Rats were submitted to either heat stress (HS groups) or anesthesia without hyperthermia (Sham groups). Prior to this procedure, animals were treated or not with chelerythrine (Sigma) as previously described [6]. Subsequently, all animals were allowed to recover for 20 min (for PKC analysis) or 24 h (for Hsp27 analysis) before their hearts were rapidly excised and stored in liquid nitrogen.
In these two major groups (sacrificed 20 min or 24 h after HS), six experimental groups (n = 4 in each) were studied:
Sham group - rats received saline 10 min prior to anesthesia;
Sham+VC group - animals received the vehicle of chelerythrine (water containing 7% ethanol, 2 ml kg−1, ip) 10 min prior to anesthesia;
Sham+Chele group - animals were treated with cheleryhtrine (5 mg kg−1 in a volume of 2 ml kg−1, ip) 10 min prior to anesthesia;
In HS, HS+VC and HS+Chele groups, rats were similarly treated prior to heat stress. The experimental protocol is summarized in Figure 1.
Figure 1.
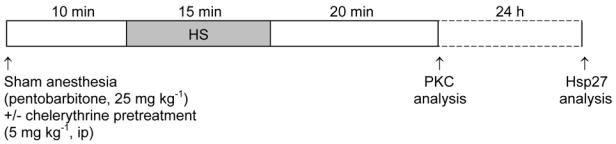
Experimental protocol.
Heat stress protocol
Heat stress was achieved by placing anesthetized (with 25 mg kg−1 ip sodium pentobarbitone) rats in an environmental chamber under an infrared light. Their body temperature, recorded with a rectal probe, was increased to 42 ± 0.2°C for 15 min. Sham control animals were anesthetized only. All rats were allowed to recover for 20 min or 24h.
Tissue sample preparation
Frozen myocardial tissue samples were minced and homogenized in a lysis buffer pH 7.4 containing 50 mM Tris-HCl, 5 mM EDTA, 1 mM NaF, 1 mM Na3VO4, l‰ microcystine and 5μl ml−1 protease inhibitors. The total cellular proteins in the homogenates were subjected to ultracentrifugation at 100,000 g for 60 min. The supernatants were collected (cytosolic fractions) and the pellets were resuspended in the above sample buffer (particulate fractions). Protein concentrations in homogenized samples were determined using BCA protein assay kit (Pierce).
Western immunoblotting analysis
The fractions were subjected to SDS-polyacrylamide gel electrophoresis on 8% polyacrylamide gels according to the method of Laemmli [16]. Proteins were then transferred to a PVDF membrane (Hybond-C, Amersham) incubated with antibodies towards alpha, delta or epsilon PKC isoforms (Transduction Laboratories) and Hsp27 (total and phosphorylated) (Upstate Biotechnology). The second antibody was horseradish peroxidase-conjugated anti-mouse IgG (Jackson ImmunoResearch Laboratories). The membrane was developed using an enhanced chemiluminescence system (Pierce) to obtain an autoradiogram. After scanning, the blots were analyzed for optical density (NIH Image for Windows) expressed in arbitrary units (a.u.). Each signal was normalized to the corresponding Ponceau signal.
Statistical analysis
All data are reported as mean±s.e.mean. Differences in optical density were analyzed using Student’s t-tests. P values <0.05 were considered significant.
Results
Effect of heat stress on subcellular distribution of myocardial PKC isoforms
Figure 2 shows representative data of Western blots indicating the subcellular distribution of PKC α, δ and ε isoforms. HS had no effect on the distribution of PKC α and δ, since these proteins were present to the same extent in the cytosolic and in the particulate fractions of both Sham and HS hearts (Figures 2B and 2D). In contrast, as shown in Figures 2E and 2F, PKCε, which was predominantly present in the cytosolic fraction of Sham hearts (5404±324 a.u. in cytosolic vs 3076±333 a.u. in particulate fraction, p=0.002), exhibited a significant translocation from the cytosolic to the particulate fraction 20 min after HS (4426±128 a.u. in cytosolic vs 6258±316 a.u. in particulate fraction, p=0.002). Pretreatment with chelerythrine, a PKC inhibitor, abolished this HS-induced translocation of PKCε (Figure 3).
