

Abstract
Aims
We assessed whether hypoosmotic swelling of cardiac myocytes activates volume-sensitive Cl− current (ICl,swell) via the angiotensin II (AngII)-reactive oxygen species (ROS) signalling cascade. The AngII-ROS pathway previously was shown to elicit ICl,swell upon mechanical stretch of β1D integrin. Integrin stretch and osmotic swelling are, however, distinct stimuli. For example, blocking Src kinases stimulates swelling-induced but inhibits stretch-induced ICl,swell.
Methods and results
ICl,swell was measured in rabbit ventricular myocytes by whole-cell voltage clamp. Swelling-induced ICl,swell was completely blocked by losartan and eprosartan, AngII type I receptor (AT1) antagonists. AT1 stimulation transactivates epidermal growth factor receptor (EGFR) kinase. Blockade of EGFR kinase with AG1478 abolished both ICl,swell and AngII-induced Cl− current, whereas exogenous EGF evoked a Cl− current that was suppressed by osmotic shrinkage. Phosphatidylinositol 3-kinase (PI-3K) is downstream of EGFR kinase, and PI-3K inhibitors LY294002 and wortmannin blocked ICl,swell. Ultimately, AngII signals via NADPH oxidase (NOX) and superoxide anion, . NOX inhibitors, diphenyleneiodonium, apocynin and gp91ds-tat, eliminated ICl,swell, whereas scramb-tat, an inactive gp91ds-tat analogue, was ineffective. rapidly dismutates to H2O2. Consistent with H2O2 being a downstream effector, catalase inhibited ICl,swell, and exogenous H2O2 overcame suppression of ICl,swell by AT1 receptor, EGFR kinase, and PI-3K blockers. H2O2-induced current was not blocked by osmotic shrinkage, however.
Conclusion
Activation of ICl,swell by osmotic swelling is controlled by the AngII-ROS cascade, the same pathway previously implicated in ICl,swell activation by integrin stretch. This in part explains why ICl,swell is persistently activated in several models of cardiac disease.
Keywords: Cl-channel, Angiotensin, NADPH oxidase, Signal transduction, Stretch/m-e coupling
1. Introduction
Volume-sensitive Cl− current, ICl,swell, is elicited by osmotic swelling and isosmotic hydrostatic pressure-induced inflation of cardiac myocytes, and the current is persistently activated in myocytes isolated from ischaemic and non-ischaemic models of dilated cardiomyopathy in which angiotensin II (AngII) levels are elevated (for review,1–3). ICl,swell influences cardiac electrical activity4,5 and cell volume regulation6 and may have a role in ischaemic preconditioning7 and apoptosis.8
ICl,swell also is activated by stretching β1D integrins with paramagnetic beads coated with anti-β1D monoclonal antibodies; integrin-bound beads are pulled towards an electromagnet placed above the cells.9–11 Integrin stretch elicits ICl,swell via Src, focal adhesion kinase (FAK), and the AngII AT1 receptor signalling cascade. AT1 signalling involves transactivation of epidermal growth factor receptor (EGFR) kinase, phosphatidylinositol 3-kinase (PI-3K), sarcolemmal NADPH oxidase (NOX), superoxide anion ( ), and ultimately H2O2.12–14 AngII is released by cardiac stretch, stretch causes AT1-dependent hypertrophy,15 and AngII activates NOX in cardiomyocytes16–19 and the vasculature.14,20 AngII and stretch also were recently found to destabilize Kv4.3 mRNA in neonatal rat myocytes via NOX-dependent production.19
Although ICl,swell is evoked both by stretching β1D integrins and by osmotic swelling, these are different stimuli and may signal by different pathways. Osmotic swelling dilutes the intracellular milieu and reduces its ionic strength, while integrin stretch is localized and does not alter the contents of the cytoplasm. Furthermore, PP2, an inhibitor of Src family tyrosine kinases, blocks ICl,swell activation upon integrin stretch9 consistent with its role as an upstream mediator of NOX activity,12 whereas PP2 augments ICl,swell in osmotically swollen myocytes.21–23
The goal of the present study was to determine whether osmotic control of ICl,swell utilizes the same AngII signalling cascade engaged by β1D integrin stretch, despite differences between the stimuli and observations that Src kinase inhibition has opposite effects on swelling- and stretch-induced ICl,swell. We found that activation of ICl,swell by osmotic swelling was abrogated by inhibition of AT1, EGFR kinase, PI-3K, or NOX and by scavenging H2O2. Moreover, exogenous epidermal growth factor (EGF) elicited ICl,swell and exogenous H2O2 overcame block of AT1 receptors, EGFR kinase, and PI-3K. In contrast, osmotic shrinkage failed to suppress H2O2-induced ICl,swell. These data argue that the AngII-ROS signaling cascade participates in the response of cardiomyocytes to osmotic swelling. AngII-dependent ICl,swell activation may modulate electrical activity and cell volume in cardiac disease.
2. Methods
2.1. Ventricular myocytes
Studies conform to Guide for the Care and Use of Laboratory Animals (NIH Publication 85-23, revised 1996). Left ventricular myocytes were isolated from anesthetized New Zealand rabbits (~3–4 kg) using collagenase (type II) and pronase (type XIV).11,22 Cardiomyocytes were washed twice and stored in modified Kraft–Brühe solution (pH 7.2; 295 mosmol/kg).11 Rod-shaped quiescent cells with clear striations and no membrane blebs were studied within 8 h of isolation.
2.2. Solutions and drugs
Bath solutions designed to isolate anion currents were isosmotic (1T; 300 mosmol/kg; T, times isosmotic), hypoosmotic (0.7T), or hyperosmotic (1.5T) and contained (mM): 90 N-methyl-D-glucamine-Cl, 3.0 MgCl2, 10 HEPES, 10 glucose, 5 CsCl, 1 BaCl2, 0.2 CdCl2, 0–250 mannitol (pH 7.4, CsOH). These solutions suppressed the time-dependence of ICl,swell at positive potentials22 and allowed osmotic challenges at constant ionic strength. Osmolarity was verified by freezing-point depression.
Stock solutions of apocynin, AG1478, diphenyleneiodonium-Cl (DPI), LY294002, and wortmannin in DMSO and EGF, losartan-K, and eprosartan mesylate in H2O were frozen (−20°C) in aliquots until use. A membrane-permeant fusion peptide inhibitor of NOX, gp91ds-tat, and a permeant but inactive scrambled analogue, scramb-tat, were synthesized.24 The inhibitor is a 9-mer that blocks NOX assembly by mimicking the gp91phox (NOX2) docking site for p47phox joined to a tat 9-mer that drives transmembrane uptake. Peptide stocks (1.2 mg/ml) were made in 150 mM NaCl plus 10 mM acetic acid and frozen (−20°C) in aliquots until use. Final diluent concentrations, 0.1–0.5%, did not alter ICl,swell.
