

Abstract
The optimization of GS39783 into potent, selective and safe positive allosteric modulators of GABAB receptors is presented.
The receptors for the major inhibitory neurotransmitter in the central nervous system, GABA, are subdivided into ionotropic GABAAand GABAC receptors and metabotropic GABAB receptors. Whereas GABAA and GABAC receptors form chloride-permeable ion channels, GABAB receptors are G-protein coupled receptors (GPCRs). These receptors were discovered in 1980 by Norman G. Bowery 1and act post and pre-synaptically to inhibit neuronal excitability and neurotransmitter release, respectively. A possible role of GABAB receptors in a large number of CNS disorders such as cognition deficits, anxiety, depression, epilepsy, pain and drug addiction has been discussed.2 Some of these diseases like anxiety, pain and drug addiction could potentially be treated by activation of GABAB receptors, which can be achieved by administration of either agonists or positive allosteric modulators. Whereas benzodiazepines are well known positive allosteric modulators of GABAA receptors, the first examples of allosteric enhancers for GABAB receptors have been described only recently.3 One of the most interesting compound found was GS39783 (Figure 1).3b However, despite an interesting in vitro and in vivo profile, 3b,4 GS39783 was found to be genotoxic probably because of its aromatic nitro group (Figure 1)55.
Figure 1.
Structure of GS39783
This communication describes our efforts towards the identification of a novel, drug-like class of compounds acting as positive allosteric modulators for GABAB receptors.
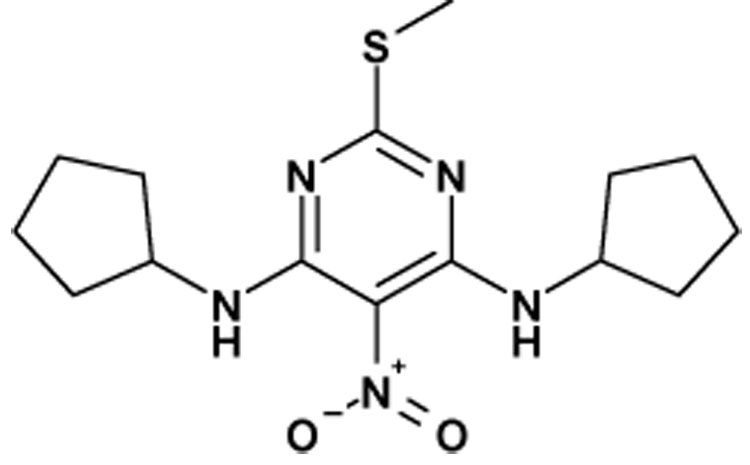
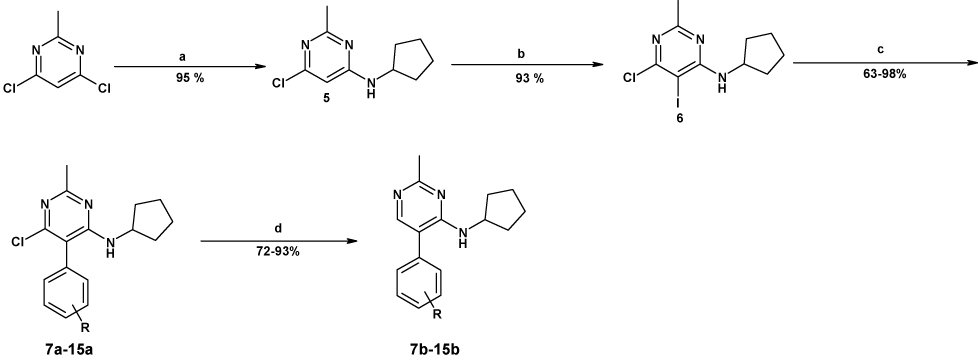
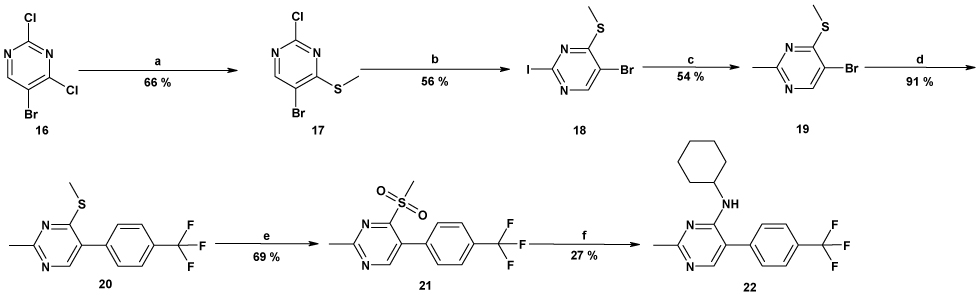
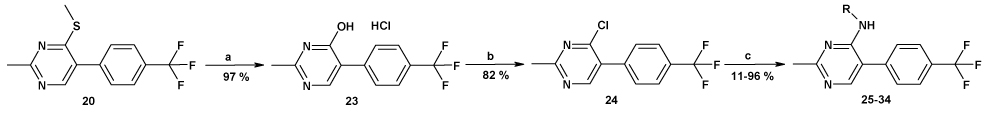
In order to introduce molecular diversity in position 5 of the pyrimidine ring, a 4 steps procedure depicted in Scheme 1 was optimized in order to obtain compounds with a chlorine or with a hydrogen in position 6. 