

Abstract
Huntington’s disease (HD) is an inherited progressive neurodegenerative disorder characterized by progressive movement, psychiatric and cognitive disturbances. Previous studies have indicated that HD pathogenesis may be mediated in part by loss of brain derived neurotrophic factor (BDNF). Antidepressants selectively blocking serotonin reuptake can increase BDNF levels, and also may increase neurogenesis. Here we report that an SSRI antidepressant, sertraline, prolongs survival, improves motor performance, and ameliorates brain atrophy in the R6/2 HD mouse model. Six-week-old R6/2 mice and nontransgenic control mice were administered either sertraline or vehicle daily. Motor function was assessed in an accelerating rotarod test and evaluated at 10 weeks. R6/2 mice exhibited reduced time on the rod. Sertraline treatment improved the motor performance in R6/2 mice, but did not affect nontransgenic mice. R6/2 mice showed significant striatal atrophy which was reduced by sertraline treatment. These beneficial effects of sertraline are associated with enhanced neurogenesis and increased BDNF levels in brain treated with sertraline. The effective serum and brain levels of sertraline are comparable to the levels achieved in human antidepressant treatment. Our findings provide evidence that sertraline is neuroprotective in this HD model. Successful treatment with sertraline in depressed HD patients has been reported; moreover, sertraline is safe and well-tolerated for long-term administration, including in HD patients. Our findings suggest that a clinical trial of SSRI treatment in order to retard disease progression in human HD may be warranted.
Keywords: SSRI, serotonin, BDNF, Huntington’s disease, neurogenesis
Introduction
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disorder characterized by progressive impairment of motor function accompanied by psychiatric disturbance and dementia (Reiner et al., 1998; Vonsattel et al., 1985; Myers et al., 1988;). The first clinical symptoms of HD are often psychiatric abnormalities, including depression and mood disturbances (Duff et al., 2007). Involuntary movements and dementia develop over the next 15–20 years. Although the mechanism of neuronal degeneration in HD is unclear, it is thought that disease onset and progression in part involve dysregulation of transcription (Cha 2000; Sugars and Rubinsztein, 2003) and deficits of neurotrophic factor brain derived neurotrophic factor (BDNF) (Zuccato et al., 2001; Zuccato et al., 2005; Zuccato and Cattaneo 2007; Ferrigno and Silver, 2000; Gauthier et al., 2004; Cowan and Raymond, 2006;) leading to cumulative neuronal loss.
Neurochemical abnormalities in HD patients include reduced levels of BDNF (Ferrer et al., 2000; Ciammola et al., 2007). This is also recapitulated in HD mouse models (Zuccato et al., 2001; Duan et al., 2003). There is a reciprocal interaction between BDNF and 5-HT signals: 5-HT stimulates the expression of BDNF, and BDNF enhances the growth and survival of 5-HT neurons. Impaired 5-HT and BDNF signaling is believed to be involved in depression and anxiety disorders, but could also play important roles in the pathogenesis of several age-related disorders including Huntington’s disease (Mattson, et al 2004). Other studies have shown that BDNF can protect neurons against insults that occur in the pathogenesis of HD (Bemelmans et al., 1999; Kells et al., 2004; Canals et al., 2004; Cepeda et al., 2004; Pineda et al., 2005; Zuccato et al., 2005; Lynch et al., 2007). These studies suggest that increasing BDNF levels is beneficial in HD. Altered neurogenesis has been reported in HD mouse models and in human postmortem brains (Phillips et al., 2006; Phillips et al., 2005; Grote et al., 2005; Gil et al., 2005; Lazic et al., 2004; Curtis et al., 2003). In addition to promoting neuronal survival, BDNF also regulates neurogenesis (Duman, 2004; Cotman and Berchtold 2002).
Selective serotonin reuptake inhibitors (SSRIs), which are widely prescribed for depression and severe anxiety disorders, may also have therapeutic potential as neuroprotective agents (Sanchez et al., 2001). Chronic but not acute administration of sertraline increased BDNF expression in rodent brain (Nibuya et al., 1995; Nibuya et al., 1996; Moltzen and Bang-Andersen, 2006) and stimulate neurogenesis (Malberg and Blendy, 2005). We previously showed that the SSRI paroxetine is neuroprotective in the N171-82Q HD mice (Duan et al., 2004). Therefore we explored another SSRI, sertraline in a widely used HD mouse model in present studies. Since neuroprotective drugs for HD will need to be given over long periods, it is important that they are safe and well-tolerated. SSRIs are well tolerated over long term administration in patients. There is currently no therapy available in the clinic to delay onset or prevent disease progression of HD. Our current findings suggest a rationale for clinical trials of sertraline and SSRIs in HD patients.
Materials and Methods
Mice and drug administration
Transgenic HD mice of R6/2 line were originally purchased from Jackson laboratories (Bar Harbor, ME) and the colony was maintained by breeding heterozygous R6/2 males with females from their background strain (F1 of CBA × C57Bl/6). Tails of the offspring were used to obtain DNA for determination of the genotype and CAG repeat size by PCR assay. The mice were housed in groups with access to food and water ad libitum and a 12-h light/dark cycle. All experimental mice were housed in cages including an orange mouse igloo and a green nylabone setting in the cage, and wet mash was provided to all R6/2 mice, starting at weaning. The CAG repeat size for all experimental R6/2 mice ranged from 103 to 112. Sertraline hydrocholoride (99% purity. Toroton Research Chemicals Inc, Canada) was administered to mice by daily intraperitoneal injection. All animal experiments were performed according to procedures approved by the Institutional Animal Care and Use Committee at Johns Hopkins University.
Behavioral test and Survival study
All mice were randomly divided into each group. Nontransgenic control mice are wild type littermates. There are 15 mice in each group at the beginning for survival study and motor behavioral analyses. We used the same set of animals for survival analyses and motor performance tests.
Survival was monitored twice a day, at early morning and late afternoon daily, by two experienced investigators (QP, WD). The mice were euthanized when HD mice were unable to right themselves after being placed on their backs and initiate movement after being gently prodded for 30 sec.
Motor behavioral performance was assessed with a rotarod apparatus (Columbus Instrument, OH) in which the time the mouse remains on the rod at accelerating speed from 4 to 40 rpm is measured. Each mouse was trained for 5 minutes and the training session was followed by a 1-hour rest period in the home cage. Mice were then placed back on the rotarod for three trials of maximal 5 minutes at accelerating speeds (4–40 rpm) separated by a 30-minute rest period. Mice were tested for three consecutive days, by which time a steady baseline level of performance was attained.
BrdU administration, immunohistochemistry, and stereological quantification
For the neurogenesis study, we used different sets of mice. There are 4 mice at each group. All mice were received twice daily injections of 50 mg/kg of BrdU (5-bromo-2-deoxyuridine; Sigma) in sterile 0.9% NaCl solution at 8 hours apart for 5 consecutive days. Animals in the cell proliferation study group were perfused by 4% buffered paraformadehyde at 3 days after the last BrdU injection, and animals in the cell survival study group were perfused by 45 paraformadehyde at 3 weeks after the last injection. BrdU immunostaining was performed as previously described (Lee et al., 2002). Briefly, coronal brain sections (40 µm) were treated with 0.6% H2O2 for 30 minutes to quench endogenous peroxidases, the sections were incubated in 2 N HCl for 30 minutes at 37 °C and washed in 0.1 M borate buffer (pH 8.5) for 10 minutes. For light microscope quantification of BrdU-labeled cells, a series of every sixth 40-µm section was used. Sections were blocked in 1 × TBS containing 3% horse serum and 0.3% Triton X-100 for 1 hour, followed by incubation in anti-BrdU antibody (1:200, BD Biosciences) or EM48 ( 1:200, rabbit polyclonal anti-polyglutamine antibody, Millipore) overnight at 4 °C. Sections were then incubated with biotinylated anti-mouse IgG or anti-rabbit (Vector Laboratory, CA) for 1 hour at room temperature. The immunoreactive product was detected by using the vectastain ABC kit enhancing system (Vector Laboratory. CA) and diaminobenzadine (DAB) as substrate. Sections were counterstained with Nissl substrate for stereological quantification.