2.3. Electrophysiology
Myocytes were placed in a poly-L-lysine-coated chamber and super-fused at ~2 ml/min (21–22°C). Pipettes (2–3 MΩ) were filled with (mM): 110 Cs-aspartate, 20 CsCl or 20 TEA-Cl, 2.5 Mg-ATP, 8 Cs2-EGTA, 0.15 CaCl2, 10 HEPES (pH 7.1, CsOH; liquid junction potential, −11.5 ±0.7 mV, n = 9).22 This gave a free-[Ca2+]i of ~60 nM (WinMAXC 2.40; http://www.stanford.edu/~cpatton). Junction potentials were corrected, and ground was a 3-M KCl agar bridge. Seal resistances of 5–30 GΩ were achieved.
Myocytes were dialyzed for 10 min before data were taken. Whole-cell currents were recorded with an Axoclamp 200B and Digidata 1322A under pClamp 8. Currents were low-pass filtered (Bessel, 2 kHz) and digitized (5 kHz). Membrane capacitance was calculated from 5-mV steps. Successive 500-ms steps were made from −60 mV to test potentials between −100 and +60 mV in +10 mV increments. I–V relationships were obtained at 1-min intervals to track responses to interventions and were plotted from quasi steady-state currents. In preliminary studies, ICl,swell fully activated in <5 min and was stable for at least 45 min. All agents were applied for sufficient time for currents to reach steady state.
Because myocytes were studied under whole-cell conditions, interventions that alter ICl,swell are not expected to significantly alter cell volume.21,25 This was confirmed in preliminary studies using video microscopy methods as previously described.6
2.4. Statistics
Percent block or activation (±SEM) were calculated using each myocyte as its own control and assessed with paired t-tests or, in one case, with a Wilcox rank sign test because the data were not normally distributed; n represents the number of cells, and P < 0.05 was taken as significant. Analysis of mean current densities (pA/pF) for each treatment group by repeated-measures ANOVA and Student–Newman–Keuls tests gave identical statistical conclusions.
3. Results
3.1. AT1 signalling
Blocking AT1 receptors with losartan inhibits ICl,swell evoked by integrin stretch, and exogenous AngII elicits a Cl− current attributed to ICl,swell.10 Figure 1A–C shows that AT1 receptors also are involved in ICl,swell activation upon osmotic swelling in 0.7T. As expected, swelling turned on an outwardly rectifying current that reversed at −44 ± 1 mV, near the Cl− equilibrium potential (ECl), −42 mV. Adding losartan (30 μM, 10–13 min) to 0.7T strongly suppressed ICl,swell. The losartan-sensitive difference current at +60 mV was 101 ±6% (P < 0.001, n = 10) of the current activated by swelling. Although currents were smaller at −100 than +60 mV, similar results were obtained at both voltages in this and subsequent protocols. Eprosartan (30 μM, 10 min), a carboxybenzylimidazol AT1 antagonist with structural features distinct from losartan, a biphenylte-trazole,26 also fully inhibited swelling-induced currents (98 ±2%; P < 0.001, n = 9) (Figure 1D and E). In contrast, block of ICl,swell by a lower concentration of losartan (10 μM, 10 min) was incomplete (68 ±4%; P < 0.001, n = 6).
Figure 1.

AT1 blockers inhibit ICl,swell during osmotic swelling, and block is reversed by H2O2. (A) Currents and (B) I–V relationships in 1T, 0.7T (10 min), after losartan (30 μM, 10 min) in 0.7T (LOS), and after H2O2 (100 μM, 10 min) in presence of losartan (H2O2). (C ) Time course of ICl,swell at +60 mV; dash line, control current in 1T. (D and E) Analogous experiments with eprosartan (EPRO; 30 μM, 10 min). ICl,swell is time-independent because bath contained Cd2+ and Ba2+.22
Integrin stretch elicits ICl,swell in cardiomyocytes via AT1 signalling and its downstream effector H2O2.9,10 If the same pathway is stimulated by osmotic swelling, exogenous H2O2 (100 μM) should overcome block of ICl,swell by losartan (Figure 1A–C). H2O2-induced current in the presence of losartan was 154 ±6% (P < 0.001, n = 6) of ICl,swell, confirming that ROS are downstream from AngII in the regulatory pathway.
3.2. EGFR kinase
EGFR kinase is a receptor protein tyrosine kinase that undergoes transactivation upon AT1 occupancy and is downstream from AT1 in the AngII-ROS cascade.12,27 Figure 2A–C demonstrates that AG1478, a specific EGFR kinase blocker (IC50 = 0.003–0.9 μM), fully inhibited ICl,swell. Addition of AG1478 (1 μM, 10–13 min) to 0.7T suppressed 98 ±3% (P < 0.001, n = 11) of the swelling-induced current at +60 mV. Similar results were obtained in human atrial21 and rabbit ventricular22 myocytes using two other EGFR kinase blockers, AG556 and PD153035. As shown for losartan, block of ICl,swell by AG1478 was overcome by exogenous H2O2. The H2O2-induced current in the presence of AG1478 was 150 ±15% (P < 0.001, n = 6) of ICl,swell, indicating that ROS are down-stream from EGFR kinase.
Figure 2.

Block of EGFR kinase suppresses ICl,swell and is reversed by exogenous H2O2; exogenous EGF activates ICl,swell. (A) Currents and (B) I–V relationships in 1T, 0.7T (10 min), after AG1478 in 0.7T (1 μM, 10 min), and after H2O2 (100 μM, 10 min) in presence of AG1478. (C) Time course of ICl,swell at +60 mV. (D) Currents and (E) I–V relationships in 1T, after EGF in 1T (3.3 nM, 10 min; 1T+EGF), and after osmotic shrinkage in 1.5T (10 min) in presence of EGF (1.5T+EGF).
The putative role of EGFR kinase suggests that exogenous EGF should elicit a volume-sensitive Cl− current under isosmotic conditions. Consistent with this prediction, Figure 2D and E shows that an outwardly rectifying Cl−current was activated by 3.3 nM EGF in 1T (also see,11). EGF (10–15 min) increased the current at +60 mV by 137 ±19% (P < 0.001, n = 6) in the absence of osmotic swelling. Moreover, osmotically shrinking myocytes in hyperosmotic 1.5T bath solution inhibited 91±7% (P < 0.001, n = 6) of the EGF-induced current in the continued presence of EGF. Thus, the EGF-induced Cl− current was volume-sensitive, and osmotic shrinkage must act at site(s) downstream to EGF.
