

Abstract
Association-mapping methods promise to overcome the limitations of linkage-mapping methods. The main objectives of this study were to (i) evaluate various methods for association mapping in the autogamous species wheat using an empirical data set, (ii) determine a marker-based kinship matrix using a restricted maximum-likelihood (REML) estimate of the probability of two alleles at the same locus being identical in state but not identical by descent, and (iii) compare the results of association-mapping approaches based on adjusted entry means (two-step approaches) with the results of approaches in which the phenotypic data analysis and the association analysis were performed in one step (one-step approaches). On the basis of the phenotypic and genotypic data of 303 soft winter wheat (Triticum aestivum L.) inbreds, various association-mapping methods were evaluated. Spearman's rank correlation between P-values calculated on the basis of one- and two-stage association-mapping methods ranged from 0.63 to 0.93. The mixed-model association-mapping approaches using a kinship matrix estimated by REML are more appropriate for association mapping than the recently proposed QK method with respect to (i) the adherence to the nominal α-level and (ii) the adjusted power for detection of quantitative trait loci. Furthermore, we showed that our data set could be analyzed by using two-step approaches of the proposed association-mapping method without substantially increasing the empirical type I error rate in comparison to the corresponding one-step approaches.
ESTIMATION of the positions and effects of quantitative trait loci (QTL) is of central importance for marker-assisted selection. In plant genetics, this has so far been accomplished by applying classical linkage-mapping methods. Besides high costs (Parisseaux and Bernardo 2004), their major limitations are a poor resolution in detecting QTL and the fact that with biparental crosses of inbred lines only two alleles at any given locus can be studied simultaneously (Flint-Garcia et al. 2003). Association-mapping methods, which have been successfully applied in human genetics to detect genes coding for human diseases (e.g., Ozaki et al. 2002), promise to overcome these limitations (Kraakman et al. 2004). Therefore, in plant genetics several attempts were made for detecting QTL by using such methods (e.g., Kraakman et al. 2004; Olsen et al. 2004).
Application of association-mapping approaches in plants is complicated by the population structure present in most germplasm sets (Flint-Garcia et al. 2003). To overcome this problem, linear models with fixed effects for subpopulations (e.g., Breseghello and Sorrells 2006) or a logistic regression-ratio test (Pritchard et al. 2000b; Thornsberry et al. 2001) can be employed. Owing to the large germplasm sets required for dissecting complex traits, the probability increases that partially related individuals are included. This applies in particular when genotypes selected from plant-breeding populations are used for association mapping (e.g., Thornsberry et al. 2001; Kraakman et al. 2004). The above-mentioned approaches fail to adhere to the nominal α-level, however, if the germplasm set under consideration comprises related individuals (cf. Thornsberry et al. 2001).
Recently, Yu et al. (2006) proposed the QK mixed-model association-mapping approach that promises to correct for linkage disequilibrium (LD) caused by population structure and familial relatedness. The authors demonstrated the suitability of their new method for association mapping in humans and maize. Besides natural populations of Arabidopsis thaliana (cf. Zhao et al. 2007), the suitability of the QK method has to be evaluated in breeding germplasm of autogamous species, because their population structure is presumably high and levels of familial relatedness are diverse (cf. Garris et al. 2005).
In contrast to coancestry coefficients calculated from pedigree records, marker-based kinship estimates may account for the effects of deviations from expected parental contributions to progeny due to selection or genetic drift (Bernardo et al. 1996). Therefore, marker-based kinship estimates underlying the studies of Yu et al. (2006) and Zhao et al. (2007) might be more appropriate for association-mapping approaches than coancestry coefficients calculated from pedigree records. A difficulty with calculation of marker-based kinship estimates arises regarding the definition of unrelated individuals (Bernardo 1993). The marker-based kinship matrix underlying the study of Yu et al. (2006) was determined on the basis of the definition that random pairs of inbreds are unrelated, whereas Zhao et al. (2007) defined pairs of inbreds that do not share any allele as unrelated. However, both definitions are rather arbitrary. Therefore, we propose to estimate the conditional probability that marker alleles are alike in state, given that they are not identical by descent (Lynch 1988), by restricted maximum likelihood (REML).
As a first step of all earlier association-mapping studies in a plant genetics context, phenotypic data were analyzed and entry means or adjusted entry means were calculated for each individual of the population under consideration. These estimates were then used in a second step for the actual association analysis. Such two-stage procedures generally account neither for heteroscedasticity (heterogeneity in experimental errors) nor for possible covariances among the adjusted entry means (Cullis et al. 1998). These problems can be overcome by applying a one-stage association-mapping approach in which the phenotypic data analysis and the association analysis are performed in one step.
The objectives of our research were to (i) evaluate various methods for association mapping in the autogamous species wheat using an empirical data set, (ii) determine a marker-based kinship matrix based on a REML estimate of the probability that two inbreds carry alleles at the same locus that are identical in state but not identical by descent, and (iii) compare the results of one- and two-stage approaches for various association-mapping methods.
MATERIALS AND METHODS
Plant materials, field experiments, and molecular marker analyses:
A total of 303 soft winter wheat (Triticum aestivum L.) inbreds developed by Lochow-Petkus (Bergen-Wohlde, Germany) were used for this study. For 194 entries, pedigree information up to the great-grandparents was available, whereas for the other 109 entries no pedigree records were available. In 2005, all 303 entries were evaluated for grain yield in a series of five breeding trials at four to six locations, with the number of entries per trial ranging from 36 to 110. The experimental design for each trial was a lattice design with two to four replications per location. Two of the 303 entries were evaluated as common entries in each lattice.
