

Abstract
Cyclooxygenase (COX) catalysis by prostaglandin H synthase (PGHS) is a key control step for regulation of prostanoid biosynthesis. Both PGHS isoforms are integral membrane proteins and their substrate fatty acids readily partition into membranes, but the impact of phospholipids and lipid membranes on COX catalysis and the actions of COX inhibitors are not well understood. We have characterized the COX kinetics and ibuprofen inhibition of the purified PGHS isoforms in the presence of phosphatidylcholine (PC) with varying acyl chain structure and physical state. PC was found to directly inhibit COX activity, with non-competitive inhibition by PC monomers binding away from the COX active site and competitive inhibition by micellar/bilayer forms of PC due to sequestration of the arachidonate substrate. Competitive inhibition by native membranes was observed in a comparison of COX kinetics in sheep seminal vesicle microsomes before and after solubilization of PGHS-1. PC liposomes significantly increase the inhibitory potency of ibuprofen against both PGHS isoforms without changing the reversible character of ibuprofen action or requiring binding of PGHS to the liposomes. These results suggest a useful conceptual framework for analyzing the complex interactions among the PGHS proteins, substrates, inhibitors and phospholipid.
INTRODUCTION
The cyclooxygenase (COX1) activity of prostaglandin H synthase (PGHS) catalyzes the first committed step in the biosynthesis of the prostanoids, bioactive lipid signaling molecules with roles in a variety of physiological and pathological processes (1). The fatty acid substrates of COX catalysis are hydrophobic and partition readily into membranes (2), and both major isoforms of the enzyme, PGHS-1 and -2, are predominantly bound to intracellular membranes of the ER and nucleus (3). In addition, purified PGHS-1 and -2 proteins spontaneously associate with phospholipid bilayers (4). Thus, COX-mediated catalysis by both PGHS isoforms takes place in the context of various membrane environments. Nevertheless, the impacts of those membrane environments on COX catalysis are just beginning to be understood. PGG2 has been found to channel between the cyclooxygenase and peroxidase active sites in microsomal PGHS-1 but not in the detergent-solubilized protein (5). Further, detergent micelles, which mimic some aspects of phospholipid membranes, are known to markedly alter the effectiveness of several COX inhibitors (6, 7), although the large variation between detergents made mechanistic interpretation difficult. PC was reported to partially inactivate COX-2 and to increase the potency of two COX inhibitors (7), but the structure of the PC was not described and the details of the mechanism were not investigated. In the present study, we used purified enzymes to study the effects of PC molecules of varying chain length and saturation on the catalytic properties of both COX isoforms. Using this approach we acquired new information on the role of phospholipid structure, aggregation state and fluidity, and of PGHS binding to phospholipid on COX-1 and -2 kinetics and the actions of a prototypical competitive COX inhibitor, ibuprofen. The results indicate that both monomeric and bilayer forms of phospholipid affect COX activity and they suggest a general conceptual framework for interpreting the effects of micellar and bilayer materials on COX catalysis.
MATERIALS AND METHODS
Materials
Pure phospholipids and the Mini-Extruder liposome extrusion apparatus were from Avanti Polar Lipids (Alabaster, AL), soybean phosphatidylcholine (Phospholipon 90G) was from American Lecithin (Oxford, CT), fatty acids were from NuChek Preps (Elysian, MN), ibuprofen was from Cayman Chemical (Ann Arbor, MI) and Optiprep solution (60% w/v iodixanol) and heme were from Sigma (St. Louis, MO). Ovine PGHS-1 was purified from sheep seminal vesicle microsomes (8). Human PGHS-2 was purified from Sf9 cells expressing the recombinant protein (9). PGHS-1 and -2 were reconstituted with heme in the presence of phenol before use.
Cyclooxygenase assay
COX activity was measured with an oxygen electrode fitted with a high sensitivity membrane in cuvettes containing 3.0 ml of 0.1 M potassium phosphate, pH 7.2, containing 1.0 mM phenol and 1.0 μM heme (8). Unless stated otherwise, the reaction temperature was 23 °C. Working solutions of ARACHIDONATE were prepared by adding the desired volume of the stock solution of the fatty acid in toluene to a glass tube, evaporating the solvent with a stream of nitrogen gas, and dissolving the residue in 0.1 M Tris, pH 8.5. Ibuprofen was dissolved in 0.1 M Tris, pH 8.5, by vigorous vortexing and transient warming to 37 °C.
Critical micelle concentration
Critical micelle concentration (CMC) values were determined from changes in absorbance at 546 nm when buffer containing rhodamine 6G (1 μM) was titrated with the amphiphile being tested (10).
Phospholipid preparations
Phospholipid suspensions were prepared fresh each day by vortexing the dried lipid in 0.1 M potassium phosphate, pH 7.2. Preparation of unilamellar vesicles of DOPC/DOPS followed the procedure of MirAfzali et al. (4). Briefly, a chloroform solution containing DOPC (21 μmol) and DOPS (8.9 μmol), with or without oleate (8.5 μmol) was evaporated under a stream of nitrogen gas, dissolved in 0.5 ml of hexane and dried again with nitrogen. The resulting film was desiccated under vacuum for 1 hr and then hydrated for 1 hr in 25 mM Tris, pH 7.4, containing 50 mM KCl to give a phospholipid concentration of 30 mM, and then extruded 25 times through a 0.1 μm pore diameter polycarbonate filter using the Avanti Mini-Extruder. Unilamellar vesicles of DMPC in 0.1 M potassium phosphate, pH 7.2, were prepared in a similar fashion at 35−40 °C. To prepare ibuprofen pre-associated with PC, a solutions of purified soy PC (Phospholipon 90G) in chloroform was evaporated with a stream of nitrogen gas. A solution of ibuprofen solution in Tris buffer was added to the tube containing the dried lipid film, and the suspension was sonicated in a bath-type sonicator (Laboratory Supplies Company, Hicksville NY) for 30 min at room temperature as previously described (11). Unilamellar liposomes were prepared from the PC/ibuprofen dispersion by extrusion as described above.
