

Abstract
The phosphatidylinositol 3-kinase (PI3K) signaling pathway modulates growth, proliferation and cell survival in diverse tissue types and plays specialized roles in the nervous system including influences on neuronal polarity, dendritic branching and synaptic plasticity. The tumor-suppressor phosphatase with tensin homology (PTEN) is the central negative regulator of the PI3K pathway. Germline PTEN mutations result in cancer predisposition, macrocephaly and benign hamartomas in many tissues, including Lhermitte-Duclos disease, a cerebellar growth disorder. Neurological abnormalities including autism, seizures and ataxia have been observed in association with inherited PTEN mutation with variable penetrance. It remains unclear how loss of PTEN activity contributes to neurological dysfunction. To explore the effects of Pten deficiency on neuronal structure and function, we analyzed several ultra-structural features of Pten-deficient neurons in Pten conditional knockout mice. Using Golgi stain to visualize full neuronal morphology, we observed that increased size of nuclei and somata in Pten-deficient neurons was accompanied by enlarged caliber of neuronal projections and increased dendritic spine density. Electron microscopic evaluation revealed enlarged abnormal synaptic structures in the cerebral cortex and cerebellum. Severe myelination defects included thickening and unraveling of the myelin sheath surrounding hypertrophic axons in the corpus callosum. Defects in myelination of axons of normal caliber were observed in the cerebellum, suggesting intrinsic abnormalities in Pten-deficient oligodendrocytes. We did not observe these abnormalities in wild-type or conditional Pten heterozygous mice. Moreover, conditional deletion of Pten drastically weakened synaptic transmission and synaptic plasticity at excitatory synapses between CA3 and CA1 pyramidal neurons in the hippocampus. These data suggest that Pten is involved in mechanisms that control development of neuronal and synaptic structures and subsequently synaptic function.
Keywords: PTEN, PI3K, brain, neurons, myelin, hypertrophy, synaptic plasticity
The tumor suppressor PTEN (phosphatase and tensin homolog, deleted on chromosome 10) is important in the modulation of cell growth, proliferation and survival. Inactivating mutations in both alleles of PTEN occur in diverse tumor types including glioblastoma multiforme (Chow and Baker, 2006). Consistent with the two-hit hypothesis of tumor suppression, inherited mutations in PTEN are associated with cancer predisposition, and the second allele of PTEN is inactivated in tumors arising in these patients. This indicates that loss of both PTEN alleles is important for tumorigenesis. Clinical manifestations of inherited PTEN mutation occur with highly variable penetrance and also include benign hamartomas in multiple tissues, macrocephaly, seizures, ataxia, mental retardation and autism in addition to cancer predisposition (Ali et al., 1999, Goffin et al., 2001, Eng, 2003, Butler et al., 2005). Changes in PTEN levels have recently been linked to Alzheimer’s disease (Zori et al., 1998, Goffin et al., 2001, Butler et al., 2005, Griffin et al., 2005, Rickle et al., 2006). While the tumor suppressor activity of Pten has been extensively studied in multiple tissue types, work relating Pten loss of function to neuronal dysfunction has only recently been studied. Kwon et al. (Kwon et al., 2006) demonstrated a link between Pten loss and abnormal social interactions and responses to sensory stimuli using a mouse model in which Pten was deleted in select post-mitotic neurons in the cerebral cortex and the hippocampus. Abnormalities in dendrites and axonal tracts were also noted. Other work has demonstrated that decreased Pten expression can increase dendritic branching of developing hippocampal neurons in vitro (Jaworski et al., 2005).
Pten is the central negative regulator of the phosphatidylinositol 3′-kinase (PI3K) pathway that transduces signals for growth, proliferation and survival (Vivanco and Sawyers, 2002, Sulis and Parsons, 2003). In response to extracellular growth signals, PI3K phosphorylates the 3′ position of the signaling lipid phosphatidylinositol 4,5 bisphosphate (PIP2) to generate phosphatidylinositol 3,4,5-triphosphate (PIP3). PTEN is a phosphatase that removes the phosphate from the 3′ position of PIP3, thus halting the signal transduced by PI3K. In addition to its role as a tumor suppressor, PTEN plays critical functions in normal development and homeostasis. This is illustrated clinically by the diverse phenotypic spectrum observed in individuals with germline PTEN mutations, as well as experimentally in a series of mouse models in which Pten is deleted in the germline, or selectively deleted in specific tissues. Germline deletion of Pten caused embryonic lethality in mice, while Pten heterozygous mice survived, but developed tumors in the endometrium, liver, prostate, adrenal gland, gastrointestinal tract, thyroid and thymus (Suzuki et al., 1998, Podsypanina et al., 1999, Raftopoulou et al., 2004). Tissue-specific deletion of Pten results in tumors in the prostate, testis, skin, liver, and mammary glands (Di Cristofano et al., 1998, Suzuki et al., 2001, Li et al., 2002, Kimura et al., 2003, Trotman et al., 2003, Wang et al., 2003, Horie et al., 2004). While selective deletion of Pten in astrocytes can contribute to tumorigenesis in brain when combined with other genetic mutations (Holland et al., 2000, Xiao et al., 2002), deletion of Pten alone did not induce brain tumors in mice. Instead, conditional loss of Pten in different cell types in the nervous system resulted in alterations in cell migration, cell number and cell size (Backman et al., 2001, Groszer et al., 2001, Kwon et al., 2001, Marino et al., 2002). Indeed, the defects in cell migration and cell size regulation observed in Pten-deficient granule neurons of the cerebellum created a mouse model with many histopathological features of human Lhermitte-Duclos disease (Backman et al., 2001, Kwon et al., 2001). Hypertrophy of Pten-deficient neurons was dependent on activity of the rapamycin-sensitive complex of the serine-threonine kinase mTor (Kwon et al., 2003).
We and others previously reported that conditional knock-out of Pten in a substantial proportion of neurons and astrocytes throughout the brain caused severe progressive macrocephaly, seizures, ataxia and premature death. Abnormalities in the brain included neuronal migration defects in the hippocampus and cerebellum, increased nuclear and soma size of both neurons and astrocytes, and a low level of proliferation of astrocytes in the adult brain with no detectable tumor growth (Fraser et al., 2004, Wei et al., 2006). To gain insights into the functional and structural abnormalities associated with disrupted Pten signaling in brain in vivo, we evaluated a number of subcellular structural features and tested synaptic transmission and synaptic plasticity in brains from mice with conditional deletion of Pten.
Materials and Methods
Transgenic Mice
GFAP-cre transgenic mice, a gift from David H. Gutmann (Washington University School of Medicine), were previously described (Bajenaru et al., 2002) and were used to drive expression of cre recombinase specifically in the nervous system of the mouse. GFAP-cre mice were bred with ROSA26R Cre reporter mice (Soriano, 1999) to identify cell types in which cre-mediated deletion occurred in vivo as previously described (Fraser et al., 2004). Cre activity was visualized by immunofluorescent detection of the β-galactosidase reporter. To obtain Pten conditional loss in the nervous system of the mouse, Ptenloxp/loxp mice (Suzuki et al., 2001), a gift from Tak Mak (University of Toronto), were crossed with the GFAP-cre mice for two generations to generate Ptenloxp/loxp;GFAP-cre mice, (hereafter referred to as Pten cKO mice) as well as littermate control mice.
Immunohistochemistry
Mice were transcardially perfused with 2% paraformaldehyde (PFA) in PBS. Tissues were post-fixed in 2% PFA for 24 hours, and then fixed in 4% PFA for a further 24 hours. Tissues were then embedded in paraffin and cut into 5-μm sections. GFAP-cre;ROSA26R mice were transcardially perfused with 2% PFA in PBS. These tissues were post fixed in 2% PFA for 24 hours and placed in 25% sucrose in PBS for a further 24 hours. Tissues were then embedded in TBS Tissue Freezing Medium (Triangle Biomedical Sciences) for frozen sectioning at 10-μm. The Ptenloxp/loxp allele expresses wild-type Pten until cre-mediated deletion. Therefore, littermate mice with the genotypes Pten+/+;GFAP-cre, Pten+/+ or Ptenloxp/loxp without Cre served as wild-type controls and Pten+/loxp;GFAP-cre mice provided Pten conditional heterozygote controls. We performed immunofluorescence with primary antibodies to β-galactosidase (ICN), CC-1 (Calbiochem), and Synaptophysin (Chemicon) and fluorescein isothio-cyanate or cyanine 3-conjugated secondary antibodies (Jackson Immuno Research). We used microwave antigen retrieval for all antibodies except for Synaptophysin. We performed immunohistochemistry with primary antibody against Pten (Cell Signaling), biotin labeled secondary antibody and detection by peroxidase-conjugated avidin (Elite ABC, Vector Laboratories) treated with 3,3′-diaminobenzidine substrate, followed by counter stain with hematoxylin (Vector Laboratories). We also performed standard hematoxylin and eosin (H&E) (Richard Allen Scientific) stain on paraffin sections.
Nucleolar size and number measurements
The Bioquant system (Bioquant Image Analysis Corp.) was used to outline and calculate the area of individual nucleoli from 100 neurons in layer 5 of the cerebral cortex of 4 control and 4 Pten cKO mouse brain tissue sections stained with H&E. The average nucleolar area per mouse was recorded and the four averages from each mouse were averaged together to produce the average area of a nucleolus in control and Pten cKO mice. The number of nucleoli per cell was counted for 125 layer 5 neurons of the cerebral cortex in 4 control and 4 Pten cKO mice on a bright field microscope at 100X magnification.