All 303 entries as well as five wheat cultivars, which are unrelated by pedigree to the 303 entries, were fingerprinted by Lochow-Petkus following standard protocols with 36 simple sequence repeat markers and one single-nucleotide polymorphism marker. The 37 marker loci were randomly distributed across 19 of the 21 wheat chromosomes. Map positions of all markers were determined on the basis of the linkage map of Lochow-Petkus (unpublished data).
Statistical analyses:
Phenotypic data analyses:
The phenotypic data were analyzed on the basis of the statistical model
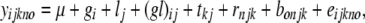 |
where yijkno was the phenotypic observation for the ith entry at the jth location in the oth incomplete block of the nth replicate of the kth trial, μ was an intercept term, gi was the genetic effect of the ith entry, lj was the effect of the jth location, tkj was the effect of the kth trial at the jth location, rnjk was the effect of the nth replicate of the kth trial at the jth location, bonjk was the effect of the oth incomplete block of the nth replication of the kth trial at the jth location, and eijkno was the residual. Error variances were assumed to be heterogeneous among locations. For estimation of variance components, all effects were considered as random.
For estimating entry means, we regarded gi as fixed and all other effects as random (Patterson 1997). Over all trials, an adjusted entry mean Mi was calculated for each of the 303 entries as
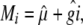 |
where 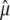 and
and 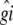 denote the generalized least-squares estimates of μ and gi, respectively.
denote the generalized least-squares estimates of μ and gi, respectively.
Two-stage association analyses:
On the basis of 10 different statistical models (summarized in Table 1), adjusted entry means Mi of the 303 entries were used to calculate a P-value for the association of each of the 37 marker loci with the phenotypic trait.
TABLE 1.
Mixed-model methods used for association mapping and the corresponding statistical models for the two-stage association approaches analyzed in this study
| Method | Statistical model | Population structure matrix D | Kinship matrix K |
|---|---|---|---|
| QK | 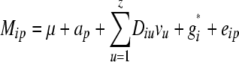 |
STRUCTURE | SPAGeDi |
| PK | 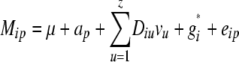 |
First eight principal components | SPAGeDi |
| K | 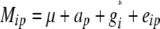 |
— | SPAGeDi |
| G | 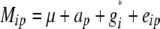 |
— | Pedigree information |
| Kunrel | 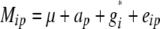 |
— | 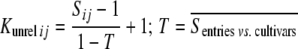 |
| QK0.70 | 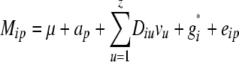 |
STRUCTURE | 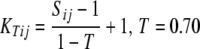 |
| PK0.70 | 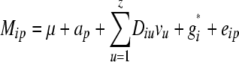 |
First eight principal components | T = 0.70 |
| K0.70 | 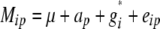 |
— | T = 0.70 |
| K0.35 | 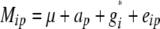 |
— | T = 0.35 |
For a detailed definition of the statistical models and description of the different methods see materials and methods.
The first model was an ANOVA model of the form
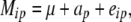 |
where Mip was the adjusted entry mean of the ith entry carrying allele p, ap the effect of allele p, and eip the residual.
The statistical model underlying our mixed-model association-mapping approaches (Table 1) was
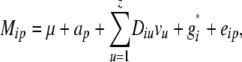 |
where vu was the effect of the uth column of the population structure matrix D and 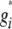 was the residual genetic effect of the ith entry. The matrix D, which comprised z linear independent columns, differed among the various association-mapping methods (Table 1) and, thus, this matrix is described in the sections detailing the individual methods. The variance of the random effects
was the residual genetic effect of the ith entry. The matrix D, which comprised z linear independent columns, differed among the various association-mapping methods (Table 1) and, thus, this matrix is described in the sections detailing the individual methods. The variance of the random effects 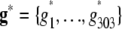 and e = {e1,1,…, e303,12} was assumed to be
and e = {e1,1,…, e303,12} was assumed to be 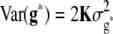 and
and 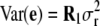 , where K was a 303 × 303 matrix of kinship coefficients that define the degree of genetic covariance between all pairs of entries.
, where K was a 303 × 303 matrix of kinship coefficients that define the degree of genetic covariance between all pairs of entries. 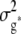 was the genetic variance and
was the genetic variance and 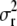 was the residual variance, both estimated by REML. For a direct comparison of our results to those of Yu et al. (2006), R1 was a 303 × 303 matrix in which the off-diagonal elements were 0 and the diagonal elements were reciprocals of the number of phenotypic observations underlying each adjusted entry mean. In a second association-mapping approach, instead of matrix R1 we used matrix R2, in which the diagonal elements were calculated as the square of the standard errors of the adjusted entry means M (Piepho and Möhring 2007).
was the residual variance, both estimated by REML. For a direct comparison of our results to those of Yu et al. (2006), R1 was a 303 × 303 matrix in which the off-diagonal elements were 0 and the diagonal elements were reciprocals of the number of phenotypic observations underlying each adjusted entry mean. In a second association-mapping approach, instead of matrix R1 we used matrix R2, in which the diagonal elements were calculated as the square of the standard errors of the adjusted entry means M (Piepho and Möhring 2007).
For the QK mixed-model method (Yu et al. 2006), the population structure matrix Q was calculated by the software STRUCTURE (Pritchard et al. 2000a), which gives for each individual under consideration the probability of membership in each of the z + 1 subpopulations. In our investigations, the set of 303 entries was analyzed by setting z from 0 to 13 in each of five repetitions. For each run of STRUCTURE, the burn-in time as well as the iteration number for the Markov chain Monte Carlo algorithm was set to 100,000, following the suggestion of Whitt and Buckler (2003).