Stereology counting was performed in serial coronal sections and done on blind-coded slides. The BrdU-positive cell numbers in the subgranule zone (SGZ) of the dentate gyrus (DG) were assessed by counting all positive cells in sections at the levels of bregma −1.34 to −2.80 by using conventional light microscopy with 40 × objective in DG region. The distance between the sections was 240 µm. The ‘optical fractionator’ was used to calculate the number of BrdU-positive cells in the DG (Schmidt-Hieber et al., 2004). The ‘optical fractionator’ technique estimates the number of cells by multiplying the sum of cells counted by the reciprocal of the fraction of the region sampled. For analysis of BrdU cells in the subventricular zone (SVZ), only the side adjacent to the striatum was quantified. On each section, the region of interest was outlined unilaterally under a 5× objective and the enclosed area was calculated. The stereology investigator software automatically identified and measured profiles. All computer-identified cell profiles were manually verified as neurons and exported to Microsoft Excel. Cross-sectional areas were analyzed by using Sigmastat statistical program.
Measurement of volume of striatum and size of lateral ventricle
The volume of the striatum and lateral ventricle measured by histological analysis was evaluated according to the principle of Cavalieri (Cyr et al., 2005) (volume = s1d1 + s2d2…sndn, where s = surface area and d = distance between two sections). The Cavalieri principle is applied to histological data because of the limited number of sections selected. We used different sets of mice for the volumetric measurement study. There are 4 mice in each group. Mice were chronically treated with sertraline (10 mg/kg/day, i.p) from age of 6 weeks, and mice were perfused by 4% paraformadehyde at 10-week-old. We considered 11 anatomical levels of the striatal sections for the estimation of the volume of each brain. Image capturing was performed by using a light microscope coupled to a color digital camera (Retina 2000R, QImaging). The surface area was calculated in each of the 11 Nissl-stained sections by contour drawing of the striatum and lateral ventricle on each brain hemisphere by using Aperio Imagescope software (Aperio Technologies). Data were expressed as the average Cavalieri volume (mm3) ± SEM of four mice per group.
Measurement of sertraline by HPLC/MS/MS
To measure the levels of sertraline in both blood and brain. Both transgenic and nontransgenic littermates (6 week-old) were used. There are 4 nontrangenic control mice and 4 HD mice used in the study. Whole brain homogenates were mixed and centrifuged at 2500 × rpm for 10 minutes at 4 °C. A volume of 100 µl of the top organic layer was transferred to a disposable borosilicate glass culture tube (13×100 mm) and 100 µl of deionized water were added to this tube, mixed vigorously for 10 s, and centrifuged at 13,000 × rpm for 5 minutes. Volumes of 20 µl were injected onto the HPLC instrument for quantitative analysis. Serum samples were prepared as previously described (Zhao et al., 2005). Chromatographic analysis was performed using a Waters X-Terra™ MS (C18 3.5 µm, 50 × 2.1 mm i.d). Separation of the analyses from potential interfering material was achieved at ambient temperature by using Waters X-Terra™ MS (C18 3.5 µm, 50 × 2.1 mm i.d) packed with a 3.5-µM ODS stationary phase, protected by a guard column packed with Waters Xterra TM (RP18, 3 µm). The mobile phase used for chromatographic separation was composed of 70% formic acid (0.1%) in CH3CN, 30% ammonium acetate (10 mM), and was isocratic at a flow rate of 0.2 ml/min. Method validation runs for serum and brain tissue calibration standard and quality controls were performed on 3 consecutive days and include a calibration curve processed in duplicate, and quality control samples, at four different concentrations, in triplicate. The accuracy and precision of the assay was assessed by the mean relative percentage derivation from the nominal concentrations and the within and between run, respectively.
ELISA analysis of BDNF protein levels
There are four mice in each group for BDNF analysis. Mice were administered sertraline (10 mg/kg/day) for 4 weeks starting from 6 week-old age. Three different brain regions including hippocampus, striatum and cortex were dissected for the assay. Brain tissues were homogenized in lysis buffer (137 mM NaCl, 20 mM Tris, 1% NP-40 detergent, 10% glycerol, 1 mM phenylmethylsulfonylfluoride, 10 mg/ml aprotinin, 1 mg/ml leupeptin, and 0.5 mM sodium orthovanadate; pH 7.2) at 4°C. Homogenates were centrifuged at 2000 × g for 20 min at 4°C, and supernatant fractions were used for ELISA analysis. BDNF protein levels were quantified by BDNF Emax Immunoassay (Promega) as described previously (Duan et al., 2003). The intra- and inter-assay variability for the BDNF ELISA was 0.55 pg and 1.24 pg, respectively. The concentrations of BDNF in each sample were determined in duplicate, and the average of the two values was used as the value for that mouse.
Double Immunofluoresent staining
Sections were quenched and blocked as described above, then incubated in mixed antibodies (rat anti-BrdU, 1:200, Serotec; mouse anti-NeuN, 1:200, or rabbit anti- GFAP, 1:1000, Chemicon Inc) overnight at 4 °C. A cocktail of secondary antibodies (FITC-anti-rat, Serotec; Cy3-anti-mouse or Cy3 anti-rabbit, Jackson Immuno Inc) was applied for 1 hour at room temperature. Images were captured and analyzed in a Zeiss LSM 510 confocal microscope.
Statistics
The data were expressed as the mean ± SEM. Statistical comparisons among groups were compared by ANOVA with Fisher LSD tests, and two-group comparison used the Standard Student’s t-test. Survival data were assessed by Kaplan-Meier analysis.
Results
Sertraline extends survival and ameliorates impaired motor function of R6/2 mice when administered after the onset of motor dysfunction
In order to determine whether sertraline might modify the course of the symptomatic phase of the disease, beginning at 6 weeks of age, R6/2 mice were injected with sertraline (5 or 10 mg/kg daily injection) or vehicle. The survival of sertraline-treated R6/2 mice was significantly and dose-dependently increased compared to that of vehicle-treated HD mice (Fig. 1a). Sertraline increased the median lifespan approximately 13% at a dose of 5 mg/kg group (91.9 ± 1.9 in sertraline group vs 84.2 ± 3.5 in vehicle group, p<0.05), and 20% (100.9 ± 2.4 in sertraline group vs 84.2 ± 3.5 in vehicle group, p<0.05) at a dose of 10 mg/kg group (Fig. 1a). R6/2 mice exhibit progressive weight loss, but sertraline administration did not significantly affect body weight loss (data not shown)
Figure 1.
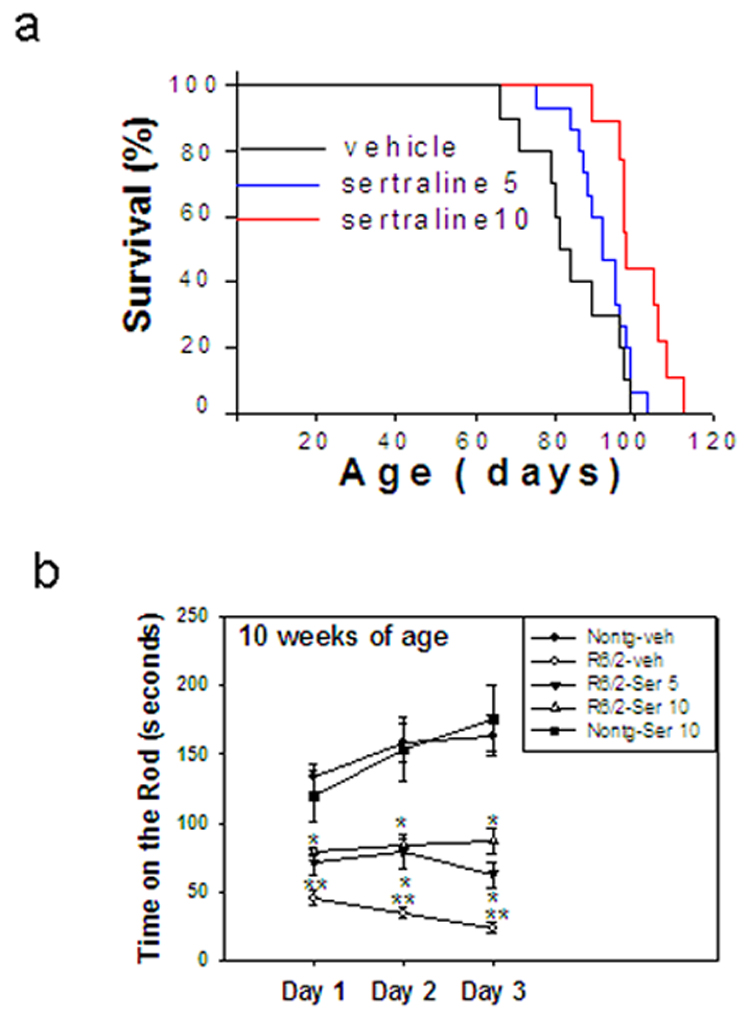
Sertraline increases survival (a), and improves motor behavioral performance (b) in R6/2 mice. Six-week-old R6/2 mice were administered sertraline at doses of 5 and 10 mg/kg. Motor behavioral performance was evaluated by an accelerating rotarod apparatus with mice at indicated ages, n=8–15. Values are the mean and SE. *p<0.05 compared to the value of HD-vehicle group; **p<0.05, compared to the value of nontransgenic-vehicle group.
