

Abstract
The cAMP fluorescent probe FlCRhR was used to monitor changes in intracellular cAMP concentration ([cAMP]i) in isolated frog ventricular myocytes. The probe was introduced into the cell through a patch pipette which allowed simultaneous recording of the whole-cell L-type Ca2+ current (ICa). Ratiometric imaging was used to monitor [cAMP]i changes in response to the β-adrenergic agonist isoprenaline (ISO) or to the direct adenylyl cyclase activator forskolin (FSK).
FlCRhR fluorescence was distributed in the cytosol in a striated pattern, with high fluorescence in the I-bands and low fluorescence in the A-bands. This pattern of distribution was mimicked by fluorescein dextran, another high molecular weight fluorescent molecule, and was therefore likely to be due to anisotropic diffusion of the probe in the cytosol due to the hindrance generated by sarcomeric proteins in the A-bands.
Introduction of FlCRhR into the cell induced a small ≈70% stimulatory effect on basal ICa, attenuating about 2-fold a subsequent response of ICa to 1-10 μm ISO (from 400 to 200%).
Brief (10 s) application of a saturating concentration of ISO (1-20 μm) to the cell induced a transient increase in both ICa and [cAMP]i. However, the [cAMP]i transient was ≈2-fold shorter in duration than the ICa transient, i.e. ICa was still strongly enhanced when [cAMP]i had already returned to control level. This indicates that hydrolysis of cAMP by phosphodiesterases is not a rate limiting step in the recovery of ICa from ISO stimulation.
When the application of ISO was maintained, ICa and [cAMP]i responses followed a similar time course, with a half-maximal response at ≈60 s. This suggests that activation of Ca2+ channels by cAMP-dependent protein kinase occurs on a much faster time scale than the rise in [cAMP]i.
When the cells were exposed to FSK (13 μm), both responses of ICa and [cAMP]i were ≈2-fold slower than with ISO. This demonstrates that the slower response of ICa to FSK is due to a slower rise in [cAMP]i rather than to some inhibitory effect of FSK on ICa or to a direct or priming effect of the stimulatory G protein Gs on Ca2+ channels.
Simultaneous measurements of [cAMP]i and ICa changes in intact cardiac myocytes opens the way to dissect the temporal sequence of events in the cAMP cascade mediating the response of the heart to a large number of hormones and inotropic agents.
The beat rate and force of contraction of the heart are under the dual control of the sympathetic and parasympathetic systems (Brodde & Michel, 1999). Both systems control reciprocally the synthesis of cAMP and, hence, the activity of cAMP-dependent protein kinase (PKA) and the phosphorylation of a multitude of regulatory proteins, including the L-type Ca2+ channel (Osterrieder et al. 1982; for review see Hartzell, 1988; McDonald et al. 1994; Striessnig, 1999), phospholamban (Simmerman & Jones, 1998), the sarcoplasmic reticulum Ca2+ release channel (Hain et al. 1995; Valdivia et al. 1995) and contractile proteins (Rapundalo, 1998). At the level of a single cardiac myocyte, a variation of intracellular cAMP level ([cAMP]i) can be assessed indirectly by measuring the changes in the activity of a PKA substrate protein. Measurement of L-type Ca2+ channel current (ICa) variations in response to cAMP-elevating agents is a prototypical example of such an approach (Kameyama et al. 1986; for review see Hartzell, 1988; McDonald et al. 1994; Striessnig, 1999). Indeed, the amplitude of ICa is increased by β-adrenergic agonists, such as isoprenaline (ISO) (Kameyama et al. 1986), by forskolin (FSK), a direct activator of adenylyl cyclase (Hartzell & Fischmeister, 1987), by phosphodiesterase inhibitors, such as 3-isobutyl-1-methylxanthine (IBMX) (Fischmeister & Hartzell, 1990), or by intracellular dialysis of cAMP (Schouten & Morad, 1989) or PKA (Osterrieder et al. 1982; White & Hartzell, 1988). All these manoeuvres converge towards the stimulation of ICa via activation of PKA, since inhibition of the enzyme with a competitive inhibitor analogue of cAMP, such as Rp-cAMPS, or with PKI, a highly selective peptide inhibitor of PKA (Walsh et al. 1990), fully abolishes the increase in ICa (Kameyama et al. 1986; Hartzell et al. 1991; Zhou et al. 1997). Similarly, ICa was used to demonstrate a decrease in [cAMP]i and PKA activity in cardiac myocytes upon activation of Gi protein-coupled receptors, such as muscarinic (Hartzell, 1988; Méry et al. 1997) or adenosine A1 receptors (Mubagwa et al. 1996). Variations in ICa were also used as an index of local [cAMP]i changes in double-microperfusion experiments in frog ventricular myocytes (Jurevicius & Fischmeister, 1996).
Combined with rapid extracellular perfusion and UV-flash photolysis of caged compounds, measurement of ICa variations was also used to estimate and compare the time course of [cAMP]i changes upon a sudden application of different cAMP-elevating agents. For instance, in guinea-pig (Yatani & Brown, 1989) and frog ventricular myocytes (Frace et al. 1993), the stimulatory effect of FSK on ICa develops with a 2- to 3-fold slower time scale than the effect of ISO at doses that are equipotent in their effects on ICa. While this difference was attributed to different kinetics in [cAMP]i changes (Jurevicius & Fischmeister, 1996), it could also result from some concomitant inhibitory effects of FSK on ICa (Boutjdir et al. 1990; Asai et al. 1996) or from the presence of an additional activatory mechanism in β-adrenergic stimulation such as a priming effect of the G protein Gs on PKA phosphorylation of the Ca2+ channel (Cavaliéet al. 1991). Clearly, the indirect assay of [cAMP]i changes via the measurement of ICa has reached its limits in trying to distinguish between these hypotheses. For this and many other applications, a direct assessment of [cAMP]i in an intact isolated cardiac myocyte is required. However, to our knowledge, this has not been technically feasible until now.
Here, we developed a novel approach which combines electrophysiological measurement of ICa and simultaneous detection of [cAMP]i changes by the fluorescent probe FlCRhR in intact single frog ventricular myocytes. While this probe has already been used in several non-excitable cells (Adams et al. 1991) and in giant invertebrate neurones (Bacskai et al. 1993; Hempel et al. 1996), it has never been used in native excitable vertebrate cells. Combined measurements of [cAMP]i and ICa allowed us to compare the time course of the responses of these parameters to a challenge of the myocyte with ISO or FSK.
A preliminary report of some of these results has been published in abstract form (Vincent et al. 2000).
METHODS
The investigation conforms with our institution’s guidelines which are defined by the European Community guiding principles in the care and use of animals and the French decree no. 87/848 of October 19, 1987. Authorisations to perform animal experiments according to this decree were obtained from the French Ministere de l’Agriculture, de la Peche et de l’Alimentation (no. 7475, May 27, 1997).
Cell dissociation
Ventricular cells were enzymatically dispersed from frog (Rana esculenta) heart, by a combination of collagenase and trypsin as described previously (Fischmeister & Jurevicius, 1996). Frogs were killed by decapitation and double pithed. The isolated cells were stored in storage Ringer solution (see below), and kept at 4°C until use (1-5 days following dissociation). Prior to the experiment, the cells were allowed to fall to the bottom of a glass chamber coated with laminin to increase cell adhesion.
