

Abstract
Congestive heart failure (CHF) affects more than five million people in the US, and results in considerable morbidity, mortality, and economic costs. Patients with Class III and IV CHF have a 40–50% probability of dying 5 years after symptom onset despite optimal therapy, a prognosis worse than many cancers. A variety of drugs and devices have improved survival – the 50% survival time in 1980 was just eighteen months – but the outlook for patients remains dismal and the prevalence of CHF continues to increase. This unmet medical need underscores the importance of developing new approaches for the treatment of CHF. This brief review focuses on data from preclinical experiments regarding the effects of increased adenylyl cyclase type 6 (AC6) expression on cellular and cardiac function, and possible mechanisms for the unexpected favorable effects of increased AC6 content on the failing heart.
Adenylyl Cyclases in β-Adrenergic Receptor Signaling
In cardiac myocytes and other cells, a cell surface β-adrenergic receptor (βAR) is occupied by agonist (norepinephrine, epinephrine) and the stimulatory GTP-binding protein, Ga s, transduces the signal to AC, the effector molecule, which leads to increased generation of cyclic adenosine monophosphate (cAMP), which then interacts with protein kinase A (PKA) and other intracellular proteins (Ishikawa and Homcy 1997; Hanoune and Defer, 2001) to initiate a wide variety of intracellular events. The first AC isoform, subsequently named AC1, was cloned from brain and found to be calmodulin sensitive. Currently, ten mammalian AC isoforms have been cloned and characterized (Sunahara and Taussig 2002). Each AC consists of two hydrophobic domains with six transmembrane spans (M1 and M2) and two cytoplasmic domains (C1 and C2), resulting in a pseudo-symmetrical protein. The cytoplasmic domain, which constitutes the catalytic site, often is subject to intracellular regulation that can be specific for AC subtype (Figure 1).
Figure 1. Adenylyl cyclase schematic structure.
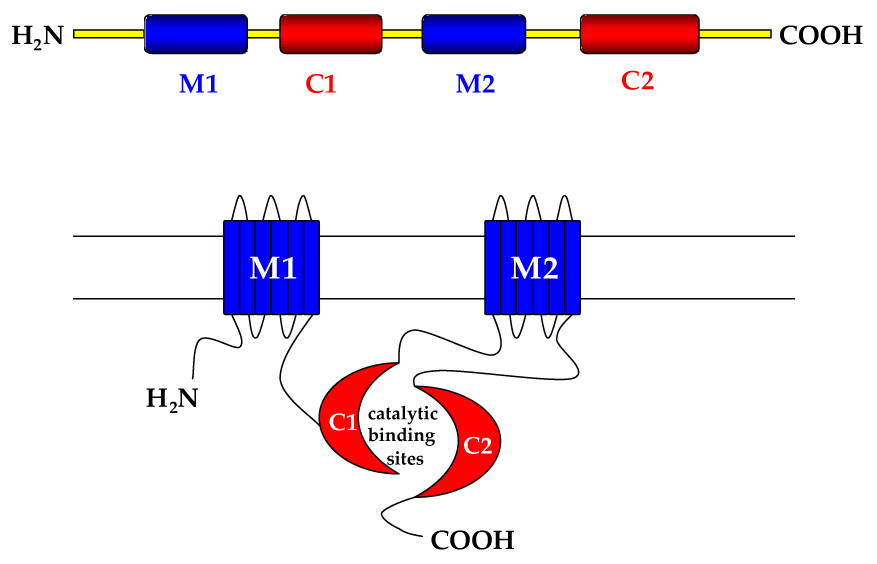
M1, M2: transmembrane domains; C1,C2: cytoplasmic domains, which form catalytic binding sites for GTP-binding proteins
In the heart, the two predominant AC isoforms, AC5 and AC6, have 65% amino acid homology. Neonatal as compared to adult rat hearts have more AC6 than AC5 mRNA, and the reverse is true with increased age (Tobise et al. 1994). In contrast, AC6 mRNA predominates in adult pig heart (Ping et al. 1997). Quantitative assessment of cardiac levels of AC5 versus AC6 protein has been impeded by the poor accessibility of type-specific antibodies. A recent report in mice with targeted deletion of AC5 found that 60% of immunodetectable AC5/6 was still present in cardiac myocytes after AC5 deletion, indicating that these two proteins are expressed at similar levels in the adult mouse heart (Tang et al. 2006; Okumura et al. 2003). Adult human left ventricle (LV) appears to express predominantly AC6 mRNA (Wang and Brown 2004), although there are no published data regarding AC isoform protein expression in adult human heart. Given their central role as effector molecules for βAR signaling and other pathways, it is surprising that quantitative data regarding cardiac AC5 and AC6 protein content – much less their specific roles and relative importance vis-à-vis cardiac function – remain to be established.
Stimulation through Ga s is the major mechanism by which AC is activated. The downstream event is the conversion of ATP to cAMP. All ACs, except possibly AC9, are also activated by the diterpene forskolin, and this commonly is used to estimate AC function independent of Ga s and βAR stimulation. ACs are variably affected by Ga i, the inhibitory GTP-binding protein. For example, Ga i acts as a noncompetitive inhibitor of AC5 and AC6 but has no effect on AC2 or AC8. Gβ? inhibits AC1 and AC8, but stimulates AC2, AC4 and AC7 (Sunahara et al. 1996). AC5 and AC6 are stimulated by Gβ? but only when Ga s or forskolin are present (Gao X et al. 2007). Furthermore, phosphorylation of ACs by PKA provides a means of negative feedback at the effector level. Both AC5 and AC6 are directly phosphorylated and inhibited by PKA; phosphorylation by PKA inhibits AC5 activity by decreasing the maximal velocity of the enzyme, whereas phosphorylation of AC6 at Ser674 interferes with the Ga s binding site, leading to inhibition of AC6 activity (Iwami et al. 1995). Finally, AC6 is inhibited, but AC5 stimulated by PKC-mediated phosphorylation (Beazely and Watts 2006), although there are differences between what occurs in cultured cells vs in intact heart. Furthermore, as alluded to above, many mediators that stimulate AC require the presence of Gas and forskolin (Beazely et al. 2005) – conditions that may not hold in the setting of increased expression of AC6.