We next evaluated motor function in R6/2 mice at 10 weeks of age, the time point when mice had received 4 weeks of sertraline administration. There was significant impairment in performance of R6/2 mice on the rotarod at 10 weeks of age, indicated by reduced time spent on the rotarod compared to nontransgenic mice. R6/2 mice treated with sertraline showed improved motor performance compared to vehicle-treated R6/2 mice. Control mice that received sertraline did not differ in motor performance compared with vehicle-treated control mice (Fig. 1b).
Sertraline treatment reduces brain atrophy
To determine whether the improved motor function and increased survival of R6/2 mice treated with sertraline resulted from a slowed progression of brain atrophy, we performed histological analyses of the brains of R6/2 mice that had been treated with sertraline or vehicle. As the disease progressed, brain atrophy occurred in the R6/2 mice, indicated by an increase in the size of the lateral ventricles and reduction of volume of the striatum and whole brain compared to those in nontransgenic mice (Fig 2a–c). The magnitude of lateral ventricular enlargement and striatal atrophy was significantly decreased in R6/2 mice that had been treated with sertraline compared to R6/2 mice treated with vehicle (Fig. 2b &c).
Figure 2.
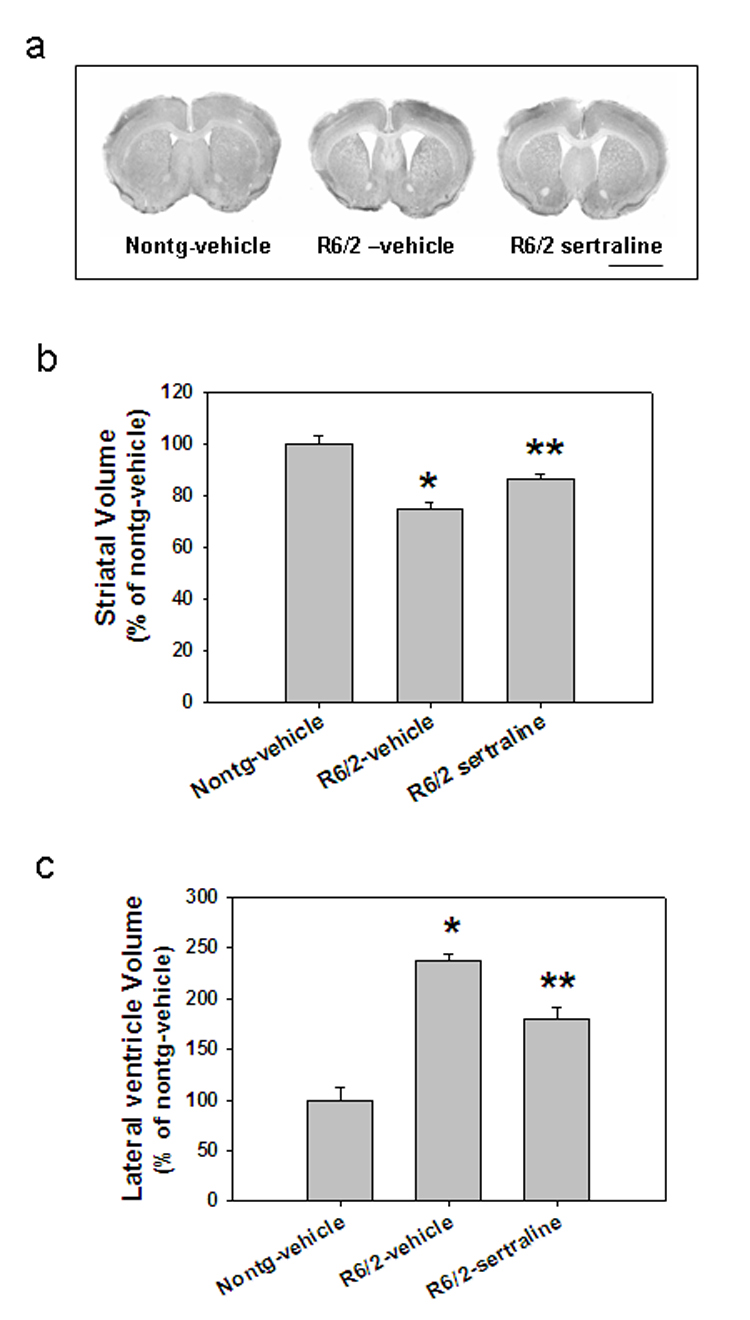
Sertraline reduces brain atrophy in R6/2 mice. (a) Representative photomicrographs of brain sections of R6/2 mice treated with sertraline or vehicle and nontransgenic mice (Nontg). Scale bar= 120 µm. (b) Quantification data of striatal volume and (c) cerebral lateral ventricle volume. n= 4 mice in each group. *p<0.05, compared to the value of Nontg vehicle group, **p<0.05 compared to the value of R6/2-vehicle group.
Sertraline does not affect htt inclusions in R6/2 mice
Intranuclear inclusions of aggregated huntingtin protein in striatal, hippocampal, and cortical neurons are present in R6/2 mice, as well as in HD patients (Ho et al., 2001). Sertraline treatment did not reduce huntingtin inclusions in these brain regions (Suppl Fig.1).
Sertraline increases basal neurogenesis in hippocampus and subventricular zone and ameliorates impaired hippocampal neurogenesis in R6/2 mice
In the hippocampus of both rodents and primates, adult-generated neuronal cells arise from progenitor cells in the subgranule zone (SGZ) and migrate into the granule cell layer, where they differentiate into granular neurons. These neurons were shown to be capable of functional integration into hippocampal circuitry. The newly generated cells in SVZ migrated into affected striatum and differentiated to medium spiny neurons in HD (Jin et al., 2005) and under ischemic conditions (Yamashita et al., 2006).
There are contrasting reports in neurogenesis between HD human brains and mouse models (Phillips et al., 2006; Phillips et al., 2005; Grote et al., 2005; Gil et al., 2005; Tattersfield et al., 2005; Lazic et al., 2004; Curtis et al., 2003). For example, reduced neurogenesis has been reported in R6/2 mice (Phillips et al., 2006; Phillips et al., 2005; Gil et al., 2005), but in humans, there was increased proliferation and neurogenesis (Curtis et al., 2003). In the current study, there was a significant decrease of cell proliferation and survival of newly generated cells, indicated by reduced number of BrdU-positive cells in the dentate gyrus (DG) of hippocampus in R6/2 mice compared to nontransgenic control mice both at 3 days and 3 weeks after the last BrdU injection (Fig 3a–d). Sertraline increased cell proliferation in DG and promoted survival of newly generated cells (Fig 3b–d). Analysis of BrdU-positive cells showed that approximately 80% of BrdU-positive cells also expressed NeuN (Fig 3e). Neither genotype nor sertraline affected the percentage of newly generated cells differentiated into neurons (Fig 3e). A few cells co-expressed GFAP with BrdU (Fig 3f).
