

Abstract
Histamine stimulation of swine carotid artery induces both contraction and actin polymerization. The importance of stimulus-induced actin polymerization is not known. Tyrosine phosphorylation of the scaffolding protein paxillin is thought to be an important regulator of actin polymerization. Noise temperature, hysteresivity, and phase angle are rheological measures of the fluidity of a tissue, i.e., whether the muscle is more a “Hookean solid” or a “Newtonian liquid.” Y118 paxillin phosphorylation, crossbridge phosphorylation, actin polymerization, noise temperature, hysteresivity, phase angle, real stiffness, and stress were measured in intact swine carotid arteries that were depolarized with high K+ or stimulated with histamine. The initial rapid force development phase of high-K+ or histamine-induced contraction was associated with increased crossbridge phosphorylation but no significant change in Y118 paxillin phosphorylation, actin polymerization, noise temperature, hysteresivity, or phase angle. This suggests that the initial contraction was caused by the increase in crossbridge phosphorylation and did not alter the tissue's rheology. Only after full force development was there a significant increase in Y118 paxillin phosphorylation and actin polymerization associated with a significant decrease in noise temperature and hysteresivity. These data suggest that some part of the sustained contraction may depend on stimulated actin polymerization and/or a transition to a more “solid” rheology. Supporting this contention was the finding that an inhibitor of actin polymerization, latrunculin-A, reduced force while increasing noise temperature/hysteresivity. Further research is needed to determine whether Y118 paxillin phosphorylation, actin polymerization, and changes in rheology could have a role in arterial smooth muscle contraction.
Keywords: cytoskeleton, hysteresivity, latch hypothesis, vascular smooth muscle
MAXIMAL STIMULATION of arterial smooth muscle typically produces a large increase in myoplasmic Ca2+, which activates myosin kinase. The activated myosin kinase phosphorylates crossbridges on Ser19 of the myosin regulatory light chain (MRLC). The resulting large increase in crossbridge phosphorylation is felt to produce rapid force development (reviewed in Ref. 23). During the sustained phase of a maximal contraction, crossbridge phosphorylation typically falls to intermediate levels while force is maintained at peak levels, a phenomenon termed “latch” (8, 26). At first, it was thought that a second regulatory system was necessary to explain the latch phenomenon (6). However, dynamic crossbridge models suggest that accumulating attached dephosphorylated crossbridges (latchbridges) can explain many of the characteristics of the latch phenomenon, specifically, maintained force despite reduced crossbridge phosphorylation, shortening velocity, and ATP consumption (14, 31, 45). These models suggest that a second regulatory system is not necessary to explain the latch phenomenon.
In other types of smooth muscle, there are data suggesting that systems beyond crossbridge phosphorylation are involved in force regulation. One system involves stimulus-induced tyrosine phosphorylation of paxillin and resultant actin polymerization (reviewed in Ref. 4 and 38). Most of the smooth muscle work on stimulus-induced paxillin phosphorylation and actin polymerization was done in airway smooth muscle (reviewed in Ref. 12). Paxillin was first found to be phosphorylated on tyrosine during acetylcholine-induced contraction of intact trachealis smooth muscle (25). The time course of paxillin tyrosine phosphorylation in trachealis was similar to the time course of contraction (41), suggesting potential regulation of contraction. Reductions in extracellular Ca2+ attenuated acetylcholine-induced increases in crossbridge phosphorylation and contraction but did not alter acetylcholine-induced increases in paxillin tyrosine phosphorylation (21), suggesting that 1) paxillin tyrosine phosphorylation is not regulated by Ca2+ or crossbridge phosphorylation and 2) paxillin phosphorylation alone is not sufficient for trachealis contraction. Carbachol increased the F-actin content of cultured human airway smooth muscle (35), a process involving G protein mediated activation of rho (16). In intact trachealis, contractile stimulation increased actin polymerization as measured by an increase in F-actin content (20). These findings are consistent with the hypothesis that stimulus-induced paxillin phosphorylation could potentially modulate trachealis contraction by altering actin polymerization.
A basal level of actin polymerization appears to be necessary for the initiation of contraction in tracheal smooth muscle. Cytochalasin B and D, agents that reduce actin polymerization, attenuated carbachol-induced increases in [Ca2+]i, crossbridge phosphorylation, and contraction in bovine trachealis (37). However, another group found that latrunculin-A, another agent that inhibits actin polymerization, reduced force without altering crossbridge phosphorylation in canine trachealis (20). These data suggest two mechanisms for short-term actin inhibition to inhibit contraction: 1) reduced [Ca2+]i/crossbridge phosphorylation and/or 2) reduced actin polymerization. On the basis of a strategy of antisense, siRNA, and mutant proteins, Gunst and colleagues (see Ref. 34 for review) showed that long-term (days) interference with some of the proteins in the actin polymerization pathway (FAK, paxillin, CrkII, Cdc42, and N-WASp) interfered with actin polymerization and contraction without altering crossbridge phosphorylation values measured 5 min after activation. These experiments showed that long-term interference with actin polymerization can attenuate contraction despite stimulus-induced increases in Ca2+ and crossbridge phosphorylation.
Less is known about stimulus-induced paxillin phosphorylation and actin polymerization in arterial smooth muscle. In cells cultured from aorta, angiotensin II induced paxillin tyrosine phosphorylation and increased actin-containing stress fibers (39), a process that may involve c-Src (17). In intact freely floating mesenteric artery, norepinephrine induced a slow increase in paxillin tyrosine phosphorylation that was maximal at 2.5 min (24, 42). This result suggests that paxillin tyrosine phosphorylation could increase slower than force; however, arterial length was not controlled and short length is known to attenuate paxillin phosphorylation in airway smooth muscle (32, 33). Cytochalasin D attenuated rat pulmonary artery contraction (1). In cerebral arteries, cytochalasin D reduced myogenic tone and decreased actin polymerization, while jasplakinolide, an agent that stabilizes F actin, increased myogenic tone and increased actin polymerization (7). Crossbridge phosphorylation was not measured in either of these studies, so the mechanism responsible for inhibition of contraction is not known. We found that sustained histamine stimulation of swine carotid artery increased F-actin content (19); however, it is not known whether histamine-induced actin polymerization was necessary for the contraction.
Oscillatory changes in muscle length during contraction can provide information about the dynamic processes present in the cytoplasm. For example, the real part of stiffness (also known as the storage modulus G') is felt to be a measure of the number of attached crossbridges (22). Fredberg et al. (5, 18) suggest that the biophysical measures hysteresivity, noise temperature, and phase angle can also provide information about the physical properties (rheology) of the cell. Noise temperature can be understood within the theory of soft glassy rheology: “glasses are substances with the structural disorder of a liquid but the stiffness of a solid” (13). Glasslike stiffness is felt to originate from interactions between neighboring molecules that tend to trap each molecule in an energy well. In a glass, “structural rearrangements become limited by long-lived microconfigurations in which the system becomes trapped” (36). Thermal energy and intermolecular interactions can cause a molecule to “hop” out of its energy well, a process that will reorganize the microconfiguration of energy wells in the system (13, 36). Noise temperature is proposed to be the quantitative level of nonthermal “random agitation (noise) (that) can excite a metastable element, causing it to hop out of its well, in turn triggering secondary rearrangements and hopping events that ripple through the system” (13). A noise temperature of 1 indicates that the system approximates a Hookean elastic solid with no internal friction, no “hopping,” and no rearrangements. A noise temperature of 2 indicates that the system approximates a Newtonian viscous fluid (18), with internal friction, frequent “hopping,” and flow, i.e., frequent rearrangement. Therefore, the noise temperature of a smooth muscle can viewed as measure of whether a tissue is more “fluid” (a high noise temperature) or “solid” (a low noise temperature). Noise temperature is closely related to phase angle and hysteresivity (9, 13). A noise temperature of 1 corresponds to a phase angle of 0 and a hysteresivity of 0.
In intact bovine trachalis, hysteresivity, a measure of the area of the length-force loop during length oscillation, was low at rest, high during the rapid phase of contraction, and low during the sustained phase of contraction (11). In isolated trachealis cells, Fredberg et al. (2, 9) measured the length-force response when magnetic beads attached to the cell membrane were oscillated in a magnetic field. Noise temperature (the slope of relation between the real part of stiffness and oscillatory frequency) and phase angle (the imaginary part of stiffness also known as the loss modulus G”) changed with treatments that altered the biophysical characteristics of these cells. Specifically, they found 1) increased noise temperature with cyclic nucleotides and agents that reduced actin polymerization and 2) decreased noise temperature with contractile agonists and agents that increased actin polymerization.