In addition, all AC isoforms are inhibited by high (nonphysiological) concentrations of calcium. AC5 and AC6, however, are inhibited by physiologically relevant concentrations of calcium (0.2 – 0.6 µM) (Ishikawa and Homcy 1997; Beazely and Watts 2006). The key functional differences between AC5 and AC6 were recently presented in an excellent review (Beazely and Watts 2006). The confluence of multiple G-protein coupled receptors with several types of transducing G proteins operating through multiple AC isoforms creates combinatorial possibilities that enable diverse physiological events and regulatory sites. The focus of the current review will be to examine the physiological consequences of altered amounts of AC6 in the heart and the implications for CHF therapy.
β-Adre2nergic Receptor Signaling in Heart Failure
The βAR signaling pathway in heart failure has been extensively studied since Bristow and colleagues first described downregulation of βAR receptors in failing human heart (Bristow et al. 1982), and was reviewed recently (Brodde et al. 2006). Here we will discuss the results of increasing the amounts of the key elements of the βAR -Ga s-AC pathway. It was shown that cardiac-directed expression of the β1AR increased left ventricular (LV) contractility in young mice (12 weeks), but caused myocardial hypertrophy and progressive heart failure by 35 weeks of age (Engelhart et al. 1999). Functional and histological abnormalities of hearts from these mice resembled those encountered in human patients with heart failure. Thus, upregulation of LV β1AR number initially increases cardiac function, but ultimately results in heart failure. Persistent activation of βAR via sustained agonist infusion, such as isoproterenol, also causes cardiac hypertrophy and cardiomyopathy. Cardiac-directed expression of the β2AR has effects that are similar to β1AR expression – initial increases in LV contractile function leading to CHF in older mice. A lower amount of β2AR expression may have sustained beneficial effects, although these studies did not examine mice over 12 months of age, so a deleterious effect in older mice was not excluded (Liggett et al. 2000).
What occurs when Gas, the transducing protein that links βAR stimulation with AC activation, is increased in the heart? Cardiac -directed Ga s expression has a deleterious effect on the heart, and is similar to the adverse effects of cardiac-directed βAR expression: initial increase in LV contractile function and heart rate, leading ultimately to severe CHF (Iwase et al. 1997). A common feature of cardiac-directed βAR and Gas expression is that both interventions lead to sustained cAMP generation and unrelenting increases in heart rate and basal LV contractile function. In this way, they have features that are similar to long term delivery of isoproterenol, which also leads to heart failure. Indeed, these animal studies have a direct correlate in clinical trials involving patients with CHF. Failing human hearts have reduced amounts of basal cAMP and impaired cAMP generation in response to agonist stimulation (Bristow et al. 1982; Feldman MD et al. 1987). It is logical to surmise that agents that might increase intracellular cAMP levels in the heart would be beneficial in treating clinical CHF. However, results of clinical trials which increase βAR stimulation (dobutamine) or cAMP content (the phosphodiesterase inhibitor milrinone) have been disappointing, perhaps because these agents would be predicted to be associated with sustained increases of intracellular cAMP, which may have provoked life-threatening cardiac arrhythmias.
Cardiac Adenylyl Cyclase and Heart Failure
A hallmark of failed myocardium in clinical settings is reduced LV cAMP and impaired AC function (Bristow et al. 1982, Feldman MD et al. 1987). Our group and others have reported reduced AC5 and AC6 mRNA (Ishikawa et al. 1994) or AC6 mRNA alone (Ping et al. 1997) in failing LV. Due to the pivotal position of AC6 in the βAR signaling pathway and its direct relationship with LV function, we asked if restoration of cardiac AC content and function would increase LV contractile function in CHF.
Transgenic mice with cardiac-directed AC6 expression were generated to determine the effects on cardiac structure and function (Gao MH et al. 1999, Gao MH et al. 2002). Cardiac-directed expression of βAR or Gas has been associated with initial sustained increases in heart rate and LV contractile function, followed eventually by LV chamber dilation, myocardial fibrosis and heart failure in older mice (Engelhardt et al. 1999; Iwase et al. 1997). In contrast, there were distinct differences in mice with cardiac-directed expression of AC6 – no increase in heart rate or LV contractile function was observed in the basal (unstimulated) state in these animals, despite 20-fold excess cardiac AC6 protein (Gao MH et al. 1999). However, hearts showed marked increases in heart rate and contractile function in response to βAR stimulation. Finally, unlike mice with cardiac-directed βAR or Gas expresssion, there was no decline in function or abnormalities in cardiac structure or histology even in mice twenty months old. Increased expression of cardiac AC6, in contrast to βAR or Gas expression, resulted in a marked increase in LV contractile reserve, and was not associated with deleterious consequences. Although the precise mechanisms for these striking differences in effect were not determined, it was noteworthy that cardiac-directed expression of βAR and Gas, but not AC6, was associated with sustained and unrelenting increases in heart rate, LV dP/dt and intracellular cAMP.
Data from these in vivo studies (Gao MH et al. 1999, Gao MH et al. 2002) emphasize a key difference between receptor and effector effects with regard to βAR signaling (Most et al. 2002), suggesting that expression of an effector molecule does not alter transmembrane signaling except when the receptors are activated, in contrast to receptor/G-protein expression, which yield continuous activation and thus negative consequences. These results were confirmed in studies conducted on cultured rat cardiac myocytes in which AC6 was increased by adenovirus-mediated gene transfer. Here it was found that the amount of AC6 protein sets limit on transmembrane βAR signaling, and that even when AC6 expression is markedly increased, basal cAMP levels are not altered (Gao MH et al. 1998).
Increased cardiac AC6 expression in otherwise normal hearts was beneficial, but if AC6 were increased in the setting of cardiomyopathy, would the consequences also be favorable? Cardiac-directed Ga q expression leads to dilated, poorly functioning hearts with impaired cAMP generation, mimicking key aspects of clinical CHF (D'Angelo et al. 1997). These mice were crossbred with mice with cardiac-directed AC6 mice and the Ga q mice compared with offspring harboring both transgenes (Ga q/AC6). Hearts and cardiac myocytes from Ga q/AC6 mice had increased LV function and restored cAMP generating capacity and βAR responsiveness (Roth et al. 1999). Subsequent longer term studies using the same crossbreeding strategy showed that expression of AC6 in this cardiomyopathic background abrogated myocardial hypertrophy and improved survival (Roth et al. 2002). When AC5 was expressed in the Gaq cardiomyopathic background, LV function was improved, but LV hypertrophy and fibrosis persisted, and no alteration in mortality was reported (Tepe NM and Liggett SB, 1999).