Golgi Stain
Mice were anesthetized and cervical dislocation was performed prior to removing the brain. Brain tissue was washed in 0.1M-phosphate buffer, pH 7.4 and then processed using the FD Rapid GolgiStain Kit according to the manufacturers protocol (FD Neuro Technologies). Brain tissue was immersed in impregnation solution and kept in the dark for one week and then placed in solution C for one week to finish the impregnation process. Tissues were snap frozen and sectioned at 100–200μm for development in a combination of solution D and E for ten minutes following a series of ethanol dehydration steps ranging from 50%, 70%, 95% and 100% and Xylenes before mounting with cover slips.
Electron Microscopy
Mice were transcardially perfused with 2% PFA in PBS. Tissue was fixed in 4% Gluteraldehyde in PBS and post-fixed in 1% osmic acid in PBS. Tissues were dehydrated in an ethanol gradient from 50% to 100% and then embedded in Spurr resin. Semi-thin sections cut at 700–900 angstroms were stained with 1% Toluidine blue (Electron Microscopy sciences) in sodium borate buffer for 1 minute at 60°C. Thin sections were stained with 2% uranyl acetate and Reynolds Lead citrate and viewed with the JOEL 1200 EX electron microscope and images were obtained on the GATAN or AMT digital camera system.
Electrophysiology
Hippocampal slices were prepared from Pten cKO and wild-type littermate mice. Slices were continuously superfused with artificial cerebrospinal fluid (ACSF) containing 125 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 2 mM MgSO4, 1.25 mM NaH2PO4, 26 mM NaHCO3, and 10 mM glucose, with 95% O2 and 5% CO2 at 30–31°C (2 ml/min). Schaffer collateral synapses were stimulated with a bipolar tungsten electrode in CA1 stratum radiatum placed 200–300 μm from the recording pipette filled with ACSF, and field excitatory postsynaptic potentials (fEPSPs) were collected using a Multiclamp 700B amplifier (Molecular Devices). In long-term potentiation (LTP) experiments, Shaffer collaterals were stimulated with a pair of stimuli (50 ms interpulse interval) at 0.033 Hz before and after induction of LTP. Stimulation intensities were chosen to evoke an fEPSP with a slope of the initial phase that is 30–35% of the slope of fEPSP evoked with a maximal stimulation. LTP was induced by a 200-Hz pulse protocol consisting of 10 trains of 200 ms stimulation at 200 Hz delivered every 5 s at baseline stimulation intensity. Data were analyzed using Clampfit 9.0 software (Molecular Devices). Results were then grouped according to mouse genotypes.
Results
Ultra-structural changes in neurons of Pten cKO mice
The GFAP-cre transgenic line (Bajenaru et al., 2002) used to drive cre activity in Pten cKO mice for this study induced conditional deletion of Pten in astrocytes throughout the brain as well as widespread deletion in neuronal populations including 80–90% of hippocampal neurons and cerebellar granule neurons, and 50–80% of pyramidal neurons in the cerebral cortex (Fraser et al., 2004). Pten-deficient neurons consistently demonstrated cell autonomous increases in the size of nuclei and somata (Fraser et al., 2004). We further evaluated the ultra-structural features of neurons from cerebral cortex of Pten cKO mice using electron microscopy. There were several obvious changes in the Pten-null pyramidal neurons in the cerebral cortex compared with controls. Consistent with our previous studies, the increased size of nuclei and somata was readily observed in neurons from Pten cKO cerebral cortex compared to controls (Fig. 1A and 1B). Interestingly, Pten cKO mitochondria also appeared substantially enlarged.
Fig. 1.
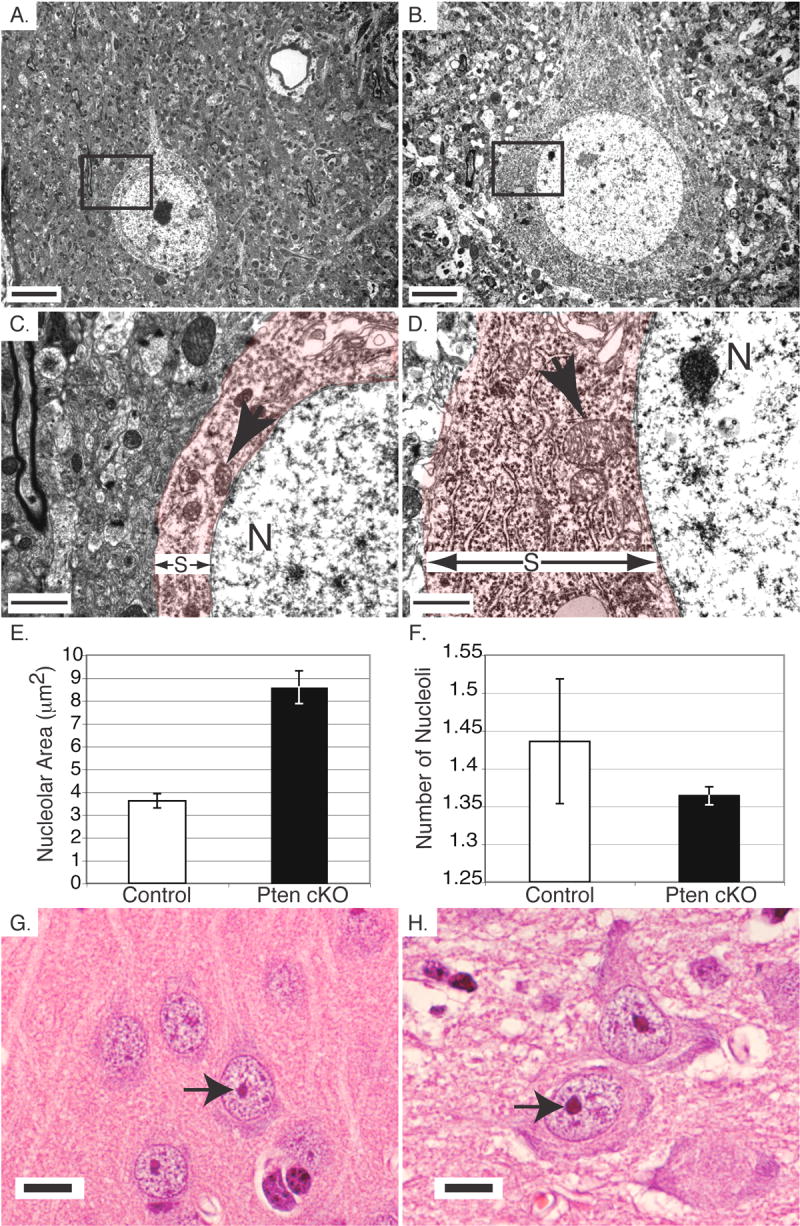
Hypertrophy of Pten cKO neurons was associated with increased ribosome density and increased nucleolar size. A,B. Electron microscopy of neurons in layer 5 of the cerebral cortex of control (A) and Pten cKO (B) mice. The boundary of the cell membrane with extra cellular matrix is less defined in the Pten cKO tissue compared with the control. C,D. Electron microscopy of the boxed areas from Figure 1A and 1B of control (C) and Pten cKO mice (D). The nucleus and mitochondria are labeled with an N and arrow, respectively. There is a dramatic increase in ribosome density and mitochondrial size in the Pten cKO (D) mice compared with the control (C). The increased soma size is indicated with red color and an S in between two arrows. E. The area of nucleoli in cerebral cortex neurons in control (white bar) and Pten cKO mice (black bar) was measured. Nucleoli were enlarged in the Pten cKO mice compared with the controls. F. The number of nucleoli per cell in control (white bar) and Pten cKO (black bar) mice were counted. There was no significant difference in the number of nucleoli per cell between control and Pten cKO mice. G,H. H&E stained sections of cerebral cortex pyramidal neurons from control (G) and Pten cKO mice (H). The increased nucleolar size is apparent in the Pten cKO mice. Scale bar for A,B=5 μm, C,D=1μm and G,H=10μm. Mice were >10 weeks old. Paired t-test P values for E=0.0108 and F=0.4773.
While the somata of control neurons were easily distinguished from the more electron-dense surrounding neuropil, cytoplasm from Pten cKO neurons was much more electron dense, therefore it was more difficult to visualize the boundaries of somata. The increased staining of Pten cKO somata, viewed at higher magnification, was due to a substantial increase in the number of ribosomes (Fig. 1C and 1D). This increased ribosome density extended into the dendritic and axonal projections proximal to the somata.
Because the nucleolus is the site of ribosome biogenesis, we compared Pten cKO neurons to controls to determine whether the increase in ribosome number was associated with a change in the size or number of nucleoli. We used the Bioquant system to measure the area of nucleoli contained in layer 5 pyramidal neurons from control and Pten cKO brains on sections stained with H&E. The average area of nucleoli in the Pten cKO mice (p<0.0001, n=400) was significantly larger than the control (n=400) (Fig. 1E). Interestingly, there did not appear to be a substantial change in the number of nucleoli per cell when comparing control to Pten cKO neurons (p=0.0863, n=500) (Fig. 1F). The dramatic increase in the size of the nucleoli is visible when comparing control and Pten cKO neurons in the cerebral cortex (Fig. 1G and 1H).