This study had two goals. The first goal was to determine whether stimulus induced Y118 paxillin phosphorylation and actin polymerization could have a role in the initial or sustained phases of swine carotid artery contraction, a result that could have implications in the latch hypothesis. The second goal was to measure noise temperature, hysteresivity, and phase angle during the initial and sustained phases of swine carotid artery contraction to evaluate the rheology of arterial smooth muscle contraction. Swine carotid arterial tissues were depolarized with high K+ or stimulated with histamine and the biochemical (Y118 paxillin polymerization, crossbridge phosphorylation, and actin polymerization) and mechanical [noise temperature, hysteresivity, phase angle, real stiffness (G'), and stress] response evaluated.
MATERIALS AND METHODS
Tissues
Physiological saline solution contained (in mM) 140 NaCl, 4.7 KCl, 2 MOPS, 1.2 Na2HPO4, 1.6 CaCl2, 1.2 MgSO4, 5.6 d-glucose, 0.02 EDTA, pH adjusted to 7.4 at 37°C. Swine common carotid arteries were obtained from an abattoir, dissected, the endothelium removed, mounted as ring or strips (detailed below), bathed in physiological saline at 37°C, and set at Lo, the optimal length for force development, as described for strips (27) and rings (43). Setting length at Lo involved two conditioning contractions with 109 mM K+ physiological saline solution where KCl was substituted stoichiometrically for NaCl. All forces were normalized to tissue cross sectional areas and therefore presented as stress in units of 105 N/m−2.
Biochemical measurements
Y118 paxillin and crossbridge (Ser19-MRLC) phosphorylation was determined in swine common carotid artery rings mounted isometrically at 1.0 Lo as described above. They were then treated for the same time as the F actin and stiffness experiments described below. Rings were then frozen in acetone dry ice, and homogenized in 1% SDS, 10% glycerol (vol/vol), and 20 mM dithiothreitol (0.22 ml/mg tissue dry wt) as described (29).
The level of Y118 paxillin phosphorylation was determined by loading tissue homogenates on two 10% SDS electrophoresis gels, followed by immunoblotting on PVDF for total paxillin (1:500 primary antibody from Zymed Laboratories no. 03−6100 and 1:15,000 secondary antibody from Amersham no. NA931V) and Y118 phosphorylated paxillin (1:1,000 primary antibody from Biosource no. 44−722G and 1:15,000 secondary antibody from Amersham, no. NA934V). In preliminary experiments, the antibody for Y31 phosphorylated paxillin (Biosource no. 44−720G) did not immunostain swine carotid artery homogenates. To minimize blotting and detection errors, all homogenates (including the unstimulated control) from the rings originating from the same artery were loaded on the same gel. Phosphorylation for each ring was calculated as the ratio of Y118 phosphorylated paxillin to total paxillin immunostaining. Phosphorylation for each ring was then normalized to the level of Y118 phosphorylation in the unstimulated control with the control value = 1.0.
The level of crossbridge (Ser19-MRLC) phosphorylation was determined by isoelectric focusing and immunoblotting as described (29). Three dilutions of homogenates were loaded to ensure that the enhanced chemiluminescence detection system was in the linear range (28). Phosphorylation is reported as mol Pi/mol protein.
Actin polymerization
The relative amount of filamentous actin (F-actin) vs. total actin in swine carotid arterial rings was determined with a commercial kit (BK037) from Cytoskeleton (Denver, CO). Lysis/F-actin stabilization buffer contained 50 mM piperazine-N,N'-bis(2-ethanesulfonic acid), 50 mM NaCl, 5 mM MgCl2, 5 mM EGTA, 5% glycerol, 0.1% Nonidet P-40, 0.1% Triton X-100, 0.1% Tween 20, and 0.1% β-mercapto-ethanol, pH 6.9, at 30°C. Immediately before use of lysis/F-actin stabilization buffer, 10 μl/ml of 100 mM ATP and 10 μl/ml of protease inhibitor cocktail, composed of (in μg/ml) 1 pepstatin, 1 leupeptin, 10 benzamidine, and 500 tosyl arginine methyl ester, was added. Arterial rings were mounted, pharmacologically treated as described above, and then frozen with tongs cooled in liquid nitrogen. The frozen carotid was weighed and pulverized in a liquid nitrogen-chilled ceramic mortar and pestle. The resultant powder was divided equally between two chilled 1.5-ml Eppendorf tubes (the assay was performed in duplicate to increase accuracy). Fifty micro-liters per milligram per tissue wet weight of Lysis/F-actin stabilization buffer with ATP and protease inhibitor cocktail (prewarmed at 30°C) was added to each tube. The tubes were then immediately vortexed and incubated at 30°C for 10 min. They were then immediately centrifuged at 16,000 g for1hat room temperature in an Eppendorf 5415C microcentrifuge. The supernatants were pipetted off the pellets, placed in empty 1.5-ml Eppendorf tubes, and then put on ice. The pellets were resuspended to the same volume as the supernatants using ice-cold nanopure water plus 1 μM cytochalasin D. The pellet samples were left on ice (with vortexing every 15 min) for 1 h to dissociate F-actin. One-half- and one-quarter-strength dilutions of each pellet and full strength supernatant were then separated on 12% SDS-polyacrylamide gels, blotted to nitrocellulose, immunostained with a rabbit commercially produced polyclonal anti-actin antibody (AAN01, Cytoskeleton) and detected with enhanced chemiluminescence. Images were analyzed with UnScanIt software. Intensities were corrected for dilution and the amount of F-actin as a percent of total actin calculated. The dilutions were corrected for offset and saturation errors as we have done with analysis of crossbridge phosphorylation (29, 40).
Stiffness, noise temperature, and hysteresivity measurements
Both ends of swine carotid tissue strips were mounted in 1.2-mm-diameter aluminum foil cylinders with cyanoacrylate glue. One cylinder was attached to an adjustable length stationary rod and the other cylinder to the lever arm of a model 310B dual mode lever operated by Dynamic Muscle Control software (Aurora Scientific, Aurora, ONT, Canada). After the tissue length was set to Lo as described above, tissues were treated pharmacologically and at various times oscillated with sine wave changes in length (0.5% Lo at 0.3, 1, 3, 10, and 30 Hz) and the resulting change in force measured (9, 10). Force values were normalized as stress values (force per cross-sectional area, in N/m−2). G' was calculated as the peak-to-peak change in stress observed with 10-Hz oscillations (this was valid because calculated phase angles were all less than 10°, i.e. cos 10° = ∼1). Noise temperature was calculated as 1 + (the least-squares regression slope of ln G' as a function of ln oscillation frequency) as described in results and in Fig. 1. Hysteresivity was calculated from plots of the change in stress as a function of change in length during 1-Hz oscillations (described in results and Fig. 2). We calculated hysteresivity as the difference between the zero length intercepts of stress during the stretch and release phases of the oscillation. Hysteresivity was then normalized to mean stress during the oscillation so that units are fractional. Phase angle (the imaginary part of stiffness also known as the loss modulus G”) was calculated using MATLAB (MathWorks, Natick, MA) from the relative delay in the zero intercept of the force oscillation compared with the zero intercept of the length oscillation (with 360° as a full cycle).
Fig. 1.
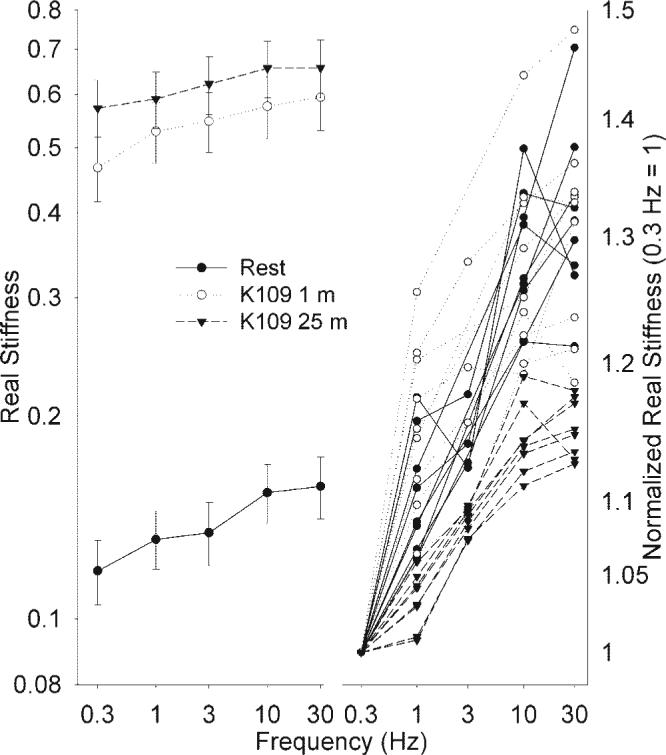
Measurement of noise temperature. Sinusoidal 0.5% length changes of swine carotid arteries were performed at 0.3, 1, 3, 10, and 30 Hz during various phases of a 109 mM K+ depolarization (●, unstimulated; ○, 1 min of depolarization; and ▼ , 25 min of depolarization). Left, ln real stiffness (G') as a function of ln frequency (mean ± 1 SE). Right, raw normalized ln G' as a function of ln frequency (G' was normalized so that real stiffness at 0.3 Hz = 1). The slope of the G'-frequency relationship, termed noise temperature, was higher in unstimulated tissues and in tissues stimulated for 1 min, compared with tissues stimulated for 25 min (▼).