Plant populations often comprise related and/or admixed entries (Camus-Kulandaivelu et al. 2007). Therefore, we used the ad hoc criterion described by Evanno et al. (2005) to estimate the number of subpopulations, as it promises to reliably detect the true number of subpopulations also in complex genetic situations. The z + 1 columns of the Q matrix add up to one and, thus, only the first z columns were used as a D matrix in the QK method to achieve linear independence. Furthermore, in accordance with Yu et al. (2006) the kinship matrix K was calculated on the basis of the 37 marker loci using the software package SPAGeDi (Hardy and Vekemans 2002), where negative kinship values between inbreds are set to 0.
The PK method was based on the same kinship matrix K as used for the QK method. Following Zhao et al. (2007), however, the first eight principal components of an allele-frequency matrix, which explain altogether 36.8% of the variance, were used as a D matrix of the PK method (Table 1).
The K and G methods were based on mixed models that do not include any vu effects (Table 1). The K method uses the same kinship matrix K as used for the QK method. For the G method, we estimated the K matrix for all 303 inbreds on the basis of the available pedigree records, according to the rules described by Falconer and Mackay (1996), and using PROC INBREED in SAS (Sas Institute 2004). The coancestry coefficient between inbreds with unknown relationship was set to 0 (Bernardo 1993).
Bernardo (1993) proposed calculating the kinship coefficient Kij between inbreds i and j (i.e., the probability that inbreds i and j carry alleles at the same locus that are identical by descent) on the basis of marker data according to
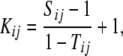 |
where Sij is the proportion of marker loci with shared variants between inbreds i and j and Tij is the average probability that a variant from one parent of inbred i and a variant from one parent of inbred j are alike in state, given that they are not identical by descent. Thus, Tij is a function of the proportion of variants common to unrelated inbreds and is specific for each pair of inbreds (Lynch 1988). In practice, the value of Tij is unknown.
Our Kunrel method uses a matrix Kunrel based on one T value for all pairs of inbreds obtained as the average Sij between each of the five wheat cultivars and the 303 entries, as proposed by Lynch (1988) and Melchinger et al. (1991).
The QKT, PKT, and KT methods were based on a matrix KT that was calculated according to
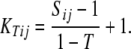 |
We examined T = 0, 0.025, …, 0.975 to obtain a REML estimate of T. Negative kinship values between inbreds were set to 0.
One-stage association analyses:
Phenotypic data analyses and association analyses were performed in one step, on the basis of the model
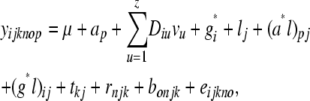 |
where, except for ap and vu, all effects were regarded as random and error variances were assumed to be heterogeneous among locations. Var(g*) and D were modeled by the same nine methods as in the two-stage analysis (Table 1).
Power simulations:
Because of the high computational effort of the one-stage association analyses, our power simulations were conducted only for the two-stage association approaches. For each of the examined methods (ANOVA, QK, PK, K, G, Kunrel, QK0.70, PK0.70, K0.70, and K0.35) the empirical type I error rate α* was calculated on the basis of the P-values observed for the 37 marker loci in a scenario without simulated QTL (α = 0.05). In our study, we examined the power to detect a QTL of interest, which (i) explained a fraction of the phenotypic variance and (ii) was in complete LD with one marker locus, as follows. The QTL effect Gr, calculated as r = 0.1 times the standard deviation of the vector of adjusted entry means M of the 303 wheat inbreds, was assigned in consecutive simulation runs to each of the detected 202 marker alleles whereas all other alleles were assigned the genotypic effect 0. In each simulation run, the genotypic value of each entry i was calculated by summing up the QTL effects of the alleles and the adjusted entry mean Mi. The above-mentioned two-stage association-mapping methods were run on the inbreds' genotypic values to determine whether the QTL can be detected. To adjust the association-mapping methods for their different empirical type I error rates α*, we calculated the adjusted power as the proportion of QTL detected at 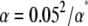 (Yu et al. 2006). In addition to r = 0.1, we examined r = 0.2, 0.3,…, 2.
(Yu et al. 2006). In addition to r = 0.1, we examined r = 0.2, 0.3,…, 2.
The percentage (π) of the total phenotypic variation explained by a QTL effect Gr was calculated as
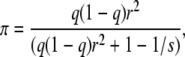 |
where s was the sample size and q the allele frequency of the QTL (Yu et al. 2006).
Measures for comparison of association-mapping methods:
Under the assumption that the random markers m = 1, 2,…, 37 in our study are unlinked to functional polymorphisms controlling yield, it is expected that the P-values observed for an association-mapping approach are uniformly distributed (cf. Yu et al. 2006). Therefore, for the P-values observed for all marker loci and association-mapping methods, expected P-values were calculated as r(xm)/37, where r(xm) is the rank of the P-value xm observed for the mth marker locus. Association-mapping methods that adhere to the nominal α-level show a uniform distribution of P-values, i.e., a diagonal line in the plot of observed vs. expected P-values. The mean of the squared difference (MSD) between observed and expected P-values of all marker loci was therefore calculated as a measure for the deviation of the observed P-values from the uniform distribution. High MSD values indicate a strong deviation of the observed P-values from the uniform distribution, which suggests that the empirical type I error rate of these approaches is considerably higher than the nominal α-level.
Computer simulations were performed to examine which difference in MSD values between two association-mapping methods could be expected purely by chance. The simulation accounted for correlation among P-values of two methods as follows. Pairs of P-values were drawn from a bivariate beta distribution (Magnussen 2004) with parameters α = β = 1 and correlation equal to the observed correlation Cobs for a pair of methods. Thus, the marginal distribution of P-values for a method was uniform, and the correlation among methods equaled Cobs. In each simulation run, the difference of the MSD value for both methods was calculated. This procedure was repeated 100,000 times and the 95% quantile of the MSD difference was determined. We investigated the following four pairs of two-stage association approaches: (i) QK/ANOVA, (ii) QK/K, (iii) QK/G, and (iv) QK/QK0.70.