Pten cKO neurons contain enlarged synapses with defective synaptic structure
To study the effects of Pten-deficient hypertrophy on the complete neuronal structure including the cell body and all of its projections, we performed a Golgi stain on adult control and Pten cKO brains. The enlarged neuronal somata demonstrated previously by Pten immunostaining (Fraser et al., 2004) and electron microscopy (Fig. 1A and 1B), were readily visible in Golgi-stained sections of cerebral cortex from Pten cKO mice compared with control mice (Fig. 2A and 2B). In addition to increased soma size, the calibers of dendritic and axonal projections were also dramatically enlarged without obvious changes in branching. At higher magnification, the enlarged dendritic processes in Pten cKO mice showed a substantial increase in the density of spines compared to control (Fig. 2C and 2D). Further, for many spines, the morphology was abnormal and lacked the distinct mushroom-shaped termini (Fiala et al., 2002) that were characteristic for the majority of spines in neurons from control mice.
Fig. 2.
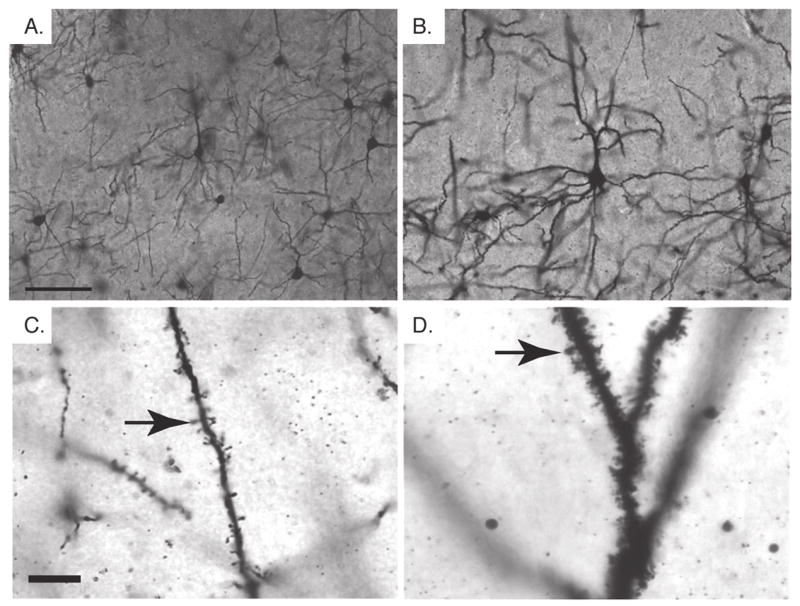
Neuronal hypertrophy in Pten cKO mice. A,B. Golgi stained neurons in the cerebral cortex of adult control (A) and Pten cKO (B) mice. Hypertrophy of Pten cKO neurons (B) involved the somata and all projections as compared to control neurons (A). C,D. At higher magnification, dendritic branches from control (C) and Pten cKO (D) mice are shown. The dendritic spines (arrow) in the control animal are spaced out along the dendrite and contain the rounded termini characteristic of mature spines. The spines (arrow) in the Pten cKO mice are densely packed and many lack the terminal rounded structure observed in the control spines. Mice were >10 weeks old. Scale bar for A,B=50μm and for C,D=10μm.
Based on the changes in dendritic spines observed by Golgi stain in Pten cKO mice, we used electron microscopy to evaluate synaptic structure in the cerebral cortex of adult mice in more detail. Normal synaptic connections, comprising a presynaptic terminal containing synaptic vesicles and the cognate postsynaptic terminal with postsynaptic density, were easily found in adult control cerebral cortex (Fig. 3A and C). In Pten cKO cortex, despite the increased density of neuronal spines, normal synaptic connections were extremely difficult to identify. Presynaptic terminals were present, although the majority were enlarged and densely packed with substantially increased numbers of synaptic vesicles. Interestingly, in greater than 60% of enlarged presynaptic terminals, the postsynaptic density was absent (Fig. 3B). Several abnormal synaptic connections were identified in which the postsynaptic density was present, but was dramatically enlarged and extended to the lateral sides of the post-synaptic terminal (Fig. 3D). A small number (<10%) of normal synapses containing both pre- and postsynaptic terminals and a normal sized postsynaptic density were detected in Pten cKO mice (data not shown). It is possible that these connections were made between neurons lacking Cre activity, in which Pten protein was still normally expressed. Enlarged presynaptic terminals with the increased number of synaptic vesicles also was consistent with increased immunofluorescent staining of the synaptic vesicle maker, synaptophysin in cerebral cortex of Pten cKO mice compared to controls (Fig. 3E and 3F).
Fig. 3.
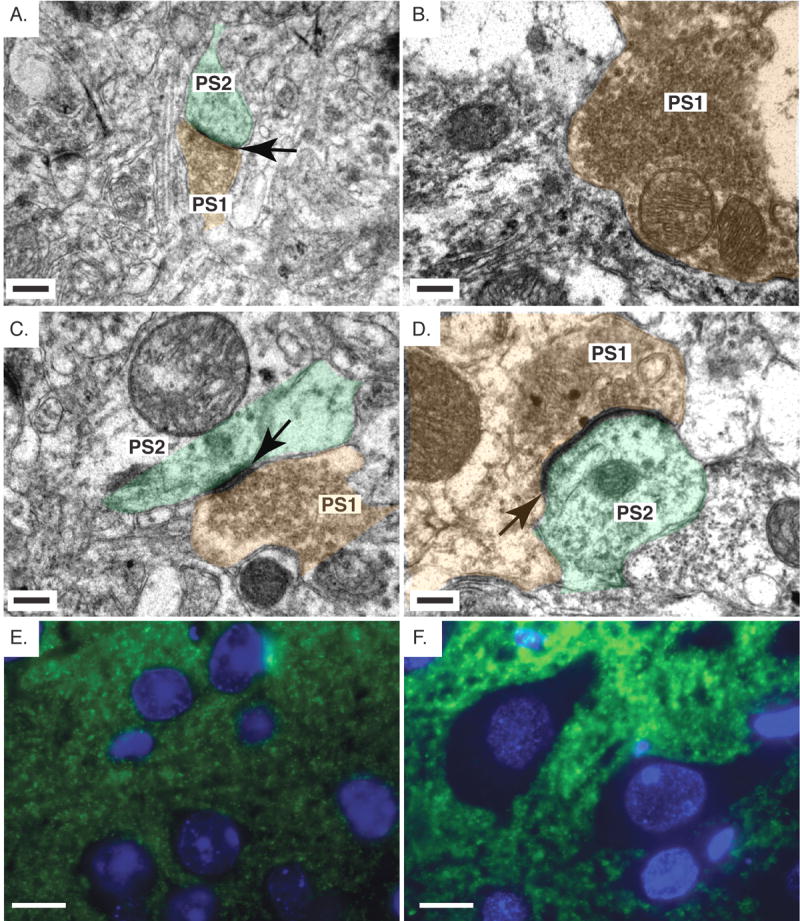
Abnormal synaptic connections in Pten cKO mice. A,B,C,D. Electron micrographs of the cerebral cortex from control (A,C) and Pten cKO mice (B,D). In the control mice, the pre- (PS1) and post- (PS2) synaptic terminals are visible. They are labeled with brown and green color, respectively. The pre-synaptic terminal is identified by the presence of synaptic vesicles. The postsynaptic density (black arrow) is also clearly visible in the control cortex. In the Pten cKO mice (B), the majority of the structures observed were enlarged pre-synaptic terminals, which contained a large number of synaptic vesicles (PS1) with no visible postsynaptic density. A second type of synapse was observed which had a postsynaptic density (black arrow) that extended past the region of the pre-synaptic terminal (D). E,F. Immunofluorescent staining with synaptophysin in the cerebral cortex of control (E) and Pten cKO mice (F) shows increased synaptophysin expression in the Pten cKO mice. Mice were >10 weeks old. Scale bar for A,B,C,D=0.3μm and E,F=10μm.
Electron microscopy revealed that Pten-null cerebellar granule neurons have enlarged nuclear and soma size and contain a dense accumulation of ribosomes in the cytoplasm, similar to changes observed in neurons of the cerebral cortex (Fig. 4A and 4B). The granule neurons have distinct synaptic alterations in the molecular layer of the Pten cKO mice. Normal synaptic junctions containing pre- and postsynaptic terminals connected at an electron-dense postsynaptic density, were easily identified in the molecular layer of the control cerebellum (Fig. 4C and 4E). In the Pten cKO molecular layer based on 66 synaptic junctions, 76% of the presynaptic terminals were significantly enlarged in size and were packed with synaptic vesicles. Six percent of the synaptic junctions appeared normal (data not shown) and they were likely made by granule neurons that have retained Pten expression. The remaining 18% appeared to be partially normal with regard to the size of the presynaptic terminal or the postsynaptic density. Of the 76% of enlarged presynaptic terminals, 18% had a normal postsynaptic density (data not shown), 30% had no visible postsynaptic density (data not shown), while 52% had a postsynaptic density that was significantly enlarged in size (Fig. 4D and 4F). More than 50% of the enlarged presynaptic terminals showed multiple synaptic junctions with multiple postsynaptic terminals (Fig. 4F). This was not observed in control cerebella.
Fig. 4.