Figure 3.
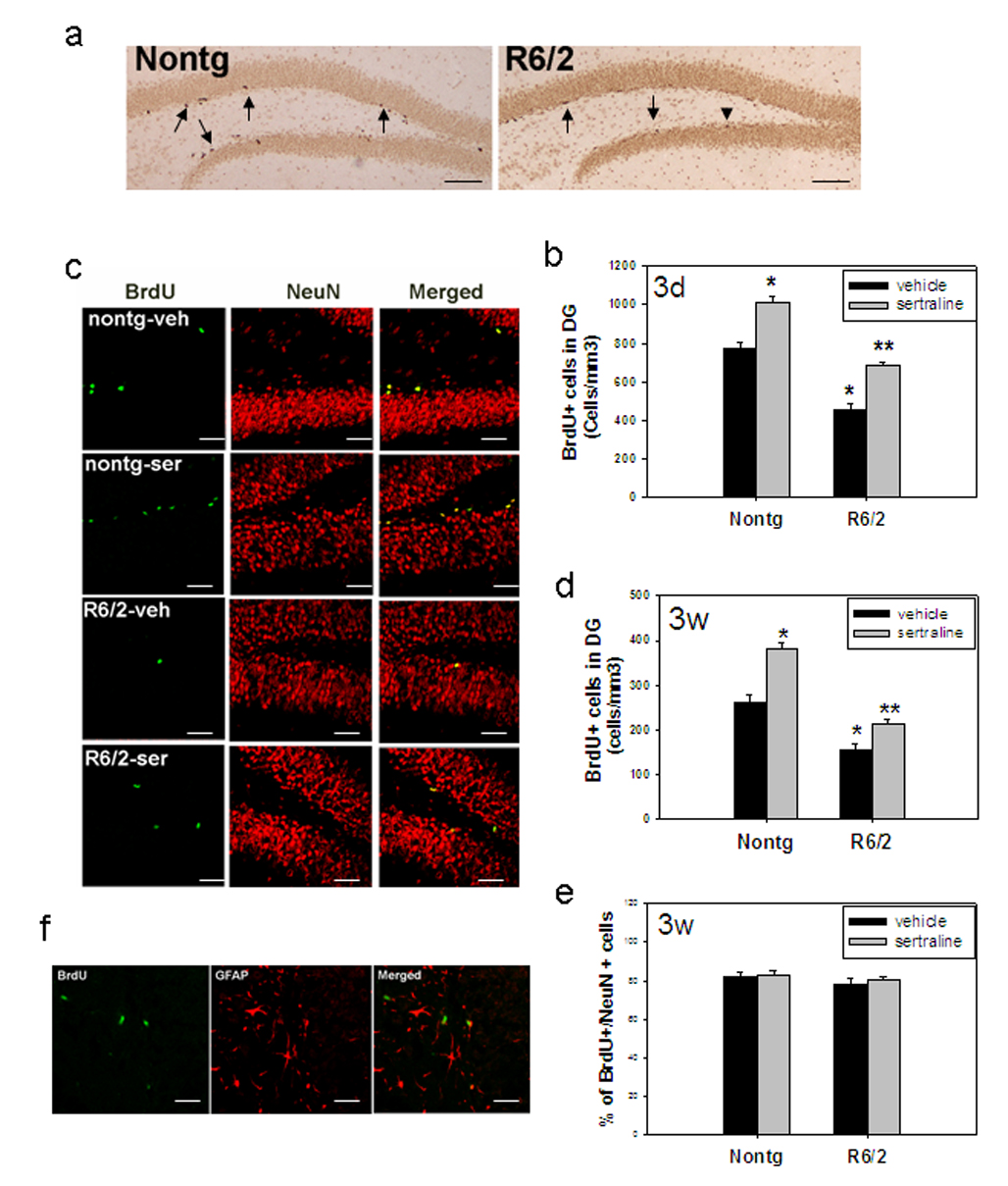
Sertraline increases neurogenesis in subgranular zone (SGZ) of dentate gyrus (DG) in R6/2 mice and control mice. Sertraline (10 mg/kg) was administered to mice at 6 weeks of age for 4 weeks. Mice then received 5-bromo-2-deoxyuridine (BrdU) and were perfused at 3 days after the last BrdU injection for cell proliferation (a and b) and 3 weeks after the last injection for quantifying survival of newly generated cells and cell type (c–e). (a) Representative pictures of BrdU staining from nontransgenic control (Nontg) and R6/2 mice 3 days after BrdU injection. Note that there are more BrdU-positive cells in DG of nontransgenic control mice compared to those in R6/2 mice. Scale bar=200 µm. (b) Quantification of BrdU cells in DG at 3 days after BrdU injection. BrdU cells indicate the new proliferating cells. (c) Representative sections were double labeled with anti-BrdU and anti-NeuN antibody (neuronal marker) to identify the phenotypes of newly generated cells. Representative images from confocal microscope are indicated by green for BrdU- positive staining and red for NeuN. Scale bar =100 µm (d) Quantification of newly generated neurons in dentate gyrus at 3 weeks after BrdU injection. (e) Percentage of cells with double labeling with both BrdU and NeuN. (f) Representative sections were double labeled with anti-BrdU antibody (green) and GFAP (red, astrocyte marker). Scale bar =100 µm. All values are mean and SE. *p<0.05, compared to the values of Nontg-vehicle group, **p<0.05 compared to the values of HD vehicle group. n=4 mice in each group.
There was no difference in cell proliferation in SVZ between HD and nontransgenic mice (Fig. 4b). These results are consistent with a previous report (Gil et al., 2005). Sertraline administration significantly increased cell proliferation in both R6/2 mice and control mice (Fig 4a–b).
Figure 4.
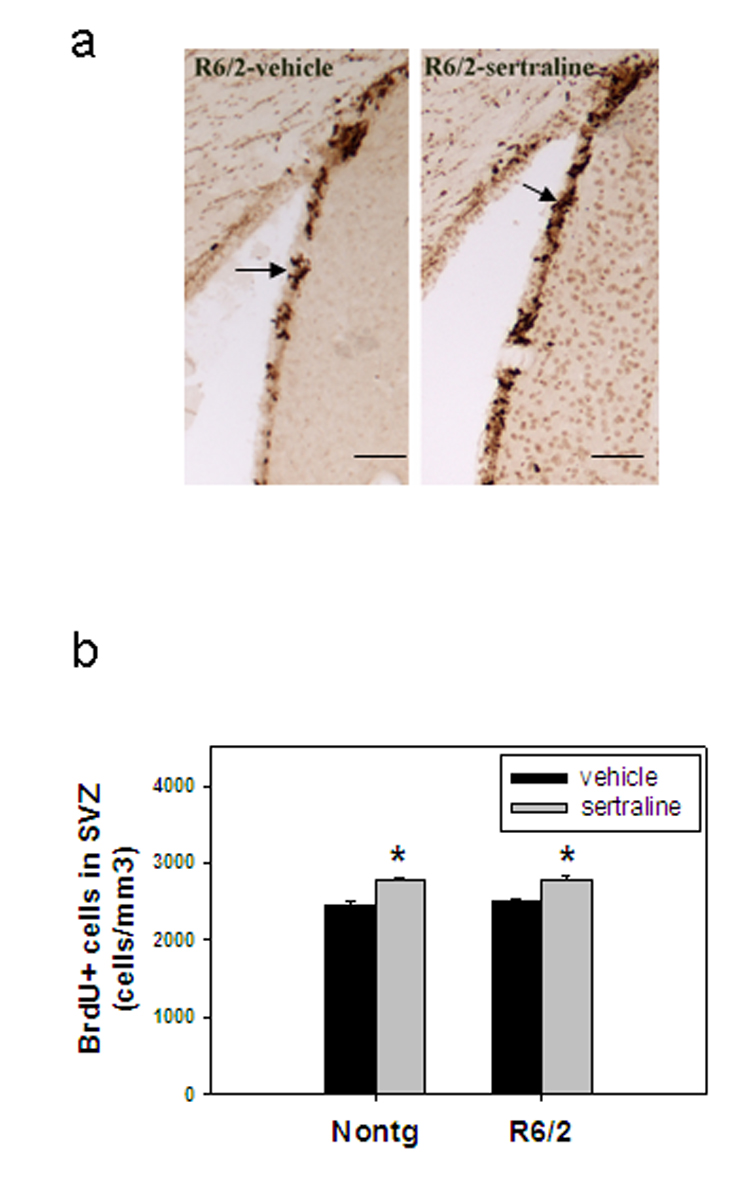
Sertraline increases cell proliferation in the subventricular zone (SVZ). Mice were administered sertraline at 10 mg/kg from 6 weeks of age for 4 weeks and perfused at 3 days after the last BrdU injection. (a) representative BrdU staining photos from each indicated group. Scale bar =100 µm. (b)There is no difference in new proliferating cells in SVZ between nontransgenic control mice and R6/2 mice; sertraline administration increased BrdU-positive cells in SVZ of both control mice and R6/2 mice. *p<0.05, compared to the values of corresponding vehicle group by Standard Student t-test.
