

Abstract
We describe a hitherto unrecognized bacterial community, inhabiting the leaf surfaces of the salt-excreting desert tree Tamarix. High temperatures, strong radiation, and very low humidity dictate a daytime existence in complete desiccation, but damp nights allow the microbial population to proliferate in a sugar-rich, alkaline, and hypersaline solution, before drying up again after sunrise. The exclusively bacterial population contains many undescribed species and genera, but nevertheless appears to be characterized by relatively limited species diversity. Sequences of 16S rRNA genes from either isolates or total community DNA place the identified members of the community in five bacterial groups (Actinobacteria, Bacteroidetes, Firmicutes, α-, and γ-Proteobacteria); in each of these, they concentrate in a very narrow branch that in most cases harbors organisms isolated from unrelated halophilic environments.
THE aerial surfaces of plants, collectively termed the phyllosphere, are considered one of earth's most prolific habitats (Morris and Kinkel 2002; Lindow and Brandl 2003). It harbors complex and relatively little studied food webs, consisting mostly of bacteria, fungi, and insects (Hirano and Upper 2000). In the study of phyllosphere microbiology, past attention was mostly focused on bacterial strains of agricultural significance such as plant pathogens or ice nucleators (Lindow and Brandl 2003). While bacterial concentrations on agricultural plants can reach high numbers, 106–107 cells/cm2, (Beattie and Lindow 1995; Andrews and Harris 2000; Hirano and Upper 2000), the leaf environment, with its fluctuating temperature, humidity, and solar irradiation levels, was nevertheless described as hostile to its microbial inhabitants (Hirano and Upper 2000). This hostility is taken to an extreme in the case of desert vegetation, and even more so for salt-excreting plant species, the surfaces of which are often encrusted with sodium chloride (Simon et al. 1994); such an environment, the leaf surfaces of the salt-excreting desert tree Tamarix aphylla, is described in this article.
A question increasingly encountered in the microbial biodiversity literature addresses the number of different microorganisms expected to be found in a specific environment; in practical terms, this often translates to the extent to which a set of samples reflects the true biodiversity of a microbial community (Hughes et al. 2001). In marine microbial communities, estimates range from hundreds to thousands of “operational taxonomic units” (OTUs) per milliliter in open water (Schloss and Handelsman 2005) and sediments (Ravenschlag et al. 1999; Kemp and Aller 2004), respectively. Sogin et al. (2006), by moving beyond the 16S rRNA genes into a hypervariable region of rRNA, revealed a much greater diversity in ocean waters, most of it occupied by representatives of the “rare biosphere,” composed of low-abundance OTUs. Microbial biodiversity estimates in plant environments have been carried out to a much more limited extent. Recent reports describe a high diversity in rhizosphere environments (Stafford et al. 2005; Edwards et al. 2006), with no attempt to estimate the total number of OTUs. In fact, very little use has been made to date of culture-independent methods for the study of phyllosphere bacterial populations and their diversity. Yang et al. (2001) identified a limited number of strains in agricultural plants and did not attempt to calculate diversity but justifiably concluded that phyllosphere communities are more complex than previously thought. Lambais et al. (2006) have surveyed bacterial diversity in the leaf canopy of a tropical Atlantic forest of Brazil. The authors estimated the number of distinct OTUs in the different trees to be ∼100–400.
In this article we show that the hostile leaf environment of the salt-excreting desert tree T. aphylla is heavily populated with a rich microbial population, much denser than that of neighboring nonsalt-excreting trees. We describe the basic structure of this population, analyze the phylogenetic affiliations of its members, and discuss its diversity.
MATERIALS AND METHODS
Site description and sampling:
Leaf and dew samples were collected from three T. aphylla trees near Sede Boker, Israel (30°52′30.20″ N, 34°47′15.61″ E), on 12 different occasions throughout 2004–2006.
To collect early morning dew condensed on T. aphylla leaves, sterile glass microscope slides were gently put into contact with the leaf tips, transferring the hanging drops onto the glass surface and then placed in sterile plastic 50-ml test tubes (Greiner, Germany). The dew was then filtered through a 0.22 μm filter (Millipore, Bedford, MA), transferred on ice to the laboratory, and kept at 4° until chemical analysis was performed.
Tamarix leaves were collected wearing gloves and kept in sterile paper bags for further analysis of the microbial populations inhabiting their surfaces.
Various protocols were investigated to maximize bacterial removal from the plant surfaces. Tested parameters included the use of differently buffered media, different durations of sonication, heating, and manual scrubbing with a fine brush. The optimized procedure included submerging the air-dried leaves in 0.1 m potassium phosphate buffer, pH 8.0 (1 g leaf/10 ml buffer) and a 20-sec sonication (Vibra-Cell, 40% intensity; Sonics, Newtown, CT). The preparations were then vortexed 6 times for 10 sec, at 5-min intervals, and the leaf wash (LW) was separated from the leaf debris by decanting. The LW was filtered on a 0.22-μm membrane filter (Millipore), and the entire filter was used for total DNA extraction. Unfiltered LW served as the source of bacterial isolates.
