

Abstract
Structural analogues of JS-K, an anti-cancer lead compound, were prepared and their in vitro anti-leukemic activity was determined. The rate of nitric oxide release from the corresponding diazeniumdiolate anions did not appear to affect the anti-leukemic activity of the prodrug forms. Two compounds with potent inhibitory activity and a potentially favorable toxicological profile were identified.
Glutathione S-transferase (GST), which is frequently over-expressed in cancer tissue, is a key phase II detoxification enzyme that catalyzes the conjugation of electrophilic xenobiotics with glutathione (GSH).1 Our laboratory has developed several diazeniumdiolate-based nitric oxide (NO) prodrugs2 that are designed as substrates for GST, notably O2-(2,4-dinitrophenyl) 1-[(4-ethoxycarbonyl)piperazin-1-yl]diazen-1-ium-1,2-diolate (JS-K, Scheme 1).3
Scheme 1.
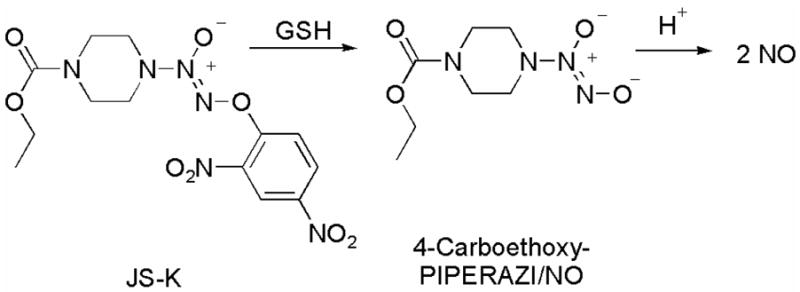
The anti-cancer lead compound JS-K, which targets GST-overexpressing cells, and its mechanism of nitric oxide release.
JS-K is a potent anti-tumor agent against HL-60 human leukemia xenografts in mice, cutting their growth rate by about half and inducing significant necrosis in the tumor mass.3a Furthermore, this drug was found to be active against: human prostate cancer xenografts in mice;3a an orthotopic model of liver cancer in rats;3d and multiple myeloma xenografts in mice.3e In this study, we synthesized numerous structural analogues of JS-K and studied their in vitro anti-leukemic activity by determining the ability of these compounds to inhibit the growth of HL-60 human leukemia cells.
It is reported that human leukemia cells are sensitive to cytotoxic effects of nitric oxide.4 While additional pathways may be operational, nitric oxide appears to be an important component of the potent JS-K cytotoxic activity.3 Nitric oxide release from JS-K is proposed to occur by nucleophilic aromatic displacement of the spontaneously NO-releasing diazeniumdiolate anion2 by glutathione (Scheme 1). In the absence of strong nucleophiles such as GSH, JS-K is relatively stable in aqueous buffer; the rate constant for hydrolysis of JS-K in pH 7.4 buffer at 37 °C was reported as 1 × 10−6 s−1, which translates to a half-life of over a week.3a In the presence of glutathione, however, JS-K nearly completely disappeared within 30 min and a second order rate constant of 1.0 M−1s−1 was reported.3a The rate constant for hydrolysis of 4-carboethoxy-PIPERAZI/NO in pH 7.4 buffer at 37 °C was reported as 1.9 × 10−3 s−1, which corresponds to a half-life of 6 min (Scheme 1).3a
Earlier, we prepared a number of JS-K structural analogues and determined their in vitro anti-leukemic activity.3a However, a systematic study of the effect of varying the half-life of the diazeniumdiolate ion decomposition on the anti-proliferative activity of the corresponding O2-aryl prodrug form was not conducted. Hence, our work was initiated by studying the effect of varying the rate of dissociation of the diazeniumdiolate ion to form NO on the cytotoxicity of the corresponding O2-(2,4-dinitrophenyl) derivative, which was determined by their ability to inhibit human leukemia HL-60 cell proliferation.