Electrophysiological experiments
The whole-cell patch-clamp technique was applied to 250-300 μm long rod-shaped Ca2+-tolerant frog ventricular myocytes using an Axopatch 200-A amplifier (Axon Instruments, Foster City, CA, USA) controlled by Axograph software (Axon Instruments). In order to facilitate optical recording, each selected cell had a few hundred micrometres stuck flat on the glass bottom of the recording chamber. The cell was usually sealed to the patch pipette (1-2 MΩ resistance) on a loose end so that the vibrations of the imaging system would not lead to seal damage (Fig. 2A). Series resistance were usually between 2.5 and 5 MΩ and electrical recordings were discarded if the series resistance was more than 10 MΩ. ICa was elicited by depolarising the cell every 5-8 s to 0 mV for 200 ms from a holding potential of -80 mV. ICa was measured as the difference between the peak inward current and the steady-state current at the end of the voltage jump. Currents were sampled at a frequency of 10 kHz using a 14-bit analog-to-digital converter. K+ currents were blocked by replacing all K+ ions with intracellular and extracellular Cs+ and tetrodotoxin was used to block the fast voltage-dependent Na+ current. All experiments were done at room temperature (22-26°C), and the temperature did not vary by more than 1°C in a given experiment.
Figure 2. Imaging of a frog ventricular myocyte perfused with FlCRhR.
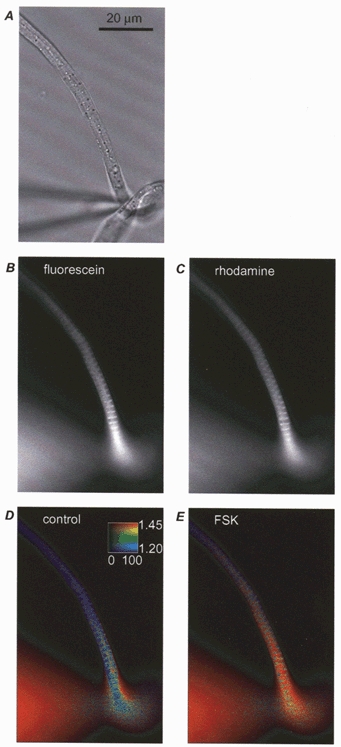
Raw images of a frog ventricular myocyte perfused with FlCRhR in transmitted light (A), fluorescence with fluorescein (B) and rhodamine (C) emission filters. Pseudocolour images in control condition (D) and with 13 μm forskolin (FSK) applied in the bath (E). Calibration square in D indicates: from bottom to top minimal (blue) to maximal (red) ratio values; from left to right, minimum and maximum intensity after corrections (see Methods) in counts per pixel per second.
Materials
For the preparation of frog ventricular myocytes, the ionic composition of Ca2+-free Ringer solution was (mm): 88.4 NaCl; 2.5 KCl; 23.8 NaHCO3; 0.6 NaH2PO4; 1.8 MgCl2; 5 mm creatine; 10 d-glucose; with 1 mg ml−1 fatty acid-free bovine serum albumin; 50 i.u ml−1 penicillin; 50 μg ml−1 streptomycin; pH 7.4 maintained with 95% O2, 5% CO2. Storage Ringer solution was Ca2+-free Ringer solution to which was added 0.9 mm CaCl2 and 10 μl ml−1 non-essential and essential amino acid and vitamin solution (100 x MEM). Dissociation medium was composed of Ca2+-free Ringer solution to which was added 0.2 mg ml−1 trypsin type XIII (Sigma, St Louis, MO, USA), 0.14 mg ml−1 collagenase (Yakult, Tokyo, Japan), and 10 μl ml−1 M199 medium (Sigma).
For electrophysiology, the control external Cs+-Ringer solution contained (mm): 107 NaCl; 10 Hepes; 20 CsCl; 4 NaHCO3; 0.8 NaH2PO4; 1.8 MgCl2; 1.8 CaCl2; 5 d-glucose; 5 sodium pyruvate; pH 7.4 adjusted with NaOH. Patch electrodes were filled with control internal solution which contained (mm): 119.8 CsCl; 5 EGTA (acid form); 4 MgCl2; 5 creatine phosphate disodium salt; 3.1 Na2ATP; 0.42 Na2GTP; 0.062 CaCl2 (pCa 8.5); 10 Hepes; pH 7.3 adjusted with CsOH. Solutions were applied focally onto the cell using a glass pipette of 180 μm tip diameter placed 600 μm away and 300 μm above the cell. Complete exchange of solution around the cell was achieved within ≈1 s, which was the time required to achieve complete block of the voltage-dependent Na+ current by tetrodotoxin (200 nm).
Tetrodotoxin was from Latoxan (Rosans, France) and all other drugs were from Aldrich or Sigma. Ten millimolar ISO stock solutions were stored at -20°C. One aliquot was used for every experiment and kept on ice until dissolved at the desired final concentration in external solutions immediately before application to the cell.
Introduction of FlCRhR in cardiomyocytes via the patch pipette
The recombinant PKA subunits Cα and RIIβ (gift from Prof. Susan S. Taylor) were fluorescently labelled (Adams et al. 1991) and the holoenzyme purified by HPLC to a final concentration of about 5 μm. Aliquots of 5 μl were kept at less than -80°C for several months without any sign of degradation. Once thawed, aliquots were kept in the dark at 4°C and used within a few days. Since small amounts of FlCRhR were obtained, all the experiments were done using the smallest possible volumes. The patch pipette contained 0.5 μl of control internal solution and a thin quartz capillary (diameter, ø= 20 μm) loaded with about 0.2 μl of FlCRhR was inserted into the patch pipette and pushed to its tip. FlCRhR was ejected into the pipette tip by a positive pressure applied to the quartz capillary.
Cyclic AMP imaging
Images were obtained on an Olympus upright microscope with a x60 0.9 NA water immersion objective using a Princeton Instruments Micromax digital camera (Roper Scientific, Trenton, NJ, USA) with 1300 x 1030 interline CCD sensor of pixels 6.7 x 6.7 μm size, cooled to -15°C. A 100 W halogen lamp was used for illumination. Images in transmitted light were obtained using 10 ms exposures with Köhler illumination, i.e. without any optical contrast enhancement. For fluorescence imaging, optical filters (Chroma Technology, Brattleboro, VT, USA) were used as follows: excitation, D480/30; dichroic, 505DCLP; emission 1 (fluorescein), D535/40; emission 2 (rhodamine), E560lp. Fluorescence images were obtained during 1 or 2 s exposures and acquired alternately at fluorescein and rhodamine wavelengths using a custom designed filter changer. Images were acquired and saved on the hard disk of a G3 computer (Apple Computers, Cuppertino, CA, USA) using IPLab software (Scannalytics, Fairfax, VA, USA). A piezo actuator (Physik Instrumente, Waldbronn, Germany) was used to correct for the slight difference in the focus of the images at the two wavelengths. Horizontal mismatch between the two images was adjusted at the level of half a pixel using brightly fluorescent spots as reference.
The ratio was measured online using IPLab scripts with background subtraction and shading correction. Corrected single wavelength images of fluorescein and rhodamine emission were ratioed pixel by pixel. Regions of interest were drawn on the image (see e.g. Fig. 4) and the average ratio in these regions was calculated as an average of all pixels (Tsien & Harootunian, 1990). Since the intensity of fluorescence depends on the morphology of the cell and the amount of fluorescent probe present in the cytosol, pseudocolour images were derived according to Tsien & Harootunian (1990) allowing simultaneous display of the intensity and ratio. A hue-saturation-value colour representation was used in which for each pixel the hue represents the ratio (vertical scale) and the value represents the intensity of fluorescence (horizontal scale, as a percentage of saturation) indicative of the amount of FlCRhR present in the cell at this position. This intensity value was calculated as the average of fluorescein and rhodamine fluorescence.