Figure 2. Heat stress induced translocation of myocardial PKCε from cytosolic to particulate fraction, but had no effect on PKCα and δ distribution.
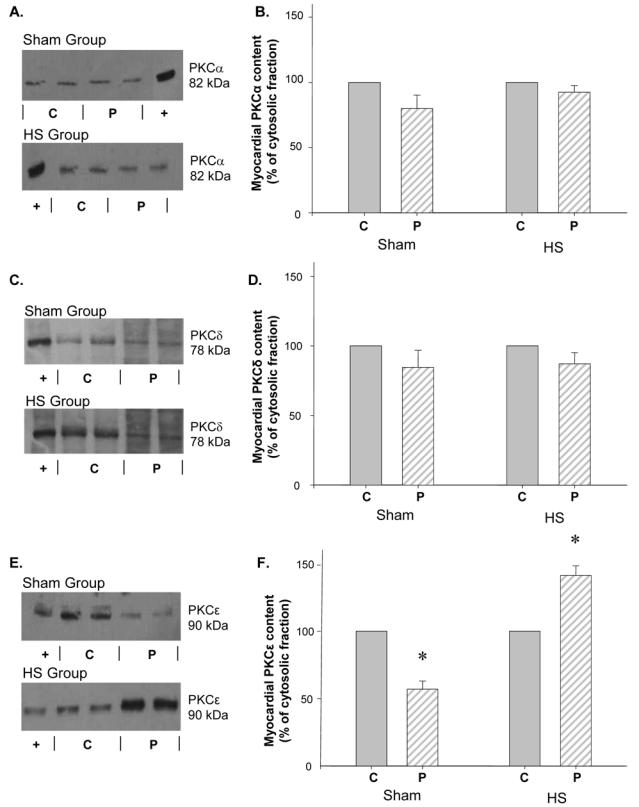
Western blots of myocardial PKCα (A.), δ (C.) and ε (E.) distribution between the cytosolic (C) and the particulate (P) fractions. (+) positive control of the different isoforms of PKC. Histograms represent the cellular distribution of PKCα (B.), δ (D.) and ε (F.), 20 min after HS. Data (mean±s.e.mean, *P<0.05 vs cytosolic fraction, n=4) are expressed as percentage of cytosolic values. Sham group: hearts excised 20 min after sham anaesthesia; HS group: hearts excised 20 min after heat stress.
Figure 3. Chelerythrine pretreatment abolished the cellular translocation of myocardial PKCε following heat stress.
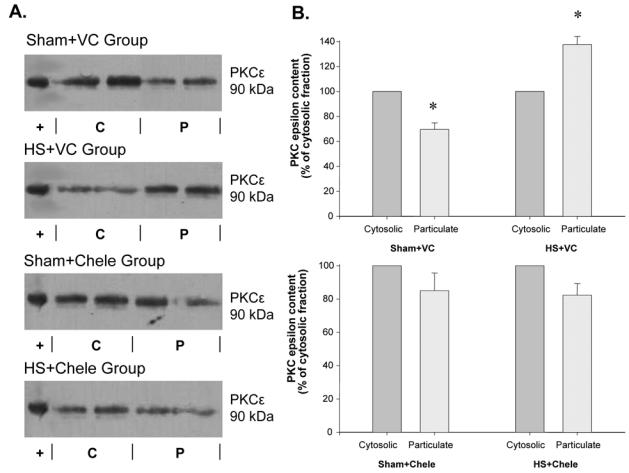
Western blots of myocardial PKCε (90 kDa) distribution (A.) between the cytosolic (C) and the particulate (P) fractions. (+): positive control of PKCε. Histograms represent the cellular distribution of PKCα (B.), 20 min after HS. Data (mean±s.e.mean, *P<0.05 vs cytosolic fraction, n=4) are expressed as percentage of cytosolic values. Sham group: hearts excised 20 min after sham anaesthesia; HS group: hearts excised 20 min after heat stress; VC: rats were pretreated with the vehicle of chelerythrine (water containing 7% ethanol, 2 ml kg−1, ip); Chele: rats were pretreated with chelerythrine (5 mg kg−1, ip).