Sertraline attenuates depletion of BDNF in R6/2 mice
BDNF protein levels were significantly decreased in different brain regions of R6/2 mice, including hippocampus, striatum, and cortex (Fig. 5a–c). Sertraline administration significantly increased BDNF protein levels in nontransgenic mice and BDNF depletion recovered in hippocampus and striatum of R6/2 mice after administration for 4 weeks (Fig. 5a–c).
Figure 5.
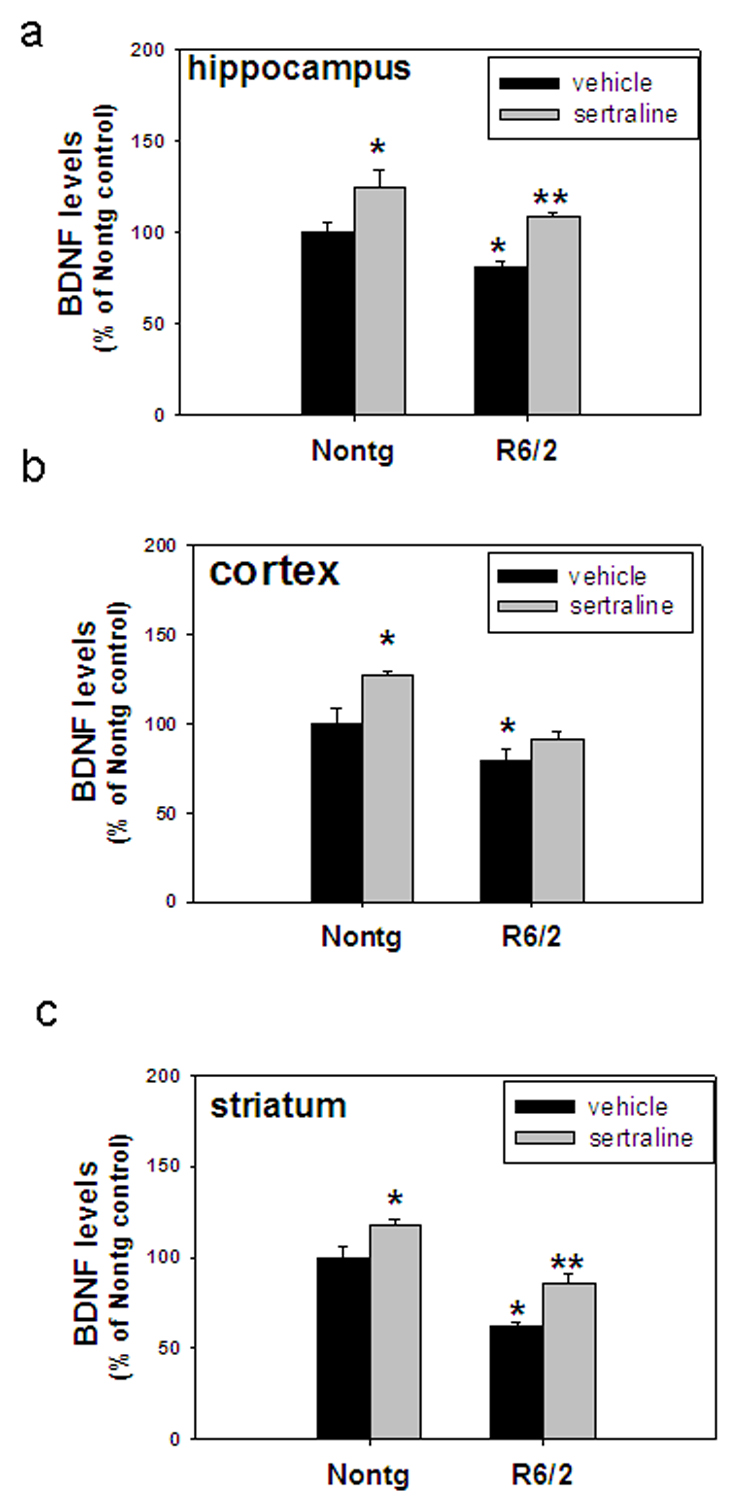
Sertraline normalizes BDNF protein levels in R6/2 mice. Six-week-old mice were administered sertraline at 10 mg/kg. Mice were euthanized 4 weeks after sertraline administration, and BDNF protein levels were measured in hippocampus (a), cortex (b) and striatum (c) *p<0.05, compared to the values of Nontg vehicle group, **p<0.05 compared to the values of R6/2-vehicle group. n=4 mice.
Effective levels of sertraline in mice are comparable to achievable levels in humans
In order to study whether effective doses in mice are comparable to doses achievable in humans, we used the LC/MS/MS method to measure the levels of sertraline both in blood and brain tissues. Sertraline was injected daily to mice for 1 week, and blood samples and brain tissues were collected for measuring drug concentrations. The blood concentrations are 23.1 ± 2.7 ng/ml (mean ± SD) in the 5 mg/kg group and 43.7 ± 2.7 (mean ± SD) ng/ml in the 10 mg/kg group. These levels are comparable to the effective levels in depressed patients who take 100–200 mg/day orally (blood concentration range around 40–60 ng/ml). Drug concentrations in brain tissues are 1100 ± 59 and 2238 ±113 ng/g wet weight, the brain/blood ratio of sertraline is over 45, indicating very high penetration of sertraline through the blood-brain barrier.
Discussion
The first clinical symptoms of HD are generally psychiatric abnormalities, most commonly depression and mood disturbances. Involuntary choreiform movements and dementia develop over the next 15–20 years, and death generally results from complications of immobility. Currently, there is no cure for HD, except temporary relief of symptoms by some treatments. Sertraline has been used to treat depressed HD patients (Ranen et al., 1996; Slaughter et al 2001), chronic administration of sertraline increased BDNF levels (Nibuya et al., 1995, 1996), whether sertraline has neuroprotective effects in HD is not known. Here we report that sertraline suppressed brain atrophy, improved motor behavioral performance, and increased the survival in R6/2 mice. The beneficial effects of sertraline in HD mice are associated with increased levels of BDNF, and stimulation of neurogenesis. The present findings provide further evidence that increasing BDNF levels can counteract the neurodegenerative process in HD.
The mechanism of how mutant huntingtin leads to the devastating course of events that culminates in cell death in HD remains elusive. It has been reported that levels of serotonin and BDNF are decreased in HD mice (Reynolds et al., 1999, Zuccato et al., 2001) and HD patients (Ferrer et al., 2000; Ciammola et al., 2007). There is a reciprocal interaction between BDNF and serotonergic signaling, in which BDNF enhances serotonin production and release, while serotonin stimulates BDNF production (Mattson et al., 2004). Moreover, increasing BDNF levels by either directly delivery of BDNF (Bemelmans et al., 1999; Kells et al., 2004; Canals et al., 2004; Cepeda et al., 2004; Pineda et al., 2005; Zuccato et al., 2005; Lynch et al., 2007) or experimental approaches increasing BDNF levels including environmental enrichment (Hockly et al., 2002; Spires et al., 2004) and dietary restriction (Duan et al., 2003) could protect neurons and alleviate symptoms in HD mice. Chronic administration of SSRIs including sertraline increased the expression of cAMP response element binding protein (CREB) and downstream gene BDNF in rodents (Nibuya, et al., 1995; 1996). These findings with our current study suggest that the sertraline may protect neurons by increase BDNF and/or 5-HT in HD, though we could not differentiate whether sertraline’s protection is directly mediated by 5-HT or indirectly by increasing BDNF levels. Nevertheless, sertraline is a safe and well-tolerated drug that can be administered for a long period of time. It is a valuable candidate for further clinical trials in HD patients.