Density gradients
Continuous iodixanol density gradients were prepared by freeze-thaw (12). Briefly, 3-ml disposable syringes were loaded with 2 ml of 1.12 g/ml iodixanol and positioned vertically. Then the syringes were subjected to two cycles of freezing (90 min at −20 °C) and thawing (4 hr at 4 °C). Reconstitution with buffer was achieved when the gradients were transferred to centrifuge tubes (13) by mixing with the contents of a syringe filled with double-strength buffer (50 mM Tris, pH 7.4, containing 100 mM KCl) using a Sage Instruments syringe pump (syringe size set at 10 ml and flow rate at 1.5 ml/min)
Density gradient analysis of PGHS-1/liposome mixtures
DOPC/DOPS or DOPC/DOPS/OA liposomes (240 μl), or the same volume of buffer, were mixed with 360 μl of 1.2 g/ml iodixanol and 30 μl of PGHS-1 apoenzyme (5.7 mg/ml) and incubated at 37 °C for 30 min. A 200 μl aliquot of each sample was loaded at the bottom of a preformed gradient before centrifugation at 27 000 rpm for 1 hr at 20 °C in a Beckman SW 60.1 Ti rotor. The gradients were fractionated from the bottom with a Beckman Fraction Recovery System connected to a Pharmacia P-1 pump. The density of each fraction was determined from its refractive index, with reference to a standard curve constructed by dilutions of stock iodixanol in the same buffer. The phospholipid level in each fraction was assayed by a modification of the procedure described by Stewart (14). Briefly, a 30 μl aliquot of each fraction was mixed with 1 ml of the 0.1 M ammonium ferrothiocyanate solution. Toluene (1.0 ml) was then added and the mixture was vortex vigorously for 30 sec. After centrifugation to separate the phases, the upper (toluene) layer was transferred to a cuvette for measurement of A467. The reliability of the ferrothiocyanate assay for measuring phospholipid in iodixanol gradient fractions was verified by parallel assays with the Phospholipid B kit (Wako Chemical). oPGHS-1 protein levels were quantitated by densitometry of the PGHS-1 band stained with SYPRO Orange (Sigma) after separation by polyacrylamide gel electrophoresis under denaturing conditions (15).
RESULTS
Actions of short chain PCs on COX-1 kinetics
COX-1 activity was examined as a function of the concentration of three short chain PCs: C7PC, C8PC and C9PC (Fig. 1). In each case, the COX-1 activity was found to decline sharply with increasing PC concentration, reaching ∼30% of control activity when the CMC was reached, and declining more slowly with further increases in PC level. These results indicate that COX-1 activity was strongly inhibited by monomers of these PCs, with less inhibition by the micellar form of the lipids.
Figure 1.

Effects of short chain PCs on cox-1 activity. PGHS-1 (15 nM) was injected into reaction mixtures containing 10 μM arachidonate and the indicated levels of short chain PC. The optimal velocity values are normalized to the control without PC. The CMC value for each PC (37) is indicated.
Effect of arachidonate concentration on COX-1 inhibition by C7PC
Conditions were sought to minimize or avoid formation of arachidonate micelles over the 0−100 μM substrate range used for COX assays. The CMC for arachidonate increased from 15 μM at pH 7.0 to 94 μM at pH 8.5 and 98 μM at pH 9.0 (Table 1). COX specific activity decreases over this same pH range, so pH 8.5 was chosen as a compromise to examine the response of COX-1 activity to changes in arachidonate concentration at two levels of C7PC, both below the phospholipid's CMC (Fig. 2). In the absence of C7PC, the Vmax was 49 units and the Km value for arachidonate was 2.7 μM. Inclusion of 0.5 mM C7PC produced a 20% decrease in Vmax without a significant change in the Km value. Increasing the C7PC level threefold to 1.5 mM resulted in a 60% decrease in Vmax with only a ∼50% increase in Km value. A similar pattern of changes in Vmax and Km with variation in C7PC was observed when the reactions were run at pH 7.2 instead of pH 8.5 (data not shown), indicating that formation of substrate micelles, which occurs at a much lower arachidonate level at the lower pH, was not affecting the outcome. The observed decreases in Vmax combined with very modest increases in apparent Km indicate that C7PC monomers do not compete with the fatty acid for binding in the COX active site.
Table 1.
Effect of pH on CMC of arachidonic acid.
| pH | CMCa (μM AA) |
|---|---|
| 7.0 | 15 |
| 7.5 | 36 |
| 8.0 | 47 |
| 8.5 | 94 |
| 9.0 | 98 |
Determined from the inflection in A546 when Rhodamine 6G (1 μM) was titrated with arachidonic acid (AA) at the desired pH. The buffers were prepared by adding HCl to 0.1 M Tris base.
Figure 2.

Effect of short chain PC on COX-1 response to arachidonate concentration. COX-1 (10 nM) was reacted with the indicated levels of arachidonate at 23 °C in 0.10 M Tris, pH 8.5 containing 1.0 mM phenol, 1.0 μM heme, and 0, 0.5 or 1.5 mM C7PC. The values of Vmax and Km were estimated by fitting the data for each C7PC level to the Michaelis-Menten equation using Kaleidagraph software.
Effects of short chain PCs on COX-1 inhibition by ibuprofen
Ibuprofen was chosen as the test inhibitor because it is well established as a simple competitive, reversible COX inhibitor under standard assay conditions (16). COX-1 inhibition by ibuprofen was examined in incubations with the three short chain PCs used for the experiment in Fig. 1. In each case, the PC concentration was fixed at a level either below or above its CMC and the ibuprofen concentration was varied to obtain an estimate of the K' value (essentially an apparent Ki for competitive inhibition) (Table 2). Each of the short chain PCs markedly increased the inhibitory potency of ibuprofen, by roughly two orders of magnitude for C7PC and C8PC at concentrations above their respective CMC values. Phospholipid micelles appeared to have a larger effect in enhancing ibuprofen's inhibitory actions than did phospholipid monomers. It may be noteworthy that the actions of micellar C9PC on ibuprofen potency were smaller than those of the other two short chain PCs.
Table 2.