Effect of heat stress on myocardial Hsp27 content, distribution and phosphorylation
Western blot analysis of this protein 24 h after HS showed a marked increase in Hsp27 expression, both in the cytosolic and in the particulate fractions (Figure 4). In addition, we observed a significant phosphorylation of Hsp27 in the particulate fraction of the HS hearts compared to that of Sham hearts (Figure 5).
Figure 4. Myocardial Hsp27 content was markedly increased in both the cytosolic and the particulate fractions, 24 h after heat stress.
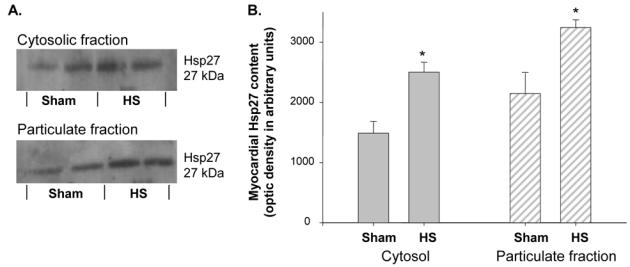
A. Western blot analysis of myocardial Hsp27 expression in the cytosolic and the particulate fractions of Sham and HS hearts. B. Histograms representing the cellular distribution and content of Hsp27, 24 h after HS. Data are expressed in arbitrary units (mean±s.e.mean, *p=0.001 vs Sham group, n=4). Sham group: hearts excised 24 h after sham anaesthesia; HS group: hearts excised 24 h after heat stress.
Figure 5. Heat stress induced a phosphorylation of myocardial Hsp27 in the particulate fraction.
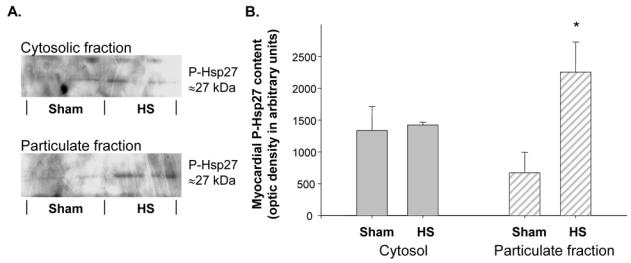
A. Western blot analysis of myocardial Hsp27 phosphorylation in the cytosolic and the particulate fractions of Sham and HS hearts. B. Histograms representing the cellular distribution and content of Phospho-Hsp27, 24 h after HS (*P=0.001 vs Sham group, n=4). Data are expressed in arbitrary units (mean±s.e.mean). Sham group: hearts excised 24 h after sham anaesthesia; HS group: hearts excised 24 h after heat stress; P-Hsp27: Phospho-Hsp27.
Discussion
The present study provides new insight into the cellular signaling pathways of the HS response. We report a significant modification in the subcellular distribution of myocardial PKC epsilon, since this protein is translocated from the cytosol to the particulate fraction, 20 min after HS. We also observed a significant increase in the expression and phosphorylation of the cytoprotective protein Hsp27, 24 h after HS. These data suggest that PKCε may act as a trigger of HS-induced preconditioning and that Hsp27 could be an end-effector of this response.
Protein kinase C and myocardial protection
We can conclude from these results that HS induces PKCε activation, since translocation from the cytosolic to the particulate fraction is known to be an important event for the activation of this isozyme [17]. Chelerythrine, a selective PKC inhibitor [18], used at a dose known to abolish the HS-induced cardioprotection against infarction in the rat myocardium [6], abolished myocardial PKCε translocation confirming that this isoform is involved in HS preconditioning and that translocation is mandatory for cardioprotection to occur. PKCε activation has also been shown to occur in other myocardial preconditionings, such as NO donor administration [19], rapid pacing [20] and ischemic preconditioning [10].
