

Abstract
Platelet adhesion, activation and fibrinogen-mediated aggregation are primary events in vascular thrombosis and occlusion. An injectable delivery system that can carry thrombolytics selectively to the sites of active platelet aggregation has immense potential in minimally invasive targeted therapy of vascular occlusion. To this end we are studying liposomes surface-modified by fibrinogen-mimetic RGD-motifs that can selectively target and bind integrin GPIIb-IIIa on activated platelets. Here we report liposome surface-modification with a conformationally constrained high-affinity cyclic RGD-motif to modulate the GPIIb-IIIa-binding capability of the liposomes. Such affinity enhancement is important for practical in vivo applications to compete with native fibrinogen towards binding GPIIb-IIIa. The platelet-binding of RGD-modified liposomes were studied by fluorescence and scanning electron microscopy, and flow cytometry, in vitro. Binding of RGD-modified liposomes was also tested in vivo in a rat carotid injury model and analyzed ex vivo by fluorescence microscopy. The results from all experiments show that cyclic RGD-liposomes bind activated platelets significantly higher compared to linear RGD-liposomes. Hence, the results establish the feasibility of modulating the platelet-targeting and binding ability of vascularly targeted liposomes by manipulating the affinity of surface-modifying ligands.
1. Introduction
Site-targeted drug delivery holds significant promise in the treatment of vascular injury-associated thrombotic and occlusive events caused by cardiovascular diseases (e.g. atherosclerosis) or interventional procedures (e.g. angioplasty and stenting) [1-3]. Current strategies for site-specific delivery are focused primarily on the local administration of therapeutic agents via trans-catheter [4] or drug eluting stent techniques [5,6]. Such techniques are usually expensive [7] and may suffer from limitations of early drug washout, reduced control on drug release kinetics and recurrence of occlusive events at sub-optimal levels of drug concentration leading to dysregulated healing and late-stage thrombosis [8,9]. Nanoparticles, surface-modified with ‘homing’ motifs for targeting to and localizing at thrombotic sites, may provide an effective alternative for cardiovascular site-specific drug delivery [10-13]. Such bioengineered nanocontainers present the advantage of protecting encapsulants from plasma interactions, thereby improving the payload circulation half-life and release profiles. In this respect, nanoscale unilamellar liposomes, with their biocompatibility, their relatively high drug encapsulation efficacy and their capability of cell-selective binding by virtue of surface-conjugated targeting motif, are attractive candidates.
Considering the mechanistic criteria and the spatio-temporal patterns of cellular and molecular distribution in thrombosis, integrin GPIIb-IIIa (αIIbβ3,) on platelets is an attractive candidate for liposome targeting [14-16]. Ligand interactions with GPIIb-IIIa have been reported extensively, particularly the arginine-glycine-aspartate (RGD) motifs located in each of fibrinogen‘s two Aαchains, and the sequence, HHLGGAKQAGDV, located within fibrinogen’s γ chains [14,16]. Based upon these mechanisms, previously, platelets themselves [17] and fibrinogen coated lipid vesicles [18] have been explored as cardiovascular drug delivery vehicles. We have previously demonstrated the feasibility of a moderate affinity linear RGD peptide as the liposome surface-modification motif for targeted delivery specifically to activated platelets [19]. For practical in vivo applications, the liposomes need to have a sufficiently high binding ability to GPIIb-IIIa, in order to compete with native ligands (e.g.fibrinogen). The liposome binding ability depends upon the specificity and affinity of the surface-modifying ligands. For peptidic ligands, specificity and affinity have been found to be dictated by the conformational constraint of the peptide sequence at the apex of a solvent exposed loop [20]. Consequently, conformationally-constrained cyclic RGD peptides have shown a higher binding affinity to receptors [21,22]. Several cyclic RGD structures having much higher specificity and affinity (compared to linear RGD structures) for GPIIb-IIIa, have been reported [23]. Following this rationale, we postulated that modification of liposome surface with GPIIb-IIIa-specific cyclic RGD motifs will result in enhancement of platelet affinity, when compared with linear RGD (lRGD) motifs. The cyclic RGD peptide, CNPRGDY(OEt)RC (terminal cysteines (C) cyclized through disulfide), has been reported to have high affinity and selectivity for GPIIb-IIIa (αIIbβ3,), compared with other RGD-recognizing receptors like αvβ3, αvβ5 and α5β1 [23]. Hence this cyclic RGD peptide (cRGD) was developed by solid phase synthesis and conjugated to lipid for incorporation into liposomes, such that the peptide ligands stay displayed on the liposome surface. Figure 1 shows a schematic model of liposomes surface-modified by fibrinogen-mimetic cRGD-peptide for active platelet-selective thrombus-targeted delivery. Liposomes surface-modified with moderate affinity linear RGD and non-specific linear RGE motifs were used as comparison controls. The interaction of the surface-modified liposomes with activated platelets was studied in vitro by microscopy and flow cytometry and ex vivo by microscopy.
Figure 1.