Previously we reported that exogenous AngII elicits a tamoxifen-sensitive Cl− current attributed to ICl,swell.10 Figure 3 shows that AngII, like swelling, signals to Cl− channels via EGFR kinase. Adding AngII (5 nM, 10 min) to 1T bathing media evoked an outwardly rectifying current that reversed near ECl, and AG1478 (1 μM, 10 min) in the continued presence of AngII inhibited 99 ±3% (P < 0.001, n = 6; +60 mV) of the AngII-induced current.
Figure 3.
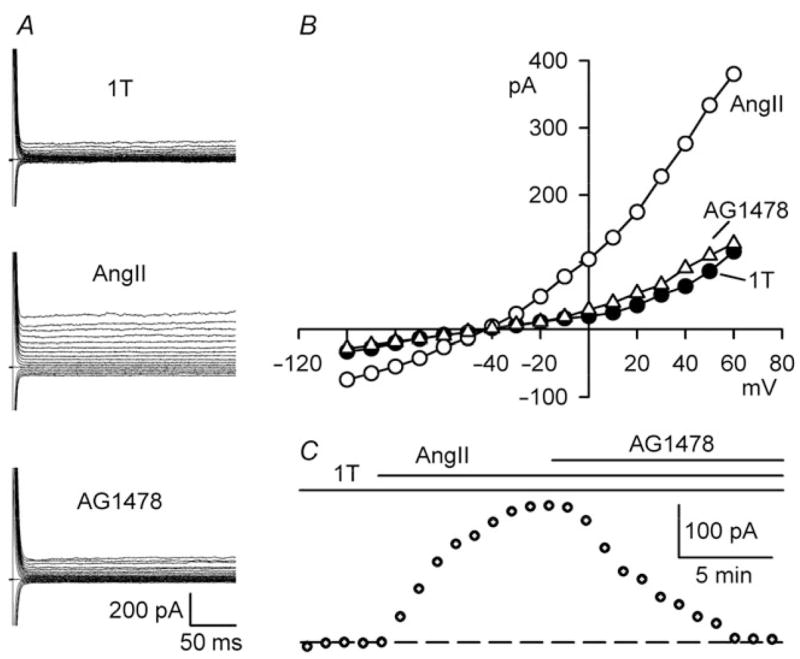
EGFR kinase inhibition blocks AngII-induced ICl,swell. (A) Currents and (B) I–V relationships in 1T, after AngII (5 nM, 10 min), and after AG1478 (1 μM, 10 min) in presence of AngII (AG1478). (C) Time course of ICl,swell at +60 mV.
3.3. Phosphatidylinositol 3-kinase
PI-3K is downstream of EGFR kinase in the AngII-ROS signalling cascade and participates in NOX activation.12,28 We tested whether PI-3K also regulates ICl,swell using selective inhibitors, LY294002 (IC50 = 10 μM) and wortmannin (IC50 = 1−10 nM). Figure 4A–C illustrates that LY294002 (100 μM, 10–15 min) fully blocked ICl,swell. Addition of LY294002 to 0.7T inhibited the swelling-activated current by 95 ±3% (P < 0.001, n = 8) at +60 mV. As expected if H2O2 is downstream of PI-3K, the suppression of ICl,swell was abrogated by exogenous H2O2. The H2O2-induced current in the presence of LY294002 was 147 ±13% (P < 0.001, n = 5) of the swelling-induced current.
Figure 4.

PI-3K blockade inhibits ICl,swell and is reversed by exogenous H2O2. (A) Currents and (B) I–V relationships in 1T, 0.7T (10 min), after LY294002 in 0.7T (100 μM, 10 min; LY), and after H2O2 (100 μM, 10 min) in presence of LY294002 (H2O2). (C ) Time course of ICl,swell at +60 mV. (D and E) Analogous experiments with wortmannin (500 nM, 13 min; WORT).
Wortmannin possesses a distinct chemical structure and mechanism of action from those of LY294002.29 After activating ICl,swell in 0.7T, adding wortmannin (500 nM, 10–15 min) reduced the current by 60 ±2% (P < 0.001, n = 6) at +60 mV (Figure 4D and E).
3.4. NADPH oxidase
AT1 activation ultimately generates ROS by initiating assembly of sarcolemmal NOX from cytoplasmic and membrane subunits,28 and NOX and ROS participate in the activation of ICl,swell in response to integrin stretch.10 Therefore, we evaluated the role of NOX in the response to osmotic swelling. Apocynin (IC50 = 80 μM) prevents NOX assembly by conjugating thiol residues.30 As shown in Figure 5A–C, apocynin (500 μM, 10 min) inhibited 84 ±12% (P = 0.002, n = 5) of ICl,swell at +60 mV. Figure 5D and E demonstrates the effect of a structural distinct NOX inhibitor, DPI (IC50 = 0.9 μM), which binds to flavins and the haem b redox centres of gp91phox and, thereby, suppresses production.31 Addition of DPI (60 μM, 10–13 min) to 0.7T completely blocked ICl,swell (116 ±16%; P = 0.002, n = 5).
Figure 5.

NOX inhibition blocks ICl,swell. (A) Currents and (B) I–V relationships in 1T, 0.7T (10 min), after apocynin in 0.7T (500 μM, 10 min; APO). (C) Time course ICl,swell at +60 mV. (D and E) Analogous experiments with DPI (60 μM, 13 min).
Although block by DPI or apocynin is widely taken as sufficient evidence to infer participation of NOX, questions can be raised regarding specificity. To address this issue, we utilized gp91ds-tat, a membrane permeant fusion peptide that is regarded as a highly selective blocker of NOX assembly.24 Figure 6A and B shows that exposure to gp91ds-tat also fully suppressed ICl,swell. Addition of gp91ds-tat (500 nM, 10–12 min) to 0.7T reduced ICl,swell by 106 ±5% (P < 0.001, n = 5) at +60 mV. To rule out non-specific actions of the inhibitor peptide, the inactive peptide scramb-tat24 was studied (Figure 6C). Exposure to scramb-tat (500 nM, 10–15 min) did not alter ICl,swell (P = ns, n = 4).
Figure 6.

Block of NOX with gp91ds-tat inhibits ICl,swell. (A) Currents and (B) I–V relationships in 1T, 0.7T (10 min), and after gp91ds-tat in 0.7T (500 nM, 10 min). (C) Analogous experiment with scramb-tat (500 nM, 10 min), a membrane permeant but inactive peptide.