For methods QKT, PKT, and KT, we profiled the deviance for T. Spearman's rank correlation was calculated between the observed P-values of one- and two-stage association-mapping approaches.
All mixed-model calculations were performed with ASReml release 2.0 (Gilmour et al. 2006).
RESULTS
For grain yield, the adjusted entry means Mi of the 303 elite inbreds varied between 7.52 and 9.60 t ha−1, with an average of 8.66 t ha−1. The genotypic variance was 0.085 t2 ha−2 and the genotype × environment variance was 0.090 t2 ha−2. For the different locations, the error variance ranged from 0.081 to 0.200 t2 ha−2.
The total number of marker alleles detected for the 37 loci was 202, with the number of alleles per marker locus ranging from 2 to 12. The average number of alleles per locus was 5.5. In principal coordinate analysis based on Rogers' distance estimates of the 303 entries as well as five wheat cultivars, the first two principal coordinates explained 14.6 and 10.7% of the molecular variance (Figure 1). With respect to these two principal coordinates, no clear grouping of inbreds could be detected. The model-based approach of STRUCTURE revealed eight subpopulations.
Figure 1.—

Principal coordinate analysis of the 303 entries as well as five wheat cultivars, which are unrelated by pedigree to the 303 entries, based on Rogers' distance estimates. Percentages in parentheses refer to the proportion of variance explained by the principal coordinate.
For the examined levels of T, the MSD between observed and expected P-values for the QKT and PKT methods ranged from 0.002 to 0.035 (Figure 2). By comparison, the MSD was higher for the KT method and varied for the various levels of T between 0.010 and 0.090. The deviances for the three methods QKT, PKT, and KT ranged from ∼ −270 to ∼ −350, with smallest values observed for T = 0.775.
Figure 2.—

Mean of the squared differences (MSD) between observed and expected P-values as well as deviance for different two-stage association-mapping methods depending on threshold T.
The MSD between observed and expected P-values of the QK and PK methods was 0.010 (Table 2), which was 10 times lower than that of the ANOVA approach (0.100). For the K, G, and Kunrel methods, the MSDs were 0.016, 0.077, and 0.013, respectively. The computer simulations that are based on the correlated beta distribution for the four pairs of association methods QK/ANOVA, QK/K, QK/G, and QK/QK0.70 resulted in a 95% quantile of MSD differences of 0.009, 0.006, 0.008, and 0.004, respectively. The trend observed for the MSD of the mixed-model approaches based on the R2 matrix was the same as that found for the approaches based on the R1 matrix.
TABLE 2.
Mean of the squared differences (MSD) between observed and expected P-values for various mixed-model association- mapping methods as well as Spearman's rank correlation coefficient ρ between the P-values of one- and two-stage association-mapping approaches
| MSD
|
|||||
|---|---|---|---|---|---|
| Two stage
|
Spearman's ρ
|
||||
| Method | R1 matrix | R2 matrix | One stage | R1 matrix | R2 matrix |
| QK | 0.010 | 0.013 | 0.088 | 0.74 | 0.79 |
| PK | 0.011 | 0.024 | 0.091 | 0.73 | 0.75 |
| K | 0.016 | 0.022 | 0.061 | 0.67 | 0.69 |
| G | 0.077 | 0.090 | 0.090 | 0.63 | 0.64 |
| Kunrel | 0.013 | 0.016 | 0.042 | 0.63 | 0.76 |
| QK0.70 | 0.003 | 0.003 | 0.002 | 0.84 | 0.93 |
| PK0.70 | 0.002 | 0.005 | 0.003 | 0.87 | 0.93 |
| K0.70 | 0.015 | 0.020 | 0.009 | 0.76 | 0.88 |
| K0.35 | 0.010 | 0.011 | 0.004 | 0.63 | 0.80 |
The adjusted power to detect QTL of all association-mapping approaches increased with increasing size of the genetic effect assigned to an allele (Figure 4). For small as well as large genetic effects the slope of the power curve was flat, whereas for genetic effects of medium size the slope was steep. For all examined sizes of genetic effects, the adjusted power of the QK0.70 and PK0.70 methods was higher than that of the QK and PK methods. In comparison with the other association-mapping methods, the ANOVA method and the G method showed the lowest adjusted power to detect QTL for all examined sizes of genetic effects.
Figure 4.—

Adjusted power to detect quantitative trait loci (QTL) for the 10 two-stage association-mapping methods depending on the size of the QTL effect Gr. The percentage of phenotypic variation explained by a QTL was calculated for an allele frequency of 0.2.
The MSD between observed and expected P-values for the one-stage association-mapping methods ranged from 0.002 (QK0.70) to 0.091 (PK). The trend observed for these approaches was similar to that found for the two-stage approaches (Table 2). Spearman's rank correlation between the P-values of one- and two-stage association analyses ranged from 0.63 to 0.87 for the nine mixed-model methods based on the R1 matrix. Likewise, the correlation ranged from 0.64 to 0.93 for association-mapping approaches based on the R2 matrix.