Role of Hsp27 in HS preconditioning
In the present study, we have also shown 24 h after HS an increased expression and phosphorylation of myocardial Hsp27, which has been identified as one of the potential end-effectors of HS-induced myocardial cardioprotection. Direct evidence of the cytoprotective properties of this protein has been demonstrated by the protection against ischemic injury seen in cardiac myocytes overexpressing Hsp27 [21]. Furthermore, phosphorylation of Hsp27 has been shown to enhance its cytoprotective activity by stabilizing the actin cytoskeleton [15]. Thus, HS-induced Hsp27 expression as well as phosphorylation could be involved in the cardioprotective effects of HS.
Hypothetical signaling pathway of the HS response
Both PKC [6] and p38 MAPK [14] are involved in HS-induced myocardial protection and it is well known that p38 MAPK, through MAPKAPK-2, induces Hsp27 phosphorylation [15]. Furthermore, a proteomic analysis reveals that PKCε-dependant cardioprotection requires development of new protein-protein interactions involving PKCε. Thus, molecules, such as MAPK or Hsp27, which have been implicated in myocardial preconditioning, are recruited to the PKCε complexes [22]. It has recently been shown that activation of PKCε enhances mitochondrial co-localisation of PKCε with MAPK, increases phosphorylation of mitochondrial MAPK and promotes the formation of mitochondrial PKCε-MAPK signalling modules, which are associated with the genesis of a cardioprotective phenotype [23].
Finally, other proteins have been implicated as potential end-effectors mediating delayed cardioprotection after HS. For instance, we and others have shown that HS-induced cardioprotection is dependent on the opening of mitochondrial ATP-sensitive potassium (mito KATP) channels [24, 25]. Accordingly, the integrity of the actin cytoskeleton has a regulatory function in gating of cardiac mito KATP channels [26, 27] and it has recently been demonstrated that ischaemic preconditioning depends on interaction between mito KATP channels and actin cytoskeleton [28]. Since the mitochondrion is a preferential location where PKCε-MAPK modules engage in signal transduction [23], we hypothesize that, in response to HS, the formation and activation of PKCε-MAPK modules in the mitochondrion induce Hsp27 phosphorylation, which may result in preservation of actin microfilaments. This in turn may maintain the opening of mito KATP channels, leading to HS-induced cardioprotection.
In conclusion, we previously reported that PKC has a pivotal role in HS-induced cardioprotection and, in the present study, we have shown that the epsilon isoform of PKC is translocated from the cytosolic to the particulate fraction, 20 min after HS. Furthermore, we have demonstrated that Hsp27 is expressed and phosphorylated following HS. We can thus suggest a role for both PKC and MAPK/Hsp27 pathways as mediators of the HS-induced cardioprotection. Further investigations are needed to assess the link between the various mediators of this HS response.