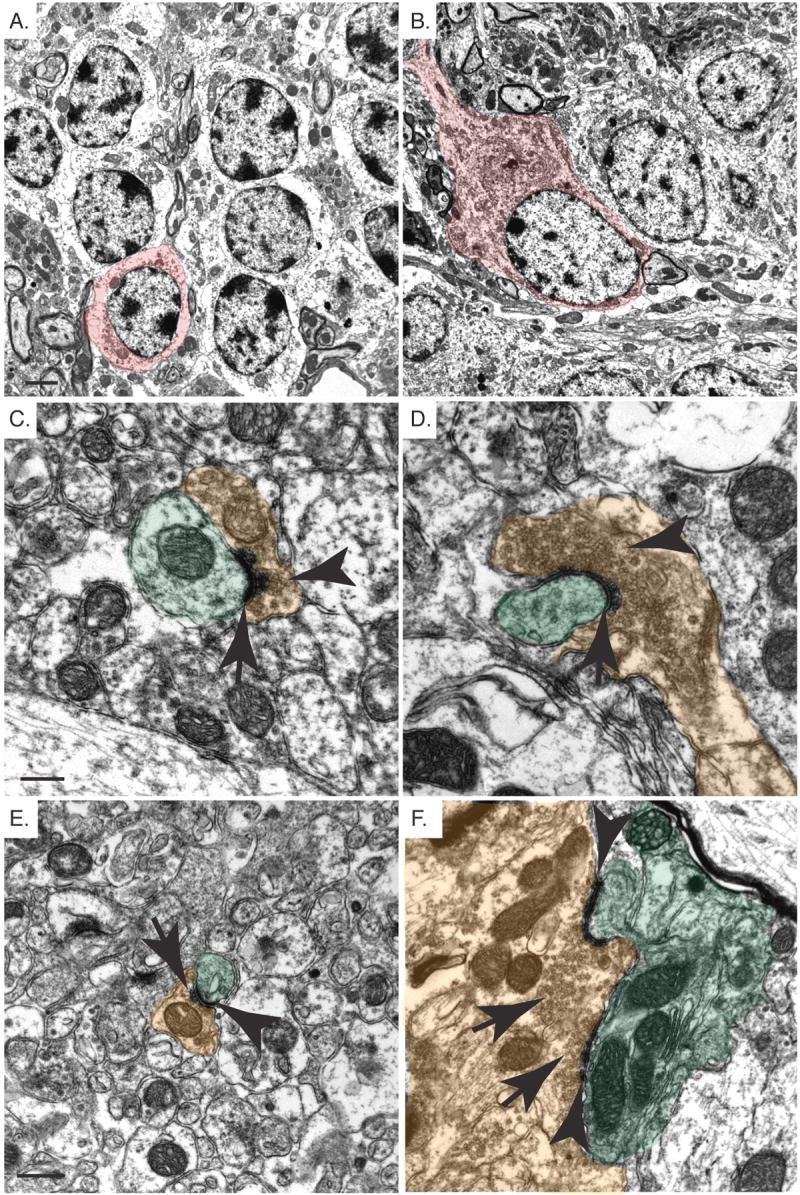
Structural abnormalities in Pten-deficient cerebellar granule neurons. A,B. Electron micrographs of the inner granule layer of control (A) and Pten cKO (B) mice. There is a significant enlargement of the granule neurons in the Pten cKO neurons compared with the control. The increased soma size is indicated with red color. These cells also have an increased ribosome density in the Pten cKO mice. C,D,E,F. Electron micrographs of the molecular layer from control (C,E) and Pten cKO (D,F) mice. Normal synapses containing a postsynaptic density (arrow) and pre-synaptic terminals containing synaptic vesicles (arrow head) are shown. The pre and post-synaptic terminals are indicated with brown and green color, respectively. The synapses in the Pten cKO (D,F) mice showed greatly enlarged pre-synaptic terminals with increased synaptic vesicles (arrow head) and postsynaptic densities (arrow) that engulfed the Purkinje cells. One pre-synaptic terminal is able to synapse with multiple post-synaptic terminals (F). Mice were >10 weeks old. Scale bar for A,B=2μm, C,D=0.3μm and E,F=0.5μm.
Pten cKO neurons show defective synaptic function
Thus, Pten cKO neurons show numerous structural abnormalities including dramatic hypertrophy of nuclei, somata, axon and dendritic caliber, enlarged presynaptic terminals and absent or enlarged postsynaptic densities. What is the functional impact of Pten deletion in brain? We addressed this question by measuring synaptic transmission at excitatory synapses between CA3 and CA1 neurons (CA3-CA1 synapses) in the hippocampus. We found that synaptic transmission was drastically decreased in Pten cKO mice (Fig. 5A). Thus, field excitatory postsynaptic potentials (fEPSPs) were significantly impaired throughout all stimulation intensities in slices from Pten cKO mice compared to that in wild-type littermates (p<0.001. Pten cKO: 28 slices, 5 mice; WT mice: 27 slices, 5 mice). To determine whether this deficit in synaptic transmission is due to a deficit in presynaptic function, we measured pair-pulse ratio (PPR), which serves as a general measure of probability of neurotransmitter release. We found that at all interpulse intervals PPR was indistinguishable between Pten cKO and wild-type littermates (Fig. 5B. p>0.05, Pten cKO mice: 28 slices, 5 mice; WT mice: 27 slices, 5 mice). These data suggest that the dramatic deficit in synaptic transmission in Pten cKO mice is not due to a deficit in neurotransmitter release from presynaptic terminals but rather postsynaptic in nature. To investigate further how conditional deletion of Pten affects synaptic plasticity, we compared long-term potentiation (LTP) at CA3-CA1 synapses in slices from Pten cKO and wild-type mice. Consistent with decreased synaptic transmission in Pten cKO mice, LTP was drastically reduced (Fig. 5C). Thus fEPSPs measured 3 hours after induction of LTP were reduced by more than 70% in slices from Pten cKO mice compared to that in wild-type littermates.
Fig. 5.
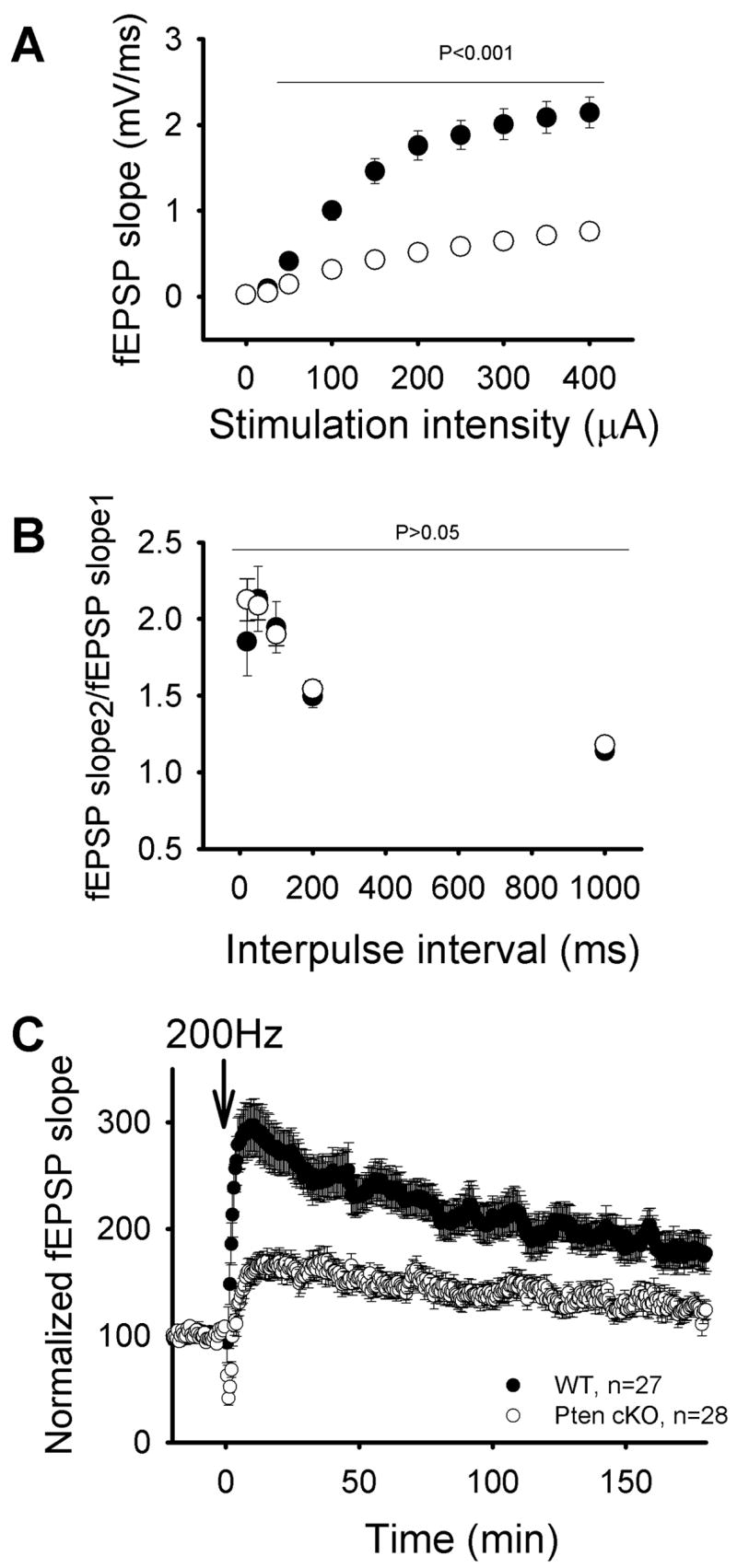
Synaptic transmission and synaptic plasticity are defective in Pten cKO mice. A. Mean fEPSPs as a function of stimulation intensity in Pten cKO mice and wild-type littermates. B. Pair-pulse ratio of two mean fEPSPs as a function of interstimulus interval measured in slices from Pten cKO and WT mice. C. Average fEPSPs vs time before and after 200 Hz tetanizations measured in Pten cKO and WT mice (downward arrows). Mice were 3.5 to 4 weeks of age.