DISCUSSION
Phenotypic data analyses in association-mapping approaches:
Previous association-mapping approaches in a plant genetics context were mostly based on entry means (e.g., Aranzana et al. 2005; Yu et al. 2006). The more complex is the phenotypic trait under consideration, however, the more elaborate are the field designs as well as phenotypic data analyses that are required. Therefore, we used an efficient method for calculating adjusted entry means Mi (Smith et al. 2001). In this analysis, error variances were assumed to be heterogeneous among locations. The statistical model is easily extended to other settings, e.g., heterogeneous block and replication variances or modeling of spatial heterogeneity at the plot level (nearest-neighbor analyses; Moreau et al. 1999).
Comparison of various association-mapping approaches:
Investigations on the adjusted power to detect QTL as well as on the type I error rate of association-mapping approaches based on empirical data require that the marker loci are unlinked to polymorphisms controlling the trait under consideration. In this study this assumption seems to be reasonable for two reasons. First, findings of Breseghello and Sorrells (2006) suggest that LD in winter wheat inbreds decays within 5 cM, which is considerably shorter than the average marker distance in our study. Second, the 37 marker loci were randomly selected from the wheat genome. Consequently, our study was based on the assumption that no polymorphisms affecting yield were present in a region of 370 cM, which corresponds to only 10% of the wheat genome (Quarrie et al. 2005). Similar to other studies comparing association-mapping approaches based on empirical data (e.g., Yu et al. 2006; Zhao et al. 2007), however, we cannot rule out the possibility that some markers might be linked to functional polymorphisms of the trait under consideration.
Similar to other studies (e.g., Yu et al. 2006; Zhao et al. 2007), we used the same markers for estimation of population structure as well as familial relatedness as were used for calculating the MSD between observed and expected P-values. Theoretical considerations suggest that by this procedure that the MSDs between observed and expected P-values are underestimated for markers that were not included in the estimation of population structure and familial relatedness. However, this issue did not influence our conclusions regarding the eligibility of various methods for association mapping, because they were compared on the basis of the same set of markers.
Our power simulations assumed a QTL that is in complete LD with one marker locus (Yu et al. 2006). This assumption maximizes the power for QTL detection. In most empirical studies, however, no markers are available that are in complete LD with the QTL. Therefore, for such studies, a lower power for QTL detection is expected depending on the extent of LD between marker and QTL. A further factor hampering the detection of the QTL of interest, which was neglected in our power simulations, is additional QTL that are linked to the QTL of interest. The incomplete LD between marker and QTL as well as additional linked QTL, however, is expected to reduce the power for QTL detection of all association-mapping methods to the same extent. Therefore, no influence on our conclusions regarding the ranking of various methods for association mapping is expected with respect to the assumptions made in our power simulations.
ANOVA approach:
A frequently used method for association mapping in a plant genetics context is the ANOVA approach (e.g., Kraakman et al. 2004; Olsen et al. 2004), which was used in the current study as a reference method. Under the assumption that the low number of random marker loci in our study is unlinked to the polymorphisms controlling grain yield, association-mapping methods that adhere to the nominal α-level show a uniform distribution of P-values. By contrast, we observed a nonuniform distribution of P-values with the ANOVA approach (Figure 3). This finding indicates that this method is inappropriate for association mapping in our germplasm set, because it results in a proportion of spurious marker–phenotype associations that is considerably higher than the nominal type I error rate.
Figure 3.—

Plot of observed vs. expected P-values for the 10 two-stage association-mapping methods.
In addition to the nonuniform distribution of P-values with the ANOVA approach, STRUCTURE revealed eight subpopulations. Consequently, absence of distinct subpopulations in the principal coordinate analysis does not necessarily imply that population structure can be neglected in the association-mapping approach. This might be explained by the fact that the current study was based on germplasm from a line-breeding program of an autogamous species. In contrast to germplasm from hybrid-breeding programs (cf. Stich et al. 2005), no distinct subpopulations are expected for such germplasm as population structure is disregarded when choosing the parents of a cross. Nevertheless, line breeding generates high levels of population structure and diverse levels of familial relatedness (cf. Garris et al. 2005).
QK approach:
Recently, Yu et al. (2006) proposed a new association-mapping approach called the QK method. The MSD between observed and expected P-values that was found for this method was ∼10 times lower than that observed for the ANOVA approach (Table 2), and this difference was considerably larger than the 95% quantile observed in our computer simulations on the correlated beta distribution. This underlines the advantage of the QK method over the ANOVA method for association mapping not only in allogamous species such as humans and maize, as suggested by the results of Yu et al. (2006), but also in the autogamous species wheat. Similar findings were reported by Zhao et al. (2007) for A. thaliana.
An association test frequently used in a plant genetics context is the logistic regression-ratio test (Pritchard et al. 2000b; Thornsberry et al. 2001). The null hypothesis of this test states that the molecular marker under consideration is associated with population structure, whereas under the alternative it is associated both with population structure and with the phenotypic variation. The logistic regression-ratio test and the EIGENSTRAT method (Price et al. 2006), recently proposed in a human genetics context, as well as linear models with fixed effects for subpopulations, however, correct only for LD caused by population stratification. The QK method, which allows the modeling of population structure and also of familial relatedness, proved to be superior to this class of association-mapping methods with respect to the adherence to the nominal α-level as well as to the adjusted power for QTL detection (e.g., Yu et al. 2006; Zhao et al. 2007). Therefore, the logistic regression-ratio test and the EIGENSTRAT method, as well as linear models with fixed effects for subpopulations, were not examined in our study.
In our study, the difference between observed and expected P-values for the QK method was slightly higher than that in the study of Yu et al. (2006). This might be explained by (i) less precise kinship estimates resulting from the lower marker density underlying our study and (ii) high levels of population structure and diverse levels of familial relatedness expected in germplasm of an autogamous species (cf. Garris et al. 2005) selected from plant-breeding programs. These issues did not influence our conclusions regarding the ranking of various methods for association mapping, because they were compared on the basis of the same data set.