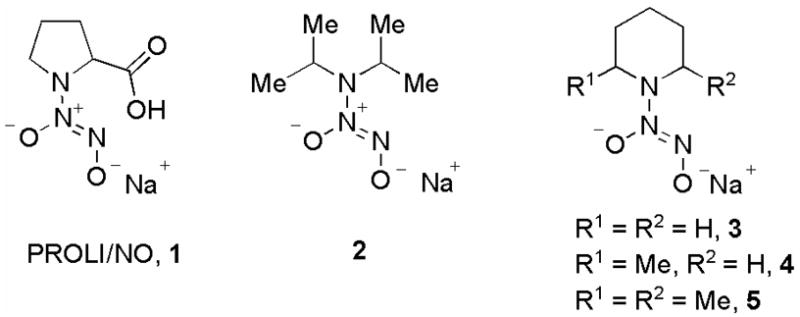
A number of diazeniumdiolate anions with a range of half-lives were identified and prepared using reported methods from the corresponding secondary amine.5 As listed in Table 1, the reported half-life of decomposition of PROLI/NO (1) is 2 s while the half-lives of decomposition of 2–5 varied from 78 s to 50 min.5 The O2-(2,4-dinitrophenyl) derivatives 6–10 of these diazeniumdiolate anions were prepared using reported methods (Scheme 2).5c,d
Table 1.
Reported half-lives of some diazeniumdiolate ions in aqueous buffer and the corresponding O2-arylated prodrug forms.
 | ||
|---|---|---|
| NO donor | Half-lifea | O2-(2,4-dinitrophenyl) derivative |
| 4-carboethoxy-PIPERAZI/NO | 6.0 min | JS-K |
| PROLI/NO, 1 | 2 s | 6 |
| 2 | 4.3 min | 7 |
| 3 | 78 s | 8 |
| 4 | 8.3 min | 9 |
| 5 | 50 min | 10 |
in pH 7.4 buffer at 37 °C.
Scheme 2.
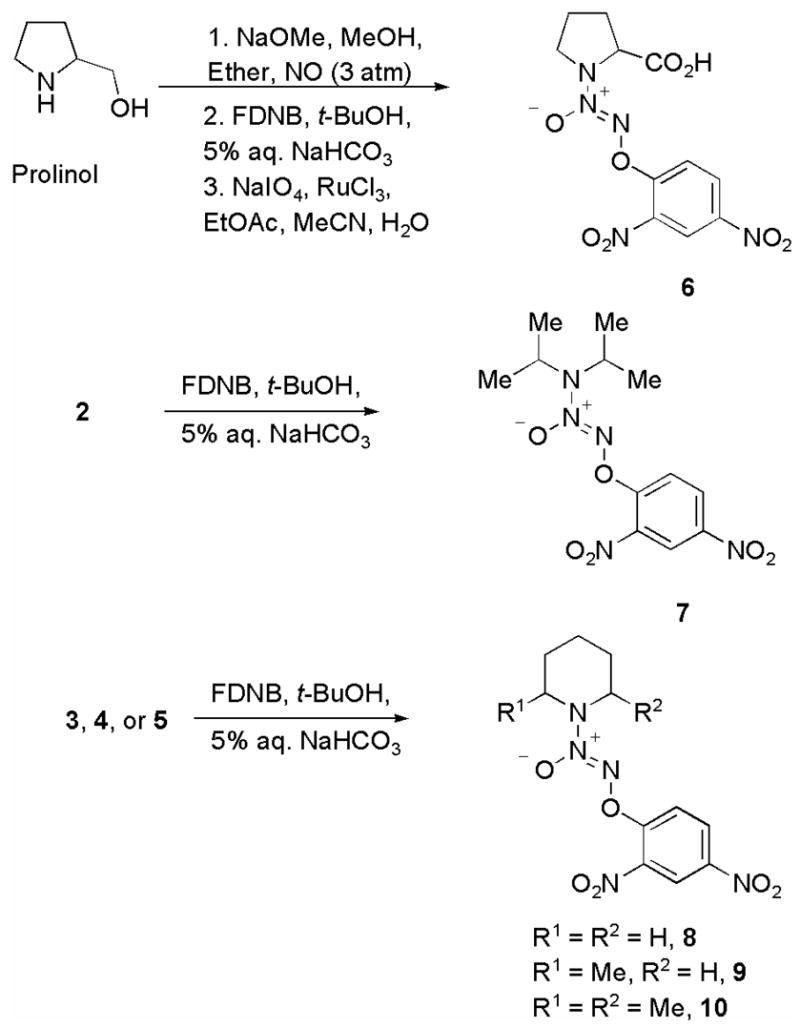
Synthesis of some O2-(2,4-dinitrophenyl) diazeniumdiolates. FDNB: 1-Fluoro-2,4-dinitrobenzene.