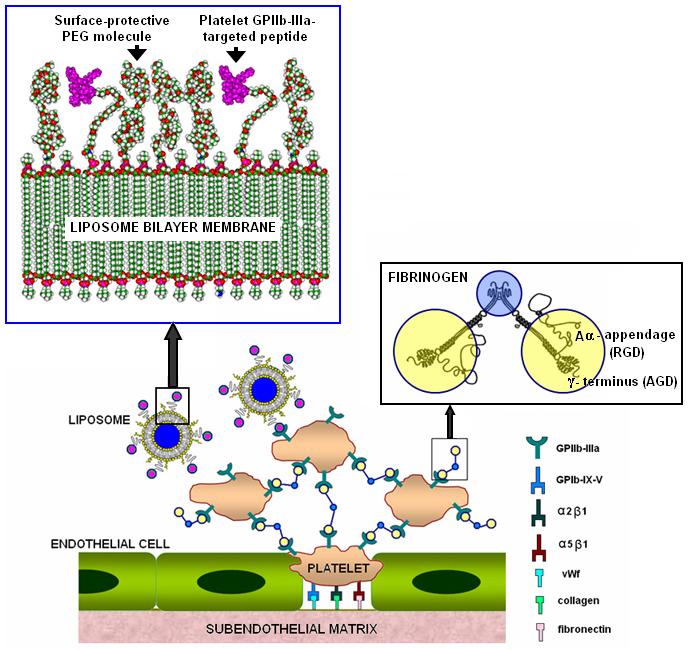
Schematic of liposomes surface-modified with fibrinogen-mimetic peptides for targeting integrin GPIIb-IIIa on activated platelets at a site of vascular injury
2. Materials and Methods
2.1. Reagents and Supplies
All amino acid derivatives and peptide synthesis reagents were purchased from Anaspec Inc. All lipids were purchased from Avanti Polar Lipids and NOF America Corp. For fluorescence studies 1-palmitoyl-2-[12-[(7-nitro-2-1,3-benzodiazol-4-yl)]amino]dodecanoyl]-sn-glycero-3-phosphocholine (PDPC-NBD, emission 534 nm, green fluorescence) was purchased from Avanti Polar Lipids and, AlexaFluor-546-tagged human fibrinogen (AlexaFluor-546-Fg, emission 573 nm, red-orange fluorescence) was purchased from Invitrogen and stored at -20° C prior to use. Cholesterol, bovine serum albumin (BSA), phosphate buffered saline (PBS) and sodium citrate were obtained from Sigma Aldrich. Mass spectrometry and microscopy supplies were used from stock at respective facilities at Case Western Reserve University.
2.2. Development of peptides and peptide-lipid conjugates
An 11-residue linear RGD-containing peptide (GSSSGRGDSPA, lRGD), a 9-residue linear precursor of cRGD peptide [CNPRGDY(OEt)RC] and an 11-residue RGE-containing negative control peptide (GSSSGRGESPA, lRGE) were synthesized using standard FMoc chemistry by solid-phase peptide synthesizer (ABI433A, Applied Biosystems) [24]. For cRGD synthesis, the terminal cysteine residues of the linear precursor were cyclized by a disulfide bond using ferricyanide-mediated oxidation process [25]. All peptides were purified by HPLC and chemical purity was confirmed by mass spectrometry (MALDI-TOF). Peptide-lipid conjugates were prepared following methods described by Wu et al [26]. For this, peptides, while still on the resin, were reacted through their N-terminal, to an N-hydroxysuccinimide (NHS)-activated polyethylene glycol carboxyester derivative of distearoylphosphatidylethanolamine (DSPE-PEG-COO-NHS, from NOF America Corporation) and the resultant lipid-peptide conjugates were then cleaved from the resin, purified by dialysis and characterized by mass spectrometry. For lipid-cRGD conjugate, the linear peptide precursor was conjugated to lipid first and the cyclization via disulphide bond was performed subsequently.
2.3. Platelet-affinity of free peptides
Affinity of free lRGD and cRGD peptides to bind activated platelets was characterized by ‘half maximal Inhibitory Concentration’ (IC50) value, which, in our assays, was the concentration of peptide required to inhibit fibrinogen-mediated platelet aggregation in platelet-rich plasma (PRP) by 50%, in an aggregometry (Bio/Data, PAP-4) set-up. PRP was prepared from citrated human whole blood by centrifugation at 800 RPM for 15 min at 25°C. 50 μl aliquots of PRP were warmed to 37°C for 2 minutes, incubated with various concentrations of lRGD and cRGD peptide in the presence of agonist adenosine di-phosphate (ADP, 10 μM) and the maximal aggregation percentage was determined while stirring for 15 min. Consequently, from the maximal aggregation percentage and comparison to aggregation without peptides, inhibition percentage was determined.
2.4. Preparation of peptide-modified liposomes
All liposomes were prepared by reverse-phase evaporation followed by extrusion through nanoporous (100 nm) Nuclepore polycarbonate membrane, as described previously for our lRGD-liposomes [19]. In all formulations, the final peptide-lipid conjugate content was kept at 1 mol %, which is lower than our previously reported lRGD-liposome formulations with 5 mol% peptide-lipid conjugate. The rationale was that if liposome surface-modification with a higher affinity peptide (e.g. cRGD) has an enhancing effect on platelet binding, the enhancement will be more sensitive and discernible at low peptide concentrations. PDPC-NBD was incorporated at 1 mol% in the formulations as a fluorescent probe. Liposome size and stability were characterized and monitored by dynamic light scattering using a Model 90Plus Brookhaven Instruments Corp Particle Size Analyzer for 30 days from the day of liposome preparation.
2.5. Microscopy analysis of liposome binding to platelets in vitro
Monolayers of platelets were adsorbed from human platelet suspension onto collagen III-coated glass coverslips following similar methods as described for our lRGD-liposome studies [19]. 3 μl of ADP (10 μM) in 120 μl PBS was added onto the platelet-adhered coverslips to ensure sufficient activation of adhered platelets. The presence of surface-adsorbed platelets was confirmed by staining with fluorescein isothiocyanate (FITC)-tagged anti-GPIIb-IIIa monoclonal antibody (FITC-anti-CD41a mAb, from BD BioSciences) and observing with a Nikon Diaphot epifluorescence microscope. The ‘activated’ state of the adherent platelets was confirmed by scanning electron microscopy (SEM) of coverslip samples. For this purpose, the coverslip-adhered platelets were fixed in 2.5 % glutaraldehyde for 2 hours at 4° C, subjected to progressive dehydration with graded series of ethanol solutions and finally critical point dried in liquid CO2. The SEM analysis was done by attaching the platelet-adhered coverslips to sample stubs and sputter coating with platinum followed by observation with a Hitachi S-4500 field emission electron microscope with an accelerating voltage of 5kV.