Although studies using the crossbreeding strategy are useful, such studies have limitations. In this approach, when heart failure is prevented, the candidate gene is said to have “rescued” the failing heart. However, this strategy does not represent treatment since heart failure is never present. Secondly, the favorable outcome may be due to interactions between transgenes during growth and development that have little to do with the treatment effect per se. A putatively therapeutic transgene that is activated for the first time in the presence of severe CHF would serve as more stringent test and be more readily applicable to the challenges posed by clinical gene transfer. Such an approach requires the use of regulated transgene expression (Fishman 1995, 1998). Using a transgenic mouse with cardiac-directed and regulated AC6 expression (Gao MH et al. 2002), coronary occlusion was performed to induce severe CHF. Five weeks after myocardial infarction, mice with severe heart failure were enrolled in the study. Half of the animals continued with no additional treatment. The remaining half underwent cardiac AC6 gene activation for the first time, which increased cardiac AC6 content 10-fold. Five weeks later there were marked improvements in measures of LV systolic and diastolic function (Lai et al. 2005). These data, published only in abstract form at this writing, indicate that increased cardiac expression of AC6 in the presence of active and severe CHF, has beneficial effects on the failing heart.
Thus far, we have focused on the transmembrane elements of βAR signaling. Protein kinase A (PKA), a downstream target of AC6 via cAMP signaling, can phosphorylate AC6 and inhibit its activity by altering its Gas binding site (Iwami et al. 1995). Cardiac-directed expression of a constitutively active catalytic subunit of PKA resulted in dilated cardiomyopathy and sudden death resembling clinical CHF, with chamber dilatation, peripheral edema, and arrhythmias (Antos et al. 2001). Further analysis showed that both ryanodine receptor 2 (RyR2) and phospholamban (PLB) were hyper-phosphorylated, suggesting that PKA functioned through phosphorylation of these two calcium regulating proteins. Thus, similar to cardiac-directed expression of βAR and Gas, cardiac-directed expression of PKA is detrimental to cardiac physiology. It therefore is difficult to assign the beneficial effects of AC6 expression solely to its activation of PKA. AC6 appears to have effects that are not replicated by PKA activation alone and which suggest unique downstream targets, or protein:protein interactions that do not directly depend on cAMP production per se, but which may influence protein phosphorylation and gene transcription.
Adenylyl Cyclase 6 and Cardiac Calcium Handling
The failing heart is beset with abnormal calcium handling (Morgan et al. 1990; Yano et al. 2005; Bers 2006), and it can be argued that even if desensitization of βAR transmembrane signaling is restored, unless calcium handling abnormalities are remedied, reduced LV contractile function will persist. Ryanodine receptors can act as calcium release channels in the sarcoplasmic reticulum (SR), and thereby regulate calcium handling. RyR2, the predominant cardiac isoform of ryanodine receptor, selectively associates with FK560-binding protein 12.6 (FKBP12.6, also known as calstabin2) (Timerman et al. 1996). The binding of FKBP12.6 to RyR2 stabilizes RyR2 channels (Lehnart et al. 2006). PKA-mediated hyperphosphorylation of RyR2 leads to its dissociation from FKBP1 2.6 and a subsequent diastolic calcium leak, which often is associated with failing LV membranes (Marx et al. 2000; Oda et al. 2005). Agents that directly influence calcium signaling, however, have not yet played a very important role in the pharmacological treatment of clinical CHF in the US.
Is there a link between cardiac AC6 content and calcium signaling that may contribute to the beneficial effects of AC in the failing heart? Mice with cardiac-directed AC6 expression were crossbred with cardiomyopathic mice (cardiac-directed Gaq). Expression of AC6 in this cardiomyopathic background was associated with increased PLB phosphorylation and restoration of impaired SR calcium uptake (Tang et al. 2004). Moreover, in contrast to cardiac-directed PKA expression where RyR2 hyperphosphorylation was observed (Antos et al. 2001), RyR2 phosphorylation was unchanged in treated mice. This suggests that AC6 may regulate RyR2 calcium channels through mechanisms not directly dependent on cAMP and PKA. In the same study, Tang and colleagues also found that PLB expression was reduced when AC6 was expressed in the cardiomyopathic background, an effect that would be predicted to increase calcium availability and LV function (Tang et al. 2004). Modification of myocardial calcium handling is recognized as a promising target for clinical gene transfer (Hajjar et al. 1998; Iwanaga et al. 2004; Hoshijima et al. 2006; Pleger et al. 2007).
Adenylyl Cyclase and Electrical Remodeling of the Heart
It might be anticipated that elevations in cardiac AC6 content would have adverse effects in the setting of myocardial infarction. The attendant increase in cAMP generation, it could be argued, would increase contractile force, exacerbate oxygen demand:supply imbalance, and have detrimental consequences by increasing border zone injury and extending infarct size. However, acute myocardial infarction (MI) of mice with cardiac-directed expression of AC6 was associated with a 45% reduction in mortality seven days after proximal left coronary occlusion (Takahashi et al. 2006). Also seen were increased LV contractility and relaxation three days after MI, and reduced incidence of high grade atrio-ventricular (AV) block (Takahashi et al. 2006). Thus, increased cardiac AC6 expression may facilitate AV conduction and thereby reduce mortality. The observation that AC6 is expressed in the atrio-ventricular node of transgenic mice supports the notion that AC6 may facilitate AV conduction (Sastry et al. 2006). Indeed, cardiac-directed expression of AC6 was shown to facilitate AV nodal anterograde and retrograde conduction without altering sinus node conduction (Sastry et al. 2006).
The effects of cardiac-directed AC6 expression on electrophysiological properties in Gaq cardiomyopathy recently were addressed (Timofeyev et al. 2006). Cardiac myocytes from Gaq mice, like failing cardiac myocytes from other models, show prolonged action potential duration (APD) with reduced Ito and IK1 current density. AC6 expression abrogated adverse electrical remodeling by normalizing APD, and increasing Ito and IK1 current density (Timofeyev et al. 2006). Prolonged APD is correlated with propensity to develop ventricular tachycardia and ventricular fibrillation in CHF (Nattel et al. 2007, Tomaselli and Marban 1999). These studies reinforce the notion that electrophysiological remodeling is an important maladaptation in cardiomyopathy, and that favorable effects of AC6 on electrophysiological remodeling may be responsible, at least in part, for its beneficial effects.