Abnormal Myelination in Pten cKO Mice
In addition to hypertrophy of the cerebral cortex and cerebellum, the corpus callosum was also grossly enlarged. Electron microscopy revealed profound defects in myelination in the corpus callosum of Pten cKO mice. Sagittal section through the corpus callosum of control mice showed a cross-sectional view of axons of varying diameters tightly wrapped in myelin sheaths (Fig. 6A and 6C). In the corpus callosum from Pten cKO mice, many axons showed tremendous thickening of the myelin sheath, with regions of unraveling myelin (Fig. 6B and 6D). The caliber of axons was also dramatically enlarged.
Fig. 6.
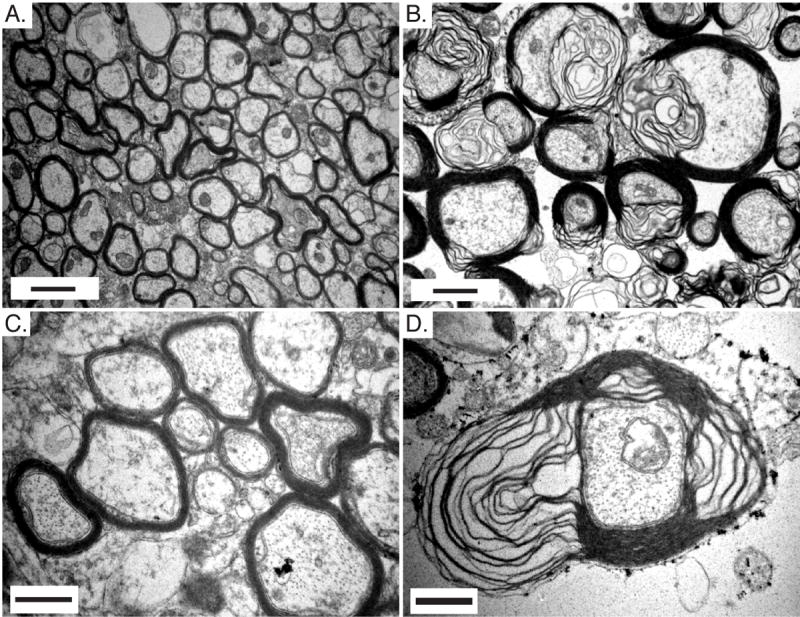
Abnormal myelination in Pten cKO mouse brains. A,B,C,D. Electron micrographs at two magnifications of the corpus callosum from adult control (A,C) and Pten cKO mice (B,D). Myelin thickness is increased and the myelin sheath is unraveling in Pten cKO mice compared with control mice. Mice were >10 weeks old. Scale bar for A,B=1μm and C,D=0.5μm.
The profound abnormalities seen in the myelin sheath could be a primary defect due to the loss of Pten in the oligodendrocytes or a secondary effect of neuronal hypertrophy. To determine whether cre-mediated recombination occurred in oligodendrocytes, we crossed the GFAP-cre mice with ROSA26R reporter mice (Soriano, 1999). The ROSA26R reporter contains a floxed stop signal upstream of the lacZ reporter gene. Upon Cre-mediated recombination, the stop signal is removed and lacZ is expressed from the ubiquitous ROSA26 promoter independent of continuing expression of Cre from the GFAP-cre mice. Evaluation of cre activity in adult GFAP-cre;ROSA26R mice showed overlapping expression of CC-1, a marker for oligodendrocytes (Murtie et al., 2005, Zai and Wrathall, 2005), and the β-Gal reporter for cre activity in the corpus callosum and the white matter tracts of the cerebellum (Fig. 7A–C and 7D–F). Oligodendrocytes in Pten cKO mice, visualized by CC-1 immunofluorescence, showed a significant increase in nuclear (data not shown) and soma size compared with controls in both the corpus callosum and cerebellar white matter tracts (Fig. 7G–H and 7I–J).
Fig. 7.
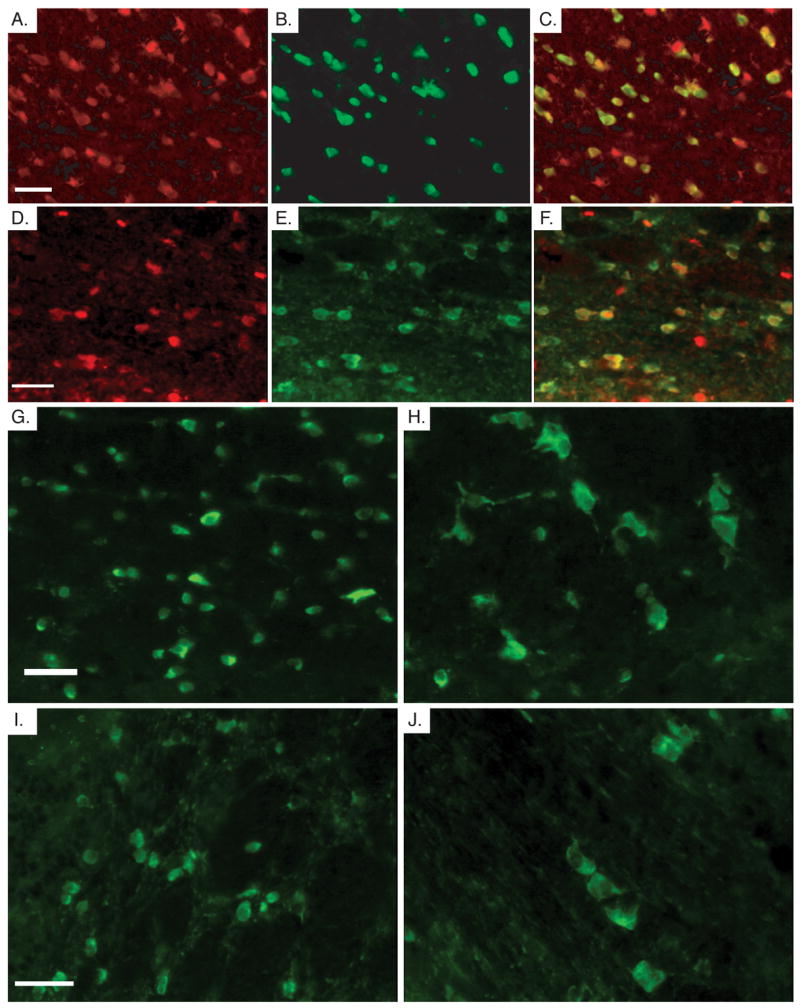
Cre activity in oligodendrocytes leads to hypertrophy. A,B,C. Immunofluorescence in the corpus callosum of adult GFAP-cre;ROSA26R mice shows β-Gal expression (A), CC-1 (B) and the overlay of the two images (C). D,E,F. Immunofluorescence in the white matter tracks of the cerebellum of adult GFAP-cre;ROSA26R brains showing β-Gal (D) and CC-1 (E), and the overlapping images (F). G,H. CC-1 immunofluorescence in the corpus callosum control (G) and Pten cKO mice (H). A population of CC-1 positive cells in the Pten cKO mice are significantly enlarged compared with the control CC-1 positive cells in the same region. I,J. CC-1 immunofluorescence in the white matter track of the cerebellum of control (I) and Pten cKO mice (J). Again, a population of oligodendrocytes are significantly enlarged in size in the Pten cKO mice. Mice were 4 weeks of age for A,B,C,D,E,F and mice were >10 weeks old for G,H,I,J. Scale bar for all=30μm.
In the cerebellum, loss of Pten occurred in the majority of the granule neurons, while Pten expression was retained in Purkinje cells (Fig. 8A and 8B), mossy and climbing fibers (data not shown). Unlike the hypertrophic axons observed in the corpus callosum, there was no substantial change in the size of the axonal diameters of the cells that extend processes into the cerebellar white matter tracts, which is consistent with Purkinje cells, mossy and climbing fibers maintaining Pten expression. However, thickened myelin sheaths as well as decreased myelin compaction were still observed in Pten cKO mice compared with the control (Fig. 8C and 8D), suggesting that myelination abnormalities may arise, at least in part, through intrinsic defects in Pten-deficient oligodendrocytes.
Fig. 8.
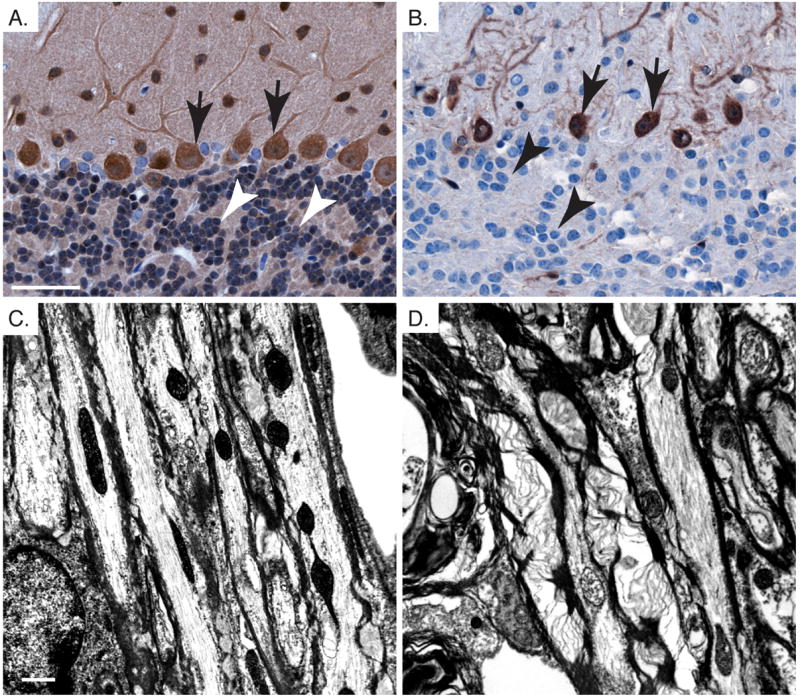
Pten loss in oligodendrocytes leads to cell autonomous defects in myelination in the cerebellum. A,B. Pten immunohistochemistry shows loss of Pten in granule neurons of the IGL (arrow head), and persistent Pten expression in Purkinje cells (arrows) from Pten cKO cerebellum (B) compared to control (A). C,D. Electron micrographs showing myelination in the cerebellar white matter tracks of control (A) and Pten cKO (B). Mice were >10 weeks old. Scale bar for A,B=50μm and C,D=1μm.