Effects of short chain lecithins on COX-1 inhibition by ibuprofen.
| Additive | Ibuprofen K′a (μM) |
|---|---|
| None | 88 ± 9 |
| C7PC (0.27 mM) | 77 ± 5 |
| C7PC (2.7 mM) | 0.9 ± 0.2 |
| C8PC (0.17 mM) | 9.5 ± 2 |
| C8PC (1.3 mM) | 0.6 ± 0.1 |
| C9PC (0.025 mM) | 33 ± 4 |
| C9PC (0.10 mM) | 6.7 ± 0.4 |
The reaction mixture contained 10 μM arachidonate in the standard reaction mixture. Reaction was started by injection of 15 nM oPGHS-1 holoenzyme and the cyclooxygenase velocity was calculated from the amount of oxygen consumption after 20 s of reaction. Fitting the data to the equation for a competitive inhibitor, v = Vmax/(1 + [Ibuprofen]/K′), with Kaleidagraph was used to obtain the apparent Ki value (K′) and the standard error of the fit.
Effect of DOPC on COX-1 kinetics
The effects of phospholipid on COX-1 activity were also assessed with a longer chain PC containing two 18 carbon polyunsaturated acyl chains, DOPC, that has an extremely low CMC and thus is essentially all in micellar form. DOPC produced a concentration-dependent inhibition of COX-1 activity, with about one third of the activity lost at DOPC levels near 150 μM (Fig. 3). The data covered a limited concentration range, but suggest an IC50 value of ∼0.7 mM.
Figure 3.

Effect of DOPC on COX-1 activity. COX-1 (15 nM) was assayed at 23 °C in reactions with 10 μM arachidonate and the indicated level of DOPC. The enzyme activity was determined from the extent of oxygen consumption after 20 s of reaction and was normalized to the control without DOPC. The line represents the fit to a two-parameter logistic equation, v = 1 / (1 + ([DOPC] / IC50)n) (38).
Effects of DOPC on characteristics of COX-1 inhibition by ibuprofen
A distinctive characteristic of many potent COX inhibitors is the conversion of the initial enzyme-inhibitor complex to a tighter complex. This behavior a hallmark of “time-dependent” or “tight-binding” inhibitors, such as indomethacin, but is not observed under standard assay conditions for simple reversible inhibitors, such as ibuprofen (16). The ability of DOPC to elicit time-dependent inhibition of COX-1 by ibuprofen was tested by preincubating the enzyme with inhibitor in the presence and absence of phospholipid before reaction was initiated with excess arachidonate (Fig. 4). Without DOPC, 50 μM ibuprofen produced ∼70% inhibition of COX-1 regardless of preincubation time, as expected for a simple reversible competitive inhibitor. The presence of 155 μM DOPC increased the inhibitory potency of ibuprofen, with about 60% inhibition at 8 μM ibuprofen, but there was no significant change in the inhibition even when the preincubation was prolonged to 240 s. Controls lacking ibuprofen demonstrated that COX-1 activity was stable during preincubation with or without DOPC. These results indicate that DOPC did not trigger time-dependent inhibition by ibuprofen.
Figure 4.
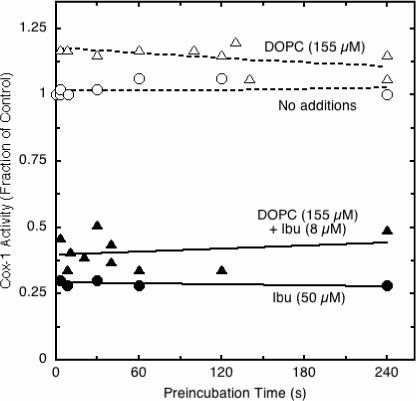
Effect of DOPC on characteristics of COX-1 inhibition by ibuprofen. PGHS-1 was preincubated at 23 °C with the indicated levels of DOPC and/or ibuprofen before reaction was started by injection of 100 μM arachidonate. COX activity was calculated from the oxygen consumed during the first 15 s and normalized to the corresponding control (no ibuprofen, reaction started by injection of enzyme).
Effects of premixing ibuprofen with phospholipid on inhibitor potency
Ibuprofen and other NSAIDs have been found to spontaneously associate with zwitterionic phospholipids such as PC due to both electrostatic and hydrophobic interactions (17, 18). Accordingly we investigated whether pre-association of ibuprofen and purified soy PC (90G) had an influence on COX activity that differed from the inhibitory effect when the same concentrations of ibuprofen and PC were added separately. For this comparison, buffer containing phenol and heme was added to the cuvette first, followed by the premixed PC/ibuprofen or the PC and ibuprofen separately. Arachidonate (10 μM) was then added and, following a two minute preincubation, the reaction was started by injection of PGHS-1 (15 nM). For the pre-associated ibuprofen/PC formulation, the IC50 value averaged 23 ± 3 μM ibuprofen in two experiments. The average IC50 value for ibuprofen added separately was 26 ± 3 μM. Thus, pre-associating ibuprofen with the PC did not significantly change its potency as a COX-1 inhibitor in vitro.
Effects of liposome bilayer fluidity of COX-1 kinetics
The relationship between liposome properties and COX inhibition was examined by measuring the inhibitory potency of DMPC over a temperature range spanning the transition temperature of the phospholipid (Fig. 5). The IC50 value for DMPC declined from almost 950 μM at 15 °C to only 250 μM at 22.5 °C and remained essentially unchanged at higher temperatures. This transition from lower to higher inhibitory potency was complete near the reported phase transition temperature for DMPC, 24 °C (19), indicating that DMPC was more inhibitory in the liquid crystalline state than in the gel state. COX-1 activity increased progressively with assay temperature from 20 to 35 °C, with no discontinuities near the transition temperature of DMPC (data not shown), confirming that the temperature-dependent change in inhibitory potency of DMPC shown in Fig. 5 reflected a change in the properties of the phospholipid and not the protein.
Figure 5.

Effect of temperature on COX-1 inhibition by DMPC. Reaction mixtures contained 10 μM arachidonate and 0−833 μM DMPC at indicated temperature. Each reaction was started by injection of 16 nM PGHS-1. The COX activity, expressed as a fraction of the control, was calculated from oxygen consumption at the time the control reaction reached 50% completion. The data for fractional activity vs. DMPC concentration at each temperature were fitted to a two-parameter logistic equation, v = 1 / (1 + ([DMPC] / IC50)n) to estimate the IC50 value and the standard error of the fit (38).