For fluorescent liposome binding studies, coverslip-adhered platelets were co-incubated with PDPC-NBD labeled lRGD, cRGD or lRGE-modified liposomes (diluted to a final concentration of 2.5 μM) and AlexaFluor-546-Fg in presence of 5 mM CaCl2, for 1 hr at room temperature in the dark. The [fibrinogen: liposome] ratio in the incubation assays was maintained at 400:1 by mole, to provide a physiologically relevant competitive environment. Subsequently the coverslips were washed with PBS, fixed in 1 % paraformaldehyde (PFA) for 30 min at 37° C, and mounted on glass slides to be imaged by the epifluorescence microscope for simultaneous detection of NBD and AlexaFluor-546 fluorescence. A 40X oil-immersion objective was used to visualize fluorescence from platelets. Images were collected in MetaMorph™ (Universal Imaging Corp.), using an exposure time of 500 ms and intensity analysis was performed in the software accordingly. Fluorescence intensity was recorded as mean intensity per pixel area for statistical analysis. All coverslips were prepared in duplicate, and at least three fields were examined for each well.
2.6. Flow cytometry analysis of liposome binding to platelets in vitro
The effect of platelet activation on liposome binding was studied by a FACScan flow cytometer (Becton Dickinson) with a 488nm laser and three color detector. For cytometry analysis whole blood aliquots with different platelet activation levels were used [27]. The level of platelet-activation was assessed by simultaneous fluorescence staining of platelet integrin GPIIb-IIIa (with FITC-anti-CD41a) and P-Selectin (with PE-anti-CD62P). Previously we have reported on detecting progressive activation of platelets in freshly drawn blood with addition of agonists (e.g. ADP) and have confirmed that binding of RGD-modified liposomes to platelets increased in an activation-dependant manner [20]. Following a similar protocol, whole blood aliquots containing predominantly activated platelets were incubated with PDPC-NBD-labeled cRGD-, lRGD-, or lRGE-modified liposomes at a final concentration of 500 μM total lipid. For the liposome incubated samples, FITC-anti-CD41a labeling was not used, while PE-anti-CD62P labeling was still used as an activated platelet marker. Simultaneous PE and NBD fluorescence, for gated platelet populations, was analyzed for ∼20,000 events (counts) per sample aliquot and fluorescence data were recorded accordingly.
2.7. In vivo interaction of surface-modifed liposomes with activated platelets and ex vivo imaging and analysis
For our in vivo model, an acute vascular injury was created by a balloon catheter-induced endothelial denudation of the luminal wall in the left common carotid of Lewis rats. After induction of anesthesia with an intraperitoneal injection of xylazine and ketamine cocktail, a midline cervical incision was made to expose the left external carotid artery. The external carotid artery was ligated, and the internal carotid artery was ligated temporarily. A 2F Fogarty balloon catheter (Baxter Healthcare Corp) was introduced through the arteriotomy site of the external carotid artery. The catheter was passed into the aortic arch, and the balloon was distended with saline until a slight frictional resistance was felt on traction. The catheter was withdrawn through the left common carotid with a slight rotational movement to ensure endothelial denudation of the luminal wall. The insertion and retraction procedure was repeated three times to induce sufficient vascular injury and hence an acute thrombotic environment. Subsequently, blood perfusion through the injury site was re-established by releasing the temporary ligation and maintained for 1hr to allow platelet adhesion, activation and aggregation. To ascertain the presence of activated thrombotic platelets, two animals were sacrificed, their injured carotid vessel sections excised and after careful sample preparation, imaged with SEM. After the 1 hr blood reperfusion period, 25 μl of NBD-labeled cRGD-, lRGD-or lRGE-liposome suspension was injected into the ascending aorta proximal to the injury site. All vessels branching from the aorta, except the injured carotid artery, were ligated to maximize the flow of liposomes along with the blood through the injury site. The blood (hence liposome) flow was maintained for 15 min, following which, the rats were euthanized, and the injured vessel was gently flushed with saline through the aorta, to remove artifacts. The injured artery section was excised, and immersed in 4% PFA solution for fixing. The fixed artery section was then cut open longitudinally and mounted on glass slides with the luminal injured liposome-exposed wall facing up. The exposed wall was imaged with a fluorescence microscope for NBD fluorescence.
2.8. Data analysis
Statistical analysis, where applicable, was performed in Microsoft Excel. A two tailed, unpaired student’s t test (assuming unequal variance) was performed to compare the difference in fluorescence intensity of platelets stained by test and control liposomes in the in vitro and in vivo assays. Significance was considered as p < 0.05. All data was noted as mean ± SD.
3. Results
3.1. Development of peptides and lipid-peptide conjugates
Figure 2 shows the peptide sequence structure and representative mass spectrometry data of the (2a) linear and (2b) cyclic RGD peptides developed for targeting GPIIb-IIIa. These peptides were conjugated to lipid (DSPE-PEO-NHS) by amide bond formation between amino terminal of peptide and activated carboxyl terminal of lipid-PEG. Figure 2c shows a representative MALDI-TOF analysis data of the product after conjugation of the cRGD peptide to DSPE-PEG-COO-NHS on resin, followed by cleavage. As evident from 2c, the mass spectrometry results of the crude product from the lipid-peptide conjugation reaction followed by cleavage from resin, showed presence of residual unconjugated peptide and a broad ‘hedgehog’ peak for the conjugated lipopeptide. The ‘hedgehog’ appearance of the peak is attributed to the polydispersity of the PEG spacer block in the commercial DSPE-PEG-COO-NHS. The lipopeptide product was purified from unconjugated free peptide by HPLC and dialysis.