Figure 4. [cAMP]i and ICa responses to isoprenaline.
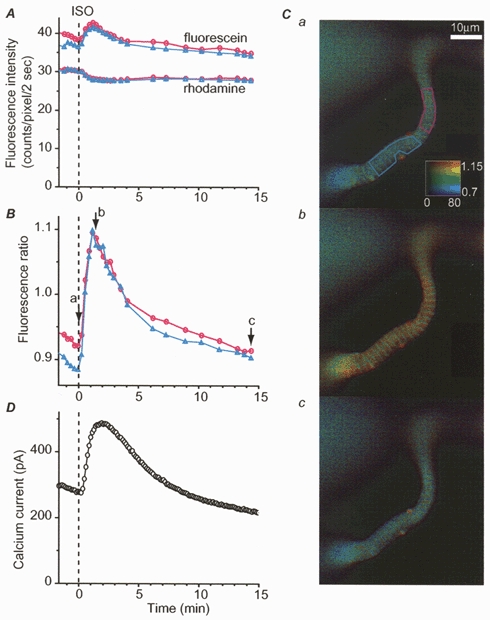
Time course of averaged fluorescence intensity (A) and ratio (B) measured in a frog ventricular myocyte in the two regions drawn with the corresponding colours indicated in Ca. A indicates the average intensity of fluorescence measured with fluorescein (top two traces) and rhodamine emission filter (two bottom traces). Values are as given by the camera, with background subtraction. Background was 75 counts pixel−1 s−1. B, ratio measurements were performed as described in Methods. D, ICa amplitude was simultaneously recorded at 0 mV. Time 0 indicates the beginning of a brief (10 s) application of isoprenaline (ISO, 10 μm). C, a, b and c: pseudocolour images obtained at the times indicated by the arrows and the corresponding letters in B.
All results are given as means ±s.e.m. for the stated number of experiments. The threshold for statistical significance was set at P < 0.05 using Student’s t test.
RESULTS
Loading cells with FlCRhR
The fluorescent probe FlCRhR is made up of PKA in which the catalytic (C) and regulatory (R) subunits are each labelled with a different fluorescent dye, respectively fluorescein and rhodamine (Adams et al. 1991). So far, microinjection has been the only way to introduce this probe into cells. Unfortunately, after several attempts, this technique turned out to be inapplicable to frog ventricular myocytes. Cell impalement in a control extracellular solution led to irreversible damage to the cell membrane, loss of cell integrity and rapid cell death. Removal of extracellular calcium or impalement of the cells bathed in an intracellular-like solution reduced somewhat the initial injury to the cells, but the cells usually died quickly when they returned to control extracellular solution.
Therefore, we decided to use the whole-cell patch-clamp technique to deliver FlCRhR into the cytosol. Since the presence of FlCRhR in the intracellular solution prevented seal formation and its large molecular mass (172 kDa) made backfilling of the pipette ineffective, FlCRhR was introduced into a thin quartz capillary (ø= 20 μm) positioned in the patch pipette at about 20 μm distance from the tip of the pipette. After the patch pipette was sealed onto the cell and a gigaohm seal resistance was obtained, a positive pressure was applied to the quartz capillary and the probe was ejected in the pipette tip. This could be done indifferently before or after breaking the patch and reaching the whole-cell configuration.
Since FlCRhR is made of active PKA, we first examined whether intracellular dialysis with FlCRhR generated spontaneous PKA enzymatic activity which could possibly affect ICa. In the absence of FlCRhR in the patch pipette, application of the non-selective β-adrenergic agonist isoprenaline (ISO, 1 μm) strongly increased ICa. When the cell was exposed only briefly (10 s) to ISO, the stimulation of ICa was transient and the current returned to a basal level within 20 min (Fig. 1A and C). Pressure ejection of FlCRhR at the tip of the patch pipette led to diffusion of the probe in the cytosol, as evidenced by the increase in fluorescence intensity (Fig. 1B). Following FlCRhR ejection, a clear increase in ICa was observed (Fig. 1A and C). However, this rise in ICa did not prevent a second response of ICa to ISO (Fig. 1A and C), although its amplitude was somewhat diminished.
Figure 1. Effect of FlCRhR diffusion on ICa in frog ventricular myocytes.
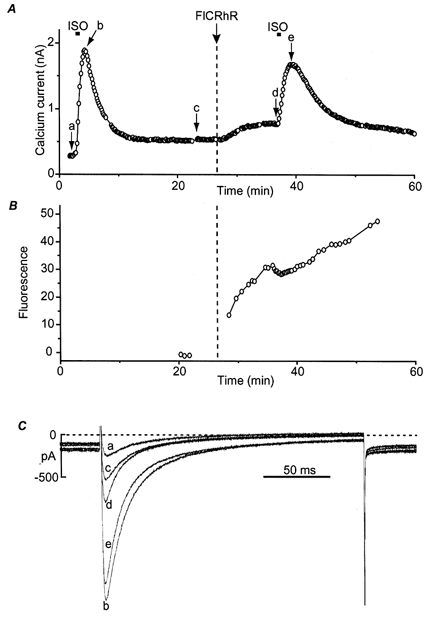
A, time course of the amplitude of ICa measured every 8 s at 0 mV (holding potential, -80 mV). The myocyte was first exposed to control intracellular and extracellular solutions. A brief (10 s) application of isoprenaline (ISO, 1 μm) induced a large transient increase in ICa. After ICa had returned to basal amplitude, FlCRhR was ejected in the pipette tip at the time indicated by the vertical dashed line, which induced a small rise in ICa. The myocyte was then exposed again briefly (10 s) to ISO. B, average fluorescein and rhodamine fluorescence intensity measured on the cell, indicating diffusion of FlCRhR into the cytosol. C, superimposed ICa current traces obtained at the times indicated by the corresponding letters in A.
On average, in nine cells, baseline ICa amplitude at 0 mV was 337 ± 63 pA (mean ±s.e.m.) and increased by 417 ± 86% to a peak response of 1627 ± 296 pA upon brief application of 1 or 10 μm ISO. Ejection of FlCRhR in the patch pipette during the recording produced a significant increase in ICa by 68 ± 16%, from a baseline value of 266 ± 41 pA to a new steady state of 441 ± 71 pA (n = 10). In the presence of FlCRhR in the patch solution, baseline ICa amplitude was 438 ± 81 pA and increased by 199 ± 33% to a value of 1255 ± 207 pA upon brief application of 1 or 10 μm ISO (n = 8). The maximum ICa amplitude obtained in response to ISO with FlCRhR was not significantly different from control responses in the absence of FlCRhR. However, the percentage ICa increase produced by ISO was significantly smaller in the presence of FlCRhR (P < 0.05). Therefore, by stimulating basal ICa without affecting the current in ISO, intracellular dialysis of FlCRhR reduced about 2-fold the relative response of ICa to ISO.
Fluorescence distribution in the cell
FlCRhR appeared to diffuse in a few minutes throughout the cytosol, up to distances > 100 μm from the point of entrance (Fig. 2B and C). However, due to the high molecular weight of the probe, its distribution was not homogeneous throughout the length of the cell. Instead, a large fluorescence gradient was observed, with high fluorescence intensities observed at both fluorescein (Fig. 2B) and rhodamine (Fig. 2C) emission wavelengths at the mouth of the patch pipette. It is conceivable that FlCRhR diffusion will depend on the size and shape of the pipette tip and the conditions of patch break leading to whole-cell configuration. While the former can be precisely controlled, the latter may vary considerably from cell to cell and even during the course of an experiment. This may explain why we observed variable levels of FlCRhR loading from cell to cell, and sometimes sudden changes in fluorescence intensity when the pipette tip unclogged.