Chemical analysis of dew samples:
Electrical conductivity (EC) was measured using a conductivity meter (S30 Seveneasy Conductivity; Mettler, Toledo, OH), and pH was determined using a pH meter equipped with a combination glass electrode (ThermoOrion, model 420, Orion).
Selected elements were determined in acid-digested samples (1 ml sample and 2 ml 65% nitric acid) by an inductively coupled plasma atomic emission spectrometer (ICP/AES; Spectroflame Modula E, Spectro GMBH, Kleve, Germany).
Anions were determined by ion chromatography, using a DX-300 ion chromatograph (Dionex Corporation, Sunnyvale, CA), with an AS4A analytical column and guard column, and an anion micromembrane suppressor.
Total organic carbon (TOC) and inorganic carbon (IC) were determined using a FormacHT Combustion TOC analyzer (Skalar Analytical B. V. Breda, The Netherlands). For TOC determination, IC was first removed by lowering the pH with phosphoric acid to <2.0, mixing and flushing with CO2-free oxygen. The instrument was calibrated with phthalate and bicarbonate standards.
Collection of isolates and DNA extraction:
Unfiltered LW samples were spread on Luria–Bertani (LB) media plates containing different NaCl concentrations (5–20%) at several pH values (7.0–11.0). The plates contained cycloheximide (100 μg/ml) to prevent fungal growth; in the absence of cycloheximide, the plates were often dominated by black yeast species. Microbial isolates were restreaked on the same medium, regrown in liquid medium, and preserved at −80° in 33% glycerol. DNA was extracted from isolates in liquid medium using an UltraClean microbial DNA extraction kit (MoBio Laboratories).
DNA was extracted from the 0.22-μm membrane filters through which the LW was filtered using the Power Soil DNA kit (MoBio Laboratories). Quality of the total DNA recovered was checked by electrophoresis in 1% agarose. DNA concentration was determined using an ND-1000 spectrophotometer (Nano-Drop). DNA was stored at −20°.
PCR amplification of 16S rRNA genes:
Genes encoding for 16S rRNA were amplified from DNA extracted from both isolates and leaf wash samples using several sets of primers (Table 1).
TABLE 1.
PCR primers used in this study for 16S rRNA gene amplification
| Application | Name | Sequence (5′–3′) | Reference |
|---|---|---|---|
| Isolates identification | 63F | CAGGCCTAACACATGCAAGTCGTT | Marchesi et al. (1998) |
| 1492R | TACGGYTACCTTGTTACGACTT | Lane (1991) | |
| Total community DNA, library construction | 8F | AGAGTTTGATCMTGGCTCAG | |
| 1387R | GGGCGGWGTGTACAAGGC | ||
| Total community DNA, DGGE analysis | 968F | GC-Clamp-AACGCGAAGAACCTTAC | Heuer et al. (1997) |
| 1378R | CGGTGTGTACAAGGCCCGGGAACG | ||
| 341F (GM5) | GC-Clamp-CCTACGGGAGGCAGCAG | Muyzer et al. (1993) | |
| 907R | CCGTCATTCMTTTRAGTTT | ||
| GC-Clamp | CGCCCGCCGCGCCCCCGCGCCGGTCCCGCCGGCCCCGCCC | ||
| Archaeal primers | ARCH344F | TCGCGCCTGCTGCICCCCGT | Giovannoni et al. (1988) |
| 1406R | TGYACACACCTCCCGT | Olsen et al. (1986) | |
| ARCH519F | GWTTTACCGCGGCKGCTG | Lane (1991) | |
| ARCH915R | GTGCTCTCCCCCGCCAATTC | Stahl and Amann (1991) |
Each 50-μl PCR mixture contained ∼15 ng of template DNA, and PCR ready-mix (ABGene, UK) was used as recommended by the manufacturer. PCR was carried out using a T-gradient thermocycler (Biometra, Germany). PCR conditions were as follows: an initial denaturation of 3 min at 94°, 25 cycles of 94°/1 min, 50°/1 min, 72°/1 min, and a final extension at 72° for 10 min. For every analysis two separate reaction vials were combined to minimize bias. PCR negative controls were prepared by replacing the template DNA with sterile water.
Clone libraries of 16S rRNA genes and diversity calculations:
Five independent 16S rRNA gene clone libraries were prepared in Escherichia coli, on the basis of LW total DNA extracts obtained from five independently collected leaf samples.