Fig. 2.
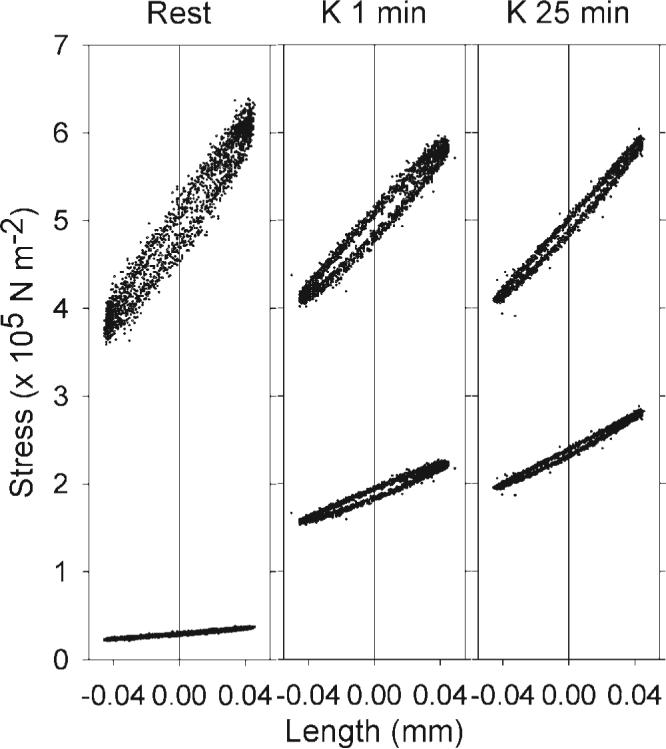
Measurement of hysteresivity. Sinusoidal 0.5% length changes of a representative swine carotid artery were performed at 1 Hz during various phases of a 109 mM K+ depolarization (left, unstimulated; middle, 1-min depolarization; right, 25-min depolarization: two full cycles of each are plotted). The bottom tracings are the measured change in stress as a function of the length change and the upper tracings are the normalized change in stress (so that mean stress was set to 5). The difference between the force tracings measured at zero length (vertical line) is the hysteresivity. Hysteresivity was higher in unstimulated tissues and in tissues stimulated for 1 min compared with tissues stimulated for 25 min.
RESULTS
Measurement of noise temperature in intact swine carotid artery
Noise temperature has not previously been measured in intact arterial smooth muscle. When a sinusoidal length change is imposed on one end of a muscle at varying frequencies, the frequency dependence of real part of stiffness (G'peak to peak change in force) can be determined. Noise temperature equals 1 + (the least-squares regression slope of ln G' as a function of ln oscillation frequency) (9). Noise temperature was measured in intact swine carotid artery tissues that were unstimulated or stimulated with 109 mM K+ for 1 or 25 min. Resting tissues had low G' and clear increases in G' at higher sinusoidal frequencies, i.e., a relatively steep slope (Fig. 1, bottom left, filled circles). Tissues depolarized with 109 mM K+ for 1 min had higher G' and clear increases in G' at higher sinusoidal frequencies, i.e., a relatively steep slope (Fig. 1, top left, open circles). Tissues depolarized with 109 mM K+ for 25 min had a slightly higher G' than that observed with 1 min of depolarization. However, 25 min of depolarization was associated with less of an increase in G' at higher sinusoidal frequencies, i.e., a flatter slope than that observed in resting tissues or after 1 min of depolarization (Fig. 1, top left, inverted triangles).
The relative slope of G' vs. frequency is best seen on the normalized plot of raw data (Fig. 1, right). Tissues at rest and those depolarized with 109 mM K+ for 1 min had a steeper dependence of normalized G' on frequency when compared with tissues depolarized for 25 min. These data show that noise temperature (slope) was higher in tissues at rest and depolarized for 1 min compared with tissues depolarized for 25 min.
Measurement of hysteresivity in intact swine carotid artery
When a sinusoidal length change is imposed on one end of a muscle, a plot of force (stress) vs. length typically exhibits an hysteresis loop. Hysteresivity can be measured as the area inside the loop or the height of the loop at its midsection (9). Figure 2 shows representative tracings from one of the tissues studied in Fig. 1. The lower tracings are the measured stress and the upper tracings are stress normalized to a value of 5, both plotted as a function of change in length. For this study, hysteresivity was measured as the height of the loop, specifically the stress during the stretch (increasing length) minus the stress during the release (decreasing length) at the midpoint (zero, the vertical line in Fig. 2) of the sinusoidal length change normalized to mean stress. Hysteresivity was higher in tissues at rest and depolarized with 109 mM K+ for 1 min compared with tissues depolarized for 25 min (Fig. 2).
Relation between noise temperature and hysteresivity
There was a significant linear dependence of hysteresivity on noise temperature in swine carotid tissues at rest, with K+ depolarization, and with histamine stimulation (Fig. 3, top; r2 = 0.36, P < 0.001). There was more variability in hysteresivity in resting tissues (Fig. 3, top, open squares). There was also a significant linear dependence of phase angle (the imaginary part of stiffness also known as the loss modulus G”) on noise temperature in swine carotid tissues at rest, with K+ depolarization, and with histamine stimulation (Fig. 3, bottom; r2 = 0.43, P < 0.001). These data suggest that noise temperature, hysteresivity, and phase angle were similar measures in swine carotid artery, as predicted by Fabry et al. (9).
Fig. 3.
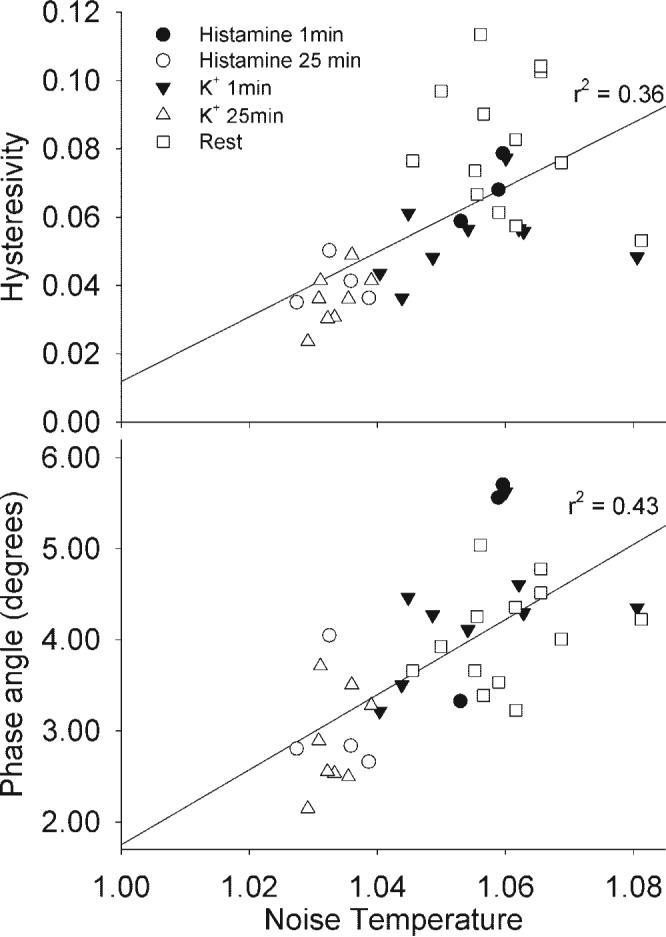
Hysteresivity and phase angle were linearly dependent on noise temperature. Hysteresivity (top) and phase angle (bottom) were plotted as a function of noise temperature for the data shown that will be shown in Fig. 5. Regression was significant with r2 = 0.36 for noise temperature and r 2 = 0.43 for phase angle, suggesting that noise temperature, hysteresivity, and phase angle are similar measures of rheology in intact smooth muscle.
Measurement of Y118 paxillin phosphorylation
Figure 4 shows a representative gel of swine carotid homogenates immunostained with antibody to total paxillin (top) and antibody to Y118 phosphorylated paxillin (bottom). High K+ depolarization for 3 and 10 min increased immunostaining for Y118 phosphorylated paxillin compared with earlier time points. There was no immunostaining with an antibody to Y31 paxillin phosphorylation with any treatment (data not shown). Total paxillin immunostaining did not significantly change with high K+ depolarization. For each tissue, Y118 paxillin phosphorylation was quantified as the ratio of Y118 paxillin immunostaining to total paxillin immunostaining. This ratio was then normalized to the Y118 paxillin immunostaining ratio observed in an unstimulated control tissue on the same gel.