As shown in Fig. 2, FlCRhR fluorescence displayed a striated pattern, which was reminiscent of the sarcomeric organisation of the myocyte. This aspect was further examined by comparing the fluorescence images of the cells with those obtained in transmitted light. As shown in Fig. 3, clear striations were observed in transmitted light (Fig. 3A, upper part), corresponding to the classical longitudinal alternation of narrow bright and larger dark bands related to I- and A-bands, respectively. A sarcomere length of 1.92 μm was calculated from the transmitted light intensity pattern (Fig. 3B). A similar succession of narrow high intensity and larger low intensity bands occurred in fluorescence images (Fig. 3A, lower part) with the same periodicity as observed in transmitted light (Fig. 3B). The two intensity profiles were quite similar, with the exception of a slight ≈3-pixel shift between them due to the presence of filters in the optical path used by fluorescent light (Fig. 3B). This strongly suggests that the striation pattern observed in FlCRhR fluorescence images is due to high fluorescence generated in the I-bands and low fluorescence generated in the A-bands.
Figure 3. Striated pattern of FlCRhR distribution in the cytosol.
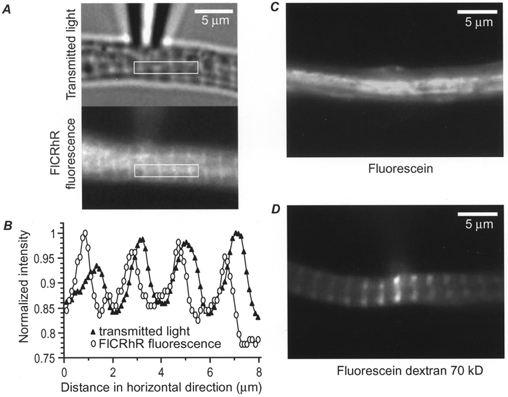
A, a frog ventricular myocyte loaded with FlCRhR is viewed in transmitted light (upper part) and in fluorescence (lower part). In the rectangular region indicated in white, the fluorescence intensities were averaged vertically and the intensity profiles are displayed in B. Intensities were normalised to the maximal value. ▴, transmitted light; ^, fluorescence. C, a frog ventricular myocyte was perfused with 5 μm fluorescein and 5 μm rhodamine added to the pipette solution. D, a frog ventricular myocyte was perfused with approximately 1 μm fluorescein dextran (70 kDa) added to the pipette solution. All three fluorescence images were obtained with the fluorescein excitation and emission filters.
To examine whether this was due to the binding of the fluorophores (fluorescein and rhodamine) or of FlCRhR to some specific cellular structures, two control experiments were performed. First, when the patch pipette was filled with a combination of free fluorescein (5 μm) and rhodamine (5 μm), the striated pattern was never observed (Fig. 3C, n = 7). This rules out the possibilities that the banding pattern observed with FlCRhR was due to (i) the refraction properties of the cytoplasm, (ii) non-specific binding of the fluorophores to the cytoplasm or (iii) dye exclusion from the cytoplasm. Second, when the pipette was perfused with fluorescein dextran, another high molecular weight (70 kDa) fluorescent molecule, clear striated labelling was also observed within a few minutes of perfusion (Fig. 3D, n = 4). With both free fluorescein and fluorescein dextran, the dye concentration was chosen so that the final concentration of fluorophore in the cytosol matched that of FlCRhR, yielding intensity values in the same range for all three types of recordings. Therefore, the striations appear to result from the high molecular weight of the probe and are unlikely to be related to binding of FlCRhR to some specific cellular structure or to some optical effects.
Adenylyl cyclase activation increases the fluorescence emission ratio
When cAMP binds to the R subunit of FlCRhR, rhodamine emission decreases and fluorescein emission increases (Adams et al. 1991). Thus, the ratio of fluorescein to rhodamine intensity can be used as an index of cAMP concentration ([cAMP]i) changes. The rationale for using a ratioing procedure was (i) to amplify the fluorescence changes observed at each individual wavelength, and (ii) to cancel the spontaneous drift in fluorescence intensity due to the continuous dialysis of FlCRhR into the cell. In our experiments, the fluorescence ratio was calculated for each pixel and is presented in pseudocolour. As illustrated in Fig. 2D and E, bath application of forskolin (FSK, 10 μm) increased the ratio in all regions of the cell, confirming the ability of this imaging method to report changes in [cAMP]i following stimulation of adenylyl cyclase in frog ventricular myocytes. As described in the following section, specific experiments were performed to characterise and compare the [cAMP]i and ICa responses to ISO and FSK.
[cAMP]i and ICa responses to a β-adrenergic stimulation
Image pairs were acquired repetitively during ISO application and single wavelength intensities and ratio were quantified. Figure 4 shows that a brief (10 s) application of ISO (10 μm), while inducing a net increase in ICa (Fig. 4D), induced opposite changes in fluorescein and rhodamine fluorescence intensities (Fig. 4A). This is the hallmark of a decrease in fluorescence energy transfer between the fluorescein and rhodamine fluorophores on the two dissociated C and R subunits of PKA upon a [cAMP]i increase (Adams et al. 1991). These changes were quite similar in two different regions of the cell (depicted in Fig. 4Ca) and resulted in a clear increase in the fluorescence ratio (Fig. 4B).
Since fluorescein is much more sensitive to bleaching than rhodamine, illumination of the cell led to a progressive ratio decrease. This decrease was found to be particularly important when pairs of images were acquired with a period shorter than 15 s. However, the fluorescence ratio returned to baseline when the illumination was stopped for more than 30 s, indicating that fresh FlCRhR diffused continuously into the cell and replaced the bleached probe. It should also be noted that the absolute fluorescence ratio value at rest was quite variable from cell to cell as well as within a cell (up to 20% ratio change in baseline condition in different regions of a cell). The ratio also drifted slowly, with a time constant of a few tens of minutes. This variability was not an imaging artefact as the baseline FlCRhR ratio measured in the pipette remained stable. For all these reasons, no attempt was made to calibrate the fluorescence ratio changes in terms of absolute [cAMP]i concentration. Instead, the use of the probe was restricted to measure short-term changes in [cAMP]i, in the range of about 15 min.