It is interesting to note that BDNF levels are significantly increased in striatum but not in cortex of R6/2 mice by sertraline administration, it suggest that sertraline may also affect BDNF transport from cortex to striatum which is compromised in HD (Gauthier et al., 2004). Another point that we would mention here is that some opposite reports about the alterations of 5-HT levels in HD patients (Reynolds and Pearson 1987; Kish et al., 1987). One possibility is that mice may not exactly represent the human condition; another possibility is that striatal atrophy that occurs in HD may have variable effects on concentrations at different times in the disease course. Nevertheless, serotonin might also have direct neuroprotective effects on striatal and cortical neurons because other signals that activate cyclic AMP and CREB have been shown to protect neurons against excitotoxic, oxidative, and metabolic insults relevant to HD (Duman 1998; Nair and Vaidya 2006; Tardito et al., 2006; Malberg and Blendy 2005). The decreased levels of BDNF in the HD brains suggest that neurons have a reduced ability to cope with stress in HD. SSRIs can increase extracellular serotonin levels and serotonin synthesis (Goggi et al., 2002), and consequently can enhance serotonergic signaling and increase BDNF expression (Schmidt and Duman 2007; Duman and Monteggia 2006; Russo-Neustadt 2003; Duman 1998; Nair and Vaidyya 2006; Tardito et al., 2006). The increase of BDNF levels in brain cells of HD mice treated sertraline is associated with an attenuation of motor dysfunction and increase in survival of the mice.
Adult neurogenesis might be part of the physiological regenerative response and might thereby alter or alleviate symptoms, but it might also become impaired by the disease mechanism and thereby contribute to the symptoms of neurodegeneration. The increased neurogenesis in human HD indicates that neurogenesis is stimulated in the disease, possibly representing an adaptive response directed toward neuronal replacement (Curtis et al., 2003; Curtis et al., 2007). However, there is a significant decrease of neurogenesis in the dentate gyrus in R6/2 mice at late stages of disease, which suggest that neurogenesis is compromised in HD mice. The compromised neurogenesis may contribute to the alteration of normal function of hippocampus, such as memory deficit in R6/2 mice (Lione et al., 1999). The decreased neurogenesis in R6/2 mice is counteracted by sertraline administration, suggesting that sertraline may restore the neuronal function by ameliorate compromised neurogenesis in HD mice. Cell loss in the hippocampus of HD patients has been reported in a few cases (Vonsattel and DiFiglia, 1998). There is an electrophysiological and behavioral evidence for hippocampal dysfunction in R6/2 mice (Murphy et al., 2000). Altered neurogenesis may be involved in the hippocampal dysfunction. The young generated neurons in hippocampus have a greater degree of plasticity than mature neurons (Schmidt-Hieber et al., 2004).
The subventricular zone (SVZ) of the forebrain that overlies the caudate nucleus is one of the principal brain regions in which neurogenesis occurs in brain. In response to the degeneration that occurs in the caudate nucleus in Huntington's disease, the SVZ increases the production of progenitor cells that migrate towards the site of the damage where they can differentiate into mature neurons and glial cells (Cutis et al., 2007). The SVZ contains three main cell types and these are progenitor cells, glial cells and migratory neuroblasts; The SVZ comprises heterogeneous cell types that are maintained in an environment that is permissive to neurogenesis and gliogenesis, and responds to neurodegenerative changes in adjacent brain regions by increasing progenitor cell proliferation and neurogenesis in an attempt to replace the cells that die as a result of neurodegeneration. At the earlier stage of disease, when the self regeneration ability of neurogenesis can cope with the neurodegeneration, cells are still functional and no severe symptoms appear. As the disease progresses, the regeneration ability may not be able to cope with the neurodegenerative process, then symptoms will start exhibit. It was reported that HD patients increased cell proliferation estimated by cell cycle marker proliferating cell nuclear antigen (PCNA) immunostaining (Curtis et al. 2003). We did not find difference in the number of proliferating cells by labeling with BrdU in the SVZ of R6/2 mice compared to that in nontransgenic control mice, which is consistent with another report (Gil et al., 2005). This discrepancy could be due to several factors: R6/2 mice have much higher numbers of CAG repeats and inclusions are observed very much earlier than in HD patients; it is also possible that there is a difference in markers for labeling proliferating cells. PCNA is expressed cells undergoing DNA repair (Tomasevic et al., 1998); finally, the different stage of disease observed in HD patients and HD mice may also contribute to the discrepancy. Further analysis of HD brains by using combinations of several markers of proliferation is therefore warranted. Although our data do not allow any firm conclusions as to the relative contributions of neurogenesis to the pathology of HD, they do suggest that the impaired neurogenesis and motor dysfunction are amenable to therapeutic intervention. The contribution of neurogenesis to functional recovery in HD is difficult to ascertain in our present study since treatment with sertraline stimulates neurogenesis and may have additional beneficial effects on cell function.
There was no effect of sertraline on neuronal inclusions in our study. Misfolding and possibly aggregation of abnormal proteins are believed to be pathogenic; however, several lines of evidence indicate that inclusions are not the main cause of toxicity, and may represent a cellular protective response. Aggregation is a complex multistep process of protein conformational change (DiFiglia., 1997; Ross and Poirier 2004 & 2005). Sertraline did not affect htt inclusions, suggesting that beneficial effects of sertraline are not mediated by inhibiting htt aggregation.
Finally, we emphasize that the effective levels of sertraline in HD mice are comparable to or even less than levels in human antidepressant treatment, and sertraline is generally safe and well-tolerated. The preclinical findings obtained in the present study may provide a rationale for clinical trials of SSRI in humans with HD, or in mutation-positive individuals who do not yet manifest the disease.