Adenylyl Cyclase 6 Effects not Solely Dependent on cAMP
One of the recurrent themes uncovered in these physiological studies of the beneficial effects of AC6 on cardiac function is the possibility that the favorable effects of increased AC6 content may not be solely dependent upon increased cAMP generation. This may be particularly germane for the failing heart, since agents that promote cAMP content tend to have adverse outcomes. Could AC6 have effects on gene transcription and protein phosphorylation that are not directly dependent on cAMP generation?
To explore the mechanism for reduced expression of PLB, seen when AC6 is co-expressed in cardiomyopathy (Tang et al. 2004), studies were conducted using cultured neonatal cardiac myocytes and adenovirus-mediated AC6 gene transfer. Increased AC6 expression reduced PLB expression via marked upregulation of activating transcriptional factor 3 (ATF-3). ATF-3, a transcriptional suppressor, was then shown to bind to and inhibit the PLB promoter. Thus, AC6 gene transfer reduces PLB expression via ATF-3 (Gao MH et al. 2004). The effects of AC6 on ATF3 expression and PLB transcription were independent of cAMP generation. Agents that increased cAMP, such as isoproterenol and forskolin, had directionally opposite effects on PLB expression compared to the effects of AC6 gene transfer. When cardiac myocytes were stimulated with isoproterenol or forskolin after AC6 gene transfer, a further reduction in PLB expression and increased PLB phosphorylation were observed (Gao MH et al. 2004). The reduced amount of PLB showed increased phosphorylation, again, not due to cAMP. Preliminary data indicate that AC6 gene transfer is associated with increased Akt phosphorylation (Gao MH et al. 2007) possibly through association with a phosphatase and subsequent prevention of dephosphorylation (Gao MH et al. 2007).
Finally, intracellular compartmentation may enable alterations of specific signaling pathways otherwise not anticipated (Head et al. 2006). For example, intracoronary AC6 gene transfer would be anticipated to increase AC6 content of both cardiac myocytes and fibroblasts. Consequently, the cardiac interstitium could be affected through alterations in fibroblast signaling (Swaney et al. 2005). Although the exact mechanisms for the beneficial effects of AC6 on the failing heart will require additional investigation, these studies indicate that AC6 gene transfer is associated with effects that are not replicated by agents that increase cAMP production, effects which may provide an insight into why AC6 has such different effects than other agents in the βAR signaling pathway (Figure 2).
Figure 2. Hypothesis for beneficial effects of AC6 gene transfer.
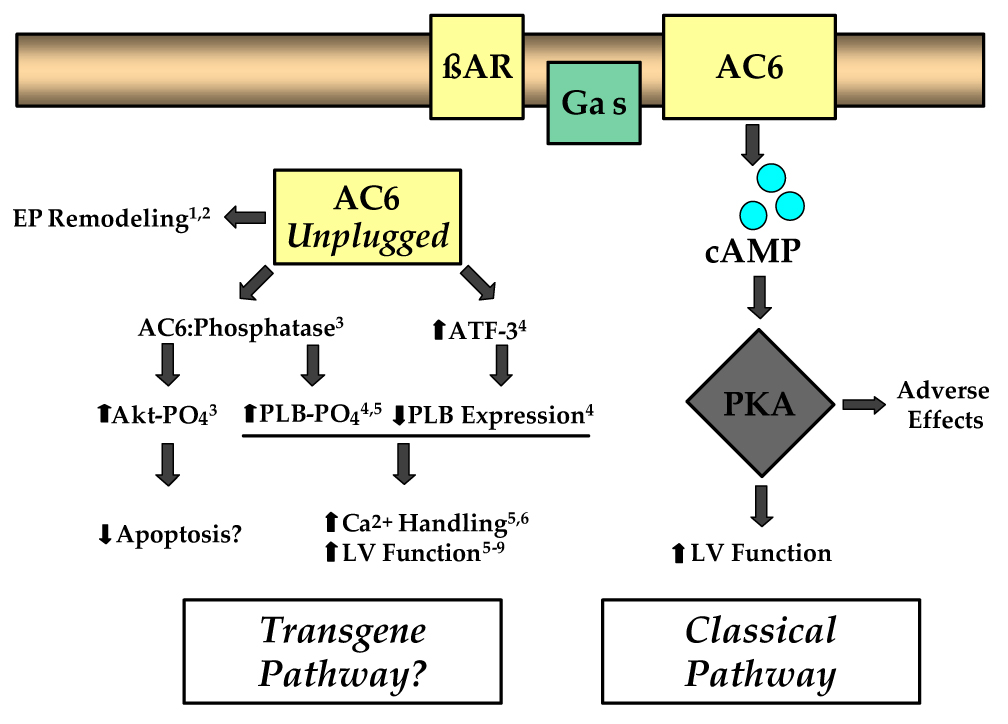
Functions of AC6 that are not dependent on β-adrenergic receptor (βAR) stimulation and cAMP generation (Transgene Pathway) may underlay the beneficial effects of AC6 gene transfer, offsetting the potentially deleterious effects associated with agents that provide excessive cAMP/PKA activation (Classical Pathway). AC6 gene transfer increases AC6 content in non-plasma membrane compartment (Gao et al., 2004). Superscripts refer to published data: 1 (Sastry et al, 2006); 2 (Timofeyev et al. 2006); 3 (Gao et al., 2007); 4 (Gao et al., 2004); 5 (Tang et al., 2004); 6 (Takahashi et al., 2006); 7 (Rebolledo et al., 2006), 8 (Roth et al., 2000); 9 (Lai et al. 2004). Gas, stimulatory GTP-binding protein; AC6, adenylyl cyclase type 6; AC6 Unplugged, transgene AC6 not associated with plasma membrane and βAR/Gas interaction; cAMP, cyclic adenosine monophosphate; PKA, protein kinase A; ATF-3, activating transcription factor-3; PLB, phospholamban; AC6:Phosphatase, postulated AC6:phosphatase interaction which may increase Akt and PLB phosphorylation in the basal (unstimulated) state
Adenylyl Cyclase 6 Gene Transfer as Therapy for Heart Failure?