Discussion
Pten function has been studied in multiple tissue types through tissue-specific knock-out mice, showing roles for Pten in proliferation, survival and cell size regulation, depending on the context (Sulis and Parsons, 2003, Chow and Baker, 2006). As a negative regulator of PI3K signaling, Pten loss of function is often associated with constitutive activation of downstream effectors in this pathway including the serine-threonine kinases Akt and mTor. A number of in vitro studies have shown specific specialized functions for PI3K signaling in aspects of neuronal development including neurite growth, growth cone dynamics, axon specification and dendritic arborization (Sanchez et al., 2001, Menager et al., 2004, Jaworski et al., 2005, Jiang et al., 2005, Kumar et al., 2005, Laurino et al., 2005, Chadborn et al., 2006). While these studies demonstrate a critical role for this pathway in neuronal development, they employ exogenous growth factor stimulation with knock-down of endogenous proteins, or constitutively active or inactive mutants. In vivo analysis of conditional knock-out mice reveals the effects of pathway deregulation in the context of physiological growth factor signaling. Several distinct brain-specific conditional knock-out models have shown hypertrophy of nuclei and somata of Pten-deficient neurons (Backman et al., 2001, Groszer et al., 2001, Kwon et al., 2001, Marino et al., 2002, Fraser et al., 2004).
Electron microscopic detection of high ribosome density in hypertrophic Pten-deficient neurons and increased nucleolar size is consistent with our previous report that hypertrophy of Pten-deficient neurons required signaling through the rapamycin-sensitive mTor complex (Kwon et al., 2003), which functions in part to increase production of translational machinery (Ruggero and Pandolfi, 2003, Fingar and Blenis, 2004, Ruggero and Sonenberg, 2005). Numerous reports have indicated a role for mTor in synaptic plasticity mediated at least in part through regulation of localized protein translation in dendrites (Tang et al., 2002, Cammalleri et al., 2003, Si et al., 2003, Takei et al., 2004, Gong et al., 2006).
The increase in spine number and alterations in synaptic structure in Pten cKO brain could be a generalized hypertrophic effect on the cell, or reflect a specific role for Pten signaling in synaptic function. In Drosophila, overexpression of PI3K or AKT induced the formation and the maintenance of supernumerary synapses in developing motor neurons and mature projection neurons respectively. However, increasing neuronal size via the epidermal growth factor did not affect synapse number or morphology, suggesting that size regulation and synaptic regulation are distinct processes influenced by the PI3K pathway (Martin-Pena et al., 2006). Similar to our observations, work by Kwon et al. (Kwon et al., 2006) supports a direct role of Pten expression in maintaining normal synapse structure. In this study, loss of Pten expression in a subset of differentiated neurons in the cerebral cortex and dentate gyrus resulted in hypertrophic axons and dendrites and significant enlargement of presynaptic terminals, which contained multiple synaptic densities and large numbers of synaptic vesicles. Evaluation of PI3K signaling in cultured hippocampal neurons also showed that combined activation of Ras and PI3K induced increased dendritic complexity while constitutively activated forms of PI3K or Akt caused increased dendritic size and disrupted dendritic shape in the absence of altered Ras signaling (Kumar et al., 2005). The tuberous sclerosis complex normally functions to inhibit mTor activity, and this inhibition can be relieved by PI3K activation through activated Akt. Loss of Tsc1 or Tsc2 caused increased soma size and elongated dendritic spines with enlarged bulbous heads in hippocampal pyramidal neurons (Tavazoie et al., 2005), reminiscent of the alterations observed in Pten cKO mice.
What is the functional consequence of the hypertrophic and abnormal synaptic structures resulting from mutation of different PI3K pathway effectors? Electrophysiological recordings from Pten cKO hippocampus indicated strong deficits in synaptic transmission, most likely due to compromised postsynaptic function. This is consistent with the structural abnormalities in postsynaptic densities revealed by electron microscopy. No general defects in presynaptic neurotransmitter release as measured by PPR were observed in Pten cKO hippocampus. Analysis of hippocampal slice cultures with conditional deletion of Tsc1 similarly showed normal presynaptic function, with no detectable abnormalities in PPR or frequency of spontaneous miniature EPSCs (mEPSCs), but abnormal postsynaptic function, shown by increased spontaneous mEPSC amplitude and an altered ratio of currents mediated by AMPA receptors and NMDA receptors (Tavazoie et al., 2005). Overexpression of PI3K in Drosophila abdominal larval motor neurons also showed a functional consequence with enhanced spontaneous and evoked neurotransmitter release (Martin-Pena et al., 2006). Pten cKO mice showed a number of profound structural defects in multiple cell types, therefore the functional abnormalities that we observed in synaptic transmission and plasticity may not be exclusively associated with cell-autonomous defects in the function of Pten-deficient neurons. Recent studies showed that conditional deletion of Tsc1, which was achieved using the same GFAP-cre transgenic mouse used in our study, caused impaired LTP and was associated with learning deficits. While these deficiencies in hippocampal function may have been the result of abnormal glutamate homeostasis in Tsc1 cKO mice, it is possible that multiple cell types lacking Tsc1 contributed to the phenotype (Zeng et al., 2007). Future studies directing Pten loss exclusively in neurons will allow the elucidation of which abnormalities are cell-autonomous. However, we clearly demonstrated that Pten deletion in brain has profound functional and structural consequences.
We also observed dramatic changes in myelination in both the corpus callosum and white matter tracts of the cerebellum in Pten cKO mice compared to controls. Enlarged oligodendrocyte somata were accompanied by increased thickness of the myelin sheath and myelin decompaction. Several lines of evidence indicate a role for PI3K signaling in myelination. IGF-1, which activates PI3K signaling, promotes the expression of myelin-associated protein markers in cultured Schwann cells. Further, introduction of a constitutively active Akt into transplanted nerve segments showed increased myelination in an in vivo transplant model (Ogata et al., 2004). Mice lacking the gene encoding the endopeptidase Bace-1 showed delayed myelination and decreased myelin thickness associated with a decrease in cleaved neuregulin and a resulting decrease in phosphorylated Akt (Hu et al., 2006).
The abnormal myelination by Pten-deficient oligodendrocytes ensheathing axons from neurons that still express Pten in the cerebellum strongly suggests that Pten loss caused cell-autonomous defects in oligodendrocytes. However, multiple factors could contribute to the profound oligodendrocyte hypertrophy and defects in myelination observed in the corpus callosum of Pten cKO mice where Pten was deleted in multiple cell types. The loss of Pten in neurons significantly increased the full neuronal structure, which could lead to increased myelin, as size and length of the dendrites and axon is known to influence myelination (Michailov et al., 2004). Also, the progressive increase in caliber of Pten-null axons could create increased pressure on the myelin sheath, potentially resulting in decompaction. Additionally, it has been demonstrated that astrocytes have the capacity to promote myelination by reacting to the action potential firing of neighboring neurons. The action potential from a neuron triggers the astrocytes to release leukemia inhibitory factor (LIF), which stimulates myelination by the oligodendrocytes (Ishibashi et al., 2006). Perhaps hypertrophic astrocytes observed in Pten cKO brain (Fraser et al., 2004) are capable of producing more LIF, leading to increased myelination.
Myelin defects in the cerebellar white matter tracts strongly suggest intrinsic defects in Pten-deficient oligodendrocytes. In this region, cre activity is observed in oligodendrocytes which are hypertrophic in Pten cKO mice and associated with increased myelin thickness and decompaction. However, the axons of the Purkinje cells, mossy fibers and climbing fibers, found in the white matter tracts, are similar in size compared with the controls due to the absence of cre activity and the retention of Pten expression in these cell types. This indicates the potential for a cell autonomous defect in myelination with the loss of Pten in oligodendrocytes. Selective deletion of Pten in oligodendrocytes would be ideal to further determine the precise role that Pten plays in myelination.
The changes in cellular ultra-structure of Pten cKO mice illustrate the spectrum of abnormalities that could occur in the absence of functional PTEN in the brain. Observations of ultrastructural defects in cerebella of Pten cKO mice are consistent with changes observed in human Lhermitte-Duclos disease (LDD), which occurs in patients with inherited PTEN mutation. In the hypertrophic lesions in LDD, the wild-type PTEN allele is inactivated resulting in the complete loss of functional PTEN (Eng and Peacocke, 1998). Electron microscopy and Golgi stain analyses demonstrated hypertrophic granule neurons accompanied by increases in ribosomes, similar to our results. The dendritic spines were thickened and pre-synaptic terminals were enlarged and contained dense regions of synaptic vesicles (Gessaga, 1980, Reznik and Schoenen, 1983, Marano et al., 1988, Ferrer et al., 1990, Hair et al., 1992). Other defects observed in Pten cKO brains reflect the spectrum of abnormalities that may result from complete Pten inactivation in different cell populations. Neurological defects in patients with inherited PTEN germline mutations may be due in part to second hits in the remaining wild-type allele in subpopulations of cells. The severity of disease would be associated with the number and types of cells that acquire the second mutation in the normal PTEN allele and the developmental stage at which such a somatic mutation is acquired. The stochastic nature of this event likely contributes to the highly variable neurological phenotype associated with inherited germline PTEN mutations.