Fig. 4.
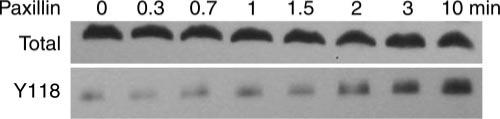
Representative immunostained blots of total paxillin (top) and Y118 phosphorylated paxillin (bottom) from swine carotid homogenates. Swine carotid tissues were either unstimulated (0) or depolarized with 109 mM K+ for 0.33, 0.67, 1, 1.5, 2, 3, and 10 min (from left to right; see Fig. 7 for mean data). Increased immunostaining for Y118 paxillin phosphorylation was clearly seen at 3 and 10 min after stimulation.
Mean biochemical and mechanical characteristics of initial and sustained phase of swine carotid artery contraction
The effect of depolarization and histamine stimulation on mean biochemical and mechanical characteristics of swine carotid artery is shown in Fig. 5. Resting tissues had low immunostaining for Y118 paxillin phosphorylation, low crossbridge phosphorylation (MRLCp), low G', and low stress along with high noise temperature, hysteresivity, and phase angle (Fig. 5, left). The initial phase of 109 mM K+ depolarization (1 min) was associated with significantly increased crossbridge phosphorylation, velocity, G', and stress; a small, but significant decrease in hysteresivity; and no significant change in immunostaining for Y118 paxillin phosphorylation, noise temperature, or phase angle (Fig. 5, column 2). The sustained phase of 109 mM K+ depolarization (25 min) was associated with intermediate levels of crossbridge phosphorylation and velocity, significantly increased immunostaining for Y118 paxillin phosphorylation, maintained high G' and stress, and significantly reduced noise temperature, hysteresivity, and phase angle (Fig. 5, column 3). Similar results were seen when tissues were stimulated with 10 μM histamine (with the exception that hysteresivity was not significantly reduced at 1 min; Fig. 5, columns 4−6).
Fig. 5.
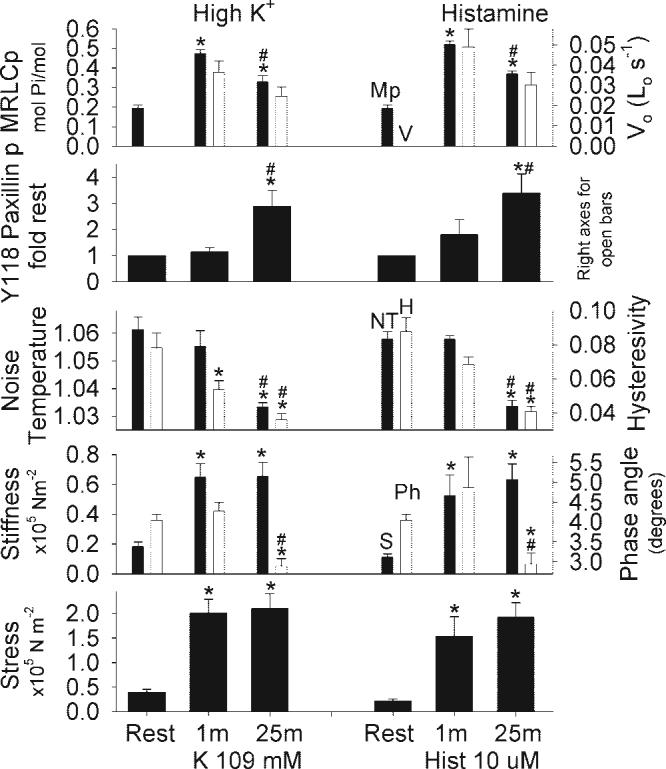
Mean biochemical and mechanical characteristics in resting and stimulated swine carotid artery tissues. Tissues were either unstimulated (column 1), depolarized with 109 mM K+ for 1 min (column 2) or 25 min (column 3), unstimulated (column 4), stimulated 10 μM histamine for 1 min (column 5) or 25 min (column 6). The panels show crossbridge phosphorylation (MRLCp, top, filled bars), unloaded shortening velocity (Vo, panel 1, open bars), Y118 paxillin phosphorylation (panel 2), noise temperature (panel 3, filled bars), hysteresivity (panel 3, open bars), real stiffness (panel 4, filled bars), phase angle (panel 4, open bars), and stress (panel 5). Data are plotted as mean ± 1 SE with n = 4−9 [the crossbridge phosphorylation measurements are from one set of tissues, the Vo measurements are from a second set of tissues previously published (27), and the remaining measurements are from a third set of tissues]. Differences were tested by ANOVA and Fisher's least-significant difference (LSD) method for individual comparisons. Y118 paxillin phosphorylation was significantly increased and noise temperature/hysteresivity/phase angle was significantly decreased only during the sustained phase of depolarization or histamine stimulation. *P < 0.001, significant difference vs. rest; #P < 0.001, significant difference vs. 1-min values.
These results show that the initial phase of high K+ or histamine-induced contraction was not associated with significant changes in noise temperature or immunostaining for Y118 paxillin phosphorylation despite large increases in crossbridge phosphorylation, velocity, G', and stress. The sustained phase of contraction was associated with significantly increased immunostaining for Y118 paxillin phosphorylation, significantly reduced noise temperature/hysteresivity/phase angle, and maintained high G'/stress.
G' is an estimate of the number of attached crossbridges. There was a significant linear dependence of G' on stress in tissues at rest, depolarized, or stimulated with histamine (r2 = 0.96, P < 0.001; Fig. 6, top). These data suggest that the amount of force per attached crossbridge was not altered during the initial or sustained phase of contraction.
Fig. 6.
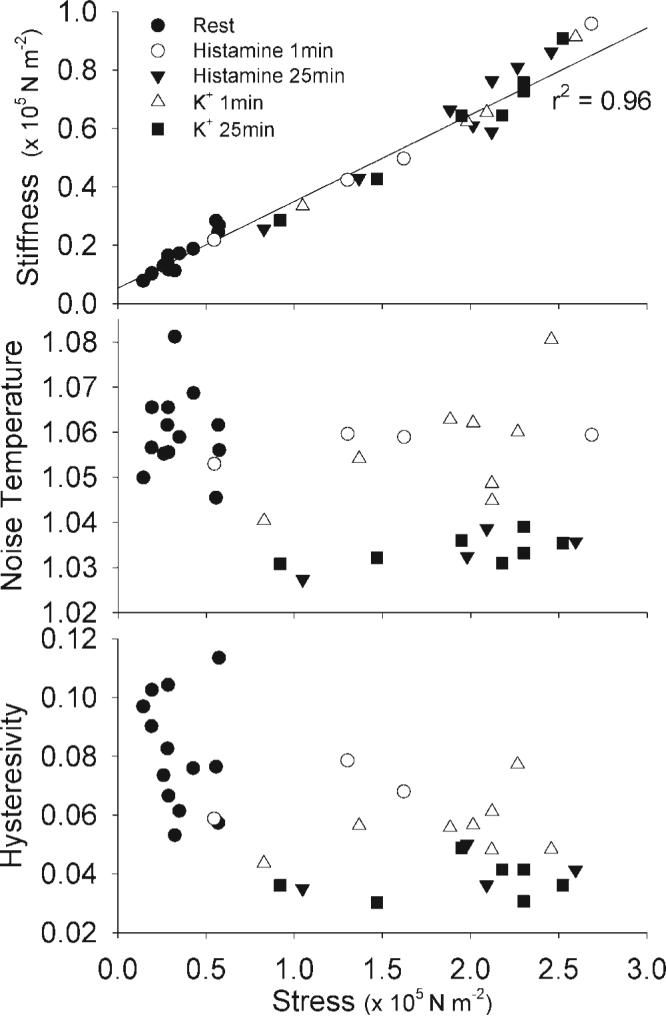
Dependence of real stiffness, noise temperature, and hysteresivity on stress (normalized force) in swine carotid artery tissues. Raw data from the experiment presented in Fig. 5 are replotted for comparison. Panel 1 (top) shows a clear and highly significant linear dependence of real stiffness on stress (r2 = 0.96). Panels 2 and 3 show a complicated dependence of noise temperature and hysteresivity on stress. Noise temperature and hysteresivity values during the initial phase of activation (open symbols) are clearly higher than those during the sustained phase of activation (▲, ■).
There was a complicated dependence of noise temperature and hysteresivity on stress (Fig. 6, bottom). The initial phase of contraction (1 min; shown as open circles and triangles) was associated with higher noise temperature and hysteresivity compared with the sustained phase of contraction (25 min, shown as filled triangles and squares), despite similar stress values.