Supplementary Material
Acknowledgements
We gratefully acknowledge the technical support for stereology analysis of Gay Rudow in Neuropathology at Johns Hopkins University and Laragen Inc. for genotyping service. This research was supported by the High Q foundation (to W. Duan) and NS NINDS 16375 (to CAR).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bemelmans AP, Horellou P, Pradier L, Brunet I, Colin P, Mallet J. Brain-derived neurotrophic factor-mediated protection of striatal neurons in an excitotoxic rat model of Huntington's disease, as demonstrated by adenoviral gene transfer. Hum Gene Ther. 1999;10(18):2987–2997. doi: 10.1089/10430349950016393. [DOI] [PubMed] [Google Scholar]
- Ciammola A, Sassone J, Cannella M, Calza S, Poletti B, Frati L, Squitieri F, Silani V. Low brain-derived neurotrophic factor (BDNF) levels in serum of Huntington's disease patients. Am J Med Genet B Neuropsychiatr Genet. 2007;144(4):574–577. doi: 10.1002/ajmg.b.30501. [DOI] [PubMed] [Google Scholar]
- Canals JM, Pineda JR, Torres-Peraza JF, Bosch M, Martin-Ibanez R, Munoz MT, Mengod G, Ernfors P, Alberch J. Brain-derived neurotrophic factor regulates the onset and severity of motor dysfunction associated with enkephalinergic neuronal degeneration in Huntington's disease. J Neurosci. 2004;24(35):7727–7739. doi: 10.1523/JNEUROSCI.1197-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Starling AJ, Wu N, Nguyen OK, Uzgil B, Soda T, Andre VM, Ariano MA, Levine MS. Increased GABAergic function in mouse models of Huntington's disease: reversal by BDNF. J Neurosci Res. 2004;78(6):855–867. doi: 10.1002/jnr.20344. [DOI] [PubMed] [Google Scholar]
- Ciammola A, Sassone J, Cannella M, Calza S, Poletti B, Frati L, Squitieri F, Silani V. Low brain-derived neurotrophic factor (BDNF) levels in serum of Huntington's disease patients. Am J Med Genet B Neuropsychiatr Genet. 2007 Jun 5;144(4):574–577. doi: 10.1002/ajmg.b.30501. [DOI] [PubMed] [Google Scholar]
- Cha JH. Transcriptional dysregulation in Huntington's disease. Trends Neurosci. 2003;23:387–392. doi: 10.1016/s0166-2236(00)01609-x. [DOI] [PubMed] [Google Scholar]
- Cotman CW, Berchtold NC. Exercise: a behavioral intervention to enhance brain health and plasticity. Trends Neurosci. 2002;25:295–301. doi: 10.1016/s0166-2236(02)02143-4. [DOI] [PubMed] [Google Scholar]
- Cowan CM, Raymond LA. Selective neuronal degeneration in Huntington's disease. Curr Top Dev Biol. 2006;75:25–71. doi: 10.1016/S0070-2153(06)75002-5. [DOI] [PubMed] [Google Scholar]
- Curtis MA, Penney EB, Pearson AG, Van Roon-Mom WMC, Butterworth NJ, Dragunow M, Connor B, Faull RLM. Increased cell proliferation and neurogenesis in the adult human Huntington's disease brain. Proc Natl Acad Sci U S A. 2003;100:9023–9027. doi: 10.1073/pnas.1532244100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MA, Eriksson PS, Faull RL. Progenitor cells and adult neurogenesis in neurodegenerative diseases and injuries of the basal ganglia. Clin. Exp Pharmacol Physiol. 2007;34:528–532. doi: 10.1111/j.1440-1681.2007.04609.x. [DOI] [PubMed] [Google Scholar]
- Cyr M, Caron MG, Johnson GA, Laakso A. Magnetic resonance imaging at microscopic resolution reveals subtle morphological changes in a mouse model of dopaminergic hyperfunction. NeuroImage. 2005;26:83–90. doi: 10.1016/j.neuroimage.2005.01.039. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intracellular inclusions and dystrophic neuritis in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- Duan W, Guo Z, Jiang H, Ware M, Li XJ, Mattson MP. Dietary restriction normalizes glucose metabolism and BDNF levels, slows disease progression, and increases survival in huntingtin mutant mice. Proc Natl Acad Sci U S A. 2003;100:2911–2916. doi: 10.1073/pnas.0536856100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan W, Guo Z, Jiang H, Ladenheim B, Xu X, Cadet JL, Mattson MP. Paroxetine retards disease onset and progression in Huntingtin mutant mice. Ann Neurol. 2004;55:590–594. doi: 10.1002/ana.20075. [DOI] [PubMed] [Google Scholar]
- Duman RS. Novel therapeutic approaches beyond the serotonin receptor. Biol Psychiatry. 1998;44(5):324–335. doi: 10.1016/s0006-3223(98)00031-6. [DOI] [PubMed] [Google Scholar]
- Duman RS. Role of neurotrophic factors in the etiology and treatment of mood disorders. Neuromolecular Med. 2004;5:11–25. doi: 10.1385/NMM:5:1:011. [DOI] [PubMed] [Google Scholar]
- Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry. 2006;59(12):1116–1127. doi: 10.1016/j.biopsych.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Goutan E, Marin C, Rey MJ, Ribalta T. Brain-derived neurotrophic factor in Huntington disease. Brain Res. 2000;866(1–2):257–261. doi: 10.1016/s0006-8993(00)02237-x. [DOI] [PubMed] [Google Scholar]
- Ferrigno P, Silver PA. Polyglutamine expansions: proteolysis, chaperones, and the dangers of promiscuity. Neuron. 2000;26:9–12. doi: 10.1016/s0896-6273(00)81132-0. [DOI] [PubMed] [Google Scholar]
- Gauthier LR, Charrin BC, Borrell-Pages M, Dompierre JP, Rangone H, Cordelieres FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, Saudou F. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118(1):127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Gil JM, Leist M, Popovic N, Brundin P, Petersen A. Asialoerythropoietin is not effective in the R6/2 line of Huntington's disease mice. BMC Neurosci. 2004;5:17. doi: 10.1186/1471-2202-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil JM, Mohapel P, Araujo IM, Popovic N, Li JY, Brundin P, Petersen A. Reduced hippocampal neurogenesis in R6/2 transgenic Huntington's disease mice. Neurobiol Dis. 2005;20:744–751. doi: 10.1016/j.nbd.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Goggi J, Pullar IA, Carney SL, Bradford HF. Modulation of neurotransmitter release induced by brain-derived neurotrophic factor in rat brain striatal slices in vitro. Brain Res. 2002;941:34–42. doi: 10.1016/s0006-8993(02)02505-2. [DOI] [PubMed] [Google Scholar]
- Grote HE, Bull ND, Howard ML, van Dellen A, Blakemore C, Bartlett PF, Hannan AJ. Cognitive disorders and neurogenesis deficits in Huntington's disease mice are rescued by fluoxetine. Eur J Neurosci. 2005;22:2081–2088. doi: 10.1111/j.1460-9568.2005.04365.x. [DOI] [PubMed] [Google Scholar]
- Ho LW, Carmichael J, Swartz J, Wyttenbach A, Rankin J, Rubinsztein DC. Differential loss of striatal projection neurons in Huntington disease. Proc. Natl. Acad. Sci. U.S.A. 1998;85:5733–5737. doi: 10.1073/pnas.85.15.5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockly E, Cordery PM, Woodman B, Mahal A, van Dellen A, Blakemore C, Lewis CM, Hannan AJ, Bates GP. Environmental enrichment slows disease progression in R6/2 Huntington's disease mice. Ann Neurol. 2002;51(2):235–242. doi: 10.1002/ana.10094. [DOI] [PubMed] [Google Scholar]
- Jin K, LaFevre-Bernt M, Sun Y, Chen S, Gafni J, Crippen D, Logvinova A, Ross CA, Greenberg DA, Ellerby LM. FGF-2 promotes neurogenesis and neuroprotection and prolongs survival in a transgenic mouse model of Huntington's disease. Proc Natl Acad Sci U S A. 2005;102:18189–18194. doi: 10.1073/pnas.0506375102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kells AP, Fong DM, Dragunow M, During MJ, Young D, Connor B. AAV-mediated gene delivery of BDNF or GDNF is neuroprotective in a model of Huntington disease. Mol Ther. 2004 May;9(5):682–688. doi: 10.1016/j.ymthe.2004.02.016. [DOI] [PubMed] [Google Scholar]
- Kish SJ, Shannak K, Hornykiewicz O. Elevated serotonin and reduced dopamine in subregionally divided Huntington's disease striatum. Ann Neurol. 1987;22(3):386–389. doi: 10.1002/ana.410220318. [DOI] [PubMed] [Google Scholar]
- Lazic SE, Grote H, Armstrong RJ, Blakemore C, Hannan AJ, van Dellen A, Barker RA. Decreased hippocampal cell proliferation in R6/1 Huntington's mice. Neuroreport. 2004 Apr 9;15(5):811–813. doi: 10.1097/00001756-200404090-00014. [DOI] [PubMed] [Google Scholar]
- Lee J, Duan W, Mattson MP. Evidence that brain-derived neurotrophic factor is required for basal neurogenesis and mediates, in part, the enhancement of neurogenesis by dietary restriction in the hippocampus of adult mice. J Neurochem. 2002;82:1367–1375. doi: 10.1046/j.1471-4159.2002.01085.x. [DOI] [PubMed] [Google Scholar]