For each sample, 16S rRNA genes were amplified using primer set 8F/1387R (Table 1); the amplified DNA was ligated into the pGEM-t Easy Vector System (Promega, Madison, WI), and the ligated plasmids were used to transform E. coli strain JM109 (Promega) by electroporation. Transformants were selected on LB medium supplemented with ampicillin (100 mg/liter), 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal; 80 mg/liter), and isopropyl-β-d-thiogalactopyranoside (IPTG; 50 μm). Plasmids were extracted from ∼60 randomly selected colonies from each library using QIAprep Miniprep (QIAGEN, Valencia, CA) as recommended by the manufacturer. Reamplification from the plasmids as templates was performed using the same primers (Table 1). The ∼300 reamplified 16S rRNA gene clones were then separated into groups according to their amplified ribosomal DNA restriction analysis (ARDRA) patterns. For this analysis, the clones were restricted using MboI (Taqara), MvaI (Fermentas), and HaeIII (Taqara) (2.5 units each, with ∼30 ng PCR template and 0.5 ml of Fermentas RX1 buffer). The products were separated in 1% agarose gels (Invitrogene, Paisley, UK) run on i-Mupid microgel electrophoresis system (CosmoBio), stained by ethidium bromide and imaged using Bio Imaging System (202D, Dinco and Rhenium).
Amplicons with identical ARDRA patterns were grouped as single OTUs, and representatives of each ARDRA pattern were sequenced. These included all of the unique patterns and duplicates of the others; all together, 89 amplicons were sequenced. Diversity indexes and rarefaction curves were constructed using EstimateS 7.5 (http://viceroy.eeb.uconn.edu/estimateS) on the basis of the ARDRA information. Richness estimators used were Chao 1, a nonparametric species-richness estimator based on the calculation of the number of singletons (Smith and van Belle 1984), and Jacknife 1 (Jack 1), which estimates total species richness from first order data (Chao 1987).
Denaturing gradient gel electrophoresis analysis:
To separate the 16S rRNA genes amplified from the LW total DNA extracts into their individual components, denaturing gradient gel electrophoresis (DGGE) was performed as previously described (Muyzer et al. 1993) for >20 samples. Briefly, 45 μl of each sample were applied to individual wells of an 8% (w/v) acrylamide gel containing a 35–70% urea gradient. Gels were run at 60° for 16 hr at 80 V in Tris-acetate-EDTA (TAE) buffer using the INGENYphorU system (INGENY). Gels were stained for 30 min in TAE containing a 1/10,000 dilution of Vistra Green (Amersham Biosciences, Piscataway, NJ), and destained for 30 min in TAE. Visualization was performed with an AlphaImager system (Alpha Innotech, San Leandro, CA) using the AlphaEaseFC standard software package (Labtrade, Hialeah, FL). Selected DGGE bands were excised from the gels and eluted into 60 μl of water at 37° overnight. DNA was reamplified with the appropriate corresponding primers without the GC clamps as follows: an initial denaturation of 5 min at 95°, followed by 25–28 cycles of 94°/1 min, 50°/45 sec, and 72°/45 sec. The reamplified products were sequenced.
Phylogenetic and cluster analyses:
Sequence analysis was carried out using >200 16S rRNA gene sequences obtained from isolates (100), excised DGGE bands (28), and from library clones (89).
Sequencing was carried out at the Center for Genomic Analysis, The Hebrew University of Jerusalem. The 16S rRNA gene sequences were compared to GenBank databases using BLAST (http://www.ncbi.nlm.nih.gov). Sequences having ≥97% similarity and matching the same GenBank sequence were assigned to the same phylotype. Only high quality sequences >900 bp were used for phylogenetic tree construction. They were introduced to the ARB database (Ludwig et al. 2004) using the parsimony algorithm of ARB, optimized by several protection filters. Construction of phylogenetic trees was done using ARB's maximum parsimony algorithm; several protection filters were employed for tree optimization.
RESULTS
Chemical environment of the Tamarix phyllosphere:
To survive, a bacterium inhabiting a Tamarix leaf (Figure 1) has to contend with a demanding combination of environmental stress factors. For most of the day the leaf surfaces are dry, necessitating bacterial preservation in a desiccated state. The only opportunity to metabolize and divide is when the relative humidity is high and dew accumulates on the hygroscopic leaf surfaces. In the Israeli Negev Desert, this occurs for over half of the nights of the year. The dew that condenses on the leaves dissolves many of the minerals on the leaf surface, thus creating a highly concentrated salt solution. The concentrations of some of the major elements and ions in early morning dew are presented in Table 2, which presents averages of 12 individual samples collected from three T. aphylla trees. Statistical differences between individual trees and different samples collected from the same tree were insignificant (not shown). The bulk of the dissolved minerals in this “growth medium” is made up of sodium and chloride, with significant contributions of magnesium, potassium, calcium, and bromide. Sulfate is present in surprisingly high concentrations, and the total dissolved salt concentration is ∼22%—>5-fold higher than seawater. The average total conductivity is 136 mS/cm, comparable to that of the Great Salt Lake (Munson et al. 2004). The pH is also high, probably due to the high concentrations of inorganic carbon (Waisel 1961, 1991). While direct pH determination yielded an average value of 8.5, the pH of a 10-fold diluted solution was 1 unit higher, pointing to both the buffer capacity of the dew as well as to the limitations of direct measurements of proton activity in a highly saline solution. Most surprising is the extremely high concentration of organic solutes, with a total organic carbon concentration of >3 g C/ml. A significant fraction of the organic carbon is made up of sugars (not shown).