A detailed time course of a maximal K+ contraction
Figure 7 shows a full characterization of the time course of a 109 mM K+ induced swine carotid artery contraction. Initially, high K+ induced rapid increases in crossbridge phosphorylation, G', and stress (crossbridge phosphorylation was significantly increased by 20 s and G'/stress by 40 s). Crossbridge phosphorylation, G', and stress reached near maximal values during the first minute of contraction. In the first minute of contraction, there were no significant changes in Y118 paxillin phosphorylation, the relative amount of F actin, or noise temperature/hysteresivity/phase angle. With sustained stimulation, i.e., >1 min, crossbridge phosphorylation decreased to intermediate values, while G' and stress remained high. There was a significant increase in Y118 paxillin phosphorylation by 3 min, a significant increase in the relative amount of F actin by 10 min, and a significant decrease in noise temperature and hysteresivity by 90 and 120 s, respectively. These data show a clear separation of force development from changes in Y118 paxillin phosphorylation, actin polymerization, noise temperature, and hysteresivity.
Fig. 7.
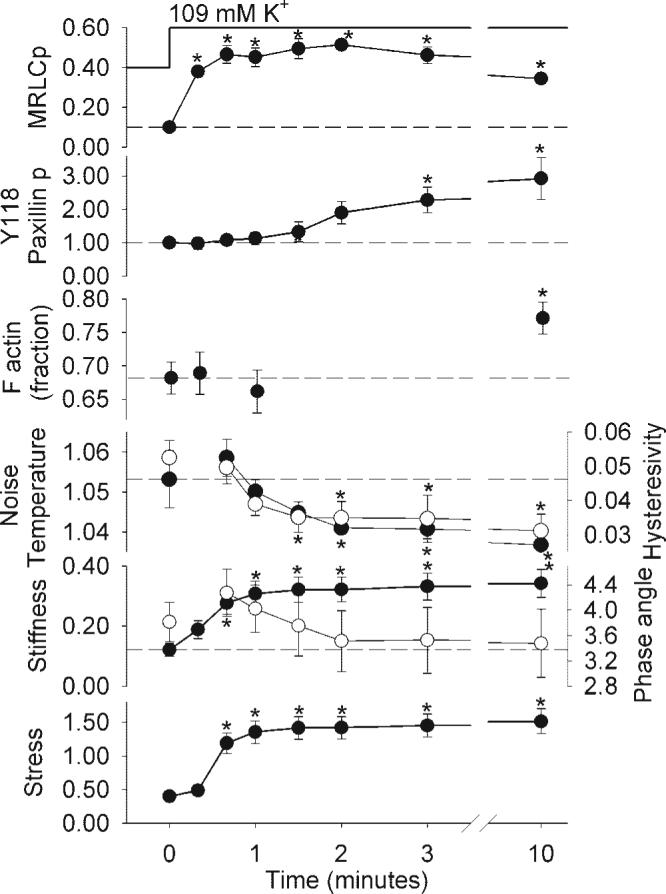
The time course of biochemical and mechanical measures in rapidly contracting swine carotid artery tissues. Tissues were depolarized with 109 mM K+ and measurements taken at 0.33, 0.67, 1, 1.5, 2, 3, and 10 min after 109 mM K+ depolarization. Data are plotted as mean ± 1 SE with n = 4−6 for the biochemical (Y118 paxillin, the relative amount of F actin, and crossbridge phosphorylation) measurements and mechanical measurements (noise temperature, hysteresivity, real stiffness, phase angle, and stress). The panels show crossbridge phosphorylation (mol Pi/mol MRLCp; panel 1), Y118 paxillin phosphorylation (fraction of the phosphorylation in the unstimulated tissue; panel 2), the relative amount of F actin (fraction of total actin, panel 3), noise temperature (panel 4; ●) and hysteresivity (○), real stiffness (× 105 N/m−2, panel 5, ●), phase angle (degrees, ○), and stress (× 105 N/m−2, panel 6). Stress is from the noise temperature data. The noise temperature, hysteresivity, and phase angle data at 20 s are not included because varying force levels during the most rapid phase of contraction prevented accurate measurement. Differences were tested by ANOVA (all except phase angle were significant overall, with the largest P value of 0.024) and Fisher's LSD method for individual comparisons; *significant difference vs. rest (0 min). One tissue had a higher phase angle both before and during contraction accounting for the large SE and lack of statistical significance for change in phase angle. Crossbridge phosphorylation was significantly increased at 20 s, real stiffness and force by 40 s, hysteresivity by 1.5 min, noise temperature by 2 min, Y118 paxillin phosphorylation by 3 min, and the relative amount of F actin at 10 min.
Steady-state relation between Y118 paxillin phosphorylation, noise temperature, and hysteresivity
To assess the steady-state relation between Y118 paxillin phosphorylation, noise temperature, hysteresivity, and contraction, swine carotid was depolarized with 25 mM K+ to produce a slow contraction. Measurements were performed as stress values were doubled, i.e., at 3%, 6%, 12%, 25%, 50%, and 100% of maximum stress. Each twofold increase in stress was associated with less than a twofold increase in Y118 paxillin phosphorylation and less than a twofold decrease in noise temperature and hysteresivity (Fig. 8). Each twofold increase in stress was associated with a twofold increase in G'. These data suggest that there was no stress threshold for K+-induced increases in Y118 paxillin phosphorylation or reductions in noise temperature and hysteresivity. The majority of the change in noise temperature/hysteresivity/phase angle occurred as force increased from resting to 50% of maximal.
Fig. 8.
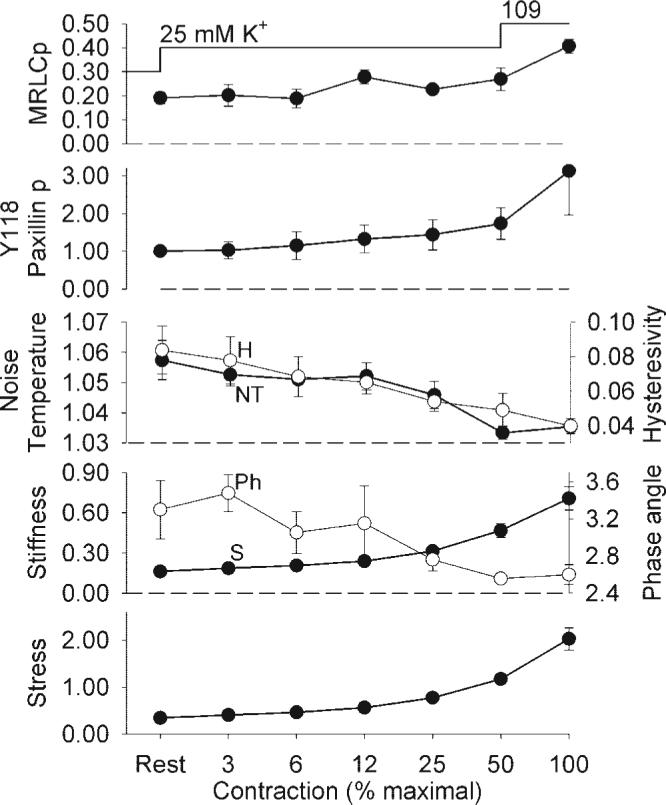
The steady-state dependence of biochemical and mechanical measures in slowly contracting swine carotid artery tissues. Tissues were depolarized with 25 mM K+ and measurements taken when the slowly contracting tissues reached 3%, 6%, 12%, 25%, and 50% of the force measured after 10 min of 109 mM K+ depolarization. If tissues stopped contracting before goal force, K+ was increased to 27.5 or 30 mM by the addition of 109 mM K+ physiological saline solution (PSS) to the bathing solution. The far left point is from unstimulated tissues and the far right point is from the prior 10 min 109 mM K+ depolarization performed to determine the percent force described above. Data are plotted as mean ± 1 SE with n = 6 for crossbridge and paxillin phosphorylation and n = 4 for noise temperature, hysteresivity, phase angle, real stiffness, and stress. The panels are identical to those in Fig. 7, except there was no F-actin measurement.
Effect of inhibitors of actin polymerization on noise temperature
Swine carotid was stimulated with 10 μM histamine and then actin polymerization inhibited with 6 μM latrunculin-A. Measurements were performed during the steady-state contraction with 10 μM histamine and with the 6 μM latrunculin-A-induced relaxation. Latrunculin-A did not significantly change crossbridge phosphorylation, significantly increased noise temperature and hysteresivity, and significantly reduced G' and stress (Table 1). These data suggest that noise temperature and hysteresivity were altered by latrunculin-A-induced inhibition of actin polymerization.
Table 1.