Figure 2.
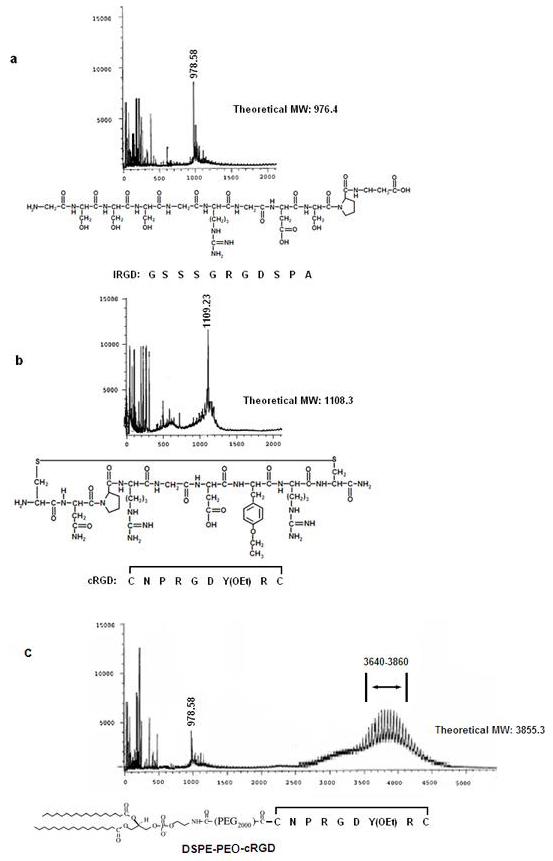
Amino acid sequence structure and corresponding mass spectral (MALDI-TOF) data for platelet GPIIb-IIIa-targeted (a) linear RGD, (b) cyclic RGD and (c) lipopeptide conjugate DSPE-PEG-cRGD.
3.2. Verification of higher affinity of the cRGD peptide
Figure 3 shows the effect of free peptide concentration (lRGD and cRGD) on the inhibition of platelet aggregation, as studied by aggregometry assays. As evident from the result, free cRGD causes 50% inhibition of platelet aggregation (dotted line in Fig 2) at a much lower peptide concentration compared to free lRGD. ‘Affinity’ is a kinetic parameter, while ‘specificity’ is a thermodynamic parameter. In our assays, both the GPIIb-IIIa-specific peptides (lRGD and cRGD) try to kinetically compete with natural ligand fibrinogen in binding active platelet GPIIb-IIIa and hence prevent fibrinogen-mediated platelet aggregation. The higher affinity peptide can outcompete fibrinogen (therefore cause 50% inhibition of platelet aggregation) at low peptide concentrations while the lower affinity peptide requires much higher concentration to gain the kinetic advantage. Hence ‘affinity’ is directly correlated with the IC50 value and the value for cRGD was found to be about 1000 times lower than that for lRGD. The significantly higher affinity of cRGD was thus established.
Figure 3.
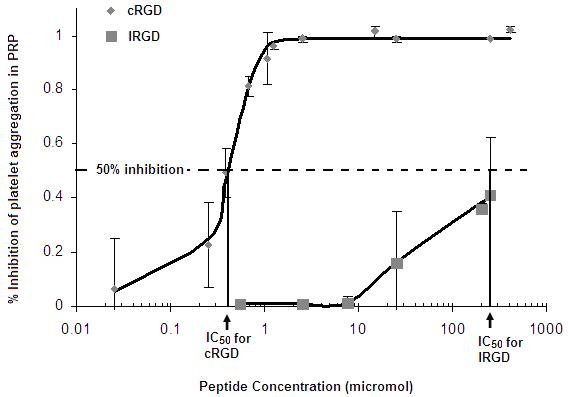
IC50 estimation by aggregometry assay for free lRGD and cRGD shows lower IC50 for cRGD by ∼ 2 orders of magnitude, compared to lRGD.
3.3. Dynamic light scattering analysis of liposome size distribution and stability
The effective diameter of liposomes in various batches was found to be ∼ 150 nm, when measured fresh after extrusion. The liposome suspensions were stored in vials at 4°C and the size distribution was monitored for 30 days using dynamic light scattering. No significant variation in effective diameter of liposomes was found for the 30 day period indicating stable non-aggregating vesicles.
3.4. Microscopy studies of platelet-liposome interaction in vitro
In order to validate our postulation that surface-modification of liposomes with higher affinity peptide provides a way to enhance liposome binding to activated platelets in a physiologically relevant competitive environment, platelet-liposome interaction was studied in vitro, where liposomes and fibrinogen were allowed to co-incubate with collagen-III adhered activated platelets. The presence of surface-adhered active platelets was confirmed by SEM, as shown in Figure 4A. The platelets co-incubated with green fluorescent liposomes and red-orange fluorescent Fg, showed dual fluorescence due to simultaneous binding. The Fg fluorescence was similar for samples co-incubated with lRGD- or cRGD-liposomes (Figs 4. B1 and C1). However, platelets co-incubated with cRGD-liposomes (Fig 4. C2) showed significantly greater (p<0.005) NBD (green) fluorescence compared to platelets co-incubated with lRGD-liposomes (Fig 4. B2). The mean NBD fluorescence intensity per pixel area of platelets incubated with cRGD-liposomes was found to be significantly higher (Fig 4. D) compared with platelets incubated with the lRGD-liposomes, for triplicate batches. This indicates that in a competitive environment cRGD-liposomes bind activated platelets at levels significantly higher than lRGD-liposomes.