4,6-dichloro-2-methylpyrimidine was first substituted by cyclopentylamine and then iodinated to lead to compound 6. This scaffold was then used in a Suzuki cross coupling6 to give very efficiently a small focused library of substituted 4-amino-6-chloro-5-phenylpyrimidines (Cpds 7a–15a) which were then hydrogenated under standard conditions to give the desired 4-amino-5-phenylpyrimidines (Cpds 7b–15b). As a means to introduce molecular diversity at the last step in position 4 of the pyrimidine ring, a versatile way of synthesis was designed (Scheme 2). Starting from the commercially available 5-bromo-2,4-dichloropyrimidine, a regioselective nucleophilic substitution was performed in position 4 exclusively to give the 4-methylthio derivative 177 which was then involved in a halogen exchange8 to give 2-iododerivative 18. The 2-methyl group and the 5-[(4-trifluoromethyl)phenyl] substituents were introduced by a Negishi cross coupling9 and a Suzuki cross coupling,10 respectively to afford 20. Our first attempt was to oxidize the methylsulfide moiety of 20 to the corresponding methylsulfonyl (21) by treatment with mCPBA11 and then to use 21 as a template. Unfortunately, only few amines reacted cleanly with 21 probably because of the steric hindrance near the leaving group. Our second attempt was to hydrolyze the methylsulfide moiety of 20 with hydrochloric acid12 to give 23 (Scheme 3). This compound was then reacted with POCl3 to give the corresponding 4-chloropyrimidine 24. This useful intermediate was submitted to nucleophilic substitutions with a collection of amines to give compounds 25–34. Finally the third library with molecular diversity in position 2 was synthesized starting from compound 16, which was substituted first with exo-2-aminonorbornane13 and then with MeSNa to give 36 (Scheme 4). The phenyl moiety was introduced by a Suzuki cross coupling to afford 3737. On one hand, a desulfurization by treatment with Ni-Raney in EtOH14 leads to 39 and on the other hand 37 can also be oxidized into the corresponding 2-methylsulfonylpyrimidine 38 which was then reacted with various nucleophiles.
Scheme 1.
Reagents and conditions: (a): cyclopentylamine (4.0 eq.), dioxane, 0°C to RT, 24h; (b): NIS, DMF, 80°C, 24h ; (c): arylboronic acid (1.05 eq.), Pd(OAc)2 (0.02 eq.), dppf (0.03 eq.), K3PO4 (2.0 eq.), DME, water, 85°C ; (d): Pd/C 10%, AcONa (1.1 eq.), EtOH, H2, RT.
Scheme 2.