Although the amount of AC6 protein has not been quantified in human failing heart, and there is debate regarding whether AC activity is impaired, there is uniform agreement that isoproterenol-stimulated cAMP production is reduced (Bristow MR, et al., 1982). Is it feasible that increased cardiac AC protein content will translate to improvement in cardiac responsiveness and symptoms in clinical heart failure? We have no experimental data of AC6 gene transfer in clinical CHF, so will speculate, based on preclinical data. First, we believe that the amount of AC (not βAR) sets a limit on a cardiac myocyte's ability to generate cAMP, as we demonstrated in gene transfer studies of AC6 in cultured cardiac myocytes (Gao MH et al., 1998). Because AC content sets the limit, it is reasonable to propose that increasing the effector (AC6) will allow increased cAMP production, and, consequently physiological responsiveness, at any given level of proximal desensitization. Indeed, that is exactly what we observe in experimentally induced heart failure in mice (Roth DM et al., 1999; Roth DM et al., 2000; Rebolledo B et al., 2006) and pigs (Lai NC et al., 2004) after increases in cardiac AC content, despite pre-treatment impairment in proximal elements of βAR signaling. Second, even when AC activity is entirely normal, increased AC6 content increases LV contractile reserve to βAR stimulation (Lai NC et al., 2000). Third, the apparent favorable effects of AC which appear to be independent of cAMP generation may important in the overall therapeutic effect of AC6 as we have previously discussed.
Might a simple medical therapy that stimulates endogenous AC have advantages over the more complicated prospect of using AC6 gene transfer? Indeed, a water soluble forskolin analog, colforsin (also known as NKH477) is used in clinical settings in Japan (Kikura M et al., 2004). Colforsin is administered by continuous intravenous infusion and has potent hemodynamic effects, and is not suited for long term therapy. In contrast, AC6 gene transfer has no acute hemodynamic effects, does not require continuous infusion, and would be anticipated, based on preclinical data, to have long term beneficial effects not limited to augmentation of cAMP production.
Exogenous gene transfer will be required if AC6 is ever to be applied in the treatment of clinical heart failure. Mice with Gaq cardiomyopathy received indirect intracoronary delivery of an adenovirus vector encoding AC6, and LV systolic and diastolic function were increased fourteen days after delivery (Rebolledo et al., 2006). However, this study used thoracotomy and aortic and pulmonary artery cross clamping to deliver the adenovirus vector, a technique that will not be applicable to patients with severe CHF. Direct intracoronary infusion of an adenovirus encoding AC6, given simultaneously with intracoronary nitroprusside, was associated with favorable effects on LV function in pigs with pacing-induced CHF (Lai et al. 2004). Previous studies had documented that the addition of intracoronary nitroprusside was associated with increased transgene delivery and cAMP-generating capacity (Roth et al. 2004). In rats, highly efficient gene transfer to cardiac myocytes was reported after only a single pass coronary delivery (Logeart et al. 2000). Nevertheless, one of many concerns regarding translation to clinical studies is gene transfer efficiency and duration of transgene expression. In pigs, direct intracoronary delivery of adenovirus provided detectable transgene expression in 39% of cells in the relevant perfusion bed (Suzuki et al. 2005); the injections were accompanied by histamine, an agent that also appears to increase gene transfer efficiency (Lai et al, 2000). It should be pointed out, however, that the dose used in these studies, >1012 virus particles (vp), is a dose that has not been used clinically, where the current maximal dose has been 3.3×1010 vp (Grines et al. 2002). Recent modifications for intracoronary delivery of virus vectors may increase cardiac gene transfer efficiency (Sasano et al. 2007). Although intracoronary AC6 gene transfer was shown to result in persistent elevations in LV cAMP generating capacity – up to eighteen weeks after delivery (Lai et al. 2000), it is generally believed that longer term expression vectors, such as adeno-associated virus, will be required for optimal treatment (Iwanaga et al. 2004; Kaspar et al. 2005). Taken together, these studies indicate that intracoronary delivery of adenovirus encoding AC6 in the setting of active heart failure may be a rational approach for the treatment of clinical CHF (Feldman AM 2002, Sesti and Kloner 2004, Nayak and Rosengart 2005).
Conclusions
AC6 is the sole element of the βAR-Gas-AC-cAMP-PKA signaling pathway that, when increased in amount, is associated with beneficial effects in the setting of heart failure. This suggests that AC6 may have effects, in addition to its classical enzymatic activity, that may serve to offset the expected negative consequences of increased cAMP content. Recently published studies suggest that some of these unanticipated effects of increased AC6 content include: favorable effects on calcium handling, electrophysiological remodeling, transcriptional regulation of genes that influence contractile function, and phosphorylation of key proteins, perhaps by protein:protein interactions that influence phosphatase activity. Finally, it seems likely that the physiological effects evoked by overexpression of pivotal proteins such as AC6, may not recapitulate classical functions of the protein, due to alterations in amounts, accessibility to unexpected intracellular microdomains and unanticipated interactions with other proteins.