Figure 1.—

A Tamarix aphylla leaf. (A) Early morning, dew-covered branches; (B) cross section emphasizing NaCl encrustation; (C) scanning electron micrograph of two adjoining stem segments; (D) environmental scanning electron micrograph of a bacterial microcolony on a leaf surface following several hours' incubation in a humid environment.
TABLE 2.
Main chemical constituents of early morning dew
| Constituent | Concentration (mg/liter) | Standard deviation (mg/liter) |
|---|---|---|
| Na+ | 38,800 | 15,704 |
| K+ | 5,964 | 2,549 |
| Ca2+ | 869 | 227 |
| Mg2+ | 8,085 | 2,510 |
| Cl− | 68,761 | 15,605 |
| Br− | 659 | 336 |
| F− | 108 | 48 |
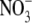 |
920 | 623 |
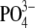 |
216 | 159 |
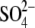 |
23,400 | 7,305 |
| TOC (mg C/liter) | 3283 | 1398 |
| IC (mg C/liter) | 315.7 | 109.3 |
| pH | 8.5 | 0.2 |
| pH of 10% dew | 9.5 | 0.1 |
| Conductivity (mS/cm) | 136.2 | 33.1 |
Phylogenetic analysis of major microbial representatives:
Direct isolation of leaf bacteria yielded a large number of colonies (∼106–107 colony-forming units/gram of dry leaves) that grew on solid media of varying salinities. As typical of other leaf systems (Lindow and Brandl 2003), most of the isolates were strongly pigmented.
The composition of the bacterial populations was analyzed by characterizing the phylogenetic affiliation of 16S rRNA gene sequences obtained from amplified PCR products from three sources: (a) DNA extracts of these isolates, (b) total DNA extracts obtained from leaf wash samples and separated by DGGE analysis (Muyzer et al. 1993), and (c) total DNA extracts obtained from leaf wash samples and segregated using E. coli clone libraries.
The phylogenetic affiliation of >200 16S rRNA gene sequences obtained by all three methods was determined using BLAST analysis on the basis of the GenBank database. The collection of sequences was grouped into 52 different genera (or less-defined deposited sequences) mostly belonging to five phylogenetic groups; two of these, Actinobacteria and Firmicutes, are gram positive and three, Bacteroidetes, α-, and γ-Proteobacteria, are gram negative (Table 3). Only two sequences seemed to be related to other phyla: one to β-Proteobacteria and the other to the Deinococcus–Thermus group. The suitability of the latter to life under extreme desiccation is obvious, although the 93% similarity indicates this leaf inhabitant is remote from its closest sequenced Deinococcus relative. One group of absentees from Table 3 deserves a special mention: to date, we have not found any archaeal isolate on the Tamarix phyllosphere, nor have we succeeded in amplifying archaeal 16S rRNA genes from DNA extracted from the leaf surfaces using any of the archaeal primers listed in Table 1. This may be surprising in view of the harsh conditions prevailing on the leaves, but in line with the observation that archaea are not a normal constituent of the phyllosphere.
TABLE 3.
Closest genera classification for bacterial Tamarix sequences, obtained from isolates, DGGE bands, and clone libraries
| Taxonomic group | Nearest relative | Similarity range (%) | Number of hits per taxon |
|---|---|---|---|
| α-Proteobacteria | “α-Proteobacterium” | 93–97 | 4 |
| “Marine bacterium” | 96–97 | 3 | |
| Oceanicola | 92–94 | 2 | |
| Rhodobacteraceae | 92–96 | 3 | |
| Roseobacterium | 93 | 1 | |
| Salipiger | 92–93 | 2 | |
| Sphingomonas | 95 | 1 | |
| γ-Proteobacteria | Chromohalobacter | 93 | 1 |
| Salinomonas | 96 | 3 | |
| Salmonella | 91 | 1 | |
| Xenorhabdus | 91 | 1 | |
| Escherichia | 97–99 | 6 | |
| “γ-Proteobacterium” | 98 | 1 | |
| Pseudomonas | 99 | 1 | |
| Shigella | 94–98 | 2 | |
| Acinetobacter | 97–98 | 2 | |
| Halomonadaceae | 94–97 | 6 | |
| Halomonas | 90–98 | 14 | |
| Bacteroidetes | “Arctic sea ice bacterium” | 95 | 1 |
| Cellulophaga | 95 | 1 | |
| Bacteroidetes | 93 | 1 | |
| Cytophaga | 94–96 | 5 | |
| Flavobacterium | 97–98 | 1 | |
| Leeuwenhoekella | 93 | 1 | |
| Salegentibacter | 95–96 | 4 | |
| Sufflavibacter | 91 | 1 | |
| “Uncultured bacterium clone” | 93–99 | 2 | |
| Actinobacteria | “Antarctic bacterium” | 98–99 | 2 |
| Arthrobacter | 97–99 | 3 | |
| Brachybacterium | 96–99 | 7 | |
| Brevibacterium | 92–99 | 7 | |
| Citricoccus | 98 | 2 | |
| “Glacial ice bacterium” | 95 | 1 | |
| Nesterenkonia | 92–99 | 4 | |
| Kocuria | 93–99 | 9 | |
| Micrococcaceae | 98 | 1 | |
| Micrococcus | 95–98 | 5 | |
| Firmicutes | Bacillus | 92–99 | 20 |
| Marinococcus | 93–99 | 16 | |
| Staphylococcus | 92–99 | 9 | |
| Staphylococcaceae | 99 | 1 | |
| “Antarctic seawater bacterium” | 99 | 1 | |
| “Blackwater bioreactor bacterium” | 96 | 1 | |
| Oceanobacillus | 98 | 2 | |
| Paenibacillus | 95–99 | 3 | |
| Paracoccus | 99 | 1 | |
| Planococcus | 94–98 | 17 | |
| Planomicrobium | 97–99 | 12 | |
| Sinococcus | 97 | 1 | |
| Salinococcus | 98 | 4 | |
| β-Proteobacteria | Variovorax | 98 | 1 |
| Deinococcus-Thermus | Deinococcus | 93 | 1 |
Quotation marks denote deposited information with no affiliation to an identified genus.