Reagents and conditions: (a): MeSNa, THF, RT; (b): HI 57%, RT; (c): MeZnCl, Pd(PPh3)4, THF, RT to 60°C; (d): 4-trifluoromethylbenzeneboronic acid, Na2CO3, Pd(PPh3)4, EtOH, Toluene, water, 110°C; (e): mCPBA, DCM, RT; (f): cyclohexylamine, DMF, 150°C, microwaves, 20 min.
Scheme 3.
Reagents and conditions: (a): HCl 37%, MeOH, reflux; (b): POCl33, one drop of DMF, 80°C; (c): R-NH2, solvent, Base, 80°C.
Scheme 4.
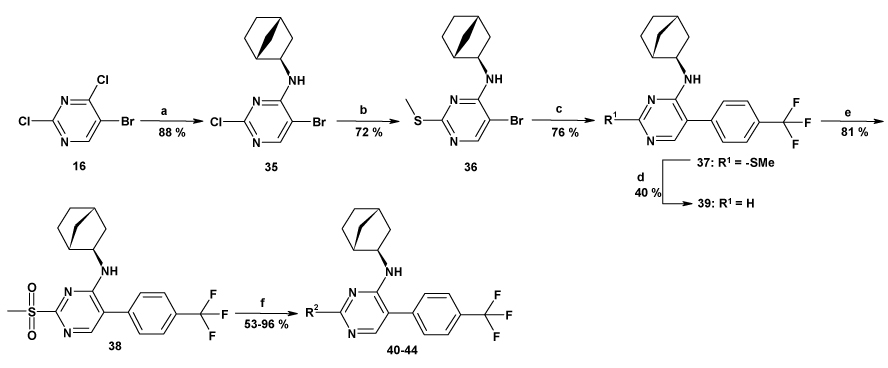
Reagents and conditions: (a): exo-2-aminonorbornane, THF, 0°C to RT; (b): MeSNa, THF, 0°C to RT; (c): 4-trifluoromethylbenzeneboronic acid, Na2CO3, Pd(PPh3)4, toluene, EtOH, water, 110°C; (d): Ni-Raney, EtOH, 60°C; (e): mCPBA, DCM, RT, 1.5h; (f): Cpd 40: KCN, DMSO, 80°C. Cpd 41: Sodium methoxide, THF, RT. Cpd 42: methylamine, EtOH, RT. Cpd 43: dimethylamine, THF, 80°C. Cpd 44: N-methylpiperazine, THF, 80°C.
Thanks to a preliminary work with nitro-mimetics (Figure 2), we found that to replace the nitro group, we should have a lipophilic substituent (see Cpd 3) with an electron-withdrawing effect (see Cpds 1 and 2). One way to combine both of these effects is to substitute the position 5 of the pyrimidine ring by a substituted phenyl. Moreover, substantial efficacy in the biological assay15 was observed for compounds with only one cyclopentylamine substituent in position 4 of the pyrimidine ring (compare 1 with 2 and 3 with 4). Furthermore, it was also shown earlier that the replacement of the 2-methylthio substituent by a 2-methyl group is not detrimental for the activity.3b After screening for GABAB receptor positive modulatory activity of the first library of compounds (Table 1), we found that the 6-chloro substituent was detrimental to the efficacy at the receptor (compare for example 8a with 8b at 25 µM of compound, 12a with 12b and 15a) with 15b. Moreover, the introduction of a second cyclopentylamino substituent in position 6 led to only weakly active or inactive compounds (data not shown) confirming our hypothesis that the space in the receptor is very limited and that a proton is the best substituent for the position 6 of the pyrimidine ring. On the other hand, as postulated before, electron-withdrawing groups on the phenyl ring such as a 4-trifluoromethyl or a 4-trifluoromethoxy showed the best results (compare 9b with 12b and 10b with 15b). Indeed, the replacement of a 4-methylphenyl (9b) or 4-methoxyphenyl (10b) by a 4-trifluoromethylphenyl (12b) or a 4-trifluoromethoxyphenyl (15b) led to more efficacious positive modulators (Table 1). Other electron-withdrawing substituents were used on the phenyl ring but none of them had increased activity at the receptor compared to the trifluoromethyl or trifluoromethoxy groups (data not shown). The introduction of a substituent in position 3 of the phenyl ring led to a decrease of activity (compare 14b with 15b). To conclude, the best nitro-mimetic group identified in this series was a 4-(trifluoromethyl)phenyl substituent. Then we focused our attention on position 4 of the pyrimidine ring (Scheme 2 and Scheme 3).