Acknowledgements
Dr. Hammond is supported by National Institutes of Health grants 5P01HL066941 and HL081741 and a Merit Review Award from the Department of Veterans Affairs. Dr. Gao is supported by a Grant in Aid from the American Heart Association, Western States Affiliate.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Antos CL, Frey N, Marx SO, Reiken S, Gaburjakova M, Richardson JA, et al. Dilated cardiomyopathy and sudden death resulting from constitutive activation of protein kinase A. Circ Res. 2001;89:997–1004. doi: 10.1161/hh2301.100003. [DOI] [PubMed] [Google Scholar]
- Beazely MA, Alan JK, Watts VJ. Protein kinase C and epidermal growth factor stimulation of Raf1 potentiates adenylyl cyclase type 6 activation in intact cells. Mol Pharmacol. 2005;67:250–259. doi: 10.1124/mol.104.001370. [DOI] [PubMed] [Google Scholar]
- Beazely MA, Watts VJ. Regulatory properties of adenylate cyclases type 5 and 6: A progress report. Eur J Pharmacol. 2006;535:1–12. doi: 10.1016/j.ejphar.2006.01.054. [DOI] [PubMed] [Google Scholar]
- Bers DM. Altered cardiac myocyte Ca regulation in heart failure. Physiology (Bethesda) 2006;21:380–387. doi: 10.1152/physiol.00019.2006. [DOI] [PubMed] [Google Scholar]
- Bristow MR, Ginsburg R, Minobe W, Cubicciotti RS, Sageman WS, Lurie K, et al. Decreased catecholamine sensitivity and beta-adrenergic-receptor density in failing human hearts. N Engl J Med. 1982;307:205–211. doi: 10.1056/NEJM198207223070401. [DOI] [PubMed] [Google Scholar]
- Brodde OE, Bruck H, Leineweber K. Cardiac adrenoceptors: physiological and pathophysiological relevance. J Pharmacol Sci. 2006;100:323–337. doi: 10.1254/jphs.crj06001x. [DOI] [PubMed] [Google Scholar]
- D'Angelo DD, Sakata Y, Lorenz JN, Boivin GP, Walsh RA, Liggett SB, et al. Transgenic Ga q overexpression induces cardiac contractile failure in mice. Proc Natl Acad Sci USA. 1997;94:8121–8126. doi: 10.1073/pnas.94.15.8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt S, Hein L, Wiesmann F, Lohse MJ. Progressive hypertrophy and heart failure in β1-adrenergic receptor transgenic mice. Proc Natl Acad Sci USA. 1999;96:7059–7064. doi: 10.1073/pnas.96.12.7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman AM. Adenylyl cyclase: a new target for heart failure therapeutics. Circulation. 2002;105:1876–1878. doi: 10.1161/01.cir.0000016965.24080.12. [DOI] [PubMed] [Google Scholar]
- Feldman MD, Copelas L, Gwathmey JK, Phillips P, Warren SE, Schoen FJ, et al. Deficient production of cyclic AMP: pharmacological evidence of an important cause of contractile dysfunction in patients with end-stage heart failure. Circulation. 1987;75:331–339. doi: 10.1161/01.cir.75.2.331. [DOI] [PubMed] [Google Scholar]
- Fishman GI. Conditional transgenics. Trends Cardiovasc Med. 1995;5:211–217. doi: 10.1016/1050-1738(95)00104-2. [DOI] [PubMed] [Google Scholar]
- Fishman GI. Timing is everything in life: conditional transgene expression in the cardiovascular system. Circ Res. 1998;82:837–844. doi: 10.1161/01.res.82.8.837. [DOI] [PubMed] [Google Scholar]
- Gao MH, Ping P, Post S, Insel PA, Tang R, Hammond HK. Increased expression of adenylylcyclase type VI proportionately increases beta-adrenergic receptor-stimulated production of cAMP in neonatal rat cardiac myocytes. Proc Natl Acad Sci USA. 1998;95:1038–1043. doi: 10.1073/pnas.95.3.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao MH, Lai NC, Roth DM, Zhou J, Zhu J, Anzai T, et al. Adenylylcyclase increases responsiveness to catecholamine stimulation in transgenic mice. Circulation. 1999;99:1618–1622. doi: 10.1161/01.cir.99.12.1618. [DOI] [PubMed] [Google Scholar]
- Gao MH, Bayat H, Roth DM, Yao Zhou J, Drumm J, Burhan J, et al. Controlled expression of cardiac-directed adenylylcyclase type VI provides increased contractile function. Cardiovasc Res. 2002;56:197–204. doi: 10.1016/s0008-6363(02)00539-4. [DOI] [PubMed] [Google Scholar]
- Gao MH, Tang T, Guo T, Sun SQ, Feramisco JR, Hammond HK. Adenylyl cyclase type VI gene transfer reduces phospholamban expression in cardiac myocytes via activating transcription factor 3. J Biol Chem. 2004;37:38797–38802. doi: 10.1074/jbc.M405701200. [DOI] [PubMed] [Google Scholar]
- Gao MH, Tang T, Guo T, Newton AC, Hammond HK. Adenylyl cyclase type VI regulates Akt activity through a PH Domain leucine-rich repeat protein phosphatase. Mol Ther. 2007;15:454. (Abstract) [Google Scholar]
- Gao X, Sadana R, Dessauer CW, Patel TB. Conditional stimulation of type V and VI adenylyl cyclases by G protein beta gamma subunits. J Biol Chem. 2007;282:294–302. doi: 10.1074/jbc.M607522200. [DOI] [PubMed] [Google Scholar]
- Grines CL, Watkins MW, Helmer G, Penny W, Brinker J, Marmur JD, et al. Angiogenic Gene Therapy (AGENT) trial in patients with stable angina pectoris. Ciruclation. 2002;105:1291–1297. doi: 10.1161/hc1102.105595. [DOI] [PubMed] [Google Scholar]
- Hajjar RJ, Schmidt U, Matsui T, Guerrero JL, Lee KH, Gwathmey JK, et al. Modulation of ventricular function through gene transfer in vivo. Proc Natl Acad Sci USA. 1998;95:5251–5256. doi: 10.1073/pnas.95.9.5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanoune J, Defer N. Regulation and role of adenylyl cyclase isoforms. Annu Rev Pharmacol Toxicol. 2001;41:145–174. doi: 10.1146/annurev.pharmtox.41.1.145. [DOI] [PubMed] [Google Scholar]
- Head BP, Patel HH, Roth DM, Murray F, Swaney JS, Niesman IR, et al. Microtubules and actin microfilaments regulate lipid raft/caveolae localization of adenylyl cyclase signaling components. J Biol Chem. 2006;281:26391–26399. doi: 10.1074/jbc.M602577200. [DOI] [PubMed] [Google Scholar]