Figure 4.
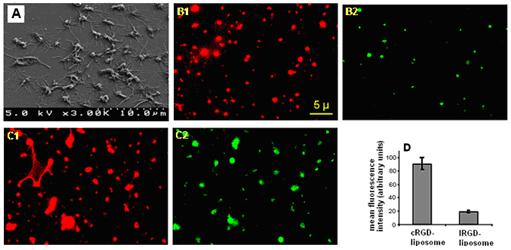
Scanning electron microscopy (SEM) image confirming presence of activated platelet monolayer on collagen-coated glass coverslip surface (4A); co-incubation of active platelets with AlexaFluor-546-labeled fibrinogen (red orange emission) and NBD-labeled peptide-modified liposomes resulted in simultaneous platelet-binding of both, and hence enabled dual fluorescence imaging of the same field of view; comparison of fluorescence images showed similar level of AlexaFluor-546 fluorescence for lRGD- (B1) and cRGD-liposome (C1) incubated platelets suggesting similar level of fibrinogen binding; NBD-fluorescence, however, was significantly enhanced for cRGD-liposome incubated sample (C2), compared to lRGD-liposome incubated sample (B2) as suggested by the mean fluorescent intensity data (D); this indicates enhanced binding of cyclic RGD-modified liposomes to activated platelets.
3.5. Flow cytometry studies of platelet-liposome interaction in vitro
The microscopy results were complimented by flow cytometry assays, where whole blood aliquots were incubated with NBD-labeled peptide-modified liposomes containing 1 mol% lipopeptide by composition. Freshly drawn blood showed only about 25% activated platelets whereas agonist-added (e.g. ADP) blood showed about 99% of the platelets to be activated, as confirmed by co-staining with GPIIb-IIIa-specific and P-selectin-specific markers (Fig 5. A and B). Blood aliquots containing predominantly resting platelets showed a minimal level of NBD fluorescence staining of platelets over background (unlabeled) when incubated with lRGD, cRGD or lRGE-liposomes (data not shown), suggesting a low degree of lipid exchange-based non-specific staining. However, for agonist-added blood aliquots, incubation with cRGD-liposomes resulted in increase of platelet-associated NBD fluorescence by about an order of magnitude higher compared to that from l-RGD liposome-incubated aliquots (Fig 5, C). This confirms that liposome modification with higher affinity cRGD peptide renders higher platelet binding ability compared to modification with moderate affinity lRGD peptide.
Figure 5.
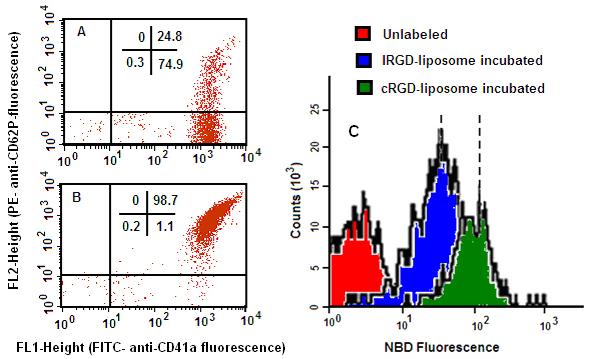
Flow cytometry studies on platelet-liposome interactions in vitro; platelet activation level was confirmed by co-labeling platelets in human whole blood aliquots with FITC-anti-CD41a (antibody to GPIIb-IIIa) and PE-antiCD62P (antibody to P-selectin), (A) before and (B) after addition of agonist ADP; incubation of activated platelets with NBD-labeled peptide-modified liposomes showed (C) enhanced NBD fluorescence for cRGD-liposome incubated sample compared to lRGD-liposome incubated sample, over background (unlabeled).
3.6. Platelet-binding of RGD-modified liposomes in vivo
SEM was used to confirm the presence of thrombotic activated platelets at the vascular injury site of the rat carotid. Figures 6A and 6B show representative SEM micrographs of the carotid artery wall in native state and after catheter-induced injury, respectively. Numerous activated platelets are visible adhered to the injured vessel wall. Figure 6C is a representative SEM micrograph of an injured artery wall after allowing blood flow for 1 hr, showing considerable thrombosis as evident from the fibrin clot ‘mesh’. Figures 6D, E, F and G show representative fluorescence microscopy images of the injured carotid section luminal surface exposed to no liposomes and, lRGE-, lRGD- and cRGD-liposomes, respectively. From analysis of mean fluorescence intensity of images from three batches of experiments, it was found that the injured carotid artery exposed to cRGD-liposomes had significantly higher (p < 0.01) NBD fluorescence than that exposed to lRGD-liposomes.
Figure 6.