Effects of liposomes on COX-1 response to arachidonate level
One potential mode of COX inhibition by PC would be sequestration of the fatty acid substrate in the bilayer. Such sequestration would lower the effective level of substrate available to the enzyme and would be manifested by an increase in the apparent Km value. Unlike the short chain PCs used to examine the effects of monomeric PCs (Fig. 2), DMPC has a predicted CMC value in the nanomolar range (20), and thus has negligible monomer present at the total DMPC concentrations used here. Accordingly, DMPC liposomes were used to evaluate the effect of PC bilayer on COX-1 response to changes in arachidonate concentration. Unsaturated fatty acids partition rapidly into DMPC vesicles above the transition temperature (21). The results (Fig. 6) show the double-reciprocal plot pattern expected for competitive inhibition. The apparent Vmax value changed little as the DMPC level was increased from 0 to 400 μM, whereas the apparent Km value increased from 4.1 μM arachidonate in the control to 31 μM arachidonate with 400 μM DMPC present. A plot of Km / Vmax vs. DMPC concentration (22) gave a straight line whose x-intercept indicated a Ki value of 55 μM DMPC (data not shown). The DMPC liposomes were prepared by extrusion through 0.1 μm pores and thus were much too large to enter the COX channel to compete directly with arachidonate binding to the protein. The competitive inhibition by the DMPC must instead reflect indirect competition by reversible sequestration of arachidonate partitioned into the liposome membrane.
Figure 6.
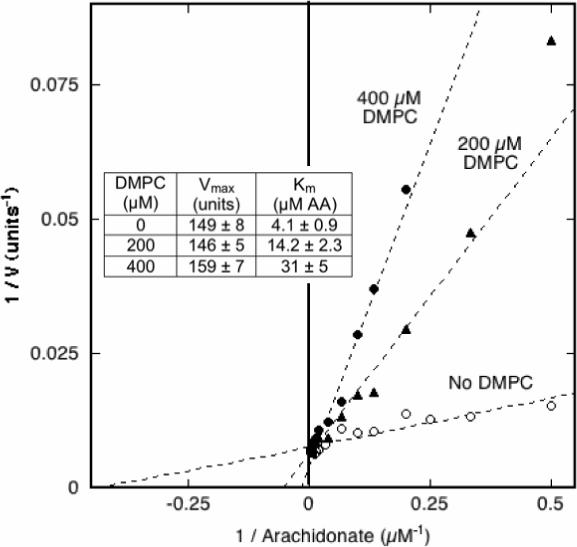
Effects of DMPC on COX-1 kinetic parameters. The COX velocity of 6.4 nM PGHS-1 was measured at 30 °C with 2−250 μM arachidonic acid (AA) and 0, 200 or 400 μM DMPC. The data are presented in double –reciprocal format. For each level of DMPC, the data were fitted to the Michaelis-Menten equation to estimate values for Vmax and Km shown in the insert (± the SD for the fit).
Responses of membrane bound and solubilized COX-1 to arachidonate level
To assess the biological significance of the COX inhibition by liposomal phospholipid observed with the purified enzyme (Fig. 6) we compared the COX-1 kinetics for the enzyme bound to microsomal membranes from sheep seminal vesicles with those of the same microsomes treated with Tween 20 to disrupt the membrane and solubilize the enzyme (Fig. 7). This level of detergent used (0.9% v/v) is enough to quantitatively extract PGHS-1 from the membranes, but the PGHS-1 was not purified, permitting direct comparison of solubilized and membrane bound enzymes in the presence of other microsomal components. The reaction pH (8.5) was chosen so that the substrate itself did not form micelles over the concentration range used (Table 1). The results shown in Fig. 7 indicate that the membrane bound enzyme had a Km value of 4.4 ± 0.3 μM arachidonate, twice that of the solubilized enzyme (2.1 ± 0.2 μM). In contrast, the Vmax values of membrane bound and solubilized enzymes were essentially the same. This competitive inhibition pattern is similar to that observed above for COX-1 and liposomes (Fig. 6) and suggests that arachidonate was sequestered by the microsomal membranes, much as it was by the liposomal membranes.
Figure 7.

Comparison of COX-1 kinetics in microsomal PGHS-1 before and after membrane disruption with detergent. The COX-1 velocity was determined at the indicated arachidonate levels for membrane bound PGHS-1 in sheep seminal vesicle microsomes and for the enzyme solubilized by addition of 0.9 % (v/v) Tween 20 to the same microsomes. The reaction buffer was 0.1 M Tris, pH 8.5, containing 0.1 mM phenol and 1 μM heme, and was thermostatted at 23 °C. Dilution of the detergent treated sample by the reaction mixture (1:375) was used to minimize the effect of the detergent itself. The activity was normalized to the protein concentration; each point represents the average of duplicate reactions. The data were fitted to the Michaelis-Menten equation to estimate values for Vmax and Km shown in the insert (± the SD for the fit).
Effect of lipid composition on PGHS-1 association with preformed liposomes
Inclusion of oleic acid in DOPC/DOPS liposomes has been reported to increase the association between the liposomes and PGHS-1 and -2 (4). To verify this effect of oleate, PGHS-1 was incubated with preformed liposomes composed of DOPC/DOPS/OA or DOPC/DOPS. Analysis of the mixtures by density gradient centrifugation (Fig. 8) showed that the inclusion of oleic acid in the liposomes markedly increased the fraction of PGHS-1 protein associated with phospholipid in lower density fractions, as expected from the earlier results (4). Interestingly, liposomal phospholipid was found in two peaks of quite different densities after incubation of PGHS-1 with the DOPC/DOPS liposomes, indicating at least two types of liposome structure were present.
Figure 8.
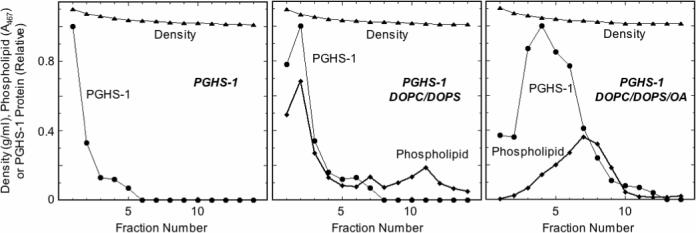
Characterization of PGHS-1 binding to liposomes. PGHS-1 was incubated with DOPC/DOPS/OA or DOPC/DOPS liposomes, or with buffer, for 30 min at 37 °C before analysis by density gradient centrifugation. Details are described in Experimental Procedures.