Comparison of the responses of [cAMP]i and ICa to ISO and FSK
In order to determine the time course of the [cAMP]i and ICa responses, ICa and the FlCRhR fluorescence ratio were measured repeatedly with 5-8 s intervals for ICa and 12 s for the fluorescence ratio. At such short image acquisition intervals, bleaching was prominent, as evidenced by the rundown of the ratio baseline trace (see e.g. Fig. 4B). To allow for comparison between the time courses of the responses of [cAMP]i and ICa to ISO and FSK, the rundown was subtracted from the experimental ratio values and the data points were normalised to their maximal amplitudes. In Fig. 5A, a saturating concentration of ISO (20 μm) was briefly (10 s) applied to the cell using the focal perfusion system. ISO induced a transient increase in ICa. Interestingly, in all cases (n = 4, 1-20 μm ISO), the response of [cAMP]i preceded that of ICa (Fig. 5A). Indeed, times for half-maximal (t1/2) and maximal response (tmax) were, respectively, 24.0 ± 8.3 and 57.0 ± 11.1 s for [cAMP]i and 35.0 ± 5.1 and 100.5 ± 22.1 s for ICa (Fig. 5B). Figure 5A also shows that the transient increase in [cAMP]i was shorter in duration than that of ICa, i.e. ICa was still strongly enhanced when [cAMP]i had already returned to control level. To examine whether this was a real phenomenon or simply a consequence of an accelerated bleaching process during ISO stimulation, the effect of a longer application of ISO was tested. In the experiment of Fig. 5C, the cell was exposed to 10 μm ISO for 5 min. Under this condition, [cAMP]i and ICa increased with a similar time course and both parameters remained stimulated during the application of ISO. Figure 5D summarises the results of three similar experiments and shows that t1/2 and tmax were not significantly different for [cAMP]i and ICa (respectively, 55.0 ± 5.6 and 129.0 ± 22.6 s for [cAMP]i and 62.3 ± 7.8 and 207.0 ± 60.1 s for ICa). Thus, the faster onset and the shorter duration of the [cAMP]i response compared to that of ICa upon a brief application of ISO were not artefactual. A final series of experiments was performed to examine the response of the cell to FSK. As shown in Fig. 5E, the response of ICa and [cAMP]i to a prolonged application of a saturating concentration of FSK (13 μm) were about 2-fold slower than with ISO, albeit a similar change in fluorescence ratio was obtained with both drugs (0.11 ± 0.045 vs. 0.16 ± 0.007, n = 4, P = 0.27). Indeed, t1/2 and tmax were, respectively, 104.5 ± 16.7 and 243.0 ± 36.5 s for [cAMP]i and 98.8 ± 12.4 and 293.0 ± 77.0 s for ICa, n = 4, Fig. 5F). While these results confirm previous findings that a direct stimulation of adenylyl cyclase with FSK leads to a 2-fold slower stimulation of ICa than that produced by activation of β-adrenergic receptors (Yatani & Brown, 1989; Hartzell et al. 1991; Frace et al. 1993), they unambiguously demonstrate that the difference in kinetics is due to a difference in the time course of [cAMP]i increase.
Figure 5. Comparison of [cAMP]i and ICa responses to isoprenaline and forskolin.
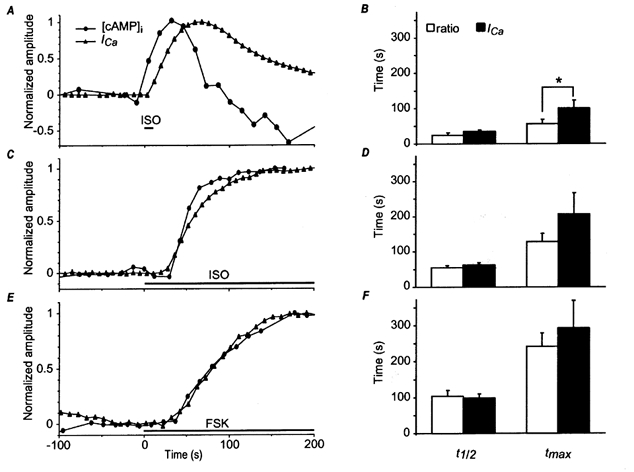
Frog ventricular myocytes were perfused with FlCRhR and the [cAMP]i and ICa responses were normalised to their maximal amplitudes. The [cAMP]i trace was corrected for the linear rundown due to bleaching by fitting a line to the last 5 data points preceding the drug application. A, focal application of 20 μm isoprenaline (ISO) during 10 s. B, average response of 4 cells. C, focal application of 0.1 μm ISO, which was then maintained throughout the rest of the experiment. D, average response of 3 cells. E, focal application of 13 μm FSK, which was then maintained throughout the rest of the experiment. F, average response of 4 cells. In B, D and F, the bars indicate mean t1/2 (time for half-maximal response) and tmax (time for maximal response) values and the lines the s.e.m. for [cAMP]i (open bars) and ICa (filled bars) responses. *Statistically significant difference at P < 0.05.
DISCUSSION
Introducing FlCRhR into the cell via the patch pipette
This report shows for the first time the use of a cAMP fluorescent indicator (FlCRhR) to measure [cAMP]i responses to stimulation of G protein-coupled receptors in native excitable cells from a vertebrate. So far, the use of this probe has been restricted to cells that could survive microinjection. These included inexcitable cells and giant neurones from invertebrates and vertebrate neurones in culture (Liu et al. 1999). However, microinjection cannot be easily applied to the small excitable cells found in vertebrates, such as cardiac myocytes. The method presented here is based on the whole-cell patch-clamp technique which allows the probe to diffuse passively from the recording pipette to the cell interior. A major advantage of this method is that it provides a simultaneous electrophysiological control of the recorded cell, allowing comparison of cAMP signals and modulation of membrane channel activity. Since most cells can be recorded using the patch-clamp technique, our approach also opens the possibility of measuring [cAMP]i in virtually any cell type.
Distribution of FlCRhR in the cytosol
The distribution of FlCRhR was not homogeneous throughout the length of the cell. Instead, a large fluorescence gradient was observed, with much higher fluorescence intensities at the mouth of the patch pipette than at the ends of the cells, even when FlCRhR diffusion lasted for several tens of minutes. FlCRhR is made of two R and two C subunits of PKA and has a molecular weight of 172 kDa. Its size can be roughly estimated from the dimensions of its subunits, which have been crystallised: 6.5 x 4.5 x 4.5 nm for the C subunit (Knighton et al. 1991) and 6.5 x 4.5 x 3.4 nm for the R subunit (Su et al. 1995). Recent studies have examined the intracellular diffusion of proteins of different sizes in striated muscle cells using modulated fringe pattern photobleaching (MFPP) of microinjected fluorescein isothiocyanate (FITC)-labelled proteins (see e.g. Arrio-Dupont et al. 1996, 2000). Using either randomly coiled dextran molecules (Arrio-Dupont et al. 1996) or globular proteins selected for their absence of interaction with the components of muscle cytoplasm (Arrio-Dupont et al. 2000), the diffusion coefficient of these proteins in the cytosol (Dcyt) was found to be a steeply decreasing function of their molecular mass and hydrodynamic radius. Thus, for a 172 kDa protein like FlCRhR, assuming a hydrodynamic radius of ≈10-15 nm, Dcyt should be in the range of 2-5 μm2 s−1 (Arrio-Dupont et al. 1996, 2000). This indicates that FlCRhR should diffuse in the cytosol up to 500 times slower than monovalent ions such as K+ or Na+ and 100 times slower than ATP or cAMP (Kushmerick & Podolsky, 1969; Arrio-Dupont et al. 1996). This slow diffusion can easily explain the large intracellular gradient of fluorescence which persisted in our experiments even > 30 min after introduction of the probe into the cytosol. Besides, according to Han & Herzfeld (1993), the diffusion of FlCRhR is likely to be anisotropic due to the high density of oriented sarcomeric proteins in the cytosol and the relatively large dimension of FlCRhR (ø≈ 10-15 nm) compared to that of actin (ø≈ 8 nm), or myosin (ø≈ 20 nm), or to the distance between actin and myosin in the A-band (≈10 nm) (Funatsu et al. 1993; Arrio-Dupont et al. 1996). Therefore, A-bands may generate a higher hindrance to FlCRhR diffusion than I-bands, which is likely to explain the distinct striation pattern observed in the fluorescence images. However, this analysis cannot unequivocally exclude the possibility that FlCRhR also binds to some specific cellular structure present in the I-bands.
Limitations of the technique
Like any other fluorescent probe, FlCRhR has its own limitations which need to be taken into account.