3.5. Role of H2O2
NOX generates by single e− reduction of molecular O2, and enzymatic and spontaneous dismutation forms H2O2, a membrane-permeant ROS, that is, a stronger oxidant.28 H2O2 is a potent agonist of cardiac ICl,swell, participates in stretch-induced ICl,swell activation,10 and as shown in Figures 1, 2 and 4, overcomes block of ICl,swell by inhibitors of AT1 receptors, EGFR kinase, and PI-3K. Therefore, we tested whether catalase, a H2O2 scavenger, would inhibit ICl,swell after osmotic swelling (Figure 7). Addition of catalase (1000 U/ml, 10–15 min) to 0.7T blocked 90 ±11% (P = 0.001, n = 5) of ICl,swell at +60 mV. This is consistent with the idea that H2O2 is a downstream mediator of ICl,swell activation.
Figure 7.
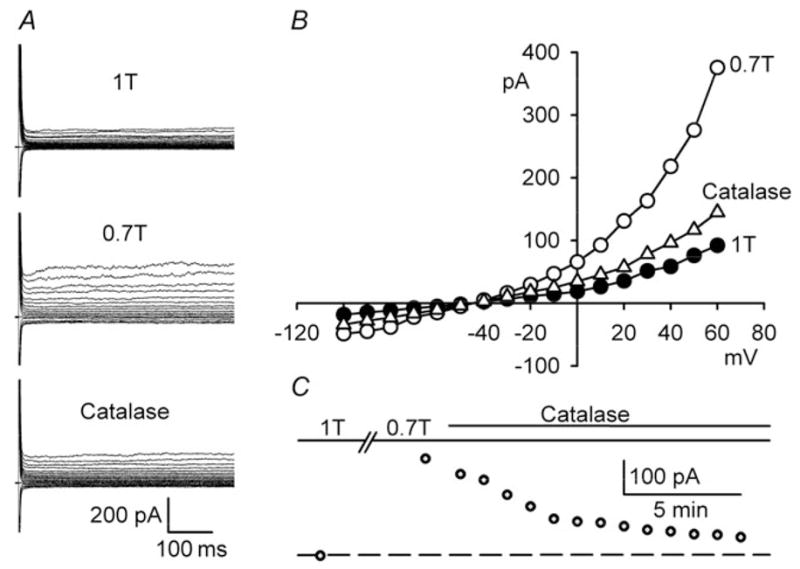
Scavenging of H2O2 by catalase inhibits ICl,swell. (A) Currents and (B) I–V relationships in 1T, 0.7 T (10 min), and after 1000 U/ml catalase to 0.7 T (10 min, Catalase). (C) Time course of ICl,swell at +60 mV.
To further characterize the site of action of H2O2, we tested whether H2O2-induced current was blocked by osmotic shrinkage. Exposure to 100 μM H2O2 in 1T increased outwardly rectifying current at +60 mV by 1.6 ±0.6 pA/pF (n = 5), but osmotic shrinkage in 1.5T in the continued presence of H2O2 failed to significantly alter the current (1.9 ±0.7 pA/pF, P = 0.34; data not shown). This suggests that H2O2 acts at sites distal to those regulated by osmotic shrinkage.
4. Discussion
We found that osmotic swelling activated ICl,swell by means of the AngII AT1 receptor signalling cascade that involves EGFR kinase, PI-3K, production of by NOX, and its dismutation to H2O2.12,14 Previously, we showed that the same pathway stimulates ICl,swell when β1D-integrins are stretched with paramagnetic beads.10,11 A scheme illustrating the results is presented in Figure 8. As expected from this scheme, osmotic activation of ICl,swell was blocked by inhibitors of AT1 receptors, EGFR kinase, PI-3K, and NOX and by scavenging H2O2 with extracellular catalase, 10,11 whereas exogenous AngII, EGF, and H2O2 elicited ICl,swell. H2O2 must act late in the cascade because it overcame the suppression of ICl,swell by AT1 receptor, EGFR kinase, and PI-3K blockade. AT1 receptor activation must be upstream from EGFR kinase because AG1478 blocked the AngII-induced current. The EGF-induced current was blocked by osmotic shrinkage, and we previously showed that it also is blocked by inhibitors of PI-3K and gp91ds-tat under different conditions.11 These data imply that EGFR kinase is upstream from both PI-3K and NOX. While osmotic shrinkage blocked the EGF-induced current, it failed to affect H2O2-induced currents. Thus, osmotic shrinkage must regulate ICl,swell at one or more sites between EGFR kinase and H2O2. Taken together, these data imply that osmotic shrinkage does not simply oppose the effect of osmotic swelling at its initial transduction site. Rather, shrinkage and swelling must be sensed by different signalling molecules.
Figure 8.
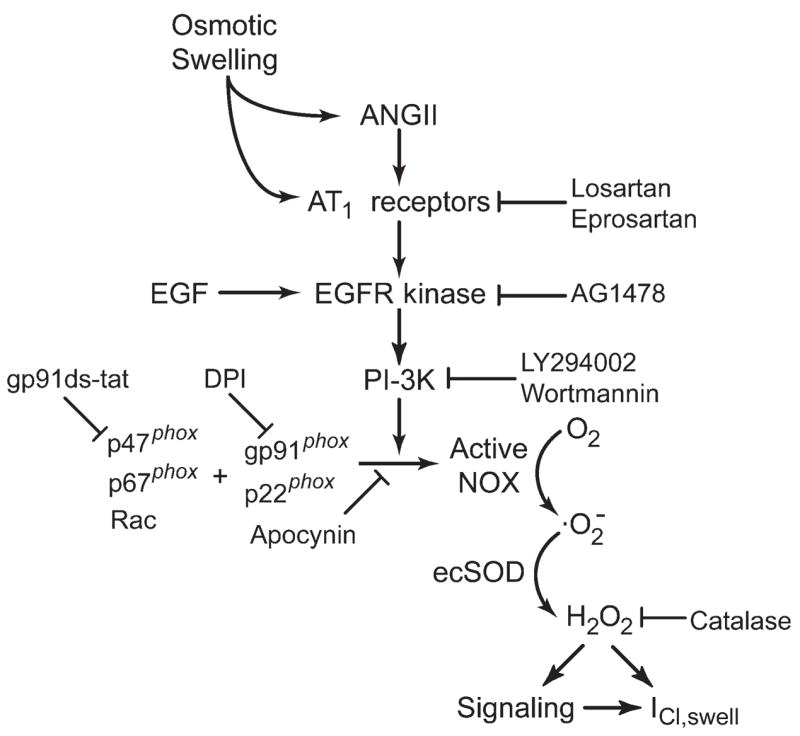
Proposed scheme for regulation of ICl,swell by AngII-ROS signalling. Osmotic swelling releases AngII and/or directly activates AT1 receptors. AT1 activation evokes downstream signalling via EGFR kinase and PI-3K, causing assembly of NOX from membrane-bound (gp91phox, p22phox) and cytoplasmic (p47phox, p67phox, Rac) components. NOX produces , which rapidly undergoes dismutation by ecSOD to membrane permeant H2O2. H2O2 may modulate ICl,swell directly or via a variety of redox sensitive kinases and phosphatases.