Effects of DOPC/DOPS and DOPC/DOPS/OA liposomes on COX-1 and -2 kinetics
The preferential binding of PGHS to DOPC/DOPS liposomes containing oleic acid (Ref. (4) and Fig. 8) provided an opportunity to test the role of PGHS-phospholipid interactions in COX inhibition by PCs. The DOPC/DOPS liposomes inhibited COX-1 activity, with a K’ value of 550 μM (Table 3), comparable to the inhibition seen with DOPC in Fig. 3. The DOPC/DOPS/OA liposomes were considerably more potent inhibitors of COX-1, with a K’ value of 178 μM (Table 3). DOPC/DOPS liposomes produced weak inhibition of COX-2 (K’ of 2.7 mM; Table 3), whereas the DOPC/DOPS/OA liposomes were stronger inhibitors (K’ of 0.96 mM; Table 3). Thus, inclusion of oleate in the liposomes resulted in a stronger inhibition of both COX-1 and -2, indicating that protein-phospholipid binding does contribute to the inhibitory effect of the phospholipid.
Table 3.
Effects of DOPC/DOPS and DOPC/DOPS/OA liposomes (lipos) on COX-1 and -2 activity.
| DOPC/DOPS | DOPC/DOPS/OA | |
|---|---|---|
| Enzyme | K′ (μM lipos)a | K′ (μM lipos)a |
| COX-1 | 550±90 (n=2) | 178±3 (n=2) |
| COX-2 | 2710±110 (n=2) | 960±480 (n=2) |
The procedures were the same as for the experiments in Table 2.
Effects of DOPC, DOPC/DOPS and DOPC/DOPS/OA liposomes on COX-1 and -2 inhibition by ibuprofen
The apparent Ki (K’) values of ibuprofen for inhibition of COX-1 and -2 were determined in the presence of DOPC, DOPC/DOPS, or DOPC/DOPS/OA (Fig. 9). Without added phospholipid, ibuprofen had K’ values of 97 and 460 μM for COX-1 and -2, respectively. All three phospholipid mixtures potentiated the inhibition of both COX-1 and -2 by ibuprofen. Inclusion of DOPS increased this effect somewhat for both COX-1 and -2, at least at the lower level of phospholipid tested. Inclusion of oleic acid had no effect on ibuprofen potency against COX-2 and slightly reduced its inhibitory potency against COX-1. Thus, DOPC/DOPS/OA and DOPC/DOPS produced similar enhancement of COX inhibition by ibuprofen even though the two classes of liposomes had marked differences in PGHS-1 binding. This suggests that phospholipid-dependent potentiation of COX-1 and -2 inhibition by ibuprofen is not due to binding of the proteins to the phospholipid.
Figure 9.

Effect of liposome composition on inhibitory potency of ibuprofen against COX-1 and -2. The apparent Ki value (K') for ibuprofen was determined for PGHS-1 (15 nM; left panel) and PGHS-2 (30 nM; right panel) in the presence of the indicated total phospholipid concentrations of DOPC, DOPC/DOPS or DOPC/DOPS/OA liposomes. The data were analyzed as described for the experiment in Table 2. The K' values plotted represent the averages ± SD for 2−4 independent experiments.
DISCUSSION
Phospholipids and COX catalysis
PGHS-1 and -2 are located primarily in the ER and nuclear membranes, and phospholipids are thus a major component of the native environment of both PGHS isoforms. The results of the present studies with various species of PC show that phospholipid can have a number of distinct effects, all inhibitory, on COX-1 and -2 activities. Conceptually, these inhibitory actions can be considered in terms of effects of monomeric or bilayer forms of phospholipid on the lipid substrate and on the proteins themselves (Fig. 10).
Figure 10.
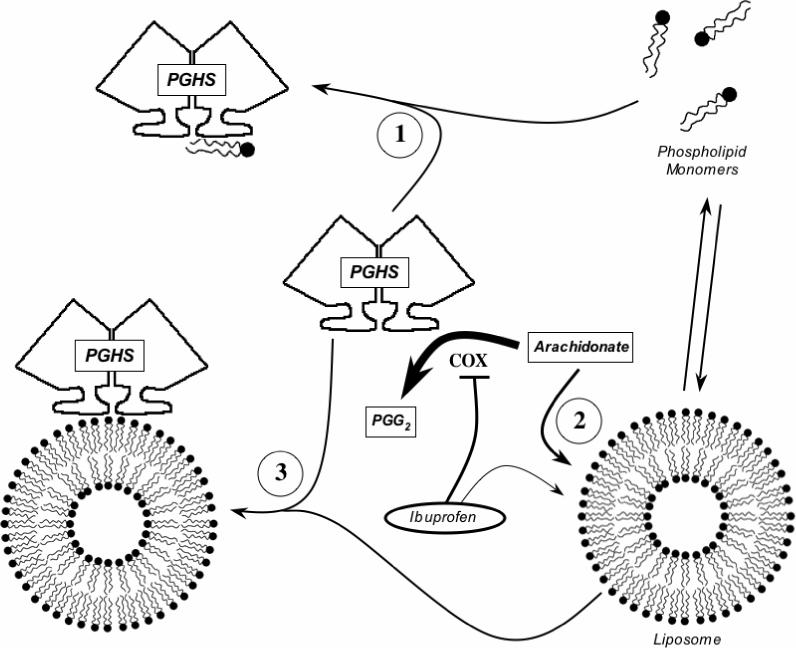
Proposed actions of phospholipid on COX activity. The hypothetical modes of interaction among the PGHS-1 or -2 dimer, monomeric and liposomal phospholipid, COX substrate (arachidonate) and COX inhibitor (ibuprofen) are depicted.
Inhibition of COX by monomeric phospholipid
All three of the short chain PCs examined showed steep dose-response curves for COX-1 inhibition below the CMC value (Fig. 1), indicating that the PC monomers were potent inhibitors (schematized as process “1” in Fig. 10). The absence of competition with arachidonate (Fig. 2) shows that the action of the PC monomers involves binding at a site distinct from the COX catalytic site. Some potential binding areas for phospholipid monomers in the membrane anchor domain have been suggested by molecular dynamics calculations (23), and by the positions of bound detergent in crystallographic structures (24, 25). COX inhibition by monomeric phospholipids is presumably not a significant factor in vivo due to the very low CMC for naturally-occurring phospholipids. Nevertheless, short chain PC monomers may be useful probes for further characterization of phospholipid interaction near the membrane anchor region.