Footnotes
The definitive version is available at www.blackwell-synergy.com
References
- 1.Currie RW, Karmazyn M, Kloc M, Mailer K. Heat-shock response is associated with enhanced postischemic ventricular recovery. CircRes. 1988;63:543–9. doi: 10.1161/01.res.63.3.543. [DOI] [PubMed] [Google Scholar]
- 2.Yellon DM, Pasini E, Cargnoni A, et al. The protective role of heat stress in the ischaemic and reperfused rabbit myocardium. J Mol Cell Cardiol. 1992;24:895–907. doi: 10.1016/0022-2828(92)91102-b. [DOI] [PubMed] [Google Scholar]
- 3.Joyeux M, Godin-Ribuot D, Patel A, et al. Infarct size-reducing effect of heat stress and alpha1 adrenoceptors in rats. Br J Pharmacol. 1998;125:645–50. doi: 10.1038/sj.bjp.0702137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arnaud C, Joyeux M, Garrel C, et al. Free-radical production triggered by hyperthermia contributes to heat stress-induced cardioprotection in isolated rat hearts. Br J Pharmacol. 2002;135:1776–82. doi: 10.1038/sj.bjp.0704619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arnaud C, Laubriet A, Joyeux M, et al. Role of nitric oxide synthases in the infarct size-reducing effect conferred by heat stress in isolated rat hearts. Br J Pharmacol. 2001;132:1845–51. doi: 10.1038/sj.bjp.0703942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Joyeux M, Baxter GF, Thomas DL, Ribuot C, Yellon DM. Protein kinase C is involved in resistance to myocardial infarction induced by heat stress. J Mol Cell Cardiol. 1997;29:3311–9. doi: 10.1006/jmcc.1997.0556. [DOI] [PubMed] [Google Scholar]
- 7.Yamashita N, Hoshida S, Nishida M, et al. Time course of tolerance to ischemia-reperfusion injury and induction of heat shock protein 72 by heat stress in the rat heart. J Mol Cell Cardiol. 1997;29:1815–21. doi: 10.1006/jmcc.1997.0416. [DOI] [PubMed] [Google Scholar]
- 8.Yoshida K, Kawamura S, Mizukami Y, Kitakaze M. Implication of protein kinase C-alpha, delta, and epsilon isoforms in ischemic preconditioning in perfused rat hearts. J Biochem (Tokyo) 1997;122:506–11. doi: 10.1093/oxfordjournals.jbchem.a021781. [DOI] [PubMed] [Google Scholar]
- 9.Kawamura S, Yoshida K, Miura T, Mizukami Y, Matsuzaki M. Ischemic preconditioning translocates PKC-delta and -epsilon, which mediate functional protection in isolated rat heart. Am J Physiol. 1998;275:H2266–71. doi: 10.1152/ajpheart.1998.275.6.H2266. [DOI] [PubMed] [Google Scholar]
- 10.Qiu Y, Ping P, Tang XL, et al. Direct evidence that protein kinase C plays an essential role in the development of late preconditioning against myocardial stunning in conscious rabbits and that epsilon is the isoform involved. J Clin Invest. 1998;101:2182–98. doi: 10.1172/JCI1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ping P, Zhang J, Qiu Y, et al. Ischemic preconditioning induces selective translocation of protein kinase C isoforms epsilon and eta in the heart of conscious rabbits without subcellular redistribution of total protein kinase C activity. Circ Res. 1997;81:404–14. doi: 10.1161/01.res.81.3.404. [DOI] [PubMed] [Google Scholar]
- 12.Ping P, Takano H, Zhang J, et al. Isoform-selective activation of protein kinase C by nitric oxide in the heart of conscious rabbits: a signaling mechanism for both nitric oxide- induced and ischemia-induced preconditioning. Circ Res. 1999;84:587–604. doi: 10.1161/01.res.84.5.587. [DOI] [PubMed] [Google Scholar]
- 13.Rafiee P, Shi Y, Kong X, et al. Activation of protein kinases in chronically hypoxic infant human and rabbit hearts: role in cardioprotection. Circulation. 2002;106:239–45. doi: 10.1161/01.cir.0000022018.68965.6d. [DOI] [PubMed] [Google Scholar]
- 14.Joyeux M, Boumendjel A, Carroll R, et al. SB 203580, a mitogen-activated protein kinase inhibitor, abolishes resistance to myocardial infarction induced by heat stress. Cardiovasc Drugs Ther. 2000;14:337–43. doi: 10.1023/a:1007847111368. [DOI] [PubMed] [Google Scholar]