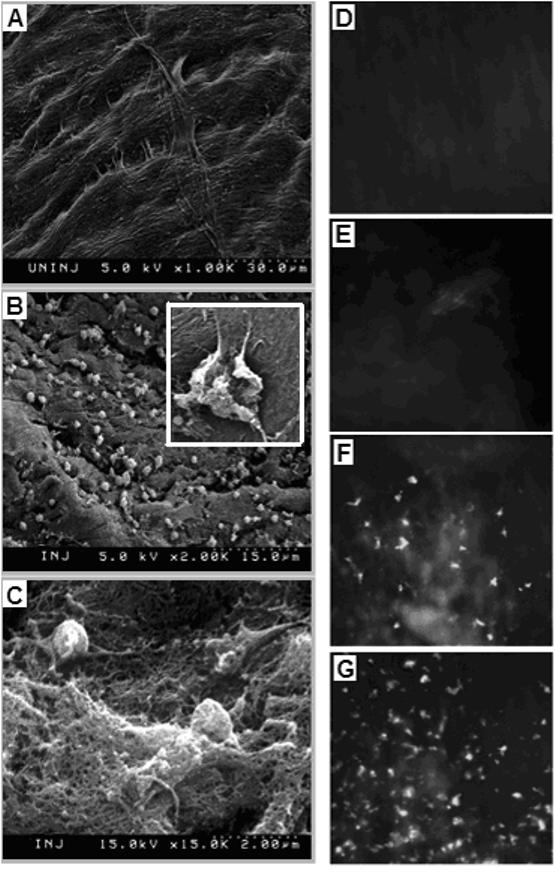
In vivo targeting of RGD-modified liposomes in a rat carotid injury model; SEM image of (A) uninjured luminal surface of carotid artery shows characteristic spindle shaped endothelial cells and extracellular matrix fibers; following catheter-induced injury, SEM image of the luminal surface shows (B) numerous adhered platelets in activated state (B, inset, magnified); repurfusion of blood through the injury site results in acute thrombotic and coagulatory event, as evidenced by (C) SEM image of the fibrin clot on the luminal surface; exposure of injured surface to NBD-labeled lRGE-, lRGD- and cRGD-liposomes via in vivo injection of respective liposome formulation, results in enhanced binding of (G) cRGD-liposomes to the injury site, compared to (E) lRGE- and (F) lRGD-liposomes, as evidenced by ex-vivo fluorescence imaging; (D) shows the fluorescence image of the luminal surface without exposure to NBD-labeled liposomes, for reference.
4. Discussion
There are recent reports on targeted delivery to thrombotic sites using fibrin-specific and tissue-factor-specific ligands as the ‘homing’ motifs [28,29]. There are also reports on ligand-modified nanoparticle-based endothelial cell targeting for vascular therapeutics, utilizing ligands and antibodies towards endothelial selectins and integrins [30,31]. Platelets are involved in primary hemostatic response which often precedes the secondary hemostatic processes of the coagulation cascade where fibrin and tissue factor are involved. Hence targeting of active platelets, may provide sensitivity towards treating and diagnosing vascular diseases in their developmental, as well as, progressive stages. In the current report, our studies show that liposome surface-modification with conformationally constrained higher affinity cRGD peptide results in enhanced binding of the liposomes to activated platelets, compared with moderate affinity lRGD modification. The enhanced affinity of the cyclic RGD results from the restricted peptide conformation, which is thermodynamically favorable for binding, and drives the kinetic equilibrium of ligand-receptor interactions towards increased ligand-receptor binding complexes.
It is to be noted that the aggregometry assays indicated the free cRGD peptide to have at least two orders of magnitude higher platelet affinity than the free lRGD peptide, whereas the liposome-platelet binding microscopy assays showed only about three-fold enhancement in fluorescence of platelet-bound cRGD-liposomes compared to lRGD-liposomes, and, the flow cytometry assays showed about one order of magnitude enhancement in platelet-binding of cRGD-liposomes. There can be several possible reasons behind such variations. Firstly, the conjugation of the peptide to a PEGylated lipid can reduce the receptor affinity of the final lipopeptide molecule compared to the free peptide. This is because, in the lipopeptide, although the peptide part itself can remain in the receptor-specific high-affinity constrained conformation, the random coil conformational behavior of the PEG spacer can affect the ligand orientation and steric environment with respect to the integrin receptor. As a result, from a single molecule perspective, some of the lipopeptides may loose the orientation necessary for specific high affinity binding to the receptor. Although a direct estimation of the average number of conjugated ligands on a single liposome, per liposome batch, was not performed in this study, theoretical approximations based on volume calculations assuming liposome average diameter of 150 nm, the liposome membrane bilayer thickness of 5 nm, the lipopeptide headgroup cross section diameter of 10 Å, the lipopeptide content of 1 mol % and the probability of at least 50% of the lipopeptides per liposomes to stay expressed on the outer surface, give the number of ligands on the liposome outer surface to be ∼850 per liposome. Due to the presence of such multiple copies of the lipopeptides on the outer leaflet of liposomes, the probability of many of the peptide molecules retaining the receptor-specific high affinity orientation is a reasonable assumption and this possibly contributes to the enhanced platelet binding of cRGD-liposomes compared to lRGD- and lRGE-liposomes. Also, the sensitivity and operational principles of the aggregometry, fluorescence and flow cytometry assays are different and hence, it is difficult to draw direct quantitative comparison between them. Nevertheless, all assays showed the feasibility of modulating platelet-binding affinity of liposomes by surface-modification with higher affinity cRGD peptide.
There are only few reports on platelet GPIIb-IIIa targeting by RGD-based peptides in the context of non-human platelets. The general consensus of these reports is that platelet GPIIb-IIIa in conventional small animals used for in vivo studies (e.g. rats, mice, rabbits) are relatively less sensitive to RGD-based peptides, compared to GPIIb-IIIa of human platelets [32,33]. Consequently, a small animal model for in vivo testing of RGD-GPIIb-IIIa interaction is rare. The few reports on RGD-interaction with rat or rabbit platelet GPIIb-IIIa are with linear GRGDS type of peptide and no reports are available regarding cyclic peptides. We decided to test the RGD-liposomes at a vascular injury site using a rat carotid injury model with the rationale that, even if the sensitivity of rat GPIIb-IIIa to RGD peptide is low, the specificity of RGD-mediated interaction will still be valid for rat GPIIb-IIIa and fibrinogen. Consequently, introduction of RGD-liposomes proximal to the vascular injury site may still provide a reasonable environment for the interaction of these liposomes with active platelets accumulating at the injury site. As evident from the ex vivo imaging studies, the model was effective in showing the enhancement in injury site binding of the cRGD-liposomes, compared to lRGD-liposomes and lRGE-liposomes. The RGD ligands for liposome modification were predominantly specific towards platelet GPIIb-IIIa, and hence the fluorescence in ex vivo images are most probably activated platelets. However, injured endothelium also expresses certain RGD-recognizing receptors like αVβ3 and α5β1, which may bind the RGD-modified liposomes to a certain degree. To determine absolute selectivity of ligand-modified liposomes to activated platelets vs. injured endothelial cells, further studies investigating binding of the free peptide, as well as, peptide-modified liposomes to activated endothelial cells would need to be performed.