Inhibition of COX by bilayer phospholipid
Concentration-dependent COX-1 inhibition was also seen with micellar forms of the short chain PCs (Fig. 1) and with DMPC, DOPC, DOPC/DOPS mixtures and soy phospholipid, each of which adopts bilayer forms with negligible monomer levels (Figs. 1, 3 and 9; Tables 2 and 3). Phospholipid bilayers are known to rapidly take up unsaturated fatty acids (21, 26), which would reduce the level of substrate available to COX (depicted as process “2” in Fig. 10) and account for the competitive kinetics observed for this mode of inhibition (Fig. 6). Inhibition by DMPC bilayers was more potent above the phase transition temperature (Fig. 5), showing that the physical state of the bilayer was important, and fits with the temperature-dependent kinetics of fatty acid binding to DMPC liposomes (21).
Association of PGHS with liposomes and COX catalysis
The membrane anchor region near the mouth of the COX channel of PGHS-1 and -2 binds to DOPC/DOPS/OA liposomes (4, 27) and any arachidonate partitioning into such liposomes might be expected to be presented efficiently to enzyme bound to the bilayer. Surprisingly, DOPC/DOPS/OA liposomes proved to be more inhibitory to COX-1 and -2 activity than the DOPC/DOPS liposomes (Table 3), which bind the proteins poorly (Ref. (4) and Fig. 8). It thus appears that PGHS binding to the DOPC/DOPS/OA liposomes actually lowers the COX activity (depicted as process “3” in Fig. 10). The mechanism of this mode of inhibition is unclear, but oleate is known to be a weak competitive inhibitor for COX-1 (28) so oleate in the bilayer might compete with arachidonate for access to the COX active site.
Preincubation of PGHS with PC for 15 min was reported earlier to lead to almost complete loss of COX-2 activity (7). In contrast, little loss of activity was observed in the present studies when COX-1 was preincubated with DOPC (Fig. 4). The reason for this difference is unclear, and the structure of the PC used in the previous study was not specified.
Differential effects of liposomes on COX-1 and -2
Partitioning of arachidonate into liposomes (process “2” in Fig. 10) will sequester the amount of substrate fatty acid regardless of which enzyme is added to the reaction mixture. DOPC/DOPS/OA liposomes have roughly equal binding capacities for PGHS-1 and -2 (process “3” in Fig. 10) (4). Yet the data in Table 3 show that liposomes have different inhibitory potencies for COX-1 and -2. This is true for DOPC/DOPS liposomes, which act by mode “2” (Fig. 10), and for DOPC/DOPS/OA liposomes, which act by modes 2 and 3 (Fig. 10). In each case, COX-2 was approximately five-fold less sensitive to inhibition than COX-1. The weaker inhibition of COX-2 by liposomes can probably be traced to the higher efficiency of feedback activation by product peroxide (PGG2) in that isoform (9): partition of a given portion of the relatively hydrophobic PGG2 into liposomes would have a stronger effect on COX-1 than on COX-2.
In vivo implications of COX inhibition by PC
The inhibitory effects of phospholipid bilayers on COX-1 and -2 activities observed in the present in vitro studies have some implications for the enzymes in vivo. The reversible partition of substrate fatty acids (arachidonate and other polyunsaturated fatty acids) into cellular membranes would lower the level of substrate available to both COX-1 and -2. This would tend to extend PGG2 generation over a longer time. Indeed, the COX reaction in cells provided excess exogenous substrate takes ∼10 min to be complete, whereas the reaction of isolated enzyme in vitro is essentially over in less than 2 min (29-32). Microsomal preparations of PGHS-1 or -2, which retain the membrane environment but have much lower local phospholipid concentrations than found in intact cells, might be expected to sequester the fatty acid substrate, and indeed the apparent Km value of microsomal COX-1 was twice that found after the membranes were disrupted with detergent (Fig. 7). Another indication of native membrane effects on COX activity is the significant increase in enzyme units encountered occasionally upon detergent solubilization of recombinant COX-2 expressed in Sf9 cells (data not shown).
Phospholipids and COX inhibitor actions
A previous extensive study of detergent effects on COX-1 and -2 inhibition by a series of anticyclooxygenase agents (7) included a limited number of COX-2 experiments with PC of an unspecified structure. The effects of the detergents on the inhibitors' potency were complex and were not interpreted in terms of a unified mechanism. The PC was reported to increase the inhibitory potency of ibuprofen and flufenamic acid, fitting the pattern observed with several nonionic detergents (7). The present results with structurally defined DOPC liposomes and a simple, reversible competitive inhibitor (ibuprofen) showed roughly 5-fold increases in the inhibitor's potency against both COX-1 and -2 (Fig. 9). This fits the pattern reported earlier, though the magnitude of the inhibitor potentiation is more modest in the present case. A remarkable (100-fold) increase in the potency of ibuprofen as a COX-1 inhibitor was observed with short chain (C7 and C8) PCs at levels above their CMC values (Table 2). Thus, condensed forms of phospholipid, whether bilayer or micellar, increased the inhibitory potency of ibuprofen. Ibuprofen appeared to retain a reversible, competitive mode of action in the presence of phospholipid (Fig. 4), in distinction with the reported conversion of the compound to a time-dependent inhibitor in the presence of some detergents (7).
Mechanism of action of phospholipid on COX inhibitor potency
The conceptual scheme used for analysis of the inhibitory actions of phospholipid on COX catalysis (Fig. 10) provides a useful framework for considering the effects of lipid bilayers on the potency of COX inhibitors. COX inhibitors, like COX substrates, tend to be hydrophobic and/or amphipathic molecules, with significant partitioning into phospholipid bilayers (process “2” in Fig. 10). Further, COX inhibitors, like COX substrates, bind at the COX active site. If the liposomes absorb a higher fraction of the arachidonate than of the inhibitor, the inhibitor will become a more effective competitor at the COX active site and its apparent affinity for the enzyme will increase. This interpretation envisages that, unlike the case of ibuprofen, some inhibitors may actually partition into phospholipid bilayers more effectively than arachidonate and thus have their potency decreased. This provides a potential explanation for the otherwise puzzling observation that detergent micelles decreased the potency of some COX inhibitors (7). There are also kinetic considerations. The equilibration of unsaturated fatty acids between bulk solution and liposomes occurs quite quickly (21) and is likely to be complete under the COX assay conditions used in the present study. However, the relative rates of partitioning of arachidonate and an NSAID into liposomes may influence the outcome if the mixture does not have time to equilibrate before the COX reaction is started.