(1) FlCRhR fluorescence ratio increases as a consequence of the dissociation of R and C subunits of PKA. Thus, a ratio increase reflects the activation of PKA by cAMP rather than an increase in [cAMP]i per se. This may not be so much of a limitation, since the binding of cAMP to the R subunit and the subsequent dissociation of the R from the C subunit occur on the time scale of milliseconds, i.e. much faster than the kinetics measured during onset. However, a decrease in fluorescence ratio reflects the re-association of C and R subunits upon a decrease in [cAMP]i as well as diffusion of FlCRhR holoenzyme from the patch pipette into the cell. Thus, it is likely that [cAMP]i may recover faster than actually measured by the decrease in fluorescence ratio.
(2) Based on the protein concentration in the stock solution and the dilution factor in the patch pipette, the amount of FlCRhR present in the cell can be estimated to be in the order of 4 μm, which is approximately 4-fold higher than the concentration of endogenous PKA measured in mammalian heart (Corbin & Keely, 1977; Corbin et al. 1977). This fairly high concentration will have two major consequences. First, FlCRhR will compete with endogenous PKA for cAMP binding. Indeed, the 4:1 ratio of concentration suggests that, upon an increase in [cAMP]i, 80% of total PKA activity will be due to FlCRhR and only 20% to endogenous PKA. This may explain why the fluorescence returns to basal levels after stimulation (Fig. 4 and 5), since the activated subunits of FlCRhR will have a 4-fold higher chance of re-associating with the fluorescently labelled subunits than with endogenous PKA subunits. Second, FlCRhR is likely to buffer free intracellular cAMP. This may partly explain why ejecting FlCRhR in the pipette produces an immediate increase in ICa (but see below). Indeed, basal cAMP production may be insufficient to activate endogenous PKA and stimulate ICa under normal conditions, but sufficient to induce some degree of phosphorylation when the total PKA concentration is increased by a factor of 5.
(3) FlCRhR may partially dissociate in C and R subunits in the absence of cAMP, leading to an artefactual increase in intracellular PKA activity. This may also contribute to the immediate increase in ICa observed when ejecting FlCRhR in the pipette (Fig. 1). However, the amount of free C subunit in the FlCRhR solution must be relatively low (considering the excess of FlCRhR over endogenous PKA, see above) since ejection of the probe leads to a rather modest increase in ICa compared to the maximal stimulation observed with an intracellular perfusion of purified C subunit (Osterrieder et al. 1982; White & Hartzell, 1988) or activation of the cAMP cascade by saturating concentrations of ISO or FSK (e.g. this study).
(4) FlCRhR is composed of PKA, which is a labile protein. The R subunit is particularly sensitive and cleaves spontaneously in the N-terminal region after several days of storage at 4°C. A minor degradation of FlCRhR subunits has no major consequence when the probe is microinjected into the cell, since the proportion of degraded probe is exactly the same from cell to cell and does not induce ratio variations, as it only decreases the dynamic range of the ratio response. However, this partial degradation of FlCRhR is more problematic with our method since free diffusion from the patch pipette to the cytosol will favour the intracellular delivery of the smallest molecules, such as free subunits or proteolysis products. The conditions of cell dialysis may also vary from cell to cell and change during the course of an experiment since it will strongly depend on the access resistance to the cell obtained after patch break. FlCRhR proteolysis could also explain why remote parts of the cell have a slightly lower baseline ratio than parts closer to the patch pipette. Indeed, remote parts are likely to be filled with the smallest breakdown products of R subunit labelled with rhodamine.
Due to the above limitations, no attempt was made to determine accurately absolute [cAMP]i values in our experiments. However, the ratiometric properties of FlCRhR allowed us to cancel the continuous changes in fluorescence due to diffusion of the probe from the pipette to the cell and to amplify the signal measured at any single wavelength. Under these conditions, stable recordings were obtained for periods of ≈15 min.
Time course of ICa and [cAMP]i responses
The introduction of FlCRhR in frog ventricular myocytes allowed us to follow at the same time changes in ICa and [cAMP]i. Although our results are still preliminary, a number of interesting and new observations were made.
(1) A continuous application of ISO induced an increase in both ICa and [cAMP]i, which followed a similar time course. Earlier studies in frog ventricular myocytes using a rapid perfusion system have shown that a rapid application of ISO leads to an increase in ICa which develops (at room temperature) with a lag time of ≈3 s, a half-time of ≈20 s and a peak response at ≈40 s (Frace et al. 1993; Méry et al. 1993). With the exception of the lag time, a very similar time course was observed when the cells were loaded with 1-(2-nitrophenyl)ethyl-cAMP (caged cAMP) and intracellular cAMP was suddenly released by an UV-light flash (Frace et al. 1993). These experiments suggested that the rate limiting steps in ICa activation were activation of adenylyl cyclase and cAMP accumulation, which were responsible for the lag time, and activation of PKA, PKA phosphorylation of the substrate (possibly a Ca2+ channel subunit: Gao et al. 1997; Bünemann et al. 1999) and the effect of the substrate on Ca2+ channel gating, which were responsible for the rising phase (Frace et al. 1993). Unfortunately, the use of a rapid perfusion system was prohibited in the present study because the cells adhered to the glass bottom of the recording chamber and unavoidable vibrations would have caused the optical recording and the stability of the patch to deteriorate. We used instead a focal perfusion system which allowed a much slower drug application (complete change in the solution around the cell within 1 s compared to 50 ms in earlier experiments (Frace et al. 1993; Méry et al. 1993). Hence, the time course of the ICa response to an application of ISO was slowed down 2- to 3-fold. Since an increase in FlCRhR fluorescence ratio actually reflects the dissociation of PKA subunits, i.e. the process of PKA activation upon binding of cAMP to the R subunit, the parallelism observed between the kinetics of ICa and fluorescence ratio suggests that activation of PKA is the rate limiting step in the rising phase of the ICa response to ISO. In other words, mechanisms downstream from PKA activation, including substrate phosphorylation and modification of Ca2+ channel gating, occur on a much faster time scale.
(2) The time course of ICa and [cAMP]i in response to an application of FSK was ≈2-fold slower than with ISO. Earlier studies performed in guinea-pig (Yatani & Brown, 1989) and frog ventricular myocytes (Frace et al. 1993) using a rapid perfusion system have shown that the stimulatory effect of FSK on ICa develops with a 2- to 3-fold slower time scale than the effect of ISO at doses that are equipotent in their effects on ICa. However, it was unclear whether this difference was due to (i) a slower rise in [cAMP]i (Jurevicius & Fischmeister, 1996), (ii) the presence of some concomitant inhibitory effects of FSK on ICa (Boutjdir et al. 1990; Asai et al. 1996), or (iii) the presence of an additional activatory mechanism in β-adrenergic stimulation such as a priming effect of the G protein Gs on PKA phosphorylation of the Ca2+ channel (Cavaliéet al. 1991). Our present results strongly favour the first hypothesis. Indeed, FSK slowed down similarly the response of ICa and the fluorescence ratio. Since both time courses remained parallel, the simplest explanation is that FSK induced a slower rise in [cAMP]i than ISO, which was responsible for a slower activation of PKA and, hence, a slower response of ICa.