Mean biochemical and mechanical characteristics with latrunculin-A treatment to disrupt actin filaments
| Histamine | Histamine+ Latrunculin-A | P Values | |
|---|---|---|---|
| Crossbridge phosphorylation | 0.369±0.016 | 0.336±0.019 | 0.22 |
| Noise temperature | 1.036±0.001 | 1.042±0.002 | 0.022* |
| Hysteresivity | 0.057±0.002 | 0.066±0.003 | 0.022* |
| Real stiffness (×105 N m−2) | 0.75±0.09 | 0.43±0.05 | <0.001* |
| Stress (×105 N m−2) | 2.24±0.24 | 1.34±0.11 | 0.001* |
Values are means ± SE. The experimental data in this paper is part of the experiment detailed in the companion paper (25a). Tissues were evaluated 1) with 10 μM histamine stimulation for 25−40 min and 2) with 10 μM histamine for 25−40 min, followed by the addition of 6 μM latrunculin-A in the continued presence of 10 μM histamine. Differences were tested by paired t-test;
statistical significance.
Dependence of actin polymerization and noise temperature on Y118 paxillin phosphorylation
There was a significant correlation between mean Y118 paxillin phosphorylation and increases in actin polymerization as measured by the relative amount of F actin (Fig. 9, top). There was also an inverse correlation between mean immunostaining intensity for Y118 paxillin phosphorylation and mean noise temperature (Fig. 9, bottom). The correlation between mean Y118 paxillin phosphorylation and mean hysteresivity/phase angle was less significant than that observed with noise temperature (data not shown).
Fig. 9.
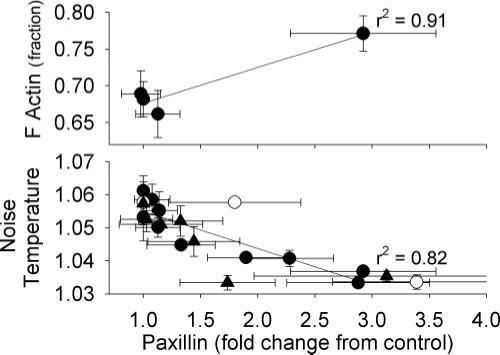
Significant dependence of actin polymerization and noise temperature on Y118 paxillin phosphorylation in swine carotid artery tissues. Data from 109 mM K+ are plotted (●; see Figs. 5 and 7), from 25 mM K+ (▲; see Fig. 8), and from histamine (○; see Fig. 5, mean ± 1 SE). Top, significant dependence of the relative amount of F-actin on mean Y118 paxillin phosphorylation. Bottom, an inverse dependence of mean noise temperature on mean Y118 paxillin phosphorylation.
DISCUSSION
Role of Y118 paxillin phosphorylation and stimulated actin polymerization in arterial smooth muscle contraction
The first goal of this study was to determine whether stimulus-induced Y118 paxillin phosphorylation and actin polymerization could have a role in the initial or sustained phase of swine carotid artery contraction. We clearly show no significant change in Y118 paxillin phosphorylation or actin polymerization during the force development phase of a maximal contraction (Figs. 5 and 7). Therefore, it appears that the increase in crossbridge phosphorylation (Figs. 5 and 7) can explain the rapid phase of force development. The sustained phase of a rapid arterial smooth contraction was associated with significant increases in Y118 paxillin phosphorylation and F actin consistent with stimulus-induced actin polymerization (Figs. 5 and 7). Our finding that sustained 109 mM K+ depolarization increased the relative amount of F actin is similar to our prior finding that sustained histamine stimulation of swine carotid artery increased the relative amount of F actin (19). When arterial smooth muscle was slowly contracted, the increase in force was paralleled by an increase in Y118 paxillin phosphorylation (Fig. 8). We also find that increases in actin polymerization correlated with increases in Y118 paxillin phosphorylation (Fig. 9), suggesting that Y118 paxillin phosphorylation could be regulating actin polymerization. These data suggest a potential role for Y118 paxillin phosphorylation-induced actin polymerization in the regulation of the sustained phase of contraction.
Our data differs from that found in airway smooth muscle, in which the time course of paxillin tyrosine phosphorylation paralleled the time course of the contraction (41). There are clear physiological differences between airway and arterial smooth muscle. Airway smooth muscle is on the extreme continuum of smooth muscle in that it does not have a fixed length-tension relation (3). In contrast, the length-tension relation of arterial smooth muscle is less plastic. It is possible that the variable length-tension relation observed in airway smooth muscle requires a more plastic actin cytoskeleton, so that a stimulus-activated actin polymerization pathway is required for the initial contraction. Such a system may not be required in arterial smooth muscle.
There are several possible hypotheses for the role of stimulus-induced actin polymerization that occurs only during the sustained phase of smooth muscle contraction. First, it is possible that actin polymerization is required only to create a cytoskeletal structure necessary for subsequent contraction. Once the cytoskeletal structure is formed, further stimulus-dependent actin polymerization can occur but is not required for the sustained phase of arterial contraction. This hypothesis suggests no role for stimulus-induced actin polymerization in the sustained contraction. Second, it is possible that stimulus-dependent actin polymerization is only required during stimulus-dependent sustained contraction. This would suggest that actin polymerization is not necessary to create the cytoskeletal structure that is required for subsequent contraction. We think this possibility is unlikely given that ∼70% of actin is polymerized (F actin) in resting smooth muscle (Fig. 7 and Ref. 19). Third, it is possible that the actin polymerization pathway is required to both to create a cytoskeletal structure necessary for subsequent contraction and for the sustained phase of stimulus-dependent contraction, i.e., latch. This hypothesis predicts that interference with stimulated actin polymerization would abolish the latch phenomenon, i.e., if stimulus-induced actin polymerization were inhibited, then force would decreased in parallel with decreasing crossbridge phosphorylation during a sustained maximal contraction. Finally, it is possible that the actin polymerization pathway is required to create a cytoskeletal structure necessary for subsequent contraction and these proteins modulate, but are not required for, the sustained phase of stimulus-dependent contraction (latch). This hypothesis would imply that force is maintained during latch by both attached dephosphorylated crossbridges, i.e., latchbridges and stimulus-dependent actin polymerization.
Measurement of noise temperature and hysteresivity in intact swine carotid artery
The second goal was to measure noise temperature, hysteresivity, and phase angle during the initial and sustained phases of swine carotid artery contraction to evaluate the rheology of arterial smooth muscle contraction. As described in the introduction, noise temperature, hysteresivity, and phase angle are rheologic measures of the “fluidity” of a tissue (18). We clearly show no significant change in noise temperature, hysteresivity, and phase angle during the initial force development phase of a maximal contraction (Figs. 5 and 7). Therefore, it appears that the increase in crossbridge phosphorylation, G', and stress observed during with initial activation were not associated with a change in smooth muscle rheology.
In contrast, the sustained phase of a rapid arterial smooth contraction was associated with significant decreases in noise temperature, hysteresivity, and phase angle (Figs. 5 and 7). These data suggest that the smooth muscle behaved more like a “solid” during the sustained phase of contraction when crossbridge phosphorylation fell to intermediate levels and actin was becoming more polymerized. When arterial smooth muscle was slowly contracted, the increase in force was paralleled by decreases in noise temperature, hysteresivity, and phase angle (Fig. 8). These data suggest that the transition to a “solid” rheology, i.e., with a low noise temperature, 1) was not associated with crossbridge interactions (no correlation with crossbridge phosphorylation or stress: Figs. 5 and 6), 2) was time dependent (Figs. 7 and 8), 3) correlated with Y118 paxillin phosphorylation (Fig. 5, 7, and 9), and 4) correlated with increased actin polymerization (Fig. 7). It is possible that stimulated actin polymerization may be partly responsible for this transition to a more “solid” rheology. Supporting this contention was the finding that an inhibitor of actin polymerization, latrunculin-A, significantly increased noise temperature and hysteresivity in histamine stimulated swine carotid artery (Table 1). Our finding that latrunculin-A increased noise temperature in intact swine carotid artery parallels the finding that cytochalasin-D increased noise temperature in multiple types of isolated cultured cells (9).
Previously, noise temperature had only been measured with beads attached to the plasma membrane of isolated cells (2, 10). This study shows that noise temperature can be measured in intact tissues. Values of noise temperature measured in intact smooth muscle (this study) were approximately one-half of those measured in isolated cells (2, 10), suggesting that measurement of noise temperature at the tissue ends is different than measurement of noise temperature at the plasma membrane (2, 10). This difference could represent contributions to noise temperature from connective tissue in the intact tissue and/or differences from measurement in the axial dimension in intact tissues vs. rotational measurements in the isolated cell. As predicted (9), noise temperature, hysteresivity, and phase angle appeared to be similar measurements in the depolarized and histamine stimulated swine carotid artery (Fig. 3). This similarity suggests these measurements are robust. There was more variability in the hysteresivity measurement in resting tissues, perhaps because stress was low in resting tissues so that force measurement errors in resting tissues were amplified with normalization, e.g., Fig. 2.
The noise temperature, hysteresivity, and phase angle response did not differ when tissues were depolarized with K+ or stimulated with histamine. This result suggests that some mechanism common to depolarization or histamine (but not crossbridge phosphorylation) is responsible for changing the rheology of smooth muscle to a more “solid”-like state during the sustained phase of contraction. Therefore, any mechanisms specific to depolarization, e.g., higher Ca2+, or specific to histamine stimulation, e.g., protein kinase C activation or myosin phosphatase inhibition, are unlikely to be responsible for the observed changes in the rheology of smooth muscle.