It is noteworthy that the diazeniumdiolate ions PROLI/NO,5a–c 2,5d and 55d are nitric oxide prodrugs with potential clinical relevance as a possible byproduct of NO release from these compounds, the corresponding nitrosamine, is reported to have minimal carcinogenicity, thus suggesting a favorable toxicological profile.6 For example, N-nitrosoproline, a possible byproduct of PROLI/NO decomposition, was not found to display any carcinogenic activity in several animal models (Scheme 3).6e–h
Scheme 3.
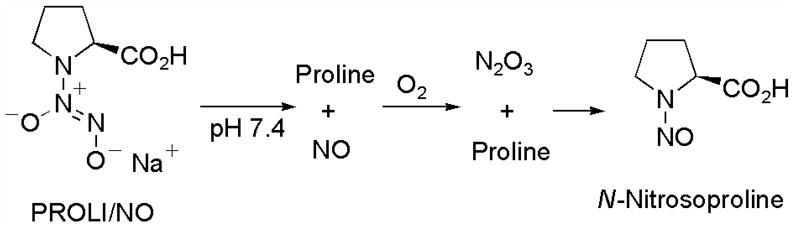
Proposed mechanism of nitrosamine formation during diazeniumdiolate ion decomposition in aqueous buffer.
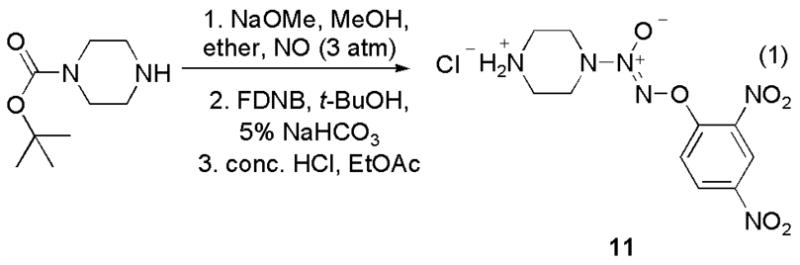
Next, we prepared the O2-aryl diazeniumdiolate 11 in three steps from N-(tert-butoxy)piperazine using a reported method (eq 1).7 This compound served as a convenient divergence point in the preparation of a variety of JS-K analogues by exposing 11 to a series of electrophiles (Scheme 4).8
Scheme 4.