Analysis of the distribution of genus-level data in Table 3 according to the methodological approach by which they have been identified revealed a significant overlap (Figure 2). Although each of the methodologies appears to be characterized by a certain bias (Curtis et al. 2002), the overlapping of the circles in Figure 2 indicates that together they describe a relatively compact grouping of a rather limited diversity. The single genus identified by all three approaches was Halomonas, which was also one of the most abundant sequences in our collection. This well-studied genus of mostly moderately halophilic species (Euzéby 2006) was previously isolated from a variety of saline environments (Mata et al. 2002; Garcia et al. 2005; Llamas et al. 2006). The ability of members of this genus to metabolize aromatic molecules (Garcia et al. 2005) may be of significance for life on a tree surface, the exudates of which may contain such compounds.
Figure 2.—

Numbers of identified (closest relatives) genera of Tamarix phyllosphere bacteria; red, isolates growing on solid media; yellow, 16S rRNA gene sequences from DGGE gels; blue, 16S rRNA gene sequences from clone libraries.
To investigate the phylogenetic affiliation of the Tamarix bacterial inhabitants, detailed phylogenetic trees were constructed for each of the five phyla that contain representatives of the phyllosphere populations (supplemental Figures 1–5). A study of these trees reveals that in each case, the Tamarix sequences group into very specific branches, displaying what appears to be a very limited local diversity. To illustrate this point, Figure 3 presents an overview of the bacterial domain and its main subdivisions, highlighting the limited areas where the Tamarix sequences congregate. For example, all Actinobacteria sequences found on Tamarix congregate in only three families (Brevibacteriaceae, Dermabacteraceae, and Micrococcaceae), grouped closely together among the 43 families in the order Actinomycetales; this order, in turn, is the only one of the six in this phylum to which we could associate Tamarix sequences. Interestingly, the nearest neighbors in practically all cases are bacterial species isolated from other saline habitats, indicating several independent halophilic origins.
Figure 3.—

A general phylogenetic tree (constructed using ARB) of the Bacteria domain, highlighting the branches where Tamarix sequences congregate. Phyla are marked in blue; the five phyla harboring Tamarix representatives were expanded to the order level, and the orders inhabited by these representatives were marked in red. For detailed phylogenetic trees of the five phyla see supplemental Figures 1–5.
Estimation of microbial diversity:
Diversity estimates of phyllosphere bacterial populations were based on five independent E. coli 16S rRNA gene clone libraries, each containing ∼50–60 samples of amplified 16S rRNA genes. The genes were restricted by three endonucleases and visually grouped according to their ARDRA patterns. For the purpose of the present analysis, these patterns were defined as our OTU. To evaluate diversity, two richness estimators were used: nonparametric Chao 1 and parametric Jacknife 1. Figure 4 shows the results of the analyses using these two estimators, with ARDRA patterns of 16 rRNA genes as the OTU. Both indexes indicate that the population is composed of ∼70–80 OTUs, of which our samples appear to cover >60%.
Figure 4.—

Diversity estimates of phyllosphere bacterial populations based on OTUs derived from 16S rRNA gene clone libraries and on amplified ribosomal DNA restriction analysis patterns.
DISCUSSION
In this article we describe for the first time the unique microbial population inhabiting the aerial surfaces of the salt-excreting desert tree Tamarix aphylla. We have shown that when humidity is high and the leaves are wet, these bacteria are exposed to a highly saline, alkaline solution. Each saline and alkaline dew drop on the leaves may thus be viewed as a miniature soda lake, but with a very significant twist: in contrast to real soda lakes, the existence of Tamarix dew is of a very transient nature, and phylloplane bacteria are exposed to repetitive wetting/drying cycles; as temperatures climb after sunrise, the dew becomes increasingly more concentrated up to total desiccation a few hours into the day.