5. Conclusion
We have designed liposomes, surface-modified with conformationally constrained cyclic RGD (cRGD) ligands having high affinity and specificity towards platelet integrin GPIIb-IIIa, and have compared their platelet-binding characteristics with liposomes bearing moderate affinity linear RGD (lRGD) ligands. The results clearly showed the cRGD-modified liposomes to have significantly higher binding to active platelets than lRGD-modified liposomes. The actual mode of delivery, after the liposomes bind their target, may be via lipid degradation, membrane fusion or internalization, as significant lipid mixing between RGD-liposomes and activated platelets has been reported [34,35]. This approach of optimizing platelet-targeting ability of ligand-modified liposomes can potentially lead to sensitive and selective delivery of therapeutic and diagnostic agents to a variety of cardiovascular diseases like atherosclerosis, thrombosis and restenosis where activated platelets play significant role in disease development, progression and outcome.
6. Acknowledgements
We would like to acknowledge Farhad Forudi for assistance in the in vivo studies and Dr. Eric H. Anderson for assistance in developing the molecular models of peptides. Facilities for in vitro studies were provided by the Center for Cardiovascular Biomaterials at Case. Facilities for in vivo studies were provided by Department of Cardiovascular Medicine at The Cleveland Clinic Foundation. The work was supported in part by NIH (research grant HL-70263).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Oberhoff M, Herdeg C, Karsch KR. Site-specific delivery of cytostatic agents. In: Camenzind E, De Scheerder IK, editors. Local Drug Delivery for Coronary Artery Disease. Taylor & Francis Ltd.; Abingdon, UK: 2005. pp. 275–280. [Google Scholar]
- 2.Chorny M, Fishbein I, Golomb G. Drug delivery systems for the treatment of restenosis. Critical Reviews in Therapeutic Drug Carrier Systems. 2000;17:249–284. [PubMed] [Google Scholar]
- 3.Helft G, Gilard M, Le Feuvre C, et al. Drug insight: antithrombotic therapy after percutaneous coronary intervention in patients with an indication for anticoagulation. Nature Clinical Practice Cardiovascular Medicine. 2006;3:673–680. doi: 10.1038/ncpcardio0712. [DOI] [PubMed] [Google Scholar]
- 4.Matsuda T. Device-directed therapeutic drug delivery systems. J Control Rel. 2002;78:125–131. doi: 10.1016/s0168-3659(01)00493-x. [DOI] [PubMed] [Google Scholar]
- 5.Ong ATL, Serruys PW. Technology insight: an overview of research in drug-eluting stents. Nature Clinical Practice Cardiovascular Medicine. 2005;2:647–658. doi: 10.1038/ncpcardio0378. [DOI] [PubMed] [Google Scholar]
- 6.Bennett MR. In-Stent Restenosis: Pathology and Implications for the Development of Drug Eluting Stents. Heart. 2003;89:218–224. doi: 10.1136/heart.89.2.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hirshfeld JW, Jr., Wilensky RL. Drug-eluting stents are here ----- now what? Implications for clinical practice and health care costs. Cleveland Clinic Journal of Medicine. 2004;71:825–828. doi: 10.3949/ccjm.71.10.825. [DOI] [PubMed] [Google Scholar]
- 8.Schwartz RS, Chronos NA, Virmani R. Preclinical Restenosis Models and Drug-Eluting Stents : Still Important, Still Much To Learn. J Am College of Cardiol. 2004;44:1373–1385. doi: 10.1016/j.jacc.2004.04.060. [DOI] [PubMed] [Google Scholar]
- 9.Muni NI, Gross TP. Problems with Drug-Eluting Coronary stents----The FDA Perspective. N Engl J Med. 2004;351:1593–1595. doi: 10.1056/NEJMp048262. [DOI] [PubMed] [Google Scholar]
- 10.Lanza G, Winter P, Cyrus T, et al. Nanomedicine opportunities in cardiology. Annals of the New York Academy of Sciences. 2006;1080:451–465. doi: 10.1196/annals.1380.034. [DOI] [PubMed] [Google Scholar]
- 11.Melo LG, Massimiliano G, Pachori AS, et al. Endothelium-targeted gene and cell-based therapies for cardiovascular disease. Athero Thromb Vasc Biol. 2004;24:1761–1774. doi: 10.1161/01.ATV.0000142363.15113.88. [DOI] [PubMed] [Google Scholar]
- 12.Wickline SA, Neubauer AM, Winter PM, et al. Molecular imaging and therapy of atherosclerosis with targeted nanoparticles. 2007;25:667–680. doi: 10.1002/jmri.20866. [DOI] [PubMed] [Google Scholar]
- 13.Guccione S, Li KCP, Bednarski MD. Vasculr-targeted naopaticles for molecular imaging and therapy. Methods in Enzymology. 2004;386:219–236. doi: 10.1016/S0076-6879(04)86010-5. [DOI] [PubMed] [Google Scholar]
- 14.Cierniewski CS, Byzova T, Papierak M, et al. Peptide ligands can bind to distinct sites in Integrin αIIbβIIIa and elicit different functional responses. J Biol Chem. 1999;274:16923–16932. doi: 10.1074/jbc.274.24.16923. [DOI] [PubMed] [Google Scholar]