(3) [cAMP]i and ICa transients lasted 2-3 min in response to a brief (10 s) application of ISO. Although, for reasons outlined above, a precise estimate of the kinetics of ISO washout was not possible in this study, ICa continued to increase for about 90 s after onset of washout. A similar conclusion was reached in earlier studies using a rapid perfusion system. Indeed, an application of ISO as short as 1 s led to a transient ICa response which lasted for about 2 min (Frace et al. 1993; Méry et al. 1993). This was not due to a slow washout of ISO since application of propranolol during the washout of ISO was without effect (Frace et al. 1993). However, the ICa transient was strongly abbreviated when the washout of ISO was followed by a sustained application of acetylcholine, to inhibit adenylyl cyclase, even when acetylcholine was applied as late as 14 s after ISO washout (Frace et al. 1993). A likely explanation of these results was that adenylyl cyclase remained activated for a few tens of seconds after dissociation of ISO from its receptor (Frace et al. 1993; Méry et al. 1993). Our present results provide a direct confirmation of this hypothesis. Indeed, [cAMP]i continued to increase for about 50 s after onset of ISO washout, suggesting that adenylyl cyclase remained activated during all that period.
(4) [cAMP]i transients were much shorter than ICa transients in response to a brief application of ISO. Indeed, upon washout of ISO, the recovery phase of ICa started about 40 s later than that of [cAMP]i (i.e. 90 s after ISO washout) and developed with a 2-fold slower time course. Several mechanisms are likely to take part in this recovery phase. These include (i) the reduction of [cAMP]i due to phosphodiesterase activity, (ii) the dephosphorylation of PKA substrate due to phosphatase activity, and (iii) recovery of basal Ca2+ channel gating, e.g. following dephosphorylation of one of its subunits (Gao et al. 1997; Bünemann et al. 1999). Since, upon arrest of adenylyl cyclase activation, the [cAMP]i recovery phase was faster than that of ICa, our results provide direct evidence that phosphodiesterase activity is not the rate limiting step in the recovery of ICa.
In summary, simultaneous measurements of [cAMP]i and ICa changes in intact isolated cardiac myocytes were possible using FlCRhR and the whole-cell patch-clamp technique. Since the cAMP cascade plays a determinant role in the hormonal response of a large number of cell types to which the patch-clamp technique can be applied, it is likely that this technique will prove to be useful in different fields of research. While still in its infancy, this technique provides a unique approach to investigating in detail the temporal sequence of events in the cAMP cascade responsible for the response of the myocardium to a large number of hormones and inotropic agents. Further studies will be devoted to addressing their alterations in pathophysiological conditions. In particular, it can be envisioned that this technique should provide insights into the mechanisms responsible for the alteration of the β-adrenergic response during heart failure (Dzimiri, 1999).
Acknowledgments
We thank Roger Y. Tsien for his help in the development of this method and Stephen R. Adams in the preparation of FlCRhR. We thank Daniel Brusciano and Florence Lefebvre for expert technical assistance, and Renée Ventura-Clapier and Jean-Luc Mazet for helpful discussions. This work was supported by a grant from the Fondation pour la Recherche Médicale. J.M.G. was a recipient of a predoctoral fellowship from the French Ministere de l’Education Nationale de la Recherche et de la Technologie.
References
- Adams SR, Harootunian AT, Buechler YJ, Taylor SS, Tsien RY. Fluorescence ratio imaging of cyclic AMP in single cells. Nature. 1991;349:694–697. doi: 10.1038/349694a0. [DOI] [PubMed] [Google Scholar]
- Arrio-Dupont M, Cribier S, Foucault G, Devaux, P F & d‘Albis A. Diffusion of fluorescently labeled macromolecules in cultured muscle cells. Biophysical Journal. 1996;70:2327–2332. doi: 10.1016/S0006-3495(96)79798-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrio-Dupont M, Foucault G, Vacher M, Devaux PF, Cribier S. Translational diffusion of globular proteins in the cytoplasm of cultured muscle cells. Biophysical Journal. 2000;78:901–907. doi: 10.1016/S0006-3495(00)76647-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai T, Pelzer S, McDonald TF. Cyclic AMP-independent inhibition of cardiac calcium current by forskolin. Molecular Pharmacology. 1996;50:1262–1272. [PubMed] [Google Scholar]
- Bacskai B, Hochner B, Mahaut-Smith M, Adams S, Kaang B, Kandel E, Tsien R. Spatially resolved dynamics of cAMP and protein kinase A subunits in Aplysia sensory neurons. Science. 1993;260:222–226. doi: 10.1126/science.7682336. [DOI] [PubMed] [Google Scholar]
- Boutjdir M, Méry PF, Hanf R, Shrier A, Fischmeister R. High affinity forskolin inhibition of L-type Ca2+ current in cardiac cells. Molecular Pharmacology. 1990;38:758–765. [PubMed] [Google Scholar]
- Brodde OE, Michel MC. Adrenergic and muscarinic receptors in the human heart. Pharmacological Reviews. 1999;51:651–690. [PubMed] [Google Scholar]
- Bunemann M, Gerhardstein BL, Gao T, Hosey MM. Functional regulation of L-type calcium channels via protein kinase A-mediated phosphorylation of the β2 subunit. Journal of Biological Chemistry. 1999;274:33851–33854. doi: 10.1074/jbc.274.48.33851. [DOI] [PubMed] [Google Scholar]
- Cavalie A, Allen TJ, Trautwein W. Role of the GTP-binding protein Gs in the β-adrenergic modulation of cardiac Ca channels. Pflügers Archiv. 1991;419:433–443. doi: 10.1007/BF00370785. [DOI] [PubMed] [Google Scholar]
- Corbin JD, Keely SL. Characterization and regulation of heart adenosine 3′:5′-monophosphate-dependent protein kinase isozymes. Journal of Biological Chemistry. 1977;252:910–918. [PubMed] [Google Scholar]
- Corbin JD, Sugden PH, Lincoln TM, Keely SL. Compartmentalization of adenosine 3′:5′-monophosphate and adenosine 3′:5′-monophosphate-dependent protein kinase in heart tissue. Journal of Biological Chemistry. 1977;252:3854–3861. [PubMed] [Google Scholar]
- Dzimiri N. Regulation of β-adrenoceptor signaling in cardiac function and disease. Pharmacological Reviews. 1999;51:465–501. [PubMed] [Google Scholar]
- Fischmeister R, Hartzell HC. Regulation of calcium current by low-Km cyclic AMP phosphodiesterases in cardiac cells. Molecular Pharmacology. 1990;38:426–433. [PubMed] [Google Scholar]