Acknowledgments
We thank Dr. Jim Morgan for helpful discussions and Dr. Richard Smeyne for help with the BioQuant system. We thank Jennifer Parris for sectioning Golgi stained tissue. We thank Linda Mann and Sharon Frase of the St Jude Children’s Research Hospital and University of Memphis, respectively for their expertise in electron microscopy. This work was supported by NIH grants NS044172 and CA096832 from the National Institutes of Health (SJB), Searle Scholars Programs and Whitehall Foundation (SSZ) and by the American Lebanese and Syrian Associated Charities.
List of Abbreviations
- Pten
phosphatase and tensin homolog, deleted on chromosome 10
- PI3K
phosphatidylinositol 3-kinase
- PIP2
phosphatidylinositol 4,5 bisphosphate
- PIP3
phosphatidylinositol 3,4,5-triphosphate
- GFAP
glial fibrillary acidic protein
- β-gal
β-galactosidase
- fEPSPs
field excitatory postsynaptic potentials
- mEPSCs
miniature excitatory postsynaptic currents
- PPR
pair-pulse ratio
- LTP
long-term potentiation
- LDD
Lhermitte-Duclos disease
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ali IU, Schriml LM, Dean M. Mutational spectra of PTEN/MMAC1 gene: a tumor suppressor with lipid phosphatase activity. J Natl Cancer Inst. 1999;91:1922–1932. doi: 10.1093/jnci/91.22.1922. [DOI] [PubMed] [Google Scholar]
- Backman SA, Stambolic V, Suzuki A, Haight J, Elia A, Pretorius J, Tsao MS, Shannon P, Bolon B, Ivy GO, Mak TW. Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte-Duclos disease. Nat Genet. 2001;29:396–403. doi: 10.1038/ng782. [DOI] [PubMed] [Google Scholar]
- Bajenaru ML, Zhu Y, Hedrick NM, Donahoe J, Parada LF, Gutmann DH. Astrocyte-specific inactivation of the neurofibromatosis 1 gene (NF1) is insufficient for astrocytoma formation. Mol Cell Biol. 2002;22:5100–5113. doi: 10.1128/MCB.22.14.5100-5113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MG, Dasouki MJ, Zhou XP, Talebizadeh Z, Brown M, Takahashi TN, Miles JH, Wang CH, Stratton R, Pilarski R, Eng C. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet. 2005;42:318–321. doi: 10.1136/jmg.2004.024646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cammalleri M, Lutjens R, Berton F, King AR, Simpson C, Francesconi W, Sanna PP. Time-restricted role for dendritic activation of the mTOR-p70S6K pathway in the induction of late-phase long-term potentiation in the CA1. Proc Natl Acad Sci U S A. 2003;100:14368–14373. doi: 10.1073/pnas.2336098100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadborn NH, Ahmed AI, Holt MR, Prinjha R, Dunn GA, Jones GE, Eickholt BJ. PTEN couples Sema3A signalling to growth cone collapse. J Cell Sci. 2006;119:951–957. doi: 10.1242/jcs.02801. [DOI] [PubMed] [Google Scholar]
- Chow LM, Baker SJ. PTEN function in normal and neoplastic growth. Cancer Lett. 2006 doi: 10.1016/j.canlet.2005.11.042. [DOI] [PubMed] [Google Scholar]
- Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998;19:348–355. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- Eng C. PTEN: one gene, many syndromes. Hum Mutat. 2003;22:183–198. doi: 10.1002/humu.10257. [DOI] [PubMed] [Google Scholar]
- Eng C, Peacocke M. PTEN and inherited hamartoma-cancer syndromes. Nat Genet. 1998;19:223. doi: 10.1038/897. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Marti E, Guionnet N, Bella R, Serrano T, Towse J, Conesa G, Isamat F. Studies with the Golgi method in central gangliogliomas and dysplastic gangliocytoma of the cerebellum (Lhermitte-Duclos disease) Histol Histopathol. 1990;5:329–336. [PubMed] [Google Scholar]
- Fiala JC, Spacek J, Harris KM. Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Brain Res Rev. 2002;39:29–54. doi: 10.1016/s0165-0173(02)00158-3. [DOI] [PubMed] [Google Scholar]
- Fingar DC, Blenis J. Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene. 2004;23:3151–3171. doi: 10.1038/sj.onc.1207542. [DOI] [PubMed] [Google Scholar]
- Fraser MM, Zhu X, Kwon CH, Uhlmann EJ, Gutmann DH, Baker SJ. Pten loss causes hypertrophy and increased proliferation of astrocytes in vivo. Cancer Res. 2004;64:7773–7779. doi: 10.1158/0008-5472.CAN-04-2487. [DOI] [PubMed] [Google Scholar]
- Gessaga EC. Lhermitte-Duclos disease (diffuse hypertrophy of the cerebellum). Report of two cases. Neurosurg Rev. 1980;3:151–158. doi: 10.1007/BF01644067. [DOI] [PubMed] [Google Scholar]
- Goffin A, Hoefsloot LH, Bosgoed E, Swillen A, Fryns JP. PTEN mutation in a family with Cowden syndrome and autism. Am J Med Genet. 2001;105:521–524. doi: 10.1002/ajmg.1477. [DOI] [PubMed] [Google Scholar]
- Gong R, Park CS, Abbassi NR, Tang SJ. Roles of glutamate receptors and the mammalian target of rapamycin (mTOR) signaling pathway in activity-dependent dendritic protein synthesis in hippocampal neurons. J Biol Chem. 2006;281:18802–18815. doi: 10.1074/jbc.M512524200. [DOI] [PubMed] [Google Scholar]
- Griffin RJ, Moloney A, Kelliher M, Johnston JA, Ravid R, Dockery P, O’Connor R, O’Neill C. Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer’s disease pathology. J Neurochem. 2005;93:105–117. doi: 10.1111/j.1471-4159.2004.02949.x. [DOI] [PubMed] [Google Scholar]
- Groszer M, Erickson R, Scripture-Adams DD, Lesche R, Trumpp A, Zack JA, Kornblum HI, Liu X, Wu H. Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science. 2001;294:2186–2189. doi: 10.1126/science.1065518. [DOI] [PubMed] [Google Scholar]
- Hair LS, Symmans F, Powers JM, Carmel P. Immunohistochemistry and proliferative activity in Lhermitte-Duclos disease. Acta Neuropathol (Berl) 1992;84:570–573. doi: 10.1007/BF00304477. [DOI] [PubMed] [Google Scholar]
- Holland EC, Li Y, Celestino J, Dai C, Schaefer L, Sawaya RA, Fuller GN. Astrocytes give rise to oligodendrogliomas and astrocytomas after gene transfer of polyoma virus middle T antigen in vivo. Am J Pathol. 2000;157:1031–1037. doi: 10.1016/S0002-9440(10)64615-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie Y, Suzuki A, Kataoka E, Sasaki T, Hamada K, Sasaki J, Mizuno K, Hasegawa G, Kishimoto H, Iizuka M, Naito M, Enomoto K, Watanabe S, Mak TW, Nakano T. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J Clin Invest. 2004;113:1774–1783. doi: 10.1172/JCI20513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Hicks CW, He W, Wong P, Macklin WB, Trapp BD, Yan R. Bace1 modulates myelination in the central and peripheral nervous system. Nat Neurosci. 2006;9:1520–1525. doi: 10.1038/nn1797. [DOI] [PubMed] [Google Scholar]
- Ishibashi T, Dakin KA, Stevens B, Lee PR, Kozlov SV, Stewart CL, Fields RD. Astrocytes promote myelination in response to electrical impulses. Neuron. 2006;49:823–832. doi: 10.1016/j.neuron.2006.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworski J, Spangler S, Seeburg DP, Hoogenraad CC, Sheng M. Control of dendritic arborization by the phosphoinositide-3′-kinase-Akt-mammalian target of rapamycin pathway. J Neurosci. 2005;25:11300–11312. doi: 10.1523/JNEUROSCI.2270-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Guo W, Liang X, Rao Y. Both the establishment and the maintenance of neuronal polarity require active mechanisms: critical roles of GSK-3beta and its upstream regulators. Cell. 2005;120:123–135. doi: 10.1016/j.cell.2004.12.033. [DOI] [PubMed] [Google Scholar]
- Kimura T, Suzuki A, Fujita Y, Yomogida K, Lomeli H, Asada N, Ikeuchi M, Nagy A, Mak TW, Nakano T. Conditional loss of PTEN leads to testicular teratoma and enhances embryonic germ cell production. Development. 2003;130:1691–1700. doi: 10.1242/dev.00392. [DOI] [PubMed] [Google Scholar]