- 15.Guay J, Lambert H, Gingras-Breton G, et al. Regulation of actin filament dynamics by p38 map kinase-mediated phosphorylation of heat shock protein 27. J Cell Sci. 1997;110 (Pt 3):357–68. doi: 10.1242/jcs.110.3.357. [DOI] [PubMed] [Google Scholar]
- 16.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–5. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 17.Balafanova Z, Bolli R, Zhang J, et al. Nitric oxide induces nitration of PKCepsilon, facilitating PKCepsilon translocation via enhanced PKCepsilon -RACK2 interactions: A novel mechanism of NO-triggered activation of PKCepsilon. J Biol Chem. 2002;11:11. doi: 10.1074/jbc.M112451200. [DOI] [PubMed] [Google Scholar]
- 18.Herbert JM, Augereau JM, Gleye J, Maffrand JP. Chelerythrine is a potent and specific inhibitor of protein kinase C. Biochem Biophys Res Commun. 1990;172:993–9. doi: 10.1016/0006-291x(90)91544-3. [DOI] [PubMed] [Google Scholar]
- 19.Vondriska TM, Zhang J, Song C, et al. Protein kinase C epsilon-Src modules direct signal transduction in nitric oxide-induced cardioprotection: complex formation as a means for cardioprotective signaling. Circ Res. 2001;88:1306–13. doi: 10.1161/hh1201.092994. [DOI] [PubMed] [Google Scholar]
- 20.Wilson S, Song W, Karoly K, et al. Delayed cardioprotection is associated with the sub-cellular relocalisation of ventricular protein kinase C epsilon, but not p42/44MAPK. Mol Cell Biochem. 1996;160–161:225–30. doi: 10.1007/BF00240053. [DOI] [PubMed] [Google Scholar]
- 21.Martin JL, Mestril R, Hilal-Dandan R, Brunton LL, Dillmann WH. Small heat shock proteins and protection against ischemic injury in cardiac myocytes. Circulation. 1997;96:4343–8. doi: 10.1161/01.cir.96.12.4343. [DOI] [PubMed] [Google Scholar]
- 22.Ping P, Zhang J, Pierce WM, Jr, Bolli R. Functional proteomic analysis of protein kinase C epsilon signaling complexes in the normal heart and during cardioprotection. Circ Res. 2001;88:59–62. doi: 10.1161/01.res.88.1.59. [DOI] [PubMed] [Google Scholar]
- 23.Baines CP, Zhang J, Wang GW, et al. Mitochondrial PKCepsilon and MAPK form signaling modules in the murine heart: enhanced mitochondrial PKCepsilon-MAPK interactions and differential MAPK activation in PKCepsilon-induced cardioprotection. Circ Res. 2002;90:390–7. doi: 10.1161/01.res.0000012702.90501.8d. [DOI] [PubMed] [Google Scholar]
- 24.Hoag JB, Qian YZ, Nayeem MA, D’Angelo M, Kukreja RC. ATP-sensitive potassium channel mediates delayed ischemic protection by heat stress in rabbit heart. Am J Physiol. 1997;273:H2458–64. doi: 10.1152/ajpheart.1997.273.5.H2458. [DOI] [PubMed] [Google Scholar]
- 25.Joyeux M, Godin-Ribuot D, Ribuot C. Resistance to myocardial infarction induced by heat stress and the effect of ATP-sensitive potassium channel blockade in the rat isolated heart. Br J Pharmacol. 1998;123:1085–8. doi: 10.1038/sj.bjp.0701710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Furukawa T, Yamane T, Terai T, Katayama Y, Hiraoka M. Functional linkage of the cardiac ATP-sensitive K+ channel to the actin cytoskeleton. Pflugers Arch. 1996;431:504–12. doi: 10.1007/BF02191896. [DOI] [PubMed] [Google Scholar]
- 27.Terzic A, Kurachi Y. Actin microfilament disrupters enhance K(ATP) channel opening in patches from guinea-pig cardiomyocytes. J Physiol. 1996;492 (Pt 2):395–404. doi: 10.1113/jphysiol.1996.sp021316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baines CP, Liu GS, Birincioglu M, et al. Ischemic preconditioning depends on interaction between mitochondrial KATP channels and actin cytoskeleton. Am J Physiol. 1999;276:H1361–8. doi: 10.1152/ajpheart.1999.276.4.H1361. [DOI] [PubMed] [Google Scholar]