- Frace AM, Méry PF, Fischmeister R, Hartzell HC. Rate-limiting steps in the β-adrenergic stimulation of cardiac calcium current. Journal of General Physiology. 1993;101:337–353. doi: 10.1085/jgp.101.3.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funatsu T, Kono E, Higuchi H, Kimura S, Ishiwata S, Yoshioka T, Maruyama K, Tsukita S. Elastic filaments in situ in cardiac muscle: deep-etch replica analysis in combination with selective removal of actin and myosin filaments. Journal of Cellular Biology. 1993;120:711–724. doi: 10.1083/jcb.120.3.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao T, Yatani A, Dell‘Acqua ML, Sako H, Green SA, Dascal N, Scott JD, Hosey MM. cAMP-dependent regulation of cardiac L-type Ca2+ channels requires membrane targeting of PKA and phosphorylation of channel subunits. Neuron. 1997;19:185–196. doi: 10.1016/s0896-6273(00)80358-x. [DOI] [PubMed] [Google Scholar]
- Hain J, Onoue H, Mayrleitner M, Fleischer S, Schindler H. Phosphorylation modulates the function of the calcium release channel of sarcoplasmic reticulum from cardiac muscle. Journal of Biological Chemistry. 1995;270:2074–2081. doi: 10.1074/jbc.270.5.2074. [DOI] [PubMed] [Google Scholar]
- Han J, Herzfeld J. Macromolecular diffusion in crowded solutions. Biophysical Journal. 1993;65:1155–1161. doi: 10.1016/S0006-3495(93)81145-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartzell HC. Regulation of cardiac ion channels by catecholamines, acetylcholine and 2nd messenger systems. Progress in Biophysics and Molecular Biology. 1988;52:165–247. doi: 10.1016/0079-6107(88)90014-4. [DOI] [PubMed] [Google Scholar]
- Hartzell HC, Fischmeister R. Effect of forskolin and acetylcholine on calcium current in single isolated cardiac myocytes. Molecular Pharmacology. 1987;32:639–645. [PubMed] [Google Scholar]
- Hartzell HC, Méry PF, Fischmeister R, Szabo G. Sympathetic regulation of cardiac calcium current is due exclusively to cAMP-dependent phosphorylation. Nature. 1991;351:573–576. doi: 10.1038/351573a0. [DOI] [PubMed] [Google Scholar]
- Hempel CM, Vincent P, Adams SR, Tsien RY, Selverston AI. Spatio-temporal dynamics of cAMP signals in an intact neural circuit. Nature. 1996;384:166–169. doi: 10.1038/384166a0. [DOI] [PubMed] [Google Scholar]
- Jurevicius J, Fischmeister R. cAMP compartmentation is responsible for a local activation of cardiac Ca2+ channels by β-adrenergic agonists. Proceedings of the National Academy of Sciences of the USA. 1996;93:295–299. doi: 10.1073/pnas.93.1.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameyama M, Hescheler J, Hofmann F, Trautwein W. Modulation of Ca current during the phosphorylation cycle in the guinea pig heart. Pflügers Archiv. 1986;407:123–128. doi: 10.1007/BF00580662. [DOI] [PubMed] [Google Scholar]
- Knighton DR, Zheng JH, Ten Eyck LF, Ashford VA, Xuong NH, Taylor SS, Sowadski JM. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science. 1991;253:407–414. doi: 10.1126/science.1862342. [DOI] [PubMed] [Google Scholar]
- Kushmerick MJ, Podolsky RJ. Ionic mobility in muscle cells. Nature. 1969;166:1297–1298. doi: 10.1126/science.166.3910.1297. [DOI] [PubMed] [Google Scholar]
- Liu CY, Jamaleddin AJ, Zhang H, Christofi FL. FlCRhR/cyclic AMP signaling in myenteric ganglia and calbindin-D28 intrinsic primary afferent neurons involves adenylyl cyclases I, III and IV. Brain Research. 1999;826:253–269. doi: 10.1016/s0006-8993(99)01269-x. [DOI] [PubMed] [Google Scholar]
- McDonald TF, Pelzer S, Trautwein W, Pelzer DJ. Regulation and modulation of calcium channels in cardiac, skeletal, and smooth muscle cells. Physiological Reviews. 1994;74:365–507. doi: 10.1152/physrev.1994.74.2.365. [DOI] [PubMed] [Google Scholar]
- Mery PF, Abi-Gerges N, Vandecasteele G, Jurevicius J, Eschenhagen T, Fischmeister R. Muscarinic regulation of the L-type calcium current in isolated cardiac myocytes. Life Sciences. 1997;60:1113–1120. doi: 10.1016/s0024-3205(97)00055-6. [DOI] [PubMed] [Google Scholar]
- Mery PF, Frace AM, Hartzell HC, Fischmeister R. A comparative analysis of the time course of cardiac Ca2+ current response to rapid applications of β-adrenergic and dihydropyridine agonists. Naunyn-Schmiedeberg’s Archives of Pharmacology. 1993;348:197–206. doi: 10.1007/BF00164799. [DOI] [PubMed] [Google Scholar]
- Mubagwa K, Mullane K, Flameng W. Role of adenosine in the heart and circulation. Cardiovascular Research. 1996;32:797–813. [PubMed] [Google Scholar]
- Osterrieder W, Brum G, Hescheler J, Trautwein W, Flockerzi V, Hofmann F. Injection of subunits of cyclic AMP-dependent protein kinase into cardiac myocytes modulates Ca2+ current. Nature. 1982;298:576–578. doi: 10.1038/298576a0. [DOI] [PubMed] [Google Scholar]
- Rapundalo ST. Cardiac protein phosphorylation: functional and pathophysiological correlates. Cardiovascular Research. 1998;38:559–588. doi: 10.1016/s0008-6363(98)00063-7. [DOI] [PubMed] [Google Scholar]
- Schouten VJA, Morad M. Regulation of Ca2+ current in frog ventricular myocytes by the holding potential, cAMP and frequency. Pflügers Archiv. 1989;415:1–11. doi: 10.1007/BF00373135. [DOI] [PubMed] [Google Scholar]
- Simmerman HK, Jones LR. Phospholamban: protein structure, mechanism of action, and role in cardiac function. Physiological Reviews. 1998;78:921–947. doi: 10.1152/physrev.1998.78.4.921. [DOI] [PubMed] [Google Scholar]
- Striessnig J. Pharmacology, structure and function of cardiac L-type Ca2+ channels. Cellular Physiology and Biochemistry. 1999;9:242–269. doi: 10.1159/000016320. [DOI] [PubMed] [Google Scholar]
- Su Y, Dostmann WR, Herberg FW, Durick K, Xuong NH, Ten Eyck L, Taylor SS, Varughese KI. Regulatory subunit of protein kinase A: structure of deletion mutant with cAMP binding domains. Science. 1995;269:807–813. doi: 10.1126/science.7638597. [DOI] [PubMed] [Google Scholar]
- Tsien RY, Harootunian AT. Practical design criteria for a dynamic ratio imaging system. Cell Calcium. 1990;11:93–109. doi: 10.1016/0143-4160(90)90063-z. [DOI] [PubMed] [Google Scholar]
- Valdivia HH, Kaplan JH, Ellisdavies GCR, Lederer WJ. Rapid adaptation of cardiac ryanodine receptors: Modulation by Mg2+ and phosphorylation. Science. 1995;267:1997–2000. doi: 10.1126/science.7701323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent P, Goaillard J-M, Fischmeister R. Combined measurements of intracellular cAMP and L-type Ca current in isolated cardiomyocytes. FASEB Journal. 2000;14:442.19. [Google Scholar]
- Walsh DA, Angelos KL, Van Patten SM, Glass DB, Garetto LP. In: Peptides and Protein Phosphorylation. Kemp BE, editor. Boca Raton, FL, USA: CRC Press; 1990. pp. 43–84. [Google Scholar]
- White RE, Hartzell HC. Effects of intracellular free magnesium on calcium current in isolated cardiac myocytes. Science. 1988;239:778–780. doi: 10.1126/science.2448878. [DOI] [PubMed] [Google Scholar]
- Yatani A, Brown AM. Rapid β-adrenergic modulation of cardiac calcium channel currents by a fast G protein pathway. Science. 1989;245:71–74. doi: 10.1126/science.2544999. [DOI] [PubMed] [Google Scholar]
- Zhou YY, Cheng H, Bogdanov KY, Hohl C, Altschuld R, Lakatta EG, Xiao RP. Localized cAMP-dependent signaling mediates β2-adrenergic modulation of cardiac excitation-contraction coupling. American Journal of Physiology. 1997;273:H1611–1618. doi: 10.1152/ajpheart.1997.273.3.H1611. [DOI] [PubMed] [Google Scholar]