Despite promising results for the QK association-mapping approach, this method has several drawbacks. Estimation of the Q matrix using STRUCTURE is computationally demanding (Balding 2006; Price et al. 2006). Even more problematic is that STRUCTURE was designed for unrelated individuals that belong to populations in Hardy–Weinberg equilibrium (Pritchard et al. 2000a). For germplasm sets of most species, however, these assumptions are not met and, thus, results of STRUCTURE demand careful interpretation (cf. Camus-Kulandaivelu et al. 2007). Because of these issues, we examined the PK mixed-model association-mapping approach in which the Q matrix from STRUCTURE was replaced by a matrix comprising the first eight principal components from the allele-frequency matrix.
PK approach:
The MSD between observed and expected P-values, which was found for this method, was similar to that observed for the QK approach (Figure 3). Furthermore, both methods yielded a similar adjusted power of QTL detection (Figure 4). In accordance with Zhao et al. (2007), these findings suggested that the PK method is a promising alternative to the QK method.
The QK method as well as the PK method is based on the integration of the fixed effects in the association-mapping model. This leads to a loss of degrees of freedom, which is mainly a problem if the number of entries is low. Furthermore, such approaches hamper the detection of loci contributing to phenotypic differences among subpopulations, because the differences between subpopulations are disregarded in the estimation of the genotypic effects of the loci under consideration. Because of these issues, we examined mixed-model association-mapping approaches that are not based on the assignment of individuals to subpopulations.
G approach:
In plant-breeding populations, extensive information about pedigree relationships is available. In our study, pedigree records were used to calculate the K matrix for the G mixed-model approach. Despite the fact that pedigree information was lacking for about one-third of all inbreds, the MSD between observed and expected P-values was slightly lower for this method than that for the ANOVA (Figure 3). The difference in MSD between these two methods was slightly higher when comparing them on the basis of a data set comprising only entries with available pedigree records (data not shown). These observations suggested that for our data set the G method is more appropriate for association mapping than the ANOVA approach.
Nevertheless, the MSD between observed and expected P-values for the G method was considerably higher than those of the QK and PK methods irrespective of whether the complete data set (Table 2) or a data set comprising only entries with available pedigree records (data not shown) was used. The opposite was true for the adjusted power of QTL detection (Figure 4). These observations suggest that in our study the G method was less appropriate for association mapping than the QK and PK methods. This might be explained by (i) incomplete or wrong pedigree records and (ii) differences between actual coancestry and coancestry computed from pedigree records due to selection and genetic drift (Bernardo 1993; Schut et al. 1997; Tams et al. 2004).
K approach:
For the mixed-model association-mapping approach K, we observed a lower value for the MSD between observed and expected P-values than that calculated for the G method irrespective of whether the complete data set (Table 2) or a data set comprising only entries with available pedigree records (data not shown) was used. This observation indicated that kinship coefficients estimated from molecular marker data are more appropriate than coancestry coefficients calculated from pedigree records. Nevertheless, for the K method the MSD was higher than that observed for the QK method as well as PK method, and our results on the correlated beta distribution suggested that this difference is considerably larger than what is expected at random. This result might be explained by the fact that the software package SPAGeDi (Hardy and Vekemans 2002), proposed in the study of Yu et al. (2006) for calculation of the kinship coefficients, assumes that random pairs of individuals of the germplasm set under consideration are unrelated and assigns them a kinship coefficient of 0.
This definition of unrelated individuals seems to be arbitrary. Furthermore, it results in a kinship matrix for which a large number of pairwise kinship estimates are negative. Yu et al. (2006) replaced these negative values by 0, arguing that such pairs of individuals are less related than random pairs of individuals. This approach ignores information on the structure of unrelated individuals, which was composed in the kinship matrix, and consequently necessitates the inclusion of the Q matrix from STRUCTURE in the mixed model. This suggests examining mixed-model association-mapping approaches that are based on K matrices calculated for different thresholds T.
Approaches based on K matrices calculated for different values of T:
For the QKT, PKT, and KT methods, the optimum value of T, which was calculated for the current data set using a REML approach, was 0.775 (Figure 2). The value of T estimated in this way was in good accordance with the optimum T identified using the MSD profiles. This observation suggested that for association-mapping approaches the optimum T value might be identified using a REML approach.
Because the REML-based deviance, used to estimate T, can be compared only among models that are based on the same set of fixed effects, we used the MSD between observed and expected P-values for comparison of the QKT, PKT, and KT method. The MSD profiles of QKT and PKT had their global minimum at T = 0.70, while that of the KT method was found for T = 0.35 (Figure 2). This observation might be explained by the fact that for an association-mapping model, which is not based on the assignment of individuals to subpopulations, lower values for T reduce the number of negative pairwise kinship estimates. Thereby, the use of information concerning the structure of unrelated individuals, which was composed in the kinship matrix KT, is improved.
The MSD observed for the K0.35 method was slightly lower than that of the QK as well as the PK method (Table 2). The opposite was true for the adjusted power of QTL detection (Figure 4). These findings suggested that the K0.35 method, which is based on the optimum KT matrix, performed slightly better than the QK and PK methods. Furthermore, the K0.35 method avoids the previously described shortcomings of association-mapping methods that are based on the assignment of individuals to subpopulations. By contrast, the MSD of methods QK0.70 and PK0.70 is considerably lower than that of the K0.35 method, whereas higher values for the adjusted power of QTL detection were observed for the former. Therefore, the QK0.70 and PK0.70 methods were the most appropriate methods for association mapping in the examined data set.