Figure 2.
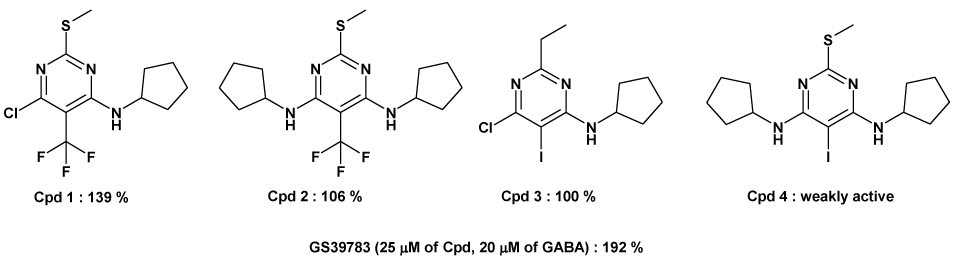
Biological activity of some GS39783 derivatives bearing nitro-mimetics in position 5. The potentiation of the GABA-induced stimulation of GTP(γ)35S binding was measured using membranes from GABABB(1b/2) expressing CHO-K1 cells as described. 3a The activities of compounds (25 µM) were determined in the presence of 20 µM GABA. The data are normalized to the maximal effect (100%) obtained with a saturating concentration of GABA (1mM). 20 µM GABA, when applied alone, stimulates to approximately EC80 levels (80%).
Table 1.
Biological activities obtained for compounds 7a–15b. The compounds were assayed as described.3,15 When applied alone, 1µM GABA stimulates to approximately EC20 levels (20% stimulation). The effects (%) are normalized to the maximal effect of a saturating concentration of GABA (1mM). Values were calculated from triplicate measurements. pEC50 and Emax values were calculated from concentration-response curves (8 concentrations) in the presence of 1µM of GABA.
| Cpd | R | 2.5 µM of Cpd, 1 µM of GABA | 25 µM of Cpd, 1 µM of GABA | 1 µM of GABA | |
|---|---|---|---|---|---|
| Effect (%) ± sem | Effect (%) ± sem | pEC50 ± sem | Emax(%) ± sem | ||
| GS39783 | - | 132 ± 4 | 153 ± 2 | 6.13 ± 0.06 | 146 ± 5 |
| 7a | 4-nButyl | 27 ± 1 | 39 ± 1 | - | - |
| 7b | 25 ± 1 | 18 ± 3 | - | - | |
| 8a | 4-Ethyl | 23 ± 2 | 39 ± 2 | - | - |
| 8b | 29 ± 1 | 61 ± 1 | - | - | |
| 9a | 4-Ethyl | 23 ± 2 | 34 ± 1 | - | - |
| 9b | 23 ± 2 | 41 ± 2 | - | - | |
| 10a | 4-Methoxy | 21 ± 1 | 29 ± 1 | - | - |
| 10b | 21 ± 1 | 38 ± 2 | - | - | |
| 11a | 3-Methyl | 25 ± 1 | 36 ± 1 | - | - |
| 11b | 21 ± 0 | 28 ± 2 | - | - | |
| 12a | 4-CF3 | 17 ± 1 | 26 ± 1 | - | - |
| 12b | 62 ± 3 | 114 ± 2 | 5.30 ± 0.03 | 127 ± 3 | |
| 13a | 4-COOEt | 14 ± 1 | 5 ± 3 | - | - |
| 13b | 26 ± 0 | 31 ± 0 | - | - | |
| 14a | 3-OCF3 | 20 ± 1 | 33 ± 1 | - | - |
| 14b | 23 ± 1 | 25 ± 1 | - | - | |
| 15a | 4-OCF3 | 19 ± 1 | 35 ± 1 | - | - |
| 15b | 48 ± 3 | 100 ± 3 | 5.34 ± 0.07 | 117 ± 7 | |
This collection of compounds shows interesting structure activity relationships (Table 2). For this series it became obvious that an increased size of the cycloalkyl led to increased efficacy. This trend was first observed with compound 29 in which the cyclopentyl substituent was replaced by a cycloheptyl (compare 12bb and 22 with 29 at 2.5 µM). Interestingly, the introduction of an additional bulky substituent on the cyclohexyl ring (30) led to a less active product (compare 30 with 22). Moreover, the introduction of an aromatic ring onto the amino group was not tolerated by the receptor (compare 31, 32, 34 with 22) therefore we thought that a hydrophobic interaction with a spatially extended substituent was necessary at this position. Surprisingly, despite its 4-tert-butylamino substituent, 33 was found to be a weak positive modulator. The needs of bulky, cyclic aliphatic side chain was then confirmed when cycloalkyl chains were used such as a norbornyl (27), an adamantyl (25),(26) or a cycloheptyl (29). A substantial increase in activity was observed when comparing 29 to 12b and to 22. Despite its good efficacy, 29 was not investigated any further because of significant binding activities to other GPCRs (data not shown). We were then interested in compounds 25 and 26 bearing an adamantyl substituent on the amino group. Both compounds