The scheme presented here does not include other molecules that participate in AngII signalling in vasculature32 and heart.33 For example, a PKC-induced activation of NOX by AngII that is independent of EGFR kinase is found in vascular tissue.32 Such a mechanism apparently does not significantly regulate ICl,swell in ventricular myocytes because EGFR kinase inhibitors fully blocked ICl,swell. Nevertheless, a more complex interplay between components of the proposed cascade cannot be excluded. Interactions with other PKC- and PKA-dependent mechanisms that regulate ICl,swell (for review,1–3) are unexplored.
Osmotic swelling also does not appear to stimulate ICl,swell by activating integrin-dependent signalling mediated by FAK and Src. Block of the Src family by PP2 or FAK by over expression of the endogenous peptide inhibitor FRNK augments swelling-induced current in rabbit22 and neonatal rat ventricular23 and human atrial21 myocytes, whereas PP2 suppresses stretch-induced ICl,swell.9 Notably, PP2 prevents Src- and integrin-dependent FAK autophosphorylation at Tyr397 and Tyr577, whereas G protein-dependent phosphorylation at Tyr397 can occur by a PP2-insensitive, Src-independent pathway.34 Src family members are differentially modulated by osmotic swelling,35 and unique Src family members may dominate the responses to swelling and stretch.
4.1. AT1 receptors
Autocrine-paracrine regulation of ventricular myocytes by stretch-induced AngII release has been described.15 Although AngII secretion was not measured, it seems likely that AngII released from numerous myocytes in the chamber upon swelling initiated autocrine-paracrine AT1 signalling, and ICl,swell blockade by losartan and eprosartan is consistent with this idea. In contrast, Zou et al.36 recently made a strong case that cardiomyocyte AT1 receptors are mechanosensors and respond to stretch by an AngII-independent mechanism that, nonetheless, is blocked by the AT1 antagonist candesartan. In the present study, complete block of ICl,swell occurred with 30 μM but not 10 μM losartan. This implies an IC50 higher than the 31 nM reported for [125I]-AngII binding to AT1 receptors in rabbit ventricular membranes at low ionic strength,37 but lower than that for AT2 receptors, 81 μM.38 Losartan is a non-competitive/surmountable antagonist,26 and its IC50 for putative block of direct AT1 activation by stretch is unknown. The involvement of AT1 receptors is strongly supported by data implicating downstream components of its signalling cascade12 in ICl,swell regulation. Nevertheless, we cannot rigorously exclude the possibility that losartan and eprosartan act at another unidentified site.
4.2. EGFR kinase
AT1 activation triggers transactivation of EGFR kinase12,27 by stimulating membrane-bound metalloproteases that clip and release heparin-binding EGF, as well as by association of AT1 receptors with EGFR kinase.13 We found that the EGFR kinase inhibitor AG1478 blocked both ICl,swell and the AngII induced Cl− current, and Cl− current elicited by exogenous EGF was inhibited by osmotic shrinkage. These data implicate EGFR kinase in the signalling pathway and further suggest that osmotic shrinkage must act at a distal site in the signalling cascade. Block of volume-sensitive, EGF-induced Cl− current by tamoxifen, an ICl,swell blocker, also was reported in studies under different experimental conditions.11 Previously, we showed that block of EGFR kinase with PD153035 and AG556 inhibit ICl,swell in rabbit ventricular22 and human atrial21 myocytes, respectively. In contrast, tyrphostin A51 (AG183), another EGFR kinase blocker, failed to suppress ICl,swell in canine atrial myocytes.25 The basis for this discrepancy is unknown. Consistent with the present results, activation of EGFR kinase by exogenous EGF or transfection with bovine papilloma virus recently were shown to potentiate ICl,swell in C127 mammary cells,39 and EGF also stimulates ICl,swell in liver-derived HTC cells,40 suggesting this is a common mode of ICl,swell regulation in several tissues.
4.3. Phosphatidylinositol 3-kinase
PI-3K is downstream of EGFR kinase12,28 and is stimulated by osmotic swelling.41,42 In turn, PI-3K produce phosphoinositol-3,4,5-triphosphate and phosphoinositol-3,4-bisphosphate,29 which activate NOX both by binding to the PX domain of p47phox 28 and by augmenting Rac-mediated nucleotide exchange.12,29 Block of ICl,swell by LY294002 and wortmannin argue that PI-3K participates in the regulation of cardiomyocyte ICl,swell. We did not investigate why block by 500 nM wortmannin was incomplete. One possibility is involvement of monomeric class II PI-3K-C2α, a cardiac isoform that is more resistant to wortmannin (IC50 = 400 nM) than dimeric PI-3K (IC50 = 1−10 nM).29 Inhibition of PI-3K also suppresses ICl,swell in hepatocytes43 and pulmonary artery smooth muscle42 and swelling-induced Na+-K+ pump stimulation in rabbit ventricular myocytes.44
4.4. Reactive oxygen species
A major component of AT1 signalling depends on generation of by NOX.14,33 The involvement of NOX and ROS in hypoosmotic activation of ICl,swell is strongly supported by findings that three distinct NOX inhibitors all blocked ICl,swell. DPI is promiscuous in binding to FAD-containing enzymes45,46 and does not distinguish between mitochondrial, microsomal, and sarcolemmal sources of ROS. Apocynin does not alter production by cardiac mitochondria,47 but it inhibits P450 monooxygenase in endothelial cells.48 In contrast, gp91ds-tat is thought to be highly NOX selective because of specific interactions between the 9-mer docking sequence and p47phox or its homologues (e.g. NOXO1, assembles with NOX1).24 NOX2 (gp91phox) and NOX4 are expressed in cardiomyocytes.17,49 The present experiments do not establish which isoform is responsible, but two factors favour involvement of NOX2. First, knockout of NOX2 fully abrogates AngII-induced, NOX-dependent, production in cardiomyocytes, indicating that NOX2 rather than NOX4 is the AngII-responsive isoform.17,50 Second, NOX4 activity in native cells and heterologous expression systems does not depend on cytoplasmic subunits including p47phox and its homologues.49 Thus, gp91ds-tat binding to p47phox-like proteins should not have suppressed NOX4 activity.