Kunrel approach:
Lynch (1988) and Melchinger et al. (1991) proposed to estimate T as the average proportion of marker loci with shared variants between two sets of genotypes: (i) the entries and (ii) genotypes that are unrelated by pedigree to the entries. The T value calculated in the current study on the basis of five wheat cultivars, which are unrelated by pedigree to the 303 entries, was 0.30. This value is in good accordance with the T value of 0.35 estimated on the basis of the MSD profile for the KT method, suggesting that this approach might be used in studies on genetic diversity where no phenotypic data are available. The MSD for K0.35, however, was lower than that of Kunrel. Furthermore, the optimum T for the QKT and PKT methods was considerably higher than that estimated on the basis of genotypes unrelated by pedigree. These observations indicated that in association-mapping studies and especially in studies requiring fixed subpopulation effects, estimation of T based on MSD or likelihood profiles are more promising than estimation based on genotypes unrelated by pedigree alone.
Comparison of one- and two-stage association-mapping approaches:
In all types of genetic mapping experiments, the one-step approach, in which the phenotypic and genotypic data analysis is performed in one step, is fully efficient (Cullis et al. 1998). Consequently, P-values calculated for the marker loci under consideration on the basis of such a statistical model are the reference values (Piepho and Pillen 2004). To our knowledge, however, only two-stage association-mapping approaches were applied in all earlier association-mapping studies with plants, i.e., entry means or adjusted entry means were calculated in the first step and then used for association mapping in the second step. Therefore, we compared one- and two-stage association-mapping approaches.
The lowest MSD values among the one-stage association-mapping approaches were observed for the QK0.70, PK0.70, K0.70, and K0.35 methods, which were also the most appropriate methods for two-step association mapping (Table 2). For these methods, the MSD of the one-stage approaches was lower than that for the corresponding two-stage association approaches, indicating that in our data set the former were more appropriate for association mapping than the latter, although the differences were rather small. Furthermore, for the association-mapping methods based on KT matrices, high correlation coefficients between P-values calculated for all marker loci on the basis of two-stage association-mapping approaches and the corresponding one-stage association approaches were found. These observations suggest that our data set could be analyzed by two-step association-mapping methods, using KT without increasing the empirical type I error rate too much in comparison to the corresponding one-step approaches.
Conclusions:
The results of our study indicate that the ANOVA approach is inappropriate for association mapping in the examined germplasm set. Furthermore, our observations suggest that the QK method is appropriate for association mapping not only in allogamous species such as humans and maize (Yu et al. 2006), but also in the autogamous species wheat, when the examined data set is similar in size compared to that of our study. Nevertheless, we recommend replacing the K matrix of the QK and PK approaches by a KT matrix, which is based on a REML estimate of the probability that two inbreds carry alleles at the same locus that are identical in state but not identical by descent and, thus, increases (i) the adherence to the nominal α-level as well as (ii) the adjusted power of QTL detection. Finally, we showed that our data set might be analyzed using the newly proposed two-step association-mapping method without increasing the empirical type I error rate too much in comparison to the corresponding one-step approaches.
Acknowledgments
We thank Lochow-Petkus for providing phenotypic and genotypic data. The authors appreciate the editorial work of J. Muminović, whose suggestions considerably improved the style of the manuscript. The authors thank the associate editor and two anonymous reviewers for their valuable suggestions. This research was conducted within the Breeding and Informatics project of the Genome Analysis of the Plant Biological System (GABI) initiative (http://www.gabi.de). E.S.B. was supported by the National Science Foundation (DBI-0321467).
References
- Aranzana, M. J., S. Kim, K. Zhao, E. Bakker, M. Horton et al., 2005. Genome-wide association mapping in Arabidopsis identifies previously known flowering time and pathogen resistance genes. PLoS Genet. 1 e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balding, D. J., 2006. A tutorial on statistical methods for population association studies. Nat. Rev. Genet. 7 781–791. [DOI] [PubMed] [Google Scholar]
- Bernardo, R., 1993. Estimation of coefficient of coancestry using molecular markers in maize. Theor. Appl. Genet. 85 1055–1062. [DOI] [PubMed] [Google Scholar]
- Bernardo, R., A. Murigneux and Z. Karaman, 1996. Marker-based estimates of identity by descent and alikeness in state among maize inbreds. Theor. Appl. Genet. 93 262–267. [DOI] [PubMed] [Google Scholar]
- Breseghello, F., and M. E. Sorrells, 2006. Association mapping of kernel size and milling quality in wheat (Triticum aestivum L.) cultivars. Genetics 172 1165–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camus-Kulandaivelu, L., J.-B. Veyrieras, B. Gouesnard, A. Charcosset and D. Manicacci, 2007. Evaluating the reliability of structure outputs in case of relatedness between individuals. Crop Sci. 47 887–892. [Google Scholar]
- Cullis, B., B. Gogel, A. Verbyla and R. Thompson, 1998. Spatial analysis of multi-environment early generation variety trials. Biometrics 54 1–18. [Google Scholar]
- Evanno, G., S. Regnaut and J. Goudet, 2005. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14 2611–2620. [DOI] [PubMed] [Google Scholar]
- Falconer, D. S., and T. F. C. Mackay, 1996. Introduction to Quantitative Genetics, Ed. 4. Longman Group, London.
- Flint-Garcia, S. A., J. M. Thornsberry and E. S. Buckler, 2003. Structure of linkage disequilibrium in plants. Annu. Rev. Plant Biol. 54 357–374. [DOI] [PubMed] [Google Scholar]
- Garris, A. J., T. H. Tai, J. Coburn, S. Kresovich and S. McCouch, 2005. Genetic structure and diversity in Oryza sativa L. Genetics 169 1631–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmour, A. R., B. J. Gogel, B. R. Cullis and R. Thompson, 2006. ASReml User Guide Release 2.0. VSN International, Hemel Hempstead, UK.