showed were potent positive allosteric modulators (Table 2) which led to the hypothesis that the substituents in position 4 of the pyrimidine ring bind into a large lipophilic pocket in the receptor. However, again these products were not considered for further evaluation despite an increased selectivity profile because of their high logPs (log P > 5.9) and low water solubilities (< 10 mg.L−1). Finally, we focused our attention on 27 which bears a norbornyl group at its amino function. Both the exo isomer (27) and the endo isomer (28) were evaluated (Table 2). Compound 27 was more efficacious, and was of similar potency and efficacy compared to GS39783. Genotoxicity16 and mutagenicity17 assays were performed and 27 was found to be safe both in the micronucleus test and in the Ames test. For that reason 27 was considered for further in vitro and in vivo evaluations and the exo-2-norbornyl substituent was kept for the optimization of the position 2.
Table 2.
Biological activities obtained for compounds 22, 25–34. The compounds were assayed as described under Table 1. Compounds 25–27, 29 and 34 have a relatively low water solubility due to their high lipophilicity. This is reflected in this assay by a similar or reduced activity at 25 µM versus 2.5 µM. Concentration-dependent increase in activity however was observed in full concentration response curves, with maximal effects at 10 µM.
| Cpd | R | 2.5 µM of Cpd, 1 µM of GABA | 25 µM of Cpd, 1 µM of GABA | 1 µM of GABA | |
|---|---|---|---|---|---|
| Effect (%) ± sem | Effect (%) ± sem | pEC50 ± sem | Emax (%) ± sem | ||
| 22 |  |
80 ±1 | 93 ± 5 | 6.06 ± 0.05 | 83 ± 4 |
| 25 |  |
90 ± 1 | 80 ± 2 | 5.56 ± 0.13 | 132 ± 14 |
| 26 | 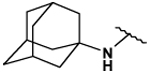 |
125 ± 4 | 141 ± 6 | 5.46 ± 0.10 | 185 ± 15 |
| 27 | 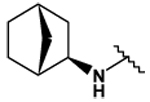 |
122 ± 3 | 110 ± 0 | 5.78 ± .03 | 183 ± 4 |
| 28 | 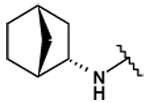 |
82 ± 5 | 52 ± 4 | - | - |
| 29 | 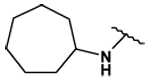 |
128 ± 4 | 70 ± 4 | 5.78 ± 0.03 | 137 ± 3 |
| 30 | 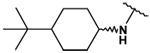 |
28 ± 1 | 32 ± 2 | - | - |
| 31 | 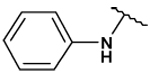 |
36 ± 2 | 69 ± 6 | - | - |
| 32 | 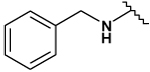 |
29 ± 1 | 39 ± 2 | - | - |
| 33 | 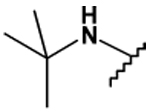 |
40 ± 3 | 21 ± 7 | - | - |
| 34 | 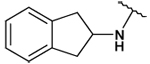 |
58 ± 11 | 59 ± 4 | - | - |
Finally the screening of the collection of compounds with molecular diversity in position 2 gave surprising results (Table 3). The replacement of the methyl moiety of 27 by an hydrogen led to a less active compound which indicated that a substitution at this position is mandatory. The introduction of a 2-SMe group (37), a methylsulfonyl (38) or a methylamino (42) were detrimental for activity (compare 37, 38 and 4242 with 27). In contrast, the replacement of a 2-methyl group by a cyano (40), a methoxy (41) or a dimethylamino (43) gave compounds with a similar potency than GS39783. Surprisingly, the introduction of a N-methylpiperazin-1-yl (44) reduced the biological activity suggesting that the space in the receptor is limited and that only small substituents are tolerated. Compounds 40, 41 and 43 are currently under evaluation for their physicochemical and pharmacokinetics properties as well as their full pharmacological characterization.