- Kumar V, Zhang MX, Swank MW, Kunz J, Wu GY. Regulation of dendritic morphogenesis by Ras-PI3K-Akt-mTOR and Ras-MAPK signaling pathways. J Neurosci. 2005;25:11288–11299. doi: 10.1523/JNEUROSCI.2284-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, Parada LF. Pten regulates neuronal arborization and social interaction in mice. Neuron. 2006;50:377–388. doi: 10.1016/j.neuron.2006.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon CH, Zhu X, Zhang J, Baker SJ. mTor is required for hypertrophy of Pten-deficient neuronal soma in vivo. Proc Natl Acad Sci U S A. 2003;100:12923–12928. doi: 10.1073/pnas.2132711100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon CH, Zhu X, Zhang J, Knoop LL, Tharp R, Smeyne RJ, Eberhart CG, Burger PC, Baker SJ. Pten regulates neuronal soma size: a mouse model of Lhermitte-Duclos disease. Nat Genet. 2001;29:404–411. doi: 10.1038/ng781. [DOI] [PubMed] [Google Scholar]
- Laurino L, Wang XX, de la Houssaye BA, Sosa L, Dupraz S, Caceres A, Pfenninger KH, Quiroga S. PI3K activation by IGF-1 is essential for the regulation of membrane expansion at the nerve growth cone. J Cell Sci. 2005;118:3653–3662. doi: 10.1242/jcs.02490. [DOI] [PubMed] [Google Scholar]
- Li G, Robinson GW, Lesche R, Martinez-Diaz H, Jiang Z, Rozengurt N, Wagner KU, Wu DC, Lane TF, Liu X, Hennighausen L, Wu H. Conditional loss of PTEN leads to precocious development and neoplasia in the mammary gland. Development. 2002;129:4159–4170. doi: 10.1242/dev.129.17.4159. [DOI] [PubMed] [Google Scholar]
- Marano SR, Johnson PC, Spetzler RF. Recurrent Lhermitte-Duclos disease in a child. Case report. J Neurosurg. 1988;69:599–603. doi: 10.3171/jns.1988.69.4.0599. [DOI] [PubMed] [Google Scholar]
- Marino S, Krimpenfort P, Leung C, van der Korput HA, Trapman J, Camenisch I, Berns A, Brandner S. PTEN is essential for cell migration but not for fate determination and tumourigenesis in the cerebellum. Development. 2002;129:3513–3522. doi: 10.1242/dev.129.14.3513. [DOI] [PubMed] [Google Scholar]
- Martin-Pena A, Acebes A, Rodriguez JR, Sorribes A, de Polavieja GG, Fernandez-Funez P, Ferrus A. Age-independent synaptogenesis by phosphoinositide 3 kinase. J Neurosci. 2006;26:10199–10208. doi: 10.1523/JNEUROSCI.1223-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menager C, Arimura N, Fukata Y, Kaibuchi K. PIP3 is involved in neuronal polarization and axon formation. J Neurochem. 2004;89:109–118. doi: 10.1046/j.1471-4159.2004.02302.x. [DOI] [PubMed] [Google Scholar]
- Michailov GV, Sereda MW, Brinkmann BG, Fischer TM, Haug B, Birchmeier C, Role L, Lai C, Schwab MH, Nave KA. Axonal neuregulin-1 regulates myelin sheath thickness. Science. 2004;304:700–703. doi: 10.1126/science.1095862. [DOI] [PubMed] [Google Scholar]
- Murtie JC, Zhou YX, Le TQ, Armstrong RC. In vivo analysis of oligodendrocyte lineage development in postnatal FGF2 null mice. Glia. 2005;49:542–554. doi: 10.1002/glia.20142. [DOI] [PubMed] [Google Scholar]
- Ogata T, Iijima S, Hoshikawa S, Miura T, Yamamoto S, Oda H, Nakamura K, Tanaka S. Opposing extracellular signal-regulated kinase and Akt pathways control Schwann cell myelination. J Neurosci. 2004;24:6724–6732. doi: 10.1523/JNEUROSCI.5520-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podsypanina K, Ellenson LH, Nemes A, Gu J, Tamura M, Yamada KM, Cordon-Cardo C, Catoretti G, Fisher PE, Parsons R. Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc Natl Acad Sci U S A. 1999;96:1563–1568. doi: 10.1073/pnas.96.4.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raftopoulou M, Etienne-Manneville S, Self A, Nicholls S, Hall A. Regulation of cell migration by the C2 domain of the tumor suppressor PTEN. Science. 2004;303:1179–1181. doi: 10.1126/science.1092089. [DOI] [PubMed] [Google Scholar]
- Reznik M, Schoenen J. Lhermitte-Duclos disease. Acta Neuropathol (Berl) 1983;59:88–94. doi: 10.1007/BF00691592. [DOI] [PubMed] [Google Scholar]
- Rickle A, Bogdanovic N, Volkmann I, Zhou X, Pei JJ, Winblad B, Cowburn RF. PTEN levels in Alzheimer’s disease medial temporal cortex. Neurochem Int. 2006;48:114–123. doi: 10.1016/j.neuint.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Ruggero D, Pandolfi PP. Does the ribosome translate cancer? Nat Rev Cancer. 2003;3:179–192. doi: 10.1038/nrc1015. [DOI] [PubMed] [Google Scholar]
- Ruggero D, Sonenberg N. The Akt of translational control. Oncogene. 2005;24:7426–7434. doi: 10.1038/sj.onc.1209098. [DOI] [PubMed] [Google Scholar]
- Sanchez S, Sayas CL, Lim F, Diaz-Nido J, Avila J, Wandosell F. The inhibition of phosphatidylinositol-3-kinase induces neurite retraction and activates GSK3. J Neurochem. 2001;78:468–481. doi: 10.1046/j.1471-4159.2001.00453.x. [DOI] [PubMed] [Google Scholar]
- Si K, Giustetto M, Etkin A, Hsu R, Janisiewicz AM, Miniaci MC, Kim JH, Zhu H, Kandel ER. A neuronal isoform of CPEB regulates local protein synthesis and stabilizes synapse-specific long-term facilitation in aplysia. Cell. 2003;115:893–904. doi: 10.1016/s0092-8674(03)01021-3. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Sulis ML, Parsons R. PTEN: from pathology to biology. Trends Cell Biol. 2003;13:478–483. doi: 10.1016/s0962-8924(03)00175-2. [DOI] [PubMed] [Google Scholar]
- Suzuki A, de la Pompa JL, Stambolic V, Elia AJ, Sasaki T, del Barco Barrantes I, Ho A, Wakeham A, Itie A, Khoo W, Fukumoto M, Mak TW. High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr Biol. 1998;8:1169–1178. doi: 10.1016/s0960-9822(07)00488-5. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Yamaguchi MT, Ohteki T, Sasaki T, Kaisho T, Kimura Y, Yoshida R, Wakeham A, Higuchi T, Fukumoto M, Tsubata T, Ohashi PS, Koyasu S, Penninger JM, Nakano T, Mak TW. T cell-specific loss of Pten leads to defects in central and peripheral tolerance. Immunity. 2001;14:523–534. doi: 10.1016/s1074-7613(01)00134-0. [DOI] [PubMed] [Google Scholar]
- Takei N, Inamura N, Kawamura M, Namba H, Hara K, Yonezawa K, Nawa H. Brain-derived neurotrophic factor induces mammalian target of rapamycin-dependent local activation of translation machinery and protein synthesis in neuronal dendrites. J Neurosci. 2004;24:9760–9769. doi: 10.1523/JNEUROSCI.1427-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci U S A. 2002;99:467–472. doi: 10.1073/pnas.012605299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci. 2005;8:1727–1734. doi: 10.1038/nn1566. [DOI] [PubMed] [Google Scholar]
- Trotman LC, Niki M, Dotan ZA, Koutcher JA, Di Cristofano A, Xiao A, Khoo AS, Roy-Burman P, Greenberg NM, Van Dyke T, Cordon-Cardo C, Pandolfi PP. Pten dose dictates cancer progression in the prostate. PLoS Biol. 2003;1:E59. doi: 10.1371/journal.pbio.0000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, Thomas GV, Li G, Roy-Burman P, Nelson PS, Liu X, Wu H. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell. 2003;4:209–221. doi: 10.1016/s1535-6108(03)00215-0. [DOI] [PubMed] [Google Scholar]
- Wei Q, Clarke L, Scheidenhelm DK, Qian B, Tong A, Sabha N, Karim Z, Bock NA, Reti R, Swoboda R, Purev E, Lavoie JF, Bajenaru ML, Shannon P, Herlyn D, Kaplan D, Henkelman RM, Gutmann DH, Guha A. High-grade glioma formation results from postnatal pten loss or mutant epidermal growth factor receptor expression in a transgenic mouse glioma model. Cancer Res. 2006;66:7429–7437. doi: 10.1158/0008-5472.CAN-06-0712. [DOI] [PubMed] [Google Scholar]
- Xiao A, Wu H, Pandolfi PP, Louis DN, Van Dyke T. Astrocyte inactivation of the pRb pathway predisposes mice to malignant astrocytoma development that is accelerated by PTEN mutation. Cancer Cell. 2002;1:157–168. doi: 10.1016/s1535-6108(02)00029-6. [DOI] [PubMed] [Google Scholar]
- Zai LJ, Wrathall JR. Cell proliferation and replacement following contusive spinal cord injury. Glia. 2005;50:247–257. doi: 10.1002/glia.20176. [DOI] [PubMed] [Google Scholar]
- Zeng LH, Ouyang Y, Gazit V, Cirrito JR, Jansen LA, Ess KC, Yamada KA, Wozniak DF, Holtzman DM, Gutmann DH, Wong M. Abnormal glutamate homeostasis and impaired synaptic plasticity and learning in a mouse model of tuberous sclerosis complex. Neurobiol Dis. 2007 doi: 10.1016/j.nbd.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zori RT, Marsh DJ, Graham GE, Marliss EB, Eng C. Germline PTEN mutation in a family with Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome. Am J Med Genet. 1998;80:399–402. [PubMed] [Google Scholar]