- Hardy, O. J., and X. Vekemans, 2002. SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population level. Mol. Ecol. Notes 2 618–620. [Google Scholar]
- Kraakman, A. T. W., R. E. Niks, P. M. M. M. Van den Berg, P. Stam and F. A. Van Eeuwijk, 2004. Linkage disequilibrium mapping of yield and yield stability in modern spring barley cultivars. Genetics 168 435–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M., 1988. Estimation of relatedness by DNA fingerprinting. Mol. Biol. Evol. 5 584–599. [DOI] [PubMed] [Google Scholar]
- Magnussen, S., 2004. An algorithm for generating positively correlated Beta-distributed random variables with known marginal distributions and a specified correlation. Comput. Stat. Data Anal. 46 397–406. [Google Scholar]
- Melchinger, A. E., M. M. Messmer, M. Lee, W. L. Woodman and K. R. Lamkey, 1991. Diversity and relationships among U.S. maize inbreds revealed by restriction fragment length polymorphisms. Crop Sci. 31 669–678. [Google Scholar]
- Moreau, L., H. Monod, A. Charcosset and A. Gallais, 1999. Marker-assisted selection with spatial analysis of unreplicated field trials. Theor. Appl. Genet. 98 234–242. [Google Scholar]
- Olsen, K. O., S. S. Halldorsdottir, J. R. Stinchcomb, C. Weinig, J. Schmitt et al., 2004. Linkage disequilibrium mapping of Arabidopsis CRY2 flowering time alleles. Genetics 167 1361–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki, K., Y. Ohnishi, A. Iida, A. Sekine, R. Yamada et al., 2002. Functional SNPs in the lymphotoxin-α gene that are associated with susceptibility to myocardial infarction. Nat. Genet. 32 650–654. [DOI] [PubMed] [Google Scholar]
- Parisseaux, B., and R. Bernardo, 2004. In silico mapping of quantitative trait loci in maize. Theor. Appl. Genet. 109 508–514. [DOI] [PubMed] [Google Scholar]
- Patterson, H. D., 1997. Analysis of series of variety trials, pp. 139–161 in Statistical Methods for Plant Variety Evaluation, edited by R. A. Kempton and P. N. Fox. Chapman & Hall, London.
- Piepho, H.-P., and J. Möhring, 2007. On weighting in two-stage analyses of series of experiments. Biul. Oceny Odmian 32 109–121. [Google Scholar]
- Piepho, H.-P., and K. Pillen, 2004. Mixed modelling for QTL × environment interaction analysis. Euphytica 137 147–153. [Google Scholar]
- Price, A. L., N. J. Patterson, R. M. Plenge, M. E. Weinblatt, N. A. Shadick et al., 2006. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38 904–909. [DOI] [PubMed] [Google Scholar]
- Pritchard, J. K., M. Stephens and P. Donelly, 2000. a Inference of population structure using multilocus genotype data. Genetics 155 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard, J. K., M. Stephens, N. A. Rosenberg and P. Donnelly, 2000. b Association mapping in structured populations. Am. J. Hum. Genet. 67 170–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarrie, S. A., A. Steed, C. Calestani, A. Semikhodskii, C. Lebreton et al., 2005. A high-density genetic map of hexaploid wheat (Triticum aestivum L.) from the cross Chinese Spring SQ1 and its use to compare QTLs for grain yield across a range of environments. Theor. Appl. Genet. 110 865–880. [DOI] [PubMed] [Google Scholar]
- SAS Institute, 2004. SAS Version 9.1. SAS Institute, Cary, NC.
- Schut, J. W., X. Qi and P. Stam, 1997. Association between relationship measures based on AFLP markers, pedigree data and morphological traits in barley. Theor. Appl. Genet. 95 1161–1168. [Google Scholar]
- Smith, A. B., B. R. Cullis and A. R. Gilmour, 2001. Analysing variety by environment data using multiplicative mixed models and adjustments for spatial field trend. Biometrics 57 1138–1147. [DOI] [PubMed] [Google Scholar]
- Stich, B., A. E. Melchinger, M. Frisch, H. P. Maurer, M. Heckenberger et al., 2005. Linkage disequilibrium in European elite maize germplasm investigated with SSRs. Theor. Appl. Genet. 111 723–730. [DOI] [PubMed] [Google Scholar]
- Tams, S. H., E. Bauer, G. Oettler and A. E. Melchinger, 2004. Genetic diversity in European winter triticale determined with SSR markers and coancestry coefficient. Theor. Appl. Genet. 108 1385–1391. [DOI] [PubMed] [Google Scholar]
- Thornsberry, J. M., M. M. Goodman, J. Doebley, S. Kresovich, D. Nielsen et al., 2001. Dwarf8 polymorphisms associate with variation in flowering time. Nat. Genet. 28 286–289. [DOI] [PubMed] [Google Scholar]
- Whitt, S. R., and E. S. Buckler, 2003. Using natural allelic diversity to evaluate gene function, pp. 123–139 in Plant Functional Genomics: Methods and Protocols, edited by E. Grotewald. Humana Press, Clifton, NJ. [DOI] [PubMed]
- Yu, J., G. Pressoir, W. H. Briggs, I. V. Bi, M. Yamasaki et al., 2006. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 38 203–208. [DOI] [PubMed] [Google Scholar]
- Zhao, K., M. J. Aranzana, S. Kim, C. Lister, C. Shindo et al., 2007. An Arabidopsis example of association mapping in structured samples. PLoS Genet. 3 71–82. [DOI] [PMC free article] [PubMed] [Google Scholar]