In partial compensation for the high and diurnal fluctuating salinity, Tamarix phyllosphere epiphytes are rewarded by very high organic carbon concentrations (>3 g C/liter), as well as by nonlimiting concentrations of phosphorus (mostly as phosphate) and nitrogen (mostly as nitrate). Macronutrient availability, therefore, is likely not to limit activity during the wetter periods of the diurnal cycle. The surprisingly high concentrations of organic compounds are mostly contributed by readily utilizable sugars, most probably excreted by insects residing on the trees (Douglas 2006). Tamarix sugars are among the modern candidates for the biblical manna (Bodenheimer 1947); they may participate in osmotic regulation as well as play a significant role in integrity and viability conservation of the bacteria during the desiccation period (Yancey 2004).
The number of CFUs we have found on the leaves, 106–107/g, is somewhat lower than that reported for agricultural plants (Lindow and Brandl 2003); since CFU numbers are often a gross underestimation of the size of a bacterial population (Rappe and Giovannoni 2003), the actual concentration of total viable bacteria on Tamarix leaves is expected to be much higher. To get a more reliable idea as to the nature of the microbial inhabitants of the leaf surfaces, we have analyzed the composition of the bacterial populations by characterizing the phylogenetic affiliation of these isolates, as well as of that of PCR-amplified 16S rRNA gene sequences derived from DGGE (Muyzer 1999) and from 16S rRNA gene clone libraries.
Our diversity estimates of Tamarix phyllosphere bacteria, performed using both parametric and nonparametric approaches (Bohannan and Hughes 2003), yielded very similar estimates, which are surprisingly close to those obtained from rain forest data (Lambais et al. 2006). Probably, a significant fraction of this population is represented in our current list of candidate leaf inhabitants, which contains 52 names of the closest distinct genera (or related deposited sequences). A phylogenetic analysis reveals that although they belong to five distinct bacterial phylogenetic groups, in each of them they are grouped rather tightly on very close branches. In most cases they are grouped near other sequences obtained from marine or other saline environments. Unsurprisingly, colonization of the Tamarix phyllosphere is more successful for bacterial species genetically predisposed to survive in similar environments (Tehei and Zaccai 2005).
The affiliation of most of the available Tamarix sequences to five specific groups may originate from both the saline nature of their environment as well as from their arboreal origin. Representatives of Bacteriodetes and γ-Proteobacteria were reported to dominate several saline habitats (Demergasso et al. 2004). Furthermore, Firmicutes, α-Proteobacteria, and Actinobacteria were reported to exist in other extreme habitats (Humayoun et al. 2003; Smith et al. 2006). The same five phyla have also been reported in other phyllosphere environments, such as in the tree canopies of the Atlantic forest (Lambais et al. 2006). While this phylogenetic similarity may stem from the known biases of the molecular techniques (Schloss and Handelsman 2004), it should be noted that our data were composed of sequences obtained from several independent sampling methods.
The large degree of overlap of the sequences obtained by the different sampling methods demonstrated in Figure 2 is initially surprising, especially in view of the relatively small number (<100) of sequences involved. A possible explanation for this may lie in the biases (Schloss and Handelsman 2004) and technical limitations of the experimental and computational methodologies used; it may also be partially due to the fact that the Tamarix phyllosphere may not have been comprehensively sampled, and that in spite our optimization efforts a fraction of the population that strongly adhered to the leaf surface was not removed into the leaf wash. However, the most likely explanation for the overlap observed in Figure 2 emerges from the statistical analysis summarized in Figure 4. The total number of OTUs that may be expected to be found in this environment is in fact small, probably not exceeding a total of 80. The 68 genera represented in Figure 2 thus represent a significant fraction of this population, and the observed overlap is a direct consequence of this limited diversity. The most probable cause for this focused diversity is the multiple stresses imposed upon the bacteria inhabiting this habitat. While it is very likely that not all the rarely occurring genera have been sampled, and that additional sampling will most probably expand the list in Table 3, it is nevertheless expected that this will not dramatically modify the limited diversity demonstrated by the data in Figures 2 and 4.
Localized habitats that we normally regard as extreme, such as saline lakes or hot springs, are often characteristic of specific geographical locations, separated from each other by large distances. Salt-excreting Tamarix trees, in contrast, the phylloplane of each harboring an extreme and fluctuating environment of the type described herein, are common worldwide, as either endemic or invasive species (the latter is particularly true for North America). This global distribution may provide an excellent opportunity to study the combined effects of geographical, climatological, and other environmental parameters on the distribution, composition, and diversity of the microbial communities of such habitats. The environmental effectors to be studied should include the effect of the chemistry of the water utilized by the trees on dew composition and hence on microbial life on the leaves, the nature and origin of the organic dew constituents and their nutritional value for the phyllosphere microorganisms, and the interactions with the insect populations inhabiting the trees, some of them known to secrete honeydew onto the leaves they inhabit.