- Lione LA, Carter RJ, Hunt MJ, Bates GP, Morton AJ, Dunnett SB. Selective discrimination learning impairments in mice expressing the human Huntington’s disease mutation. J. Neurosci. 1999;19:10428–10437. doi: 10.1523/JNEUROSCI.19-23-10428.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch G, Kramar EA, Rex CS, Jia Y, Chappas D, Gall CM, Simmons DA. Brain-derived neurotrophic factor restores synaptic plasticity in a knock-in mouse model of Huntington's disease. J Neurosci. 2007 Apr 18;27(16):4424–4434. doi: 10.1523/JNEUROSCI.5113-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malberg JE, Blendy JA. Antidepressant action: to the nucleus and beyond. Trends Pharmacol Sci. 2005;26:631–638. doi: 10.1016/j.tips.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Maudesley S, Martin B. A neural signaling triumvirate that influences ageing and age-related disease: insulin/IGF-1, BDNF and serotonin. Ageing Res Rev. 2004;3(4):445–464. doi: 10.1016/j.arr.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Murphy KP, Carter RJ, Lione LA, Mangiarini L, Mahal A, Bates GP, Dunnett SB, Morton AJ. Abnormal synaptic plasticity and impaired spatial cognition in mice transgenic for exon 1 of the human Huntington's disease mutation. J Neurosci. 2000;20(13):5115–5123. doi: 10.1523/JNEUROSCI.20-13-05115.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers RH, Vonsattel JP, Stevens TJ, Cupples LA, Richardson EP, Martin JB, Bird ED. Clinical and neuropathologic assessment of severity in Huntington's disease. Neurology. 1988;38(3):341–347. doi: 10.1212/wnl.38.3.341. [DOI] [PubMed] [Google Scholar]
- Nair A, Vaidya VA. Cyclic AMP response element binding protein and brain-derived neurotrophic factor: molecules that modulate our mood? J Biosci. 2006;31(3):423–434. doi: 10.1007/BF02704114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci. 1995;15(11):7539–7547. doi: 10.1523/JNEUROSCI.15-11-07539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nibuya M, Nestler EJ, Duman RS. Chronic antidepressant administration increases the expression of cAMP response element binding protein (CREB) in rat hippocampus. J Neurosci. 1996;16(7):2365–2372. doi: 10.1523/JNEUROSCI.16-07-02365.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips W, Jennifer Morton A, Barker RA. Limbic neurogenesis/plasticity in the R6/2 mouse model of Huntington's disease. Neuroreport. 2006;17:1623–1627. doi: 10.1097/01.wnr.0000236855.85962.f6. [DOI] [PubMed] [Google Scholar]
- Phillips W, Morton AJ, Barker RA. Abnormalities of neurogenesis in the R6/2 mouse model of Huntington's disease are attributable to the in vivo microenvironment. J Neurosci. 2005;25:11564–11576. doi: 10.1523/JNEUROSCI.3796-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda JR, Canals JM, Bosch M, Adell A, Mengod G, Artigas F, Ernfors P, Alberch J. Brain-derived neurotrophic factor modulates dopaminergic deficits in a transgenic mouse model of Huntington's disease. J Neurochem. 2005;93(5):1057–1068. doi: 10.1111/j.1471-4159.2005.03047.x. [DOI] [PubMed] [Google Scholar]
- Ranen NG, Lipsey JR, Treisman G, Ross CA. Sertraline in the treatment of severe aggressiveness in Huntington’s disease. J Neuropsychiatry Clin Neurosci. 1996;8:338–340. doi: 10.1176/jnp.8.3.338. [DOI] [PubMed] [Google Scholar]
- Reiner A, Albin RL, Anderson KD, D'Amato CJ, Penney JB, Young AB. Differential loss of striatal projection neurons in Huntington disease. Proc. Natl. Acad. Sci. U.S.A. 1998;85:5733–5737. doi: 10.1073/pnas.85.15.5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds GP, Pearson SJ. Decreased glutamic acid and increased 5-hydroxytryptamine in Huntington's disease brain. Neurosci Lett. 22. 1987;78(2):233–238. doi: 10.1016/0304-3940(87)90639-2. [DOI] [PubMed] [Google Scholar]
- Reynolds GP, Dalton CF, Tillery CL, Mangiarini L, Davies SW, Bates GP. Brain neurotransmitter deficits in mice transgenic for the Huntington's disease mutation. J Neurochem. 1999;72:1773–1776. doi: 10.1046/j.1471-4159.1999.721773.x. [DOI] [PubMed] [Google Scholar]
- Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10 Suppl:S10–S17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- Ross CA, Poirier MA. Opinion: What is the role of protein aggregation in neurodegeneration? Nat Rev Mol Cell Bio. 2005;6:891–898. doi: 10.1038/nrm1742. [DOI] [PubMed] [Google Scholar]
- Russo-Neustadt A. Brain-derived neurotrophic factor, behavior, and new directions for the treatment of mental disorders. Semin Clin Neuropsychiatry. 2003;8(2):109–118. doi: 10.1053/scnp.2003.50014. [DOI] [PubMed] [Google Scholar]
- Schmidt HD, Duman RS. The role of neurotrophic factors in adult hippocampal neurogenesis, antidepressant treatments and animal models of depressive-like behavior. Behav Pharmacol. 2007;18(5–6):391–418. doi: 10.1097/FBP.0b013e3282ee2aa8. [DOI] [PubMed] [Google Scholar]
- Schmidt-Hieber C, Jonas P, Bischofberger J. Enhanced synaptic plasticity in newly generated granule cells of the adult hippocampus. Nature. 2004;429:184–187. doi: 10.1038/nature02553. [DOI] [PubMed] [Google Scholar]
- Spires TL, Grote HE, Varshney NK, Cordery PM, van Dellen A, Blakemore C, Hannan AJ. Environmental enrichment rescues protein deficits in a mouse model of Huntington's disease, indicating a possible disease mechanism. J Neurosci. 2004;24(9):2270–2276. doi: 10.1523/JNEUROSCI.1658-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugars KL, Rubinsztein DC. Transcriptional abnormalities in Huntington disease. Trends Genet. 2003;19:233–238. doi: 10.1016/S0168-9525(03)00074-X. [DOI] [PubMed] [Google Scholar]
- Slaughter JR, Martens MP, Slaughter KA. Depression and Huntington’s disease: prevelance, clinical manifestations, etiology, and treatment. CNS Spectr. 2001;6:306–326. doi: 10.1017/s109285290002201x. [DOI] [PubMed] [Google Scholar]
- Tardito D, Perez J, Tiraboschi E, Musazzi L, Racagni G, Popoli M. Signaling pathways regulating gene expression, neuroplasticity, and neurotrophic mechanisms in the action of antidepressants: a critical overview. Pharmacol Rev. 2006;58(1):115–134. doi: 10.1124/pr.58.1.7. [DOI] [PubMed] [Google Scholar]
- Tomasevic G, Kamme F, Wieloch T. Changes in proliferating cell nuclear antigen, a protein involved in DNA repair, in vulnerable hippocampal neurons following global cerebral ischemia. Brain Res Mol Brain Res. 1998;60(2):168–176. doi: 10.1016/s0169-328x(98)00173-9. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP., Jr Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol. 1985;44(6):559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57(5):369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Ninomiya M, Hernandez Acosta P, Garcia-Verdugo JM, Sunabori T, Sakaguchi M, Adachi K, Kojima T, Hirota Y, Kawase T, Araki N, Abe K, Okano H, Sawamoto K. Subventricular zone-derived neuroblasts migrate and differentiate into mature neurons in the post-stroke adult striatum. J Neurosci. 2006;26:6627–6636. doi: 10.1523/JNEUROSCI.0149-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M, Hartke C, Jimeno A, Li J, He P, Zabelina Y, Hidalgo M, Baker SD. Specific method for determination of gefitinib in human plasma, mouse plasma and tissues using high performance liquid chromatography coupled to tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;819:73–80. doi: 10.1016/j.jchromb.2005.01.027. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Cattaneo E. Role of brain-derived neurotrophic factor in Huntington's disease. Prog Neurobiol. 2007;81(5–6):294–330. doi: 10.1016/j.pneurobio.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E. Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science. 2001;293(5529):493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Liber D, Ramos C, Tarditi A, Rigamonti D, Tartari M, Valenza M, Cattaneo E. Progressive loss of BDNF in a mouse model of Huntington's disease and rescue by BDNF delivery. Pharmacol Res. 2005 Aug;52(2):133–139. doi: 10.1016/j.phrs.2005.01.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.