The significant dependence of the relative amount of F actin and noise temperature on Y118 paxillin phosphorylation (Fig. 9) are consistent with the hypothesis that sustained depolarization and histamine stimulation increased Y118 paxillin phosphorylation which then caused actin polymerization, a process associated with a “solidification” of swine arterial smooth muscle. While this correlation existed for these experiments, it must be emphasized that rheology can be altered by multiple other factors that alter intermolecular interactions which then change the energy microconfiguration of the muscle. Additional research is required to further understand both the importance and regulation of stimulus-induced changes in noise temperature and hysteresivity in arterial smooth muscle.
Implications of noise temperature results for the latch phenomenon
As noted, these data suggest a potential role for Y118 paxillin phosphorylation-induced actin polymerization and/or a “solid” rheology in the sustained (latch) phase of contraction. In intact bovine trachalis, Fredberg et al. (11) found that hysteresivity, a measure of the area of the length-force loop during length oscillation, was low at rest, high during the rapid phase of contraction, and low during the sustained phase of contraction. These results led Fredberg et al. to hypothesize that tracheal smooth muscle was “solid” at rest when crossbridge cycling was low, became more “fluid” during force generation when crossbridge cycling was fast, and then became more “solid” during the sustained contraction when crossbridge cycling was intermediate. Their results differ from our results in which noise temperature, hysteresivity, and phase angle were high at rest, high during the rapid phase of contraction, and only low during the sustained phase of contraction (Figs. 5 and 7). Our data suggests that arterial smooth muscle was “fluid” both at rest and during rapid force generation, suggesting that the increase in crossbridge interactions associated with force development was not associated with a change in the microconfiguration of energy wells in the system. Arterial smooth muscle only became “solid” during the sustained contraction. This difference suggests a clear difference in the rheology of tracheal and arterial smooth muscle.
Do these findings suggest that the latch phenomenon is caused by Y118 paxillin phosphorylation-induced actin polymerization and development of a “solid” rheology? The latch phenomenon is characterized by 1) maintained force with reduced crossbridge phosphorylation, velocity, and ATP utilization (14, 23, 31, 45), 2) an invariance of stiffness and yield stress despite different crossbridge phosphorylation values (Fig. 5 and 7 and Ref. 31), and 3) a requirement for crossbridge interactions for force development, i.e., myosin kinase inhibitors abolish contraction despite partly preserved Ca2+ transients (44). The current dynamic crossbridge model of latch predicts that accumulating dephosphorylated crossbridges also explain all of these characteristics of the latch phenomenon (14, 15, 30). If regulated actin polymerization were to explain the latch phenomenon, it would also need to explain these characteristics of the latch phenomenon. Therefore, we cannot yet conclude that stimulated actin polymerization is able to explain the latch phenomenon. We plan further studies to test the importance of stimulus-dependent actin polymerization and changes in tissue rheology in the sustained phase of contraction.
ACKNOWLEDGMENTS
The arteries were donated by Smithfield, a division of Gwaltney (Smithfield, VA).
GRANTS
This research was supported by National Heart, Lung, and Blood Institute Grant HL-71191.
REFERENCES
- 1.Adler KB, Krill J, Alberghini TV, Evans JN. Effect of cytochalasin-D on smooth-muscle contraction. Cell Motil Cytoskeleton. 1983;3:545–551. doi: 10.1002/cm.970030521. [DOI] [PubMed] [Google Scholar]
- 2.An SS, Laudadio RE, Lai J, Rogers RA, Fredberg JJ. Stiffness changes in cultured airway smooth muscle cells. Am J Physiol Cell Physiol. 2002;283:C792–C801. doi: 10.1152/ajpcell.00425.2001. [DOI] [PubMed] [Google Scholar]
- 3.Bai TR, Bates JHT, Brusasco V, Camoretti-Mercado B, Chitano P, Deng LH, Dowell M, Fabry B, Ford LE, Fredberg JJ, Gerthoffer WT, Gilbert SH, Gunst SJ, Hai CM, Halayko AJ, Hirst SJ, James AL, Janssen LJ, Jones KA, King GG, Lakser OJ, Lambert RK, Lauzon AM, Lutchen KR, Maksym GN, Meiss RA, Mijailovich SM, Mitchell HW, Mitchell RW, Mitzner W, Murphy TM, Pare PD, Schellenberg RR, Seow CY, Sieck GC, Smith PG, Smolensky AV, Solway J, Stephens NL, Stewart AG, Tang DD, Wang L. On the terminology for describing the length-force relationship and its changes in airway smooth muscle. J Appl Physiol. 2004;97:2029–2034. doi: 10.1152/japplphysiol.00884.2004. [DOI] [PubMed] [Google Scholar]
- 4.Brown MC, Turner CE. Paxillin: adapting to change. Physiol Rev. 2004;84:1315–1339. doi: 10.1152/physrev.00002.2004. [DOI] [PubMed] [Google Scholar]
- 5.Bursac P, Fabry B, Trepat X, Lenormand G, Butler JP, Wang N, Fredberg JJ, An SS. Cytoskeleton dynamics: fluctuations within the network. Biochem Biophys Res Commun. 2007;355:324–330. doi: 10.1016/j.bbrc.2007.01.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chatterjee M, Murphy RA. Calcium-dependent stress maintenance without myosin phosphorylation in skinned smooth muscle. Science. 1983;221:464–466. doi: 10.1126/science.6867722. [DOI] [PubMed] [Google Scholar]
- 7.Cipolla MJ, Gokina NI, Osol G. Pressure-induced actin polymerization in vascular smooth muscle as a mechanism underlying myogenic behavior. FASEB J. 2002;16:72–76. doi: 10.1096/cj.01-0104hyp. [DOI] [PubMed] [Google Scholar]
- 8.Dillon PF, Aksoy MO, Driska SP, Murphy RA. Myosin phosphorylation and the cross-bridge cycle in arterial smooth muscle. Science. 1981;211:495– 497. doi: 10.1126/science.6893872. [DOI] [PubMed] [Google Scholar]
- 9.Fabry B, Maksym GN, Butler JP, Glogauer M, Navajas D, Fredberg JJ. Scaling the microrheologoy of living cells. Physiol Rev Lett. 2001;87:148102. doi: 10.1103/PhysRevLett.87.148102. [DOI] [PubMed] [Google Scholar]
- 10.Fabry B, Maksym GN, Shore SA, Moore PE, Panettieri RA, Butler JP, Fredberg JJ. Signal transduction in smooth muscle–selected contribution: time course and heterogeneity of contractile responses in cultured human airway smooth muscle cells. J Appl Physiol. 2001;91:986–994. doi: 10.1152/jappl.2001.91.2.986. [DOI] [PubMed] [Google Scholar]
- 11.Fredberg JJ, Inouye D, Miller B, Nathan M, Jafari S, Raboudi SH, Butler JP, Shore SA. Airway smooth muscle, tidal stretches, and dynamically determined contractile states. Am J Respir Crit Care Med. 1997:1752–1759. doi: 10.1164/ajrccm.156.6.9611016. [DOI] [PubMed] [Google Scholar]
- 12.Gerthoffer WT. Actin cytoskeletal dynamics in smooth muscle contraction. Can J Physiol Pharmacol. 2005;83:851–856. doi: 10.1139/y05-088. [DOI] [PubMed] [Google Scholar]
- 13.Gunst SJ, Fredberg JJ. The first three minutes: smooth muscle contraction, cytoskeletal events, and soft glasses. J Appl Physiol. 2003;95:413–425. doi: 10.1152/japplphysiol.00277.2003. [DOI] [PubMed] [Google Scholar]
- 14.Hai CM, Murphy RA. Crossbridge phosphorylation and regulation of the latch state in smooth muscle. Am J Physiol Cell Physiol. 1988;254:C99–C106. doi: 10.1152/ajpcell.1988.254.1.C99. [DOI] [PubMed] [Google Scholar]
- 15.Hai CM, Murphy RA. Regulation of shortening velocity by cross-bridge phosphorylation in smooth muscle. Am J Physiol Cell Physiol. 1988;255:C86–C94. doi: 10.1152/ajpcell.1988.255.1.C86. [DOI] [PubMed] [Google Scholar]