- Hoshijima M, Knoll R, Pashmforoush M, Chien KR. Reversal of calcium cycling defects in advanced heart failure toward molecular therapy. J Am Coll Cardiol. 2006;48 9 Suppl 1:A15–A23. doi: 10.1016/j.jacc.2006.06.070. [DOI] [PubMed] [Google Scholar]
- Ishikawa Y, Homcy CJ. The adenylyl cyclases as integrators of transmembrane signal transduction. Circ Res. 1997;80:297–304. doi: 10.1161/01.res.80.3.297. [DOI] [PubMed] [Google Scholar]
- Ishikawa Y, Sorota S, Kiuchi K, Shannon RP, Komamura K, Katsushika S, et al. Downregulation of adenylylcyclase types V and VI mRNA levels in pacing-induced CHF in dogs. J Clin Invest. 1994;93:2224–2229. doi: 10.1172/JCI117219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwami G, Kawabe J, Ebina T, Cannon PJ, Homcy CJ, Ishikawa Y. Regulation of adenylyl cyclase by protein kinase A. J Biol Chem. 1995;270:12481–12484. doi: 10.1074/jbc.270.21.12481. [DOI] [PubMed] [Google Scholar]
- Iwanaga Y, Hoshijima M, Gu Y, Iwatate M, Dieterle T, Ikeda Y, et al. Chronic phospholamban inhibition prevents progressive cardiac dysfunction and pathological remodeling after infarction in rats. J Clin Invest. 2004;113:727–736. doi: 10.1172/JCI18716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwase M, Uechi M, Vatner DE, Asai K, Shannon RP, Kudej RK, et al. Cardiomyopathy induced by cardiac Gsα overexpression. Am J Physiol Heart Circ Physiol. 1997;272:585–589. doi: 10.1152/ajpheart.1997.272.1.H585. [DOI] [PubMed] [Google Scholar]
- Kaspar BK, Roth DM, Lai NC, Drumm JD, Erickson DA, McKirnan MD, et al. Myocardial gene transfer and long-term expression following intracoronary delivery of adeno-associated virus. J Gene Med. 2005;7:316–324. doi: 10.1002/jgm.665. [DOI] [PubMed] [Google Scholar]
- Kikura M, Morita K, Sato S. Pharmacokinetics and a simulation model of colforsin daropate, new forskolin derivative inotropic vasodilator, in patients undergoing coronary artery bypass grafting. Pharmacol Res. 2004;49:275–281. doi: 10.1016/j.phrs.2003.09.012. [DOI] [PubMed] [Google Scholar]
- Lai NC, Roth DM, Gao MH, Fine SA, Head BP, Zhu J, et al. Intracoronary delivery of adenovirus encoding adenylyl cyclase VI increases left ventricular function and cAMP-generating capacity. Circulation. 2000;102:2396–2401. doi: 10.1161/01.cir.102.19.2396. [DOI] [PubMed] [Google Scholar]
- Lai NC, Roth DM, Gao MH, Tang T, Dalton N, Lai YY, et al. Intracoronary adenovirus encoding adenylyl cyclase VI increases left ventricular function in heart failure. Circulation. 2004;110:330–336. doi: 10.1161/01.CIR.0000136033.21777.4D. [DOI] [PubMed] [Google Scholar]
- Lai NC, Saito M, Tang T, et al. Activation of cardiac adenylyl cyclase type VI expression in the presence of severe heart failure increases LV systolic and diastolic function. Circulation. 2005;112 (Suppl):II-64. (Abstract) [Google Scholar]
- Lehnart SE, Terrenoire C, Reiken S, Wehrens XHT, Song L-S, Tillman EJ, et al. Stabilization of cardiac ryanodine receptor prevents intracellular calcium leak and arrhythmias. Proc Natl Acad Sci USA. 2006;103:7906–7910. doi: 10.1073/pnas.0602133103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liggett SB, Tepe NM, Lorenz JN, Canning AM, Jantz TD, Mitarai S, et al. Early and delayed consequences of β2-adrenergic receptor overexpression in mouse hearts: critical role for expression level. Circulation. 2000;101:1707–1714. doi: 10.1161/01.cir.101.14.1707. [DOI] [PubMed] [Google Scholar]
- Logeart D, Hatem SN, Rucker-Martin C, Chossat N, Néco N, Haddada H, et al. Highly efficient adenovirus-mediated gene transfer to cardiac myocytes after single-pass coronary delivery. Hum Gene Ther. 2000;11:1015–1022. doi: 10.1089/10430340050015329. [DOI] [PubMed] [Google Scholar]
- Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, et al. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- Morgan JP, Erny RE, Allen PD, Grossman W, Gwathmey JK. Abnormal intracellular calcium handling, a major cause of systolic and diastolic dysfunction in ventricular myocardium from patients with heart failure. Circulation. 1990;81:III21–III32. [PubMed] [Google Scholar]
- Most P, Remppis A, Katus HA. Conditional AC type VI expression in the heart: relevant insights into function of inducible target gene expression. Cardiovasc Res. 2002;56:181–183. doi: 10.1016/s0008-6363(02)00653-3. [DOI] [PubMed] [Google Scholar]
- Nattel S, Maguy A, Le Bouter S, Yeh YH. Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocar dial infarction, and atrial fibrillation. Physiol Rev. 2007;87:425–456. doi: 10.1152/physrev.00014.2006. [DOI] [PubMed] [Google Scholar]
- Nayak L, Rosengart TK. Gene therapy for heart failure. Semin Thorac Cardiovasc Surg. 2005;17:343–347. doi: 10.1053/j.semtcvs.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Oda T, Yano M, Yamamoto T, Tokuhisa T, Okuda S, Doi M, et al. Defective regulation of interdomain interactions within ryanodine receptor plays a key role in the pathogenesis of heart failure. Circulation. 2005;111:3400–3410. doi: 10.1161/CIRCULATIONAHA.104.507921. [DOI] [PubMed] [Google Scholar]
- Okumura S, Tskagi G, Kawabe J, Yang G, Lee MC, Hong C, et al. Disruption of type 5 adenylyl cyclase gene preserves cardiac function against pressure overload. Proc Natl Acad Sci USA. 2003;100:9986–9990. doi: 10.1073/pnas.1733772100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping P, Anzai T, Gao M, Hammond HK. Adenylyl cyclase and G protein receptor kinase expression during development of heart failure. Am J Physiol Heart Circ Physiol. 1997;273:707–717. doi: 10.1152/ajpheart.1997.273.2.H707. [DOI] [PubMed] [Google Scholar]