NOX transfers e− from intracellular NADPH to extracellular O2 via FAD bound to its cytoplasmic C-terminal domain and two haem complexes in its membrane-spanning domains.28 Consequently, NOX produces at the extracellular membrane face, where it rapidly undergoes dismutation to H2O2 by extracellular SOD (SOD-3) anchored to proteoglycans.51 H2O2 is a longer-lived species and stronger oxidant than and is membrane permeant.28 Consistent with a role for H2O2, scavenging H2O2 by adding catalase to the bath suppressed ICl,swell in osmotically swollen myocytes. Previously, we showed that tamoxifen-sensitive ICl,swell is activated by exogenous H2O2 (EC50 = 8 μM) in rabbit ventricular myocytes.10 Recent reports indicate that exogenous H2O2 also elicits ICl,swell in HeLa (200–500 μM) and HTC (EC50 = 100 μM) cell lines.40,52 Both of these groups detected ROS production attributed to NOX upon swelling. Expression of the dominant negative p47S379A NOX subunit suppresses activation of ICl,swell in HeLa cells,40 as does the non-selective flavin-inhibitor DPI in both HeLa and HTC cells.40,52 It remains to be seen whether the ultimate target of H2O2 is the Cl− channel itself or redox-sensitive signalling pathways.33,53 Cysteines with an acidic pKa are particularly sensitive to oxidation by H2O2. Such cysteines are found in the active site of all protein tyrosine phosphatases, including PTEN, which opposes PI-3K by dephosphorylating inositols.53 Nevertheless, we cannot rule out the possibility that other reactive species contribute to the regulation of ICl,swell.
4.5. Implications
ICl,swell is persistently activated in models of dilated cardiomyopathy,2 but the underlying mechanism is obscure. The present results suggest that upregulation of the AngII cascade, which occurs in ventricular hypertrophy, heart failure, hypertension, atherosclerotic coronary artery disease, hypercholesterolemia, and diabetes, may in part explain persistent activation of ICl,swell in dilated cardiomyopathy and predicts similar findings in other settings with elevated AngII or NOX activity. For example, NOX is upregulated by subpressor doses of AngII and aortic banding,16,17 after myocardial infarction,54 during the progression from hypertrophy to failure,55 and in atrial fibrillation.56 Because ICl,swell outwardly rectifies, its upregulation in cardiac disease promotes reduction of action potential duration, which favours reentrant tachyarrhythmias by decreasing the minimum wavelength for a reentrant circuit.
Acknowledgments
Funding
National Institutes of Health (HL46764 and HL65435 to C.M.B.).
Footnotes
Conflict of interest: none declared.
References
- 1.Hume JR, Duan D, Collier ML, Yamazaki J, Horowitz B. Anion transport in heart. Physiol Rev. 2000;80:31–81. doi: 10.1152/physrev.2000.80.1.31. [DOI] [PubMed] [Google Scholar]
- 2.Baumgarten CM, Browe DM, Ren Z. Swelling- and stretch-activated chloride channels in the heart: regulation and function. In: Kamkin A, Kiseleva I, editors. Mechanosensitivity in Cells and Tissues. Moscow: Academia Publishing House Ltd; 2005. pp. 79–102. [PubMed] [Google Scholar]
- 3.Duan DY, Liu LL, Bozeat N, Huang ZM, Xiang SY, Wang GL, et al. Functional role of anion channels in cardiac diseases. Acta Pharmacol Sin. 2005;26:265–278. doi: 10.1111/j.1745-7254.2005.00061.x. [DOI] [PubMed] [Google Scholar]
- 4.Du XY, Sorota S. Cardiac swelling-induced chloride current depolarizes canine atrial myocytes. Am J Physiol Heart Circ Physiol. 1997;272:H1904–H1916. doi: 10.1152/ajpheart.1997.272.4.H1904. [DOI] [PubMed] [Google Scholar]
- 5.Vandenberg JI, Bett GC, Powell T. Contribution of a swelling-activated chloride current to changes in the cardiac action potential. Am J Physiol Cell Physiol. 1997;273:C541–C547. doi: 10.1152/ajpcell.1997.273.2.C541. [DOI] [PubMed] [Google Scholar]
- 6.Clemo HF, Stambler BS, Baumgarten CM. Swelling-activated chloride current is persistently activated in ventricular myocytes from dogs with tachycardia-induced congestive heart failure. Circ Res. 1999;84:157–165. doi: 10.1161/01.res.84.2.157. [DOI] [PubMed] [Google Scholar]
- 7.Batthish M, Diaz RJ, Zeng HP, Backx PH, Wilson GJ. Pharmacological preconditioning in rabbit myocardium is blocked by chloride channel inhibition. Cardiovasc Res. 2002;55:660–671. doi: 10.1016/s0008-6363(02)00454-6. [DOI] [PubMed] [Google Scholar]
- 8.d’Anglemont de Tassigny A, Souktani R, Henry P, Ghaleh B, Berdeaux A. Volume-sensitive chloride channels IClvol mediated doxorubicin-induced apoptosis through apoptotic volume decrease in cardiac myocytes. Fundam Clin Pharmacol. 2004;18:531–538. doi: 10.1111/j.1472-8206.2004.00273.x. [DOI] [PubMed] [Google Scholar]
- 9.Browe DM, Baumgarten CM. Stretch of β1 integrin activates an outwardly rectifying chloride current via FAK and Src in rabbit ventricular myocytes. J Gen Physiol. 2003;122:689–702. doi: 10.1085/jgp.200308899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Browe DM, Baumgarten CM. Angiotensin II (AT1) receptors NADPH oxidase regulate Cl− current elicited by β1 integrin stretch in rabbit ventricular myocytes. J Gen Physiol. 2004;124:273–287. doi: 10.1085/jgp.200409040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Browe DM, Baumgarten CM. EGFR kinase regulates volume-sensitive chloride current elicited by integrin stretch via PI-3K and NADPH oxidase in ventricular myocytes. J Gen Physiol. 2006;127:237–251. doi: 10.1085/jgp.200509366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seshiah PN, Weber DS, Rocic P, Valppu L, Taniyama Y, Griendling KK. Angiotensin II stimulation of NAD(P)H oxidase activity: upstream mediators. Circ Res. 2002;91:406–413. doi: 10.1161/01.res.0000033523.08033.16. [DOI] [PubMed] [Google Scholar]