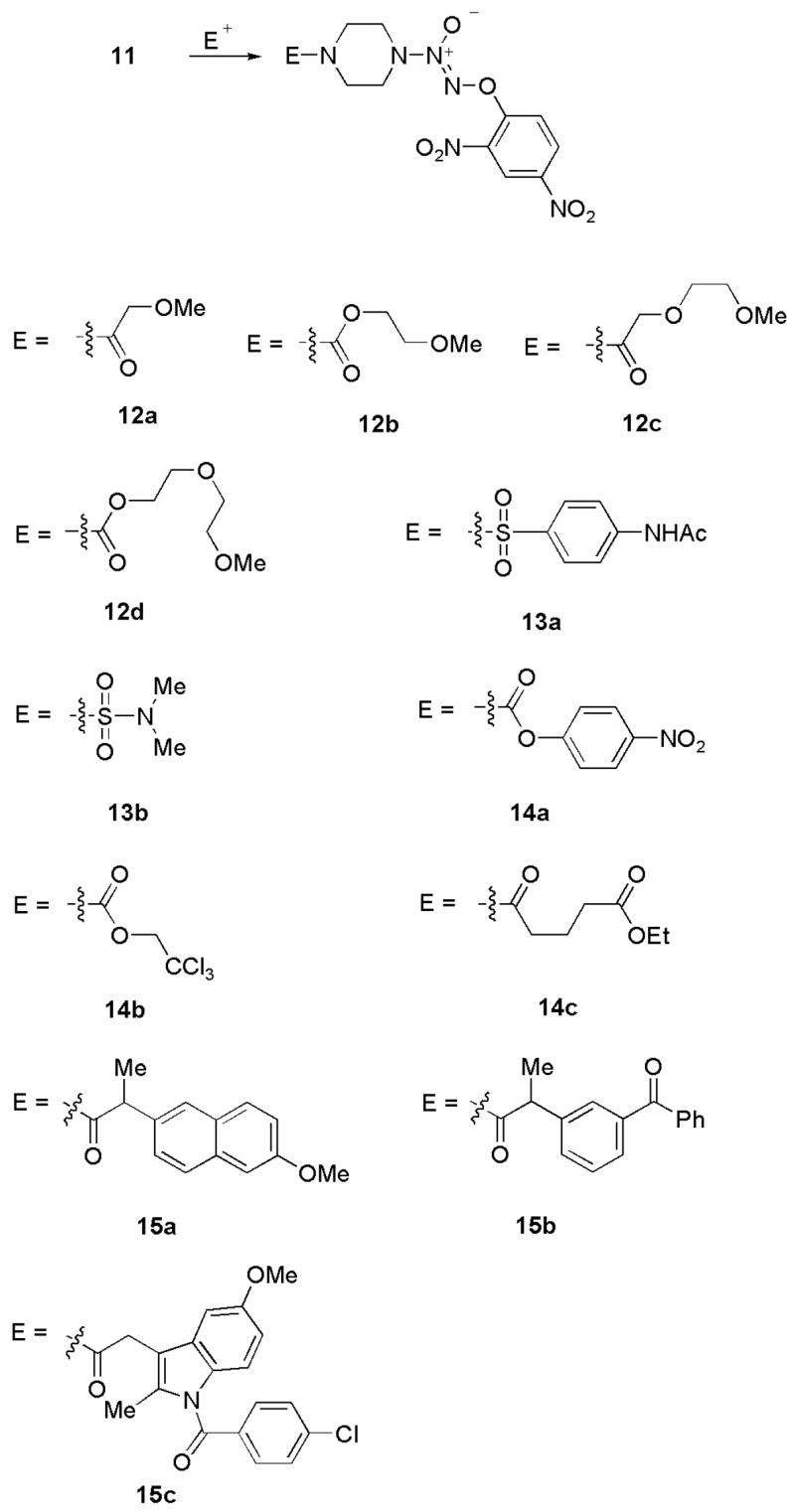
Synthesis of some structural analogues of JS-K from 11.
The methyl ethers 12a–12d were prepared by treating 11 with the corresponding acyl chloride. The carbamate portion of JS-K was replaced by a sulfamate functionality in compounds 13a and 13b. Analogues 14a–14c, where the ester group of the carbamate of JS-K was altered, were prepared in a similar fashion by reaction of the corresponding acyl chloride with 11.
Diazeniumdiolate-based nitric oxide donors conjugated with non-steroidal anti-inflammatory drugs (NSAIDs) are assuming importance as multi-faceted therapeutic agents.9 Thus, we prepared several JS-K-type NSAID-conjugates 15a–15c using the precursor 11.
The in vitro anti-leukemic activity of these compounds was studied using the HL-60 human leukemia cell line by a reported procedure.3a The IC50 of JS-K was reported as 0.2–0.5 μM (Table 2, entry 1).3a
Table 2.