Table 3.
Biological activities obtained for compounds 37-44. The compounds were assayed as described under Table 1.
| Cpd | R | 2.5 µM of Cpd, 1 µM of GABA | 25 µM of Cpd, 1 µM of GABA | 1 µM of GABA | |
|---|---|---|---|---|---|
| Effect (%) ± sem | Effect (%) ± sem | pEC50 (µM) ± sem | Emax(%) ± sem | ||
| 37 | -SMe | 82 ± 7 | 91 ± 4 | - | - |
| 38 | Me-SO2- | 24 ± 7 | 30 ± 12 | - | - |
| 39 | H | 48 ± 3 | 63 ± 4 | - | - |
| 40 | -CN | 117 ± 3 | 127 ± 1 | 5.92 ± 0.11 | 198 ± 21 |
| 41 | -OMe | 115 ± 2 | 131 ± 6 | 5.69 ± 0.13 | 191 ± 21 |
| 42 | -NHMe | 20 ± 3 | 25 ± 8 | - | - |
| 43 | -NMe2 | 94 ± 4 | 126 ± 7 | 5.78 ± 0.08 | 167 ± 16 |
| 44 | N-methylpiperazine | 26 ± 8 | 112 ± 7 | - | - |
Positive allosteric modulators of GABAB receptors represent an interesting class of therapeutic agents for the treatment of anxiety and drug addiction. Despite its good in vitro and in vivo potency, GS39783 was only useful as a pharmacological tool because of its genotoxicity. We report herein a new class of positive allosteric modulators of GABAB receptors derived from GS39783 with an increased drug-likeness, a decreased toxicity and an excellent selectivity profile. Some in vivo investigations are currently on going in anxiety and drug addiction models and the results of these studies will be reported in due course.