In vivo implications of COX inhibitor potentiation by PC
In humans, pre-associating PC with aspirin or other NSAIDs produced less gastric damage than aspirin alone, without decreasing the COX inhibition (17, 18, 33), and formulations of NSAID with PC are being considered as one approach to lowering gastrointestinal side effects of these agents (34). The protective action of PC in this setting has been attributed to actions at the surface of gastric mucosal cells that is distinct from COX inhibition (35). However, there are indications that presentation of NSAID pre-associated with PC to cultured cells in vitro can increase the intracellular actions of the agents, including their COX-inhibitory activity (11, 36). Whether this effect has any connection to the potentiation of COX inhibition by PC observed here is yet to be determined. In addition to their potential use as a mechanism of delivering NSAIDs across the mucosal barrier without surface injury, PC liposomes and/or micelles may have additional utility for in vitro screening of COX inhibitors. Only a subset of COX inhibitors have their potency increased by detergents (6, 7) and the same is likely true of phospholipids. Screening NSAIDs increased inhibitory potency in the presence of PC would be a rapid and inexpensive way of selecting agents for further testing in combined formulations with PC.
ACKNOWLEDGEMENTS
We thank Dr. Corina Rogge for preparation of the purified human PGHS-2 and Ms. Pei-Yung Hsu for assistance in purification of ovine PGHS-1.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
This work was supported in part by NIH grant GM52170 (RJK) and DK063882 (LML)
The abbreviations used are: COX, cyclooxygenase; PGHS-1 and -2, prostaglandin H synthase isoform-1 and -2; PC, phosphatidylcholine; C7PC, diheptanoyl PC; C8PC, dioctanoyl PC; C9PC, dinonanoyl PC; DOPC, dioleoyl PC; DOPS, dioleoyl phosphatidylserine; OA, oleic acid; CMC, critical micelle concentration; DMPC, dimyristoyl PC.
REFERENCES
- 1.Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem. 2000;69:145. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- 2.Hamilton JA. Fatty acid transport: difficult or easy? J Lipid Res. 1998;39:467. [PubMed] [Google Scholar]
- 3.Simmons DL, Botting RM, Hla T. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev. 2004;56:387. doi: 10.1124/pr.56.3.3. [DOI] [PubMed] [Google Scholar]
- 4.MirAfzali Z, Leipprandt JR, McCracken JL, DeWitt DL. Fast, efficient reconstitution of the cyclooxygenases into proteoliposomes. Arch Biochem Biophys. 2005;443:60. doi: 10.1016/j.abb.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 5.Eling TE, Glasgow WC, Curtis JF, Hubbard WC, Handler JA. Studies on the reduction of endogenously generated prostaglandin G2 by prostaglandin H synthase. J Biol Chem. 1991;266:12348. [PubMed] [Google Scholar]
- 6.Johnson AR, Marletta MA, Dyer RD. Slow-binding inhibition of human prostaglandin endoperoxide synthase-2 with darbufelone, an isoform-selective antiinflammatory di-tert-butyl phenol. Biochemistry. 2001;40:7736. doi: 10.1021/bi002343f. [DOI] [PubMed] [Google Scholar]
- 7.Ouellet M, Falgueyret JP, Percival MD. Detergents profoundly affect inhibitor potencies against both cyclo-oxygenase isoforms. Biochem J. 2004;377:675. doi: 10.1042/BJ20030969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kulmacz R, Lands W. In: Prostaglandins and Related Substances: A Practical Approach. Benedetto C, McDonald-Gibson R, Nigam S, Slater T, editors. IRL Press; Oxford: 1987. p. 209. [Google Scholar]
- 9.Kulmacz RJ, Wang LH. Comparison of hydroperoxide initiator requirements for the cyclooxygenase activities of prostaglandin H synthase-1 and -2. J Biol Chem. 1995;270:24019. doi: 10.1074/jbc.270.41.24019. [DOI] [PubMed] [Google Scholar]
- 10.Bonsen PP, de Haas GH, Pieterson WA, van Deenen LL. Studies on phospholipase A and its zymogen from porcine pancreas. IV. The influence of chemical modification of the lecithin structure on substrate properties. Biochim Biophys Acta. 1972;270:364. doi: 10.1016/0005-2760(72)90200-7. [DOI] [PubMed] [Google Scholar]
- 11.Lichtenberger LM, Romero JJ, de Ruijter WM, et al. Phosphatidylcholine association increases the anti-inflammatory and analgesic activity of ibuprofen in acute and chronic rodent models of joint inflammation: relationship to alterations in bioavailability and cyclooxygenase-inhibitory potency. J Pharmacol Exp Ther. 2001;298:279. [PubMed] [Google Scholar]
- 12.Haff LA. Production of Ficoll, Percoll, and albumin gradients by the freeze-thaw method. Prep Biochem. 1979;9:149. doi: 10.1080/00327487908061680. [DOI] [PubMed] [Google Scholar]
- 13.Cooper AJ, Smallwood JA, Morgan RA. The preparation of freeze-thaw density gradients with homogeneous solute concentrations. J Immunol Methods. 1984;71:259. doi: 10.1016/0022-1759(84)90072-3. [DOI] [PubMed] [Google Scholar]
- 14.Stewart JC. Colorimetric determination of phospholipids with ammonium ferrothiocyanate. Anal Biochem. 1980;104:10. doi: 10.1016/0003-2697(80)90269-9. [DOI] [PubMed] [Google Scholar]
- 15.Guo Q, Chang S, Diekman L, Xiao G, Kulmacz RJ. Comparison of prostaglandin H synthase isoform structures using limited proteolytic digestion. Arch Biochem Biophys. 1997;344:150. doi: 10.1006/abbi.1997.0192. [DOI] [PubMed] [Google Scholar]