- 16.Hirshman CA, Emala CW. Actin reorganization in airway smooth muscle cells involves Gq and Gi-2 activation of Rho. Am J Physiol Lung Cell Mol Physiol. 1999;277:L653–L661. doi: 10.1152/ajplung.1999.277.3.L653. [DOI] [PubMed] [Google Scholar]
- 17.Ishida T, Ishida M, Suero J, Takahashi M, Berk BC. Agonist-stimulated cytoskeletal reorganization and signal transduction at focal adhesions in vascular smooth muscle cells require c-Src. J Clin Invest. 1999;103:789–797. doi: 10.1172/JCI4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lenormand G, Fredberg JJ. Deformability, dynamics, and remodeling of cytoskeleton of the adherent living cell. Biorheology. 2006;43:1–30. [PubMed] [Google Scholar]
- 19.Meeks M, Ripley ML, Jin Z, Rembold CM. Heat shock protein 20-mediated force suppression in forskolin-relaxed swine carotid artery. Am J Physiol Cell Physiol. 2005;288:C633–C639. doi: 10.1152/ajpcell.00269.2004. [DOI] [PubMed] [Google Scholar]
- 20.Mehta D, Gunst SJ. Actin polymerization stimulated by contractile activation regulates force development in canine tracheal smooth muscle. J Physiol. 1999;519:829–840. doi: 10.1111/j.1469-7793.1999.0829n.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mehta D, Wang ZL, Wu MF, Gunst SJ. Relationship between paxillin and myosin phosphorylation during muscarinic stimulation of smooth muscle. Am J Physiol Cell Physiol. 1998;274:C741–C747. doi: 10.1152/ajpcell.1998.274.3.C741. [DOI] [PubMed] [Google Scholar]
- 22.Murphy RA. Handbook of Physiology. The Cardiovascular System. Vascular Smooth Muscle. II. Am. Physiol. Soc.; Bethesda, MD: 1980. Mechanics of vascular smooth muscle. pp. 325–351. sect. 2. chapt. 13. [Google Scholar]
- 23.Murphy RA, Rembold CM. The latch-bridge hypothesis of smooth muscle contraction. Can J Physiol Pharmacol. 2005;83:857–864. doi: 10.1139/y05-090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ohanian V, Gatfield K, Ohanian J. Role of the actin cytoskeleton in G-protein-coupled receptor activation of PYK2 and paxillin in vascular smooth muscle. Hypertension. 2005;46:93–99. doi: 10.1161/01.HYP.0000167990.82235.3c. [DOI] [PubMed] [Google Scholar]
- 25.Pavalko FM, Adam LP, Wu MF, Walker TL, Gunst SJ. Phosphorylation of dense-plaque proteins talin and paxillin during tracheal smooth muscle contraction. Am J Physiol Cell Physiol. 1995;268:C563–C571. doi: 10.1152/ajpcell.1995.268.3.C563. [DOI] [PubMed] [Google Scholar]
- 25a.Rembold CM. Force suppression and the crossbridge cycle in swine carotid artery. Am J Physiol Cell Physiol. 2007 May 23; doi: 10.1152/ajpcell.00091.2007. doi: 10.1152/ajpcell.00091.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rembold CM, Murphy RA. Myoplasmic calcium, myosin phosphorylation, and regulation of the crossbridge cycle in swine arterial smooth muscle. Circ Res. 1986;58:803–815. doi: 10.1161/01.res.58.6.803. [DOI] [PubMed] [Google Scholar]
- 27.Rembold CM, Murphy RA. Myoplasmic [Ca2+] determines myosin phosphorylation in agonist-stimulated swine arterial smooth muscle. Circ Res. 1988;63:593–603. doi: 10.1161/01.res.63.3.593. [DOI] [PubMed] [Google Scholar]
- 28.Rembold CM, O'Connor MJ. Caldesmon and heat shock protein 20 in nitroglycerin- and magnesium induced relaxation of swine carotid artery. Biochim Biophys Acta. 2000;1500:257–264. doi: 10.1016/s0925-4439(99)00112-x. [DOI] [PubMed] [Google Scholar]
- 29.Rembold CM, O'Connor MJ, Clarkson M, Wardle RL, Murphy RA. HSP20 phosphorylation in nitroglycerin- and forskolin-induced sustained reductions in swine carotid media tone. J Appl Physiol. 2001;91:1460–1466. doi: 10.1152/jappl.2001.91.3.1460. [DOI] [PubMed] [Google Scholar]
- 30.Rembold CM, Wardle RL, Wingard CJ, Batts TW, Etter EF, Murphy RA. Cooperative attachment of cross bridges predicts regulation of smooth muscle force by myosin phosphorylation. Am J Physiol Cell Physiol. 2004;287:C594–C602. doi: 10.1152/ajpcell.00082.2004. [DOI] [PubMed] [Google Scholar]
- 31.Singer HA, Murphy RA. Maximal rates of activation in electrically stimlated swine carotid media. Circ Res. 1987;60:438–445. doi: 10.1161/01.res.60.3.438. [DOI] [PubMed] [Google Scholar]
- 32.Smith PG, Garcia R, Kogerman L. Mechanical strain increases protein tyrosine phosphorylation in airway smooth muscle cells. Exp Cell Res. 1998;239:353–360. doi: 10.1006/excr.1997.3905. [DOI] [PubMed] [Google Scholar]
- 33.Sul D, Baron CB, Broome R, Coburn RF. Smooth muscle length-dependent PI(4,5)P-2 synthesis and paxillin tyrosine phosphorylation. Am J Physiol Cell Physiol. 2001;281:C300–C310. doi: 10.1152/ajpcell.2001.281.1.C300. [DOI] [PubMed] [Google Scholar]
- 34.Tang DD, Zhang WW, Gunst SJ. The adapter protein CrkII regulates neuronal Wiskott-Aldrich syndrome protein, actin polymerization, and tension development during contractile stimulation of smooth muscle. J Biol Chem. 2005;280:23380–23389. doi: 10.1074/jbc.M413390200. [DOI] [PubMed] [Google Scholar]
- 35.Togashi H, Emala CW, Hall IP, Hirshman CA. Carbachol-induced actin reorganization involves Gi activation of Rho in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 1998;275:L803–L809. doi: 10.1152/ajplung.1998.274.5.L803. [DOI] [PubMed] [Google Scholar]
- 36.Trepat X, Deng LH, An SS, Navajas D, Tschumperlin DJ, Gerthoffer WT, Butler JP, Fredberg JJ. Universal physical responses to stretch in the living cell. Nature. 2007;447:592–597. doi: 10.1038/nature05824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tseng S, Kim R, Kim T, Morgan KG, Hai CM. F-actin disruption attenuates agonist-induced [Ca2+], myosin phosphorylation, and force in smooth muscle. Am J Physiol Cell Physiol. 1997;272:C1960–C1967. doi: 10.1152/ajpcell.1997.272.6.C1960. [DOI] [PubMed] [Google Scholar]
- 38.Turner CE. Paxillin and focal adhesion signalling. Nat Cell Biol. 2000;2:E231–E236. doi: 10.1038/35046659. [DOI] [PubMed] [Google Scholar]
- 39.Turner CE, Pietras KM, Taylor DS, Molloy CJ. Angiotensin-II stimulation of rapid paxillin tyrosine phosphorylation correlates the formation of focal adhesions in rat aortic smooth muscle cells. J Cell Sci. 1995;108:333–342. doi: 10.1242/jcs.108.1.333. [DOI] [PubMed] [Google Scholar]
- 40.Walker JS, Walker LA, Etter EF, Murphy RA. A dilution immunoassay to measure myosin regulatory light chain phosphorylation. Anal Biochem. 2000;284:173–182. doi: 10.1006/abio.2000.4704. [DOI] [PubMed] [Google Scholar]
- 41.Wang ZL, Pavalko FM, Gunst SJ. Tyrosine phosphorylation of the dense plaque protein paxillin is regulated during smooth muscle contraction. Am J Physiol Cell Physiol. 1996;270:C1594–C1602. doi: 10.1152/ajpcell.1996.271.5.C1594. [DOI] [PubMed] [Google Scholar]
- 42.Ward D, Alder A, Ohanian J, Ohanian V. Noradrenaline-induced paxillin phosphorylation, ERK activation and MEK-regulated contraction in intact rat mesenteric arteries. J Vasc Res. 2002;39:1–11. doi: 10.1159/000048988. [DOI] [PubMed] [Google Scholar]
- 43.Wingard CJ, Browne AK, Murphy RA. Dependence of force on length at constant cross-bridge phosphorylation in the swine carotid media. J Physiol. 1995;488:729–739. doi: 10.1113/jphysiol.1995.sp021004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wingard CJ, Murphy RA. Inhibition of Ca2+-dependent contraction in swine carotid artery by myosin kinase inhibitors. Gen Pharmacol. 1999;32:483–494. doi: 10.1016/s0306-3623(98)00289-4. [DOI] [PubMed] [Google Scholar]
- 45.Wingard CJ, Paul RJ, Murphy RA. Dependence of ATP consumption on cross-bridge phosphorylation in swine carotid smooth muscle. J Physiol. 1994;481:111–117. doi: 10.1113/jphysiol.1994.sp020422. [DOI] [PMC free article] [PubMed] [Google Scholar]