In vitro anti-leukemic activity of JS-K and its structural analogues.
| entry | compound | IC50, μM |
|---|---|---|
| 1 | JS-K | 0.2–0.5 |
| 2 | 6 | >50 |
| 3 | 7 | 3.2 |
| 4 | 8 | 1.7 |
| 5 | 9 | 2.5 |
| 6 | 10 | 2.0 |
| 7 | 11 | 5.0 |
| 8 | 12a | 2.3 |
| 9 | 12b | 7.2 |
| 10 | 12c | 4.5 |
| 11 | 12d | 8.8 |
| 12 | 13a | 6.6 |
| 13 | 13b | 6.8 |
| 14 | 14a | 7.4 |
| 15 | 14b | 8.3 |
| 16 | 14c | 6.0 |
| 17 | 15a | 9.8 |
| 18 | 15b | 7.4 |
| 19 | 15c | 9.1 |
Although the aqueous solubility of 6 was much higher than any of the other JS-K analogues prepared in this study, its cytotoxicity was diminished (Table 2, entry 2). The other prodrugs 7–10 were tested in this assay and the half-life of the diazeniumdiolate anion in buffer did not appear to affect the cytotoxicity. The derivative 7 and the piperidine analogues 8–10 displayed potent but comparable activity suggesting that the differences in the rate of NO release from the corresponding diazeniumdiolate ions 2–5 did not affect potency of the diazeniumdiolate prodrug (Table 2, entries 3–6).
In an earlier study, it was reported that diazeniumdiolate anions with extended durations of nitric oxide release were more effective at inhibiting the growth of this human leukemia cell line than their counterparts with lesser durations of NO release.4 In this study, three compounds, DETA/NO with a half-life of NO release of 20 h, PAPA/NO, which has a half-life of 30 min, and MAHMA/NO, whose half-life is 2 min, were tested for their anti-proliferative activity using the HL-60 human leukemia cell line.4 Among these compounds, it was reported that the best inhibitor of cell proliferation was DETA/NO (ID50 = 50 μM), followed by PAPA/NO (ID50 = 100 μM), and MAHMA/NO, which was found to be inactive. In contrast, our study indicates that there were no observable effects of altering the rate of NO release on the cytotoxicity of these prodrugs, suggesting a role for nitric oxide-independent cytotoxic pathways.3 Furthermore, it is worth noting that, by releasing NO spontaneously, DETA/NO, PAPA/NO, and MAHMA/NO act on leukemia cells by a bystander effect while the compounds described in this study get activated to release NO intracellularly. Thus, the rate of NO release intracellularly may not be as important for cytotoxicity as when it is released extracellularly.
Earlier, we reported that 2 and 5 decomposed in aqueous buffer to release nearly quantitative amounts of NO suggesting that nitrosamine formation is a minor process.5d Due to their potent cytotoxicity and the potentially favorable toxicological profile of their corresponding parent diazeniumdiolate anions, 7 and 10 are noteworthy; further in vitro and in vivo testing of their anti-cancer activity will be carried out in due course.
The ether derivatives 12a–12d were all found to display lower potency than JS-K but comparable to that of 11; the highest cytotoxicity was shown by 12a, whose IC50 was determined as 2.3 μM (Table 2, entries 8–11). Changing the carbamate functionality to a sulfamate group (13a and 13b) or varying the ester portion of the carbamate group (14a–14c) resulted in diminished anti-proliferative activity (Table 2, entries 12–16). The IC50 values of all NSAID derivatives were closer to 10 μM (Table 2, entries 17–19). These results are consistent with those of our earlier study, wherein we observed that minor structural modifications led to substantial decrease in potency.3d For example, when the ethyl carbamate of JS-K was changed to a methyl carbamate, the potency of the resulting compound diminished from IC50 0.2–0.5 μM to 3.5 μM.3d
While the molecular mechanism of the potent anti-leukemic activity of JS-K remains to be fully determined, this study further reinforces the unique features of this compound that result in a diminution of potency with structural perturbation or conjugation of other potential pharmacophores, leading us to speculate that more than one cytotoxic pathway may be involved; the nature of such alternate pathways is currently being explored in our laboratories.