- 16.Rome LH, Lands WE. Structural requirements for time-dependent inhibition of prostaglandin biosynthesis by anti-inflammatory drugs. Proc Natl Acad Sci U S A. 1975;72:4863. doi: 10.1073/pnas.72.12.4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giraud MN, Motta C, Romero JJ, Bommelaer G, Lichtenberger LM. Interaction of indomethacin and naproxen with gastric surface-active phospholipids: a possible mechanism for the gastric toxicity of nonsteroidal anti-inflammatory drugs (NSAIDs). Biochem Pharmacol. 1999;57:247. doi: 10.1016/s0006-2952(98)00303-7. [DOI] [PubMed] [Google Scholar]
- 18.Lichtenberger LM, Wang ZM, Romero JJ, et al. Non-steroidal anti-inflammatory drugs (NSAIDs) associate with zwitterionic phospholipids: insight into the mechanism and reversal of NSAID-induced gastrointestinal injury. Nat Med. 1995;1:154. doi: 10.1038/nm0295-154. [DOI] [PubMed] [Google Scholar]
- 19.Koynova R, Caffrey M. Phases and phase transitions of the phosphatidylcholines. Biochim Biophys Acta. 1998;1376:91. doi: 10.1016/s0304-4157(98)00006-9. [DOI] [PubMed] [Google Scholar]
- 20.Weschayanwiwat P, Scamehorn J, Reilly P. Surfactant properties of low molecular weight phospholipids. Journal of Surefactants and Detergents. 2005;8:65. [Google Scholar]
- 21.Rogerson ML, Robinson BH, Bucak S, Walde P. Kinetic studies of the interaction of fatty acids with phosphatidylcholine vesicles (liposomes). Colloids Surf B Biointerfaces. 2006;48:24. doi: 10.1016/j.colsurfb.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 22.Cornish-Bowden A, editor. Portland Press; London: 2004. [Google Scholar]
- 23.Fowler PW, Coveney PV. A computational protocol for the integration of the monotopic protein prostaglandin H2 synthase into a phospholipid bilayer. Biophys J. 2006;91:401. doi: 10.1529/biophysj.105.077784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kiefer JR, Pawlitz JL, Moreland KT, et al. Structural insights into the stereochemistry of the cyclooxygenase reaction. Nature. 2000;405:97. doi: 10.1038/35011103. [DOI] [PubMed] [Google Scholar]
- 25.Gupta K, Selinsky BS, Loll PJ. 2.0 angstroms structure of prostaglandin H2 synthase-1 reconstituted with a manganese porphyrin cofactor. Acta Crystallogr D Biol Crystallogr. 2006;62:151. doi: 10.1107/S0907444905036309. [DOI] [PubMed] [Google Scholar]
- 26.Thomas RM, Baici A, Werder M, Schulthess G, Hauser H. Kinetics and mechanism of long-chain fatty acid transport into phosphatidylcholine vesicles from various donor systems. Biochemistry. 2002;41:1591. doi: 10.1021/bi011555p. [DOI] [PubMed] [Google Scholar]
- 27.MirAfzali Z, Leipprandt JR, McCracken JL, DeWitt DL. Topography of the prostaglandin endoperoxide H2 synthase-2 in membranes. J Biol Chem. 2006;281:28354. doi: 10.1074/jbc.M605206200. [DOI] [PubMed] [Google Scholar]
- 28.Lands W, LeTellier P, Rome L, Vanderhoek J. Inhibition of prostaglandin biosynthesis. Adv Biosci. 1973;9:15. [Google Scholar]
- 29.Ornberg R, Koki A. Visualization and quantitation of cyclooxygenase-1 and -2 activity by digital fluorescence microscopy. Adv Exp Med Biol. 1999;469:131. doi: 10.1007/978-1-4615-4793-8_20. [DOI] [PubMed] [Google Scholar]
- 30.Cook HW, Lands WE. Evidence for an activating factor formed during prostaglandin biosynthesis. Biochem Biophys Res Commun. 1975;65:464. doi: 10.1016/s0006-291x(75)80170-7. [DOI] [PubMed] [Google Scholar]
- 31.Miyamoto T, Ogino N, Yamamoto S, Hayaishi O. Purification of prostaglandin endoperoxide synthetase from bovine vesicular gland microsomes. J Biol Chem. 1976;251:2629. [PubMed] [Google Scholar]
- 32.Van der Ouderaa FJ, Buytenhek M, Nugteren DH, Van Dorp DA. Purification and characterisation of prostaglandin endoperoxide synthetase from sheep vesicular glands. Biochim Biophys Acta. 1977;487:315. doi: 10.1016/0005-2760(77)90008-x. [DOI] [PubMed] [Google Scholar]
- 33.Anand BS, Romero JJ, Sanduja SK, Lichtenberger LM. Phospholipid association reduces the gastric mucosal toxicity of aspirin in human subjects. Am J Gastroenterol. 1999;94:1818. doi: 10.1111/j.1572-0241.1999.01211.x. [DOI] [PubMed] [Google Scholar]
- 34.Lane ME, Kim MJ. Assessment and prevention of gastrointestinal toxicity of nonsteroidal anti-inflammatory drugs. J Pharm Pharmacol. 2006;58:1295. doi: 10.1211/jpp.58.10.0001. [DOI] [PubMed] [Google Scholar]
- 35.Lichtenberger LM, Romero JJ, Dial EJ. Surface phospholipids in gastric injury and protection when a selective cyclooxygenase-2 inhibitor (Coxib) is used in combination with aspirin. Br J Pharmacol. 2007;150:913. doi: 10.1038/sj.bjp.0707176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dial EJ, Doyen JR, Lichtenberger LM. Phosphatidylcholine-associated nonsteroidal anti-inflammatory drugs (NSAIDs) inhibit DNA synthesis and the growth of colon cancer cells in vitro. Cancer Chemother Pharmacol. 2006;57:295. doi: 10.1007/s00280-005-0048-x. [DOI] [PubMed] [Google Scholar]
- 37.Hauser H. Short-chain phospholipids as detergents. Biochim Biophys Acta. 2000;1508:164. doi: 10.1016/s0304-4157(00)00008-3. [DOI] [PubMed] [Google Scholar]
- 38.DeLean A, Munson PJ, Rodbard D. Simultaneous analysis of families of sigmoidal curves: application to bioassay, radioligand assay, and physiological dose-response curves. Am J Physiol. 1978;235:E97. doi: 10.1152/ajpendo.1978.235.2.E97. [DOI] [PubMed] [Google Scholar]