Acknowledgments
This work was supported by National Institutes of Mental Health/National Institute on Drug Abuse Grant U01 MH69062.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Bowery NG, Hill DR, Hudson AL, Doble A, Middlemiss DN, Shaw J, Turnbull M. Nature. 1980;283:92. doi: 10.1038/283092a0. [DOI] [PubMed] [Google Scholar]
- 2.(a) Marshall FH. J. Mol. Neurosci. 2005;26(2–3):169. doi: 10.1385/JMN:26:2-3:169. [DOI] [PubMed] [Google Scholar]; (b) Cryan JF, Kaupmann K. Trends Pharmacol. Sci. 2005;26(1):36. doi: 10.1016/j.tips.2004.11.004. [DOI] [PubMed] [Google Scholar]; (c) Bettler B, Kaupmann K, Mosbacher J, Gassmann M. Physiol. Rev. 2004;84:835. doi: 10.1152/physrev.00036.2003. [DOI] [PubMed] [Google Scholar]
- 3.(a) Urwyler S, Mosbacher J, Lingenhoehl K, Heid J, Hofstetter K, Froestl W, Bettler B, Kaupmann K. Mol. Pharmacol. 2001;60:963. [PubMed] [Google Scholar]; (b) Urwyler S, Pozza MF, Lingenhoehl K, Mosbacher J, Lampert C, Froestl W, Koller M, Kaupmann K. J. Pharmacol. Exp. Ther. 2003;307(1):322. doi: 10.1124/jpet.103.053074. [DOI] [PubMed] [Google Scholar]
- 4.Cryan JF, Kelly PH, Chaperon F, Gentsch C, Mombereau C, Lingenhoel K, Froestl W, Bettler B, Kaupmann K, Spooren WPJM. J. Pharmacol. Exp. Ther. 2004;310(3):952. doi: 10.1124/jpet.104.066753. [DOI] [PubMed] [Google Scholar]
- 5.(a) Purohit V, Basu AK. Chem. Res. Toxicol. 2000;13:673. doi: 10.1021/tx000002x. [DOI] [PubMed] [Google Scholar]; (b) Tocher JH. Gen. Pharmacol. 1997;28:485. doi: 10.1016/s0306-3623(96)00283-2. [DOI] [PubMed] [Google Scholar]
- 6.Richardson ML, Stevens MFG. J. Chem. Res., Synop. 2002:482. [Google Scholar]
- 7.Strekowski L. Bull. Pol. Acad. Sci., Chem. 1976;24(1):17. [Google Scholar]
- 8.Vlad G, Horvath IT. J. Org. Chem. 2002;67:6550. doi: 10.1021/jo0255781. [DOI] [PubMed] [Google Scholar]
- 9.Hocek M, Votruba I, Dvorakova H. Tetrahedron. 2003;59:607. [Google Scholar]
- 10.Hannah DR, Sherer EC, Davies RC, Titman RB, Laughton CA, Stevens MFG. Bioorg. Med. Chem. 2000;8:739. doi: 10.1016/s0968-0896(00)00017-1. [DOI] [PubMed] [Google Scholar]
- 11.Herrera A, Martinez-Alvarez R, Chioua R, Benabdelouahab F, Chioua M. Tetrahedron. 2004;60:5475. [Google Scholar]
- 12.Strekowski L, Harden D, Watson RA. Synthesis. 1988;70 [Google Scholar]
- 13.Brumby T, Jautelat R, Prien O, Schaefer M, Siemeister G, Luecking U, Huwe C. 2002096888. WO. 2002
- 14.Morimoto H, Shimadzu H, Kushiyama E, Kawanishi H, Hosaka T, Kawase Y, Yasuda K, Kikkawa K, Yamauchi-Kohno R, Yamada K. J. Med. Chem. 2001;44:3355. doi: 10.1021/jm0102304. [DOI] [PubMed] [Google Scholar]
- 15.GTP(γ)35S binding was used as functional assay for GABAB receptor activity (see reference 3b). To assay for positive modulatory activity the compounds were co-applied with GABA. 1µM GABA stimulates GABAB receptors to approximately EC20 values, 20µM GABA stimulates to approximately EC80 values. If co-application of the test compounds with GABA significantly increased the signal above EC20 (at 1µM GABA) or EC80 (at 20µM GABA) we concluded that the compound positively modulated the GABA response. In control experiments the test compounds were assayed in the absence of GABA. For selected compounds, concentration-response curves were generated and the EC50 values and maximal stimulations at a GABA concentration of 1µM determined.
- 16.(a) Fenech M. Mutat. Res. 2000;455(1–2):81. doi: 10.1016/s0027-5107(00)00065-8. [DOI] [PubMed] [Google Scholar]; (b) Miller B, Albertini S, Locher F, Thybaud V, Lorge E. Mutat. Res. 1997;392(1–2):45. doi: 10.1016/s0165-1218(97)00044-x. [DOI] [PubMed] [Google Scholar]
- 17.Flamand N, Meunier J-R, Meunier P-A, Agapakis-Caussé C. Toxicology in vitro. 2001;15:105. doi: 10.1016/s0887-2333(01)00003-0. [DOI] [PubMed] [Google Scholar]