- PLeger ST, Most P, Boucher M, Soltys S, Chuprun JK, Pleger W, et al. Stable myocardial-specific AAV6-S100A1 gene therapy results in chronic functional heart failure rescue. Circulation. 2007;115:2506–2515. doi: 10.1161/CIRCULATIONAHA.106.671701. [DOI] [PubMed] [Google Scholar]
- Rabinowitz JE, Samulski J. Adeno-associated virus expression systems for gene transfer. Curr Opin Biotech. 1998;9:470–475. doi: 10.1016/s0958-1669(98)80031-1. [DOI] [PubMed] [Google Scholar]
- Rebolledo B, Lai NC, Gao MH, Takahashi T, Roth DM, Baird SB, et al. Adenylylcyclase gene transfer increases function of the failing heart. Hum Gene Ther. 2006;17:1043–1048. doi: 10.1089/hum.2006.17.1043. [DOI] [PubMed] [Google Scholar]
- Roth DM, Gao MH, Lai NC, Drumm J, Dalton N, Zhou JY, et al. Cardiac-directed adenylyl cyclase expression improves heart function in murine cardiomyopathy. Circulation. 1999;99:3099–3102. doi: 10.1161/01.cir.99.24.3099. [DOI] [PubMed] [Google Scholar]
- Roth DM, Bayat H, Drumm JD, Gao MH, Swaney JS, Ander A, et al. Adenylyl cyclase increases survival in cardiomyopathy. Circulation. 2002;105:1989–1994. doi: 10.1161/01.cir.0000014968.54967.d3. [DOI] [PubMed] [Google Scholar]
- Roth DM, Lai NC, Gao MH, Fine S, McKirnan MD, Roth DA, et al. Nitroprusside increases gene transfer associated with intracoronary delivery of adenovirus. Hum Gene Ther. 2004;15:989–994. doi: 10.1089/hum.2004.15.989. [DOI] [PubMed] [Google Scholar]
- Sasano T, Kikuchi K, McDonald AD, Lai S, Donahue JK. Targeted high-efficiency, homogeneous myocardial gene transfer. J Mol Cell Cardiol. 2007;42:954–961. doi: 10.1016/j.yjmcc.2007.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastry A, Arnold E, Gurji H, Iwasa A, Bui H, Hassankhani A, et al. Cardiac-directed expression of adenylyl cyclase VI facilitates atrioven tricular nodal conduction. J Amer Coll Card. 2006;48:559–565. doi: 10.1016/j.jacc.2006.01.082. [DOI] [PubMed] [Google Scholar]
- Sesti C, Kloner RA. Gene therapy in congestive heart failure. Circulation. 2004;110:242–243. doi: 10.1161/01.CIR.0000137593.62669.67. [DOI] [PubMed] [Google Scholar]
- Sunahara RK, Dessauer CW, Gilman AG. Complexity and diversity of mammalian adenylyl cyclases. Annu Rev Pharmacol Toxicol. 1996;36:461–480. doi: 10.1146/annurev.pa.36.040196.002333. [DOI] [PubMed] [Google Scholar]
- Sunahara RK, Taussig R. Isoforms of mammalian adenylyl cyclase: multiplicities of signaling. Mol Interv. 2002;2:168–184. doi: 10.1124/mi.2.3.168. [DOI] [PubMed] [Google Scholar]
- Suzuki G, Lee TC, Fallavollita JA, Canty JM. Adenoviral gene transfer of FGF-5 to hibernating myocardium improves function and stimulates myocytes to hypertrophy and reenter the cell cycle. Circ Res. 2005;96:767–775. doi: 10.1161/01.RES.0000162099.01268.d1. [DOI] [PubMed] [Google Scholar]
- Swaney JS, Roth DM, Olson ER, Naugle JE, Meszaros JG, Insel PA. Inhibition of cardiac myofibroblast formation and collagen synthesis by activation and overexpression of adenylyl cyclase. Proc Natl Acad Sci. 2005;102:437–442. doi: 10.1073/pnas.0408704102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahasi T, Tang T, Lai NC, Roth DM, Rebolledo B, Saito M, et al. Increased cardiac adenylyl cyclase expression is associated with increased survival after myocardial infarction. Circulation. 2006;114:388–396. doi: 10.1161/CIRCULATIONAHA.106.632513. [DOI] [PubMed] [Google Scholar]
- Tang T, Gao MH, Roth DM, Guo T, Hammond HK. Adenylyl cyclase type VI corrects cardiac sarcoplasmic reticulum calcium uptake defects in cardiomyopathy. Am J Physiol Heart Circ Physiol. 2004;287:1906–1912. doi: 10.1152/ajpheart.00356.2004. [DOI] [PubMed] [Google Scholar]
- Tang T, Lai NC, Roth DM, Drumm J, Guo T, Lee KW, et al. Adenylyl cyclase type V deletion increases basal left ventricular function and reduces left ventricular contractile responsiveness to β-adrenergic stimulation. Basic Res Cardiol. 2006;101:117–126. doi: 10.1007/s00395-005-0559-y. [DOI] [PubMed] [Google Scholar]
- Tepe NM, Liggett SB. Transgenic replacement of type V adenylyl cyclase identifies a critical mechanism of beta-adrenergic receptor dysfunction in the G alpha q overexpressing mouse. FEBS Lett. 1999;458:236–240. doi: 10.1016/s0014-5793(99)01147-3. [DOI] [PubMed] [Google Scholar]
- Timerman AP, Onoue H, Xin HB, Barg S, Copello J, Wiederrecht G, et al. Selective binding of FKBP12.6 by the cardiac ryanodine receptor. J Biol Chem. 1996;271:20385–20391. doi: 10.1074/jbc.271.34.20385. [DOI] [PubMed] [Google Scholar]
- Timofeyev V, He Y, Tuteja D, Zhang Q, Roth DM, Hammond HK, et al. Cardiac-directed expression of adenylyl cyclase reverses electrical remodeling in cardiomyopathy. J Mol Cell Card. 2006;41:170–181. doi: 10.1016/j.yjmcc.2006.04.008. [DOI] [PubMed] [Google Scholar]
- Tobise K, Ishikawa Y, Holmer SR, Im MJ, Newell JB, Yoshi H, et al. Changes in type VI adenylyl cyclase isoform expression correlate with a decreased capacity for cAMP generation in the aging ventricle. Circ Res. 1994;74:596–603. doi: 10.1161/01.res.74.4.596. [DOI] [PubMed] [Google Scholar]
- Tomaselli GF, Marban E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res. 1999;42:270–283. doi: 10.1016/s0008-6363(99)00017-6. [DOI] [PubMed] [Google Scholar]
- Yano M, Ikeda Y, Matsuzaki M. Altered intracellular Ca2+ handling in heart failure. J Clin Invest. 2005;115:556–564. doi: 10.1172/JCI24159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Brown MJ. Differential expression of adenylyl cyclase subtypes in human cardiovascular system. Mol Cell Endocrinol. 2004;223:55–62. doi: 10.1016/j.mce.2004.05.012. [DOI] [PubMed] [Google Scholar]