Supplementary Material
Supplementary Material
Analytical data for all new compounds. Supplementary data associated with this article can be found, in the online version, at [to be inserted later].
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, as well as by National Cancer Institute contract N01-CO-12400 to SAIC-Frederick.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.(a) Townsend DM, Tew KD. Oncogene. 2003;22:7369. doi: 10.1038/sj.onc.1206940. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Armstrong RN. Chem Res Toxicol. 1991;4:131. doi: 10.1021/tx00020a001. [DOI] [PubMed] [Google Scholar]; (c) Armstrong RN. Chem Res Toxicol. 1997;10:2. doi: 10.1021/tx960072x. [DOI] [PubMed] [Google Scholar]; (d) Zhao G, Wang X. Curr Med Chem. 2006;13:1461. doi: 10.2174/092986706776872934. [DOI] [PubMed] [Google Scholar]
- 2.(a) Scatena R, Bottoni P, Martorana GE, Giardina B. Expert Opin Investig Drugs. 2005;14:835. doi: 10.1517/13543784.14.7.835. [DOI] [PubMed] [Google Scholar]; (b) Keefer LK. Curr Top Med Chem. 2005;5:625. doi: 10.2174/1568026054679380. [DOI] [PubMed] [Google Scholar]; (c) Keefer LK. Annu Rev Pharmacol Toxicol. 2003;43:585. doi: 10.1146/annurev.pharmtox.43.100901.135831. [DOI] [PubMed] [Google Scholar]; (d) Hrabie JA, Keefer LK. Chem Rev. 2002;102:1135. doi: 10.1021/cr000028t. [DOI] [PubMed] [Google Scholar]; (e) Keefer LK, Nims RW, Davies KM, Wink DA. Methods Enzymol. 1996;268:281. doi: 10.1016/s0076-6879(96)68030-6. [DOI] [PubMed] [Google Scholar]
- 3.(a) Shami PJ, Saavedra JE, Wang LY, Bonifant CL, Diwan BA, Singh SV, Gu Y, Fox SD, Buzard GS, Citro ML, Waterhouse DJ, Davies KM, Ji X, Keefer LK. Mol Cancer Ther. 2003;2:409. [PubMed] [Google Scholar]; (b) Ren Z, Kar S, Wang Z, Wang M, Saavedra JE, Carr BI. J Cell Physiol. 2003;197:426. doi: 10.1002/jcp.10380. [DOI] [PubMed] [Google Scholar]; (c) Liu J, Li C, Qu W, Leslie E, Bonifant CL, Buzard GS, Saavedra JE, Keefer LK, Waalkes MP. Mol Cancer Ther. 2004;3:709. [PubMed] [Google Scholar]; (d) Shami PJ, Saavedra JE, Bonifant CL, Chu J, Udupi V, Malaviya S, Carr BI, Kar S, Wang M, Jia L, Ji X, Keefer LK. J Med Chem. 2006;49:4356. doi: 10.1021/jm060022h. [DOI] [PubMed] [Google Scholar]; (e) Kiziltepe T, Hideshima T, Ishitsuka K, Ocio EM, Raje N, Catley L, Li CQ, Trudel LJ, Yasui H, Vallet S, Kutok JL, Chauhan D, Mitsiades CS, Saavedra JE, Wogan GN, Keefer LK, Shami PJ, Anderson KC. Blood. 2007;110:709. doi: 10.1182/blood-2006-10-052845. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Udupi V, Yu M, Malaviya S, Saavedra JE, Shami PJ. Leukemia Res. 2006;30:1279. doi: 10.1016/j.leukres.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 4.Shami PJ, Sauls DL, Weinberg JB. Leukemia. 1998;12:1461. doi: 10.1038/sj.leu.2401131. [DOI] [PubMed] [Google Scholar]