- 15.Pytela R, Piersbacher MD, Ginsberg MH, et al. Platelet membrane glycoprotein IIb/IIIa: member of a family of Arg-Gly-Asp-specific adhesion receptors. Science. 1986;231:1559–1562. doi: 10.1126/science.2420006. [DOI] [PubMed] [Google Scholar]
- 16.Plow EF, D’Souza SE, Ginsberg MH. Ligand binding to GPIIb-IIIa: a status report. Semin. Thromb. Hemost. 1992;18:324–332. doi: 10.1055/s-2007-1002571. [DOI] [PubMed] [Google Scholar]
- 17.Crawford N, Chronos N. Electro-encapsulating drugs within blood platelets: local delivery to injured arteries during angioplasty. Semin Interv Cardiol. 1996;1:91–102. [PubMed] [Google Scholar]
- 18.DeAnglis AP, Retzinger GS. Preparation and characterization of fibrinogen-coated, reversibly adhesive, lecithin/cholesterol vesicles. J Pharm Sci. 1995;84:399–403. doi: 10.1002/jps.2600840404. [DOI] [PubMed] [Google Scholar]
- 19.Sen Gupta A, Huang G, Lestini B, et al. RGD modified liposomes targeted to activated platelets as a potential vascular drug delivery system. Thromb Haemost. 2005;93:106–114. doi: 10.1160/TH04-06-0340. [DOI] [PubMed] [Google Scholar]
- 20.Torshin IY. Structural criteria of biologically active RGD-sites for analysis of protein cellular function: a bioinformatics study. Med Sci Monit. 2002;8:301–312. [PubMed] [Google Scholar]
- 21.Lee G, Chan W, Hurle MR, et al. Strong inhibition of fibrinogen binding to platelet receptor αIIbβ3 by RGD sequences installed into a presentation scaffold. Protein Engineering. 1993;6:745–754. doi: 10.1093/protein/6.7.745. [DOI] [PubMed] [Google Scholar]
- 22.Mousa SA, Bozarth JM, Naik UP, et al. Platelet GPIIb/IIIa binding characteristic of small molecule RGD mimetic: distinct binding profile for Roxifiban. British J Pharm. 2001;133:331–336. doi: 10.1038/sj.bjp.0703943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng S, Craig WS, Mullen D, et al. Design and synthesis of novel cyclic RGD-containing peptides as highly potent and selective intetrin αIIbβ3 antagonists. J Med Chem. 1994;37:1–8. doi: 10.1021/jm00027a001. [DOI] [PubMed] [Google Scholar]
- 24.Fields GB, Noble RL. Solid phase peptide synthesis utilizing 9-fluorenylmethoxycarbonyl amino acids. Int J Peptide Protein Res. 1990;35:161–214. doi: 10.1111/j.1399-3011.1990.tb00939.x. [DOI] [PubMed] [Google Scholar]
- 25.Chaturvedi DD, Srivastava N, Sarkar J, et al. A kinetic study of the oxidation reaction of L-cysteine by potassium hexacyanoferrate(III) in aqueous acidic medium. Journal of the Indian Chemical Society. 1996;73:159–161. [Google Scholar]
- 26.Wu S-K, Chang T-G, Tseng C-L, Chen L-J, Shih K-S. Solid phase method for the synthesis of peptide-spacer-lipid conjugates and targeted liposomes containing such conjugates. 7153933 US Patent.
- 27.Schmitz G, Rothe G, Ruf A, et al. European working group on clinical cell analysis: consensus protocol for the flow cytometric characterization of platelet function. Thromb Haemost. 1998;79:885–896. [PubMed] [Google Scholar]
- 28.Wickline SA, Lanza GM. Molecular imaging, targeted therapeutics and nanoscience. J Cellular Biochem Suppl. 2002;39:90–97. doi: 10.1002/jcb.10422. [DOI] [PubMed] [Google Scholar]
- 29.Tiukinhoy-Laing SD, Huang S, Klegerman M, et al. Ultrasound-facilitated thrombolysis using tissue-plasminogen activator-loaded echogenic liposomes. Thrombosis Research. 2007;119:777–784. doi: 10.1016/j.thromres.2006.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spragg DD, Alford DR, Greferath R, et al. Immunotargeting of liposomes to activated vascular endothelial cells: A strategy for site-selective delivery in the cardiovascular system. PNAS. 1997;94:8795–8800. doi: 10.1073/pnas.94.16.8795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weissleder R, Kelly K, Sun EY, et al. Cell-specific targeting of nanoparticles by multivalent attachment of small molecules. Nature Biotechnology. 2005;23:1418–1423. doi: 10.1038/nbt1159. [DOI] [PubMed] [Google Scholar]
- 32.Harfenist EJ, Packham MA, Mustard JF. Effects of cell adhesion peptide Arg-Gly-Asp-Ser on responses of washed platelets from humans, rabbits and rats. Blood. 1988;71:132–136. [PubMed] [Google Scholar]
- 33.Bennett JS. Platelet-fibrinogen interactions. Annals of the NY Academy of Sciences. 2001;936:340–354. doi: 10.1111/j.1749-6632.2001.tb03521.x. [DOI] [PubMed] [Google Scholar]
- 34.Male R, Vannier WE, Baldeschwieler JD. Phagocytosis of liposomes by human platelets. Proc Natl Acad Sci. USA. 1992;89:9191–9195. doi: 10.1073/pnas.89.19.9191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nishiya T, Sloan S. Interaction of RGD liposomes with platelets. Biochemical and Biophysical Research Communications. 1996;224:242–245. doi: 10.1006/bbrc.1996.1014. [DOI] [PubMed] [Google Scholar]