Acknowledgments
We thank R. D. Simon for introducing us to the topic many years ago; without him, this project would never have materialized. The assistance of N. Ben-Eliyahu with microscopy is gratefully acknowledged. Financial support was provided by the Bridging the Rift Foundation in the framework of the “Microbial biodiversity of the Dead Sea region” project.
References
- Andrews, J. H., and R. F. Harris, 2000. The ecology and biogeography of microorganisms on plant surfaces. Annu. Rev. Phytopathol. 38 145–180. [DOI] [PubMed] [Google Scholar]
- Beattie, G. A., and S. E. Lindow, 1995. The secret life of foliar bacterial pathogens on leaves. Annu. Rev. Phytopathol. 33 145–172. [DOI] [PubMed] [Google Scholar]
- Bodenheimer, F. S., 1947. The manna of Sinai. Biblic. Archeol. 10 1–6. [Google Scholar]
- Bohannan, B. J., and J. Hughes, 2003. New approaches to analyzing microbial biodiversity data. Curr. Opin. Microbiol. 6 282–287. [DOI] [PubMed] [Google Scholar]
- Chao, A., 1987. Estimating the population size for capture-recapture data with unequal catchability. Biometrics 43 783–791. [PubMed] [Google Scholar]
- Curtis, T. P., W. T. Sloan and J. W. Scannell, 2002. Estimating prokaryotic diversity and its limits. Proc. Natl. Acad. Sci. USA 99 10494–10499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demergasso, C., E. O. Casamayor, G. Chong, P. Galleguillos, L. Escudero et al., 2004. Distribution of prokaryotic genetic diversity in athalassohaline lakes of the Atacama Desert, Northern Chile. FEMS Microbiol. Ecol. 48 57–69. [DOI] [PubMed] [Google Scholar]
- Douglas, A. E., 2006. Phloem-sap feeding by animals: problems and solutions. J. Exp. Bot. 57 747–754. [DOI] [PubMed] [Google Scholar]
- Edwards, I. P., H. Burgmann, C. Miniaci and J. Zeyer, 2006. Variation in microbial community composition and culturability in the rhizosphere of Leucanthemopsis alpina (L.) Heywood and adjacent bare soil along an alpine chronosequence. Microb. Ecol. 52 679–692. [DOI] [PubMed] [Google Scholar]
- Euzéby, J. P., 2006. List of prokaryotic names with standing in nomenclature—genus Halomonas. http://www.bacterio.cict.fr/h/halomonas.html.
- Garcia, M. T., A. Ventosa and E. Mellado, 2005. Catabolic versatility of aromatic compound-degrading halophilic bacteria. FEMS Microbiol. Ecol. 54 97–109. [DOI] [PubMed] [Google Scholar]
- Giovannoni, S. J., E. F. DeLong, G. J. Olsen and N. R. Pace, 1988. Phylogenetic group-specific oligodeoxynucleotide probes for identification of single microbial cells. J. Bacteriol. 170 720–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuer, H., M. Krsek, P. Baker, K. Smalla and E. M. Wellington, 1997. Analysis of actinomycete communities by specific amplification of genes encoding 16S rRNA and gel-electrophoretic separation in denaturing gradients. Appl. Environ. Microbiol. 63 3233–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano, S. S., and C. D. Upper, 2000. Bacteria in the leaf ecosystem with emphasis on Pseudomonas syringae-a pathogen, ice nucleus, and epiphyte. Microbiol. Mol. Biol. Rev. 64 624–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, J. B., J. J. Hellmann, T. H. Ricketts and B. J. Bohannan, 2001. Counting the uncountable: statistical approaches to estimating microbial diversity. Appl. Environ. Microbiol. 67 4399–4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humayoun, S. B., N. Bano and J. T. Hollibaugh, 2003. Depth distribution of microbial diversity in Mono Lake, a meromictic soda lake in California. Appl. Environ. Microbiol. 69 1030–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp, P. F., and J. Y. Aller, 2004. Bacterial diversity in aquatic and other environments: what 16S rDNA libraries can tell us. FEMS Microbiol. Ecol. 47 161–177. [DOI] [PubMed] [Google Scholar]
- Lambais, M. R., D. E. Crowley, J. C. Cury, R. C. Bull and R. R. Rodrigues, 2006. Bacterial diversity in tree canopies of the Atlantic forest. Science 312 1917. [DOI] [PubMed] [Google Scholar]
- Lane, D. J., 1991. 16S/23S rRNA Sequencing. John Wiley & Sons, Chichester, UK.