- 5.(a) Saavedra JE, Southan GJ, Davies KM, Lundell A, Markou C, Hanson SR, Adrie C, Hurford WE, Zapol WM, Keefer LK. J Med Chem. 1996;39:4361. doi: 10.1021/jm960616s. [DOI] [PubMed] [Google Scholar]; (b) Waterhouse DJ, Saavedra JE, Davies KM, Citro ML, Xu X, Powell DA, Grimes GJ, Potti GK, Keefer LK. J Pharm Sci. 2006;95:108. doi: 10.1002/jps.20486. [DOI] [PubMed] [Google Scholar]; (c) Chakrapani H, Showalter BM, Kong L, Keefer LK, Saavedra JE. Org Lett. 2007;9:3409. doi: 10.1021/ol701419a. [DOI] [PubMed] [Google Scholar]; (d) Chakrapani H, Showalter BM, Citro ML, Keefer LK, Saavedra JE. Org Lett. 2007 doi: 10.1021/ol7019636. in press. [DOI] [PubMed] [Google Scholar]
- 6.(a) Lijinsky W. Chemistry and Biology of N-Nitroso Compounds. Cambridge University Press; Cambridge: 1992. [Google Scholar]; (b) Lijinsky W. Cancer Metastasis Rev. 1987;6:301. doi: 10.1007/BF00144269. [DOI] [PubMed] [Google Scholar]; (c) Lijinsky W. In: Chemical Carcinogenesis. Feo F, Pani P, Columbano A, Garcea R, editors. Plenum Publishing Corp; New York: 1988. p. 639. [Google Scholar]; (d) Lijinsky W. In: Genotoxicology of N-Nitroso Compounds. Rao TK, Lijinsky W, Epler JL, editors. Plenum Publishing Corp; New York: 1984. p. 192. [Google Scholar]; (e) Brunnemann KD, Enzmann HG, Perrone CE, Iatropoulos MJ, Williams GM. Arch Toxicol. 2002;76:606. doi: 10.1007/s00204-002-0380-4. [DOI] [PubMed] [Google Scholar]; (f) Negishi T, Shiotani T, Fujikawa K, Hayatsu H. Mutat Res. 1991;252:119. doi: 10.1016/0165-1161(91)90012-w. [DOI] [PubMed] [Google Scholar]; (g) Mirvish SS, Bulay O, Runge RG, Patil K. J Natl Cancer Inst. 1980;64:1435. doi: 10.1093/jnci/64.6.1435. [DOI] [PubMed] [Google Scholar]; (h) Nixon JE, Wales JH, Scanlan RA, Bills DD, Sinnhuber RO. Food Cosmetics Toxicol. 1976;14:133. doi: 10.1016/s0015-6264(76)80257-x. [DOI] [PubMed] [Google Scholar]
- 7.Saavedra JE, Booth MN, Hrabie JA, Davies KM, Keefer LK. J Org Chem. 1999;64:5124. doi: 10.1021/jo9901539. [DOI] [PubMed] [Google Scholar]
- 8.General procedure. A solution of 11 in DCM was reacted with a solution of one equivalent of the electrophile and two equivalents of triethylamine. The reaction mixture was diluted with DCM, washed with dil. HCl and aq. sodium bicarbonate. The organic layer was separated, dried (MgSO4), filtered, and the solvent was removed under reduced pressure to form a solid. This was then triturated with ether, filtered and dried under vacuum.
- 9.(a) Velázquez CA, Praveen Rao PN, Citro ML, Keefer LK, Knaus EE. Bioorg Med Chem. 2007;15:4767. doi: 10.1016/j.bmc.2007.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Velazquez CA, Praveen Rao PN, Knaus EE. J Med Chem. 2005;48:4061. doi: 10.1021/jm050211k. [DOI] [PubMed] [Google Scholar]; (c) Velazquez CA, Praveen Rao PN, McDonald R, Knaus EE. Bioorg Med Chem. 2005;13:2749. doi: 10.1016/j.bmc.2005.02.034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Analytical data for all new compounds. Supplementary data associated with this article can be found, in the online version, at [to be inserted later].