- 13.Shah BH, Catt KJ. A central role of EGF receptor transactivation in angiotensin II-induced cardiac hypertrophy. Trends Pharmacol Sci. 2003;24:239–244. doi: 10.1016/S0165-6147(03)00079-8. [DOI] [PubMed] [Google Scholar]
- 14.Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2006;292:C82–C97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 15.Sadoshima J, Izumo S. The cellular and molecular response of cardiac myocytes to mechanical stress. Annu Rev Physiol. 1997;59:551–571. doi: 10.1146/annurev.physiol.59.1.551. [DOI] [PubMed] [Google Scholar]
- 16.Bendall JK, Cave AC, Heymes C, Gall N, Shah AM. Pivotal role of a gp91phox-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation. 2002;105:293–296. doi: 10.1161/hc0302.103712. [DOI] [PubMed] [Google Scholar]
- 17.Byrne JA, Grieve DJ, Bendall JK, Li JM, Gove C, Lambeth JD, et al. Contrasting roles of NADPH oxidase isoforms in pressure-overload versus angiotensin II-induced cardiac hypertrophy. Circ Res. 2003;93:802–805. doi: 10.1161/01.RES.0000099504.30207.F5. [DOI] [PubMed] [Google Scholar]
- 18.Nakagami H, Takemoto M, Liao JK. NADPH oxidase-derived superoxide anion mediates angiotensin II-induced cardiac hypertrophy. J Mol Cell Cardiol. 2003;35:851–859. doi: 10.1016/s0022-2828(03)00145-7. [DOI] [PubMed] [Google Scholar]
- 19.Zhou C, Ziegler C, Birder LA, Stewart AF, Levitan ES. Angiotensin II and stretch activate NADPH oxidase to destabilize cardiac Kv4.3 channel mRNA. Circ Res. 2006;98:1040–1047. doi: 10.1161/01.RES.0000218989.52072.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cave AC, Brewer AC, Narayanapanicker A, Ray R, Grieve DJ, Walker S, et al. NADPH oxidases in cardiovascular health and disease. Antioxid Redox Signal. 2006;8:691–728. doi: 10.1089/ars.2006.8.691. [DOI] [PubMed] [Google Scholar]
- 21.Du XL, Gao Z, Lau CP, Chiu SW, Tse HF, Baumgarten CM, et al. Differential effects of tyrosine kinase inhibitors on volume-sensitive chloride current in human atrial myocytes: evidence for dual regulation by Src and EGFR kinases. J Gen Physiol. 2004;123:427–439. doi: 10.1085/jgp.200409013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ren Z, Baumgarten CM. Antagonistic regulation of swelling-activated chloride current in rabbit ventricle by Src and EGFR protein tyrosine kinases. Am J Physiol Heart Circ Physiol. 2005;288:H2628–H2636. doi: 10.1152/ajpheart.00992.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walsh KB, Zhang J. Regulation of cardiac volume-sensitive chloride channel by focal adhesion kinase and Src kinase. Am J Physiol Heart Circ Physiol. 2005;289:H2566–H2574. doi: 10.1152/ajpheart.00292.2005. [DOI] [PubMed] [Google Scholar]
- 24.Rey FE, Cifuentes ME, Kiarash A, Quinn MT, Pagano PJ. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular and systolic blood pressure in mice. Circ Res. 2001;89:408–414. doi: 10.1161/hh1701.096037. [DOI] [PubMed] [Google Scholar]
- 25.Sorota S. Tyrosine protein kinase inhibitors prevent activation of cardiac swelling-induced chloride current. Pflugers Arch. 1995;431:178–185. doi: 10.1007/BF00410189. [DOI] [PubMed] [Google Scholar]
- 26.Timmermans PB. Pharmacological properties of angiotensin II receptor antagonists. Can J Cardiol. 1999;15(Suppl F):26F–28F. [PubMed] [Google Scholar]
- 27.Frank GD, Eguchi S. Activation of tyrosine kinases by reactive oxygen species in vascular smooth muscle cells: significance and involvement of EGF receptor transactivation by angiotensin II. Antioxid Redox Signal. 2003;5:771–780. doi: 10.1089/152308603770380070. [DOI] [PubMed] [Google Scholar]
- 28.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 29.Oudit GY, Sun H, Kerfant BG, Crackower MA, Penninger JM, Backx PH. The role of phosphoinositide-3 kinase and PTEN in cardiovascular physiology and disease. J Mol Cell Cardiol. 2004;37:449–471. doi: 10.1016/j.yjmcc.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 30.‘t Hart BA, Simons JM. Metabolic activation of phenols by stimulated neutrophils: a concept for a selective type of anti-inflammatory drug. Biotechnol Ther. 1992;3:119–135. [PubMed] [Google Scholar]
- 31.Doussiere J, Gaillard J, Vignais PV. The heme component of the neutrophil NADPH oxidase complex is a target for aryliodonium compounds. Biochemistry. 1999;38:3694–3703. doi: 10.1021/bi9823481. [DOI] [PubMed] [Google Scholar]
- 32.Cai H, Griendling KK, Harrison DG. The vascular NAD(P)H oxidases as therapeutic targets in cardiovascular diseases. Trends Pharmacol Sci. 2003;24:471–478. doi: 10.1016/S0165-6147(03)00233-5. [DOI] [PubMed] [Google Scholar]
- 33.Das DK, Maulik N, Engelman RM. Redox regulation of angiotensin II signaling in the heart. J Cell Mol Med. 2004;8:144–152. doi: 10.1111/j.1582-4934.2004.tb00270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Salazar EP, Rozengurt E. Src family kinases are required for integrin-mediated but not for G protein-coupled receptor stimulation of focal adhesion kinase autophosphorylation at Tyr-397. J Biol Chem. 2001;276:17788–17795. doi: 10.1074/jbc.M100984200. [DOI] [PubMed] [Google Scholar]
- 35.Cohen DM. SRC family kinases in cell volume regulation. Am J Physiol Cell Physiol. 2005;288:C483–C493. doi: 10.1152/ajpcell.00452.2004. [DOI] [PubMed] [Google Scholar]
- 36.Zou Y, Akazawa H, Qin Y, Sano M, Takano H, Minamino T, et al. Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat Cell Biol. 2004;6:499–506. doi: 10.1038/ncb1137. [DOI] [PubMed] [Google Scholar]
