

Abstract
Kinin B1-receptors are induced by various inflammatory stimuli. Since myocardial ischaemia-reperfusion results in inflammation, we questioned whether it could induce B1-receptor-dependent responses to des-Arg9-bradykinin (DBK).
Thirty-six rabbits were submitted either to a 30 min coronary occlusion followed by a 3 h reperfusion or to a sham operation. The response to DBK was then tested in vivo on mean arterial pressure (MAP) and in vitro on isolated hearts and arterial rings.
DBK induced a dose-dependent decrease in MAP in the ischaemia-reperfusion group (DBK, 10 μg kg−1, intra-arterial: -12 ± 2 vs. -5 ± 2 mmHg in the sham group, P < 0.02), which was significantly antagonised by [Leu8]-des-Arg9-bradykinin (LBK), a B1-receptor antagonist. Following ischaemia-reperfusion, isolated hearts responded to DBK by a decrease in coronary perfusion pressure greater than that of the sham group. DBK dose-dependently decreased the isometric force of isolated carotid rings (DBK, 10−5m: -9 ± 2 vs. -1 ± 2 % in the sham group, P < 0.02) and mesenteric arteries (DBK, 10−6m: -38 ± 7 %vs. -3 ± 2 % in the sham group, P < 0.001). The vascular effects of DBK seen after ischaemia-reperfusion were significantly antagonised by LBK. The presence of B1-receptors in ischaemia-reperfusion animals was confirmed by immunolocalisation and Western blot analysis.
This study demonstrates that myocardial ischaemia-reperfusion induces a global induction of functional kinin B1-receptors in the endothelium.
B1-receptors are inducible receptors. They were first described by Regoli et al. (1977) who showed that large rabbit arteries, which normally do not respond to des-Arg9-bradykinin (DBK), gradually increased their sensitivity to this B1-receptor agonist when incubated in vitro in normal conditions. These authors also demonstrated that intravenous administration of bacterial lipopolysaccharide (LPS) 5 or 20 h earlier is able to induce hypotension or vasorelaxation in response to DBK by promoting the de novo formation of B1-receptors (Regoli et al. 1981). Various inflammatory stimuli are able to induce B1-receptors (Marceau et al. 1997). In particular, cytokines such as interleukin-1 (IL-1) have been shown to enhance the development of the response to DBK (de Bloiset al. 1988) and could be one of the mediators of the effect of LPS. The induction of B1-receptors is also enhanced by angiotensin converting enzyme (ACE) inhibition or bradykinin (BK) administration, possibly because of the inflammatory effect of the increase in kinin concentrations (Nwator et al. 1989).
Ischaemia-reperfusion is known to induce an inflammatory response that contributes to tissue injury; it is associated with the activation of monocytes and neutrophils, and the production of cytokines (Entman et al. 1991). In particular, IL-1 is released by endothelial cells upon hypoxia-reoxygenation (Ala et al. 1992), and its mRNA expression is increased in ischaemic myocardium (Herskowitz et al. 1995). Also, kinins are released during ischaemia in different species (Hashimoto et al. 1978; Matsuki et al. 1987; Baumgarten et al. 1993) and could contribute to the inflammatory response. Thus, the aim of this study was to investigate whether myocardial ischaemia-reperfusion could induce B1-receptor-dependent responses. The effect of DBK following myocardial ischaemia-reperfusion was evaluated in vivo in anaesthetised rabbits and in vitro on isolated rabbit hearts and arterial rings. The presence of cardiovascular B1-receptors was also confirmed by Western blot analysis and immunohistochemical localisation.
METHODS
Ischaemia-reperfusion model
Surgical preparation
This investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication No. 85-23, revised 1985) and was given local ethical commitee approval.
Male New Zealand White rabbits (n = 36%) weighing between 2.5 and 3.5 kg were used in this study. They were anaesthetised with intravenous pentobarbital sodium (40 mg kg−1) administered via the marginal ear vein. Additional doses were given as required (10 mg kg−1 h−1). Positive pressure respiration with room air was maintained by a pump (Roche-Kontron 3100S) connected to an endotracheal tube. The ventilation rate was 35 breaths min−1 and tidal volume was 30 ml. The animals were placed on a heating pad. Sterile solutions were used in order to limit the inflammatory reaction to the surgical preparation.
The right common carotid and right femoral artery for the in vivo study were cannulated with polyethylene catheters (internal and external diameter of 1 and 2 mm, respectively) inserted into the aorta. Arterial pressure was measured, using a pressure transducer (Baxter 33-260, Healthcare Corp., UK). A left thoracotomy was performed at the 4th intercostal space and the pericardium was opened. A 3/0 silk thread was then placed around the first marginal branch of the circumflex artery, and passed through a small polyethylene tube. Lead II of the surface electrocardiogram, heart rate and arterial pressure were continuously recorded on a computer (Macintosh Power PC) using a data acquisition system (MacLab 8 channels, ADInstruments). Monophasic action potentials were measured with a Franz epicardial electrode (model 225, EP Technologies). An intravenous injection of 2500 i.u. of heparin (Choay) was performed at the beginning of the protocol.
Experimental protocol
After the surgical preparation, rabbits were allowed a 15 min stabilisation period to reach a steady state. Coronary occlusion was produced in the ischaemia-reperfusion group by clamping the snare around the artery. Myocardial ischaemia was confirmed by elevation of the ST segment of the electrocardiogram and by monophasic action potential shortening. If ventricular fibrillation occurred, animals were defibrillated by myocardial massage. After 30 min ischaemia, the ligation was loosened and there was a 3 h reperfusion period. Rabbits from the sham group were submitted to the same surgical and experimental protocol but ischaemia was not performed. All animals survived the experimental protocol and were included in the study. Thereafter, animals from both groups were submitted to either of the following protocols for the assessment of B1-receptors.
B1-receptor assessment
In vivo protocol
The presence of B1-receptors was evaluated in vivo by measuring the arterial pressure response to several drug injections. DBK (2.5, 5 and 10 μg kg−1, Bachem) was administered through the femoral artery. The effects of DBK (10 μg kg−1) were also assessed 10 min after the beginning of an intra-arterial perfusion of 10 μg kg−1 min−1 of [Leu8]-des-Arg9-bradykinin (LBK) (Bachem), a B1-antagonist, and 10 min after an intravenous injection of 2 μg kg−1 of HOE 140 (Icatiban, Bachem), a B2-antagonist.
Isolated perfused heart protocol
Hearts were removed, placed in cold perfusion medium and rapidly perfused retrogradely in the Langendorff mode at a constant flow using a pump (Minipuls 3, Gilson). The flow was adjusted at the beginning of the experiment to achieve an initial perfusion pressure of 80 mmHg measured by a pressure transducer placed just before the heart. After adjustment, coronary flow was similar in both groups (37 ± 3 vs. 41 ± 3 ml min−1 in the sham group). The perfusion medium was a modified Krebs-Henseleit solution containing (mm): NaCl 118, NaHCO3 25, KCl 4.7, MgSO4 1.22, KH2PO4 1.2, CaCl2 1.8, glucose 11, gassed with 95% O2, 5% CO2 and adjusted to pH 7.4. The cardiac temperature was measured continuously by a thermometer probe (model T200KC, Digitron) inserted into the left ventricle and adjusted to 37°C. A water-filled latex balloon (No. 12. Hugo Sachs) connected to a pressure transducer was placed through the left atrium into the left ventricle, and adjusted to a left ventricular end-diastolic pressure of 10 mmHg. Heart rate (HR) and left ventricular developed pressure (LVDP) were recorded continuously.
After a 15 min stabilisation period, variations in perfusion pressure were measured following increasing concentrations of DBK from 10−9 to 10−6m added to the perfusion medium. The heart was then perfused with DBK (10−6m) along with LBK (10−6m) and finally with acetylcholine (10−6m) (Sigma) and sodium nitroprusside (10−4m) (Sigma). All drugs were perfused for 10 min, a time sufficient to attain a stable response.
Thereafter, the coronary artery was re-occluded and the area at risk and infarct size induced by the ischaemia-reperfusion protocol were determined using standard coloration techniques with blue dye (Unisperse blue, Ciba Geigy) and triphenyl tetrazolium chloride (1%, Sigma). The area at risk and infarct size were measured using a computerised planimetric technique (Minichromax, Biolab) and expressed as percentage of left ventricle and area at risk, respectively.
Isolated arteries protocol
Isolated carotid and aortic rings
Portions of the left carotid artery and thoracic aorta were removed and placed in cold Krebs solution. The arteries were dissected free of connective tissue, cut into 3 mm wide rings and mounted in 10 ml tissue baths containing buffered Krebs solution (mm: NaCl 130, NaHCO3 14.9, KCl 3.7, CaCl2 1.6, KH2PO4 1.2, MgSO4 1.2, glucose 11, Hepes 2.38) at 37°C and gassed with 95% O2 and 5% CO2. In some arteries, the vascular endothelium was rubbed off with a polyethylene tube introduced into the vessel. Changes in isometric force were measured with a transducer (model UF-1, Pioden) and recorded continuously. Carotid and aortic rings were stretched gradually to a passive force of 2 g and 4 g, respectively (tensions at which the response to KCl, 60 mm, was maximal). After a 1 h equilibration period, arterial rings were contracted with phenylephrine (5 x 10−7 to 5 x10−6m, Sigma) in order to reach 40-60 % of the maximal contraction. The response to cumulative concentrations of DBK (10−8 to 10−5m) was then recorded followed by the effect of LBK (10−5m). Finally, acetylcholine (10−6m) was added to verify the functionality of the endothelium, and sodium nitroprusside (10−4m) to evaluate the maximal vasodilatation.
Isolated mesenteric arteries
Segments of second-order mesenteric arteries (approx. 200 μm external diameter) were trimmed free of fat and adhering connective tissue, and mounted in a myograph (Marty Technology) containing buffered Krebs solution, according to the technique of Mulvany & Halpern (1977). Isometric force was recorded as previously described. Each vessel was placed under an optimal stretch, equivalent to an in vivo arterial blood pressure of approximately 50 mmHg. Vessels were allowed to equilibrate for 30 min. Tissue contractility was then assessed by three exposures to KCl (60 mm). Vessels were contracted with phenylephrine as previously described. The response to cumulative concentrations of DBK (10−10 to 10−6m) and the effect of LBK (10−5m), acetylcholine (10−6m) and sodium nitroprusside (10−4m) were then recorded.
Immunological assays
Peptide synthesis and production of anti B1-receptor antisera
Peptides derived from the various intracellular (ID) ID2-ID4 and extracellular (ED) ED1-ED3 domains of the human B1-receptor sequence were synthesised by the solid phase method using fluorenylmethyloxycarbonyl (Fmoc) chemistry (Table 1). The peptides were used for immunisation without prior conjugation to carrier protein except for ENI-17 and SLR-22, which were previously coupled to keyhole limpet haemocyanin by 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (Abd Alla et al. 1993). Balb/c mice were immunised using standard procedures. For Western blotting and immunocytochemistry, a mixture of equal volumes from each of the corresponding antisera was used.
Table 1.
Synthetic peptides used for the production of antibodies to the B1 receptor
| Antigena | Domainb | Positions | Conjugationc |
|---|---|---|---|
| ELQ-30 | ED1 | 9–38 | nil |
| ENI-17 | ED2 | 95–111 | KLH, EDC |
| RSI-30 | ED3 | 176–205 | nil |
| SQD-27 | ID2 | 132–158 | nil |
| SLR-22 | ID3 | 228–249 | KLH, EDC |
| RFL-29 | ID4 | 303–331 | nil |
Peptides are identified by their three terminal amino acid residues using the one-letter code, followed by the total number of residues constituting the peptide.
ED, extracellular domain; ID, intracellular domain.
Unconjugated peptides were used for immunisations except for ENI-17 and SLR-22 which were previously coupled to keyhole limpet haemocyanin (KLH) by 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC).
Western blot assay
Vessels and heart segments were sampled at the end of the ischaemia-reperfusion protocol as well as 5 h after LPS treatment (10 μg kg−1i.v.) to use as a positive control. The tissues were frozen and pulverised under liquid nitrogen. The powders were re-suspended in ice-cold lysis buffer (20 mm Tris-HCl, pH 7.5, 5 mm EGTA, 150 mm NaCl, 20 mm glycerophosphate, 10 mm NaF, 1 mm sodium orthovanadate, 1% Triton X-100, 0.1% Tween 20, 1 μg ml−1 aprotinin, 1 mm phenylmethylsulfonyl fluoride, 0.5 mmN-tosyl-l-phenylalanine chloromethyl ketone, 0.5 mmN(-)p-tosyl-l-lysine chloromethyl ketone) at a ratio of 0.3 ml/10 mg wet weight. Extracts were incubated on ice for 15 min and then centrifuged (12 000 g, 15 min, 4°C). The detergent-soluble supernatant fractions were retained.
Lysates were subjected to electrophoresis on a 9% polyacrylamide gel and transferred to nitrocellulose membranes. Membranes were blocked with 10% non-fat dry milk in TBST (20 mm Tris, pH 8.0, 150 mm NaCl, and 0.1% Tween 20) for 20 min and were then incubated with mouse anti-B1-receptor antibodies at a dilution of 1:2000 in TBST for 1 h. An enhanced chemiluminescence system (ECL plus, Amersham) was used as the detection method. Blots were washed and subjected to autoradiography. Molecular weights of proteins were estimated by using pre-stained markers (Bio-Rad, 161-0324).
Immunohistochemistry analysis
Segments of the aorta, carotid and mesenteric arteries were sampled at the end of the ischaemia-reperfusion protocol. They were mounted in embedding medium (OCT 4583, Sakura), frozen in isopentane previously cooled in liquid nitrogen and stored at -80°C. Transverse cross-sections (7 μm thin) were obtained at -20°C from the frozen embedded segments. Sections were incubated for 30 min at 37°C with the mouse anti-B1-receptor antibody previously described, and then with an anti-mouse antibody conjugated to biotin (Amersham) which was amplified by streptavidin-Texas red reagent (Amersham). Nuclei were also stained (Hoescht bis-benzidine, Sigma). Fluorescence staining was visualised using a microscope (Leica) equipped with an epifluorescence system. The anti-B1-receptor antibody was used at a dilution of 1:200. Control experiments were performed in which the second or the first antibody alone were used. In both cases, no autofluorescence was observed.
Statistical analysis of data
Kolmogorov-Smirnov tests (with Lilliefors’ correction) were used in order to assess normality prior to statistical analysis. Data were normalised when necessary (perfusion pressure in isolated hearts and isometric force of isolated mesenteric arteries) using the arcsin square root computation. Data from control and ischaemia-reperfusion groups were compared using repeated-measures two-way analyses of variance followed by Tukey's post hoc multiple comparison tests (SigmaStat statistical software v. 2.0, SPSS Science). Statistical significance was set at P < = 0.05.
RESULTS
Myocardial ischaemia-reperfusion
In our experimental conditions, occlusion-reperfusion of the proximal branch of the circumflex artery induced an area at risk of 40 ± 6% of the left ventricle and an infarct size of 45 ± 8% of the area at risk (n = 5%).
In vivo measurements
At the end of the protocol the mean arterial pressure was 52 ± 4 mmHg in the ischaemia-reperfusion group vs. 69 ± 4 mmHg in the sham group (P < 0.05); the heart rate was similar in the two groups (284 ± 14 vs. 303 ± 18 beats min−1, respectively).
Intra-arterial injection of DBK caused significant hypotension and bradycardia in both groups. The response observed in the ischaemia-reperfusion group was greater than that of the sham group (Fig. 1). The effects observed with DBK (10 μg kg−1) were antagonised by LBK in the ischaemia-reperfusion group only (