- Lindow, S. E., and M. T. Brandl, 2003. Microbiology of the phyllosphere. Appl. Environ. Microbiol. 69 1875–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llamas, I., A. del Moral, F. Martinez-Checa, Y. Arco, S. Arias et al., 2006. Halomonas maura is a physiologically versatile bacterium of both ecological and biotechnological interest. Antonie Van Leeuwenhoek 89 395–403. [DOI] [PubMed] [Google Scholar]
- Ludwig, W., O. Strunk, R. Westram, L. Richter, H. Meier et al., 2004. ARB: a software environment for sequence data. Nucleic Acids Res. 32 1363–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchesi, J. R., T. Sato, A. J. Weightman, T. A. Martin, J. C. Fry et al., 1998. Design and evaluation of useful bacterium-specific PCR primers that amplify genes coding for bacterial 16S rRNA. Appl. Environ. Microbiol. 64 795–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata, J. A., J. Martinez-Canovas, E. Quesada and V. Bejar, 2002. A detailed phenotypic characterisation of the type strains of Halomonas species. Syst. Appl. Microbiol. 25 360–375. [DOI] [PubMed] [Google Scholar]
- Morris, C. E., and L. Kinkel, 2002. Fifty years of phyllosphere microbiology: significant contributions to research in related fields, pp. 365–375 in Phyllosphere Microbiology, edited by S. E. Lindow, E. I. Hect-Poinar and V. J. Elliot. APS Press, St. Paul, MN.
- Munson, B., R Axler, C Hagley, G Host, G Merrick et al., 2004. Monitoring Minnesota lakes on the internet and training water science technicians for the future:a national on-line curriculum using advanced technologies and real-time data. University of Minnesota, Duluth, MN. http://waterontheweb.org/under/waterquality/conductivity.html.
- Muyzer, G., 1999. DGGE/TGGE a method for identifying genes from natural ecosystems. Curr. Opin. Microbiol. 2 317–322. [DOI] [PubMed] [Google Scholar]
- Muyzer, G., E. C. de Waal and A. G. Uitterlinden, 1993. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl. Environ. Microbiol. 59 695–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen, G. J., D. J. Lane, S. J. Giovannoni, N. R. Pace and D. A. Stahl, 1986. Microbial ecology and evolution: a ribosomal RNA approach. Annu. Rev. Microbiol. 40 337–365. [DOI] [PubMed] [Google Scholar]
- Rappe, M. S., and S. J. Giovannoni, 2003. The uncultured microbial majority. Annu. Rev. Microbiol. 57 369–394. [DOI] [PubMed] [Google Scholar]
- Ravenschlag, K., K. Sahm, J. Pernthaler and R. Amann, 1999. High bacterial diversity in permanently cold marine sediments. Appl. Environ. Microbiol. 65 3982–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss, P. D., and J. Handelsman, 2004. Status of the microbial census. Microbiol. Mol. Biol. Rev. 68 686–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss, P. D., and J. Handelsman, 2005. Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl. Environ. Microbiol. 71 1501–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon, R. D., A. Abeliovich and S. Belkin, 1994. A novel terrestrial halophilic environment: the phylloplane of Atriplex halimus, a salt-excreting plant. FEMS Microbiol. Ecol. 14 99–109. [Google Scholar]
- Smith, E. P., and G. van Belle, 1984. Nonparametric estimation of species richness. Biometrics 40 119–129. [Google Scholar]
- Smith, J. J., L. A. Tow, W. Stafford, C. Cary and D. A. Cowan, 2006. Bacterial diversity in three different Antarctic cold desert mineral soils. Microb. Ecol. 51 413–421. [DOI] [PubMed] [Google Scholar]
- Sogin, M. L., H. G. Morrison, J. A. Huber, D. M. Welch, S. M. Huse et al., 2006. Microbial diversity in the deep sea and the underexplored “rare biosphere.” Proc. Natl. Acad. Sci. USA 103 12115–12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl, D. A., and R. I. Amann, 1991. Development and application of nucleic acid probes in bacterial systematics, pp. 205–248 in Sequencing and Hybridization Techniques in Bacterial Systematics, edited by E. Stackebrandt and M. Goodfellow. John Wiley & Sons, Chichester, UK.
- Stafford, W. H., G. C. Baker, S. A. Brown, S. G. Burton and D. A. Cowan, 2005. Bacterial diversity in the rhizosphere of Proteaceae species. Environ. Microbiol. 7 1755–1768. [DOI] [PubMed] [Google Scholar]
- Tehei, M., and G. Zaccai, 2005. Adaptation to extreme environments: macromolecular dynamics in complex systems. Biochim. Biophys. Acta. 1724 404–410. [DOI] [PubMed] [Google Scholar]
- Waisel, Y., 1961. Ecological studies on Tamarix aphylla (L.) Karst. III. Plant Soil 13 356–364. [Google Scholar]
- Waisel, Y., 1991. The glands of Tamarix aphylla: A system for salt secretion or for carbon concentration? Physiol. Plant. 83 506. [Google Scholar]
- Yancey, P. H., 2004. Compatible and counteracting solutes: protecting cells from the Dead Sea to the deep sea. Sci. Prog. 87 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, C. H., D. E. Crowley, J. Borneman and N. T. Keen, 2001. Microbial phyllosphere populations are more complex than previously realized. Proc. Natl. Acad. Sci. USA 98 3889–3894. [DOI] [PMC free article] [PubMed] [Google Scholar]