- 37.Rogg H, Schmid A, de Gasparo M. Identification and characterization of angiotensin II receptor subtypes in rabbit ventricular myocardium. Biochem Biophys Res Commun. 1990;173:416–422. doi: 10.1016/s0006-291x(05)81074-5. [DOI] [PubMed] [Google Scholar]
- 38.Scott AL, Chang RS, Lotti VJ, Siegl PK. Cardiac angiotensin receptors: effects of selective angiotensin II receptor antagonists, DUP 753 and PD 121981, in rabbit heart. J Pharmacol Exp Ther. 1992;261:931–935. [PubMed] [Google Scholar]
- 39.Abdullaev IF, Sabirov RZ, Okada Y. Upregulation of swelling-activated Cl− channel sensitivity to cell volume by activation of EGF receptors in murine mammary cells. J Physiol. 2003;549:749–758. doi: 10.1113/jphysiol.2003.039784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Varela D, Simon F, Riveros A, Jorgensen F, Stutzin A. NAD(P)H oxidase- derived H2O2 signals chloride channel activation in cell volume regulation cell proliferation. J Biol Chem. 2004;279:13301–13304. doi: 10.1074/jbc.C400020200. [DOI] [PubMed] [Google Scholar]
- 41.Tilly BC, Edixhoven MJ, Tertoolen LG, Morii N, Saitoh Y, Narumiya S, et al. Activation of the osmo-sensitive chloride conductance involves P21rho and is accompanied by a transient reorganization of the F-actin cytoskeleton. Mol Biol Cell. 1996;7:1419–1427. doi: 10.1091/mbc.7.9.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang GX, McCrudden C, Dai YP, Horowitz B, Hume JR, Yamboliev IA. Hypotonic activation of volume-sensitive outwardly rectifying chloride channels in cultured PASMCs is modulated by SGK. Am J Physiol Heart Circ Physiol. 2004;287:H533–H544. doi: 10.1152/ajpheart.00228.2003. [DOI] [PubMed] [Google Scholar]
- 43.Feranchak AP, Roman RM, Doctor RB, Salter KD, Toker A, Fitz JG. The lipid products of phosphoinositide 3-kinase contribute to regulation of cholangiocyte ATP and chloride transport. J Biol Chem. 1999;274:30979–30986. doi: 10.1074/jbc.274.43.30979. [DOI] [PubMed] [Google Scholar]
- 44.Bewick NL, Fernandes C, Pitt AD, Rasmussen HH, Whalley DW. Mechanisms of Na+-K+ pump regulation in cardiac myocytes during hyposmolar swelling. Am J Physiol Cell Physiol. 1999;276:C1091–C1099. doi: 10.1152/ajpcell.1999.276.5.C1091. [DOI] [PubMed] [Google Scholar]
- 45.Li Y, Trush MA. Diphenyleneiodonium, an NAD(P)H oxidase inhibitor, also potently inhibits mitochondrial reactive oxygen species production. Biochem Biophys Res Commun. 1998;253:295–299. doi: 10.1006/bbrc.1998.9729. [DOI] [PubMed] [Google Scholar]
- 46.McGuire JJ, Anderson DJ, McDonald BJ, Narayanasami R, Bennett BM. Inhibition of NADPH-cytochrome P450 reductase and glyceryl trinitrate biotransformation by diphenyleneiodonium sulfate. Biochem Pharmacol. 1998;56:881–893. doi: 10.1016/s0006-2952(98)00216-0. [DOI] [PubMed] [Google Scholar]
- 47.Hool LC, Di Maria CA, Viola HM, Arthur PG. Role of NAD(P)H oxidase in the regulation of cardiac L-type Ca2+ channel function during acute hypoxia. Cardiovasc Res. 2005;67:624–635. doi: 10.1016/j.cardiores.2005.04.025. [DOI] [PubMed] [Google Scholar]
- 48.Pietersma A, de Jong N, de Wit LE, Kraak-Slee RG, Koster JF, Sluiter W. Evidence against the involvement of multiple radical generating sites in the expression of the vascular cell adhesion molecule-1. Free Radic Res. 1998;28:137–150. doi: 10.3109/10715769809065800. [DOI] [PubMed] [Google Scholar]
- 49.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 50.Bendall JK, Heymes C, Wright TJ, Wheatcroft S, Grieve DJ, Shah AM, et al. Strain-dependent variation in vascular responses to nitric oxide in the isolated murine heart. J Mol Cell Cardiol. 2002;34:1325–1333. doi: 10.1006/jmcc.2002.2083. [DOI] [PubMed] [Google Scholar]
- 51.Brahmajothi MV, Campbell DL. Heterogeneous basal expression of nitric oxide synthase and superoxide dismutase isoforms in mammalian heart: implications for mechanisms governing indirect and direct nitric oxide-related effects. Circ Res. 1999;85:575–587. doi: 10.1161/01.res.85.7.575. [DOI] [PubMed] [Google Scholar]
- 52.Shimizu T, Numata T, Okada Y. A role of reactive oxygen species in apoptotic activation of volume-sensitive Cl− channel. Proc Natl Acad Sci USA. 2004;101:6770–6773. doi: 10.1073/pnas.0401604101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rhee SG, Lee S-R, Yang K-S, Kwon J, Kang SW. Hydrogen peroxide as intracellular messenger: identification of protein tyrosine phosphatases and PTEN As H2O2 target. In: Forman HJ, Fukuto J, Torres M, editors. Signal Transduction by Reactive Oxygen and Nitrogen Species: Pathways and Chemical Principles. Dordrect: Kluwer; 2003. pp. 167–179. [Google Scholar]
- 54.Fukui T, Yoshiyama M, Hanatani A, Omura T, Yoshikawa J, Abe Y. Expression of p22-phox and gp91-phox, essential components of NADPH oxidase, increases after myocardial infarction. Biochem Biophys Res Commun. 2001;281:1200–1206. doi: 10.1006/bbrc.2001.4493. [DOI] [PubMed] [Google Scholar]
- 55.Li JM, Gall NP, Grieve DJ, Chen M, Shah AM. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension. 2002;40:477–484. doi: 10.1161/01.hyp.0000032031.30374.32. [DOI] [PubMed] [Google Scholar]
- 56.Dudley SC, Jr, Hoch NE, McCann LA, Honeycutt C, Diamandopoulos L, Fukai T, et al. Atrial fibrillation increases production of superoxide by the left atrium and left atrial appendage: role of the NADPH and xanthine oxidases. Circulation. 2005;112:1266–1273. doi: 10.1161/CIRCULATIONAHA.105.538108. [DOI] [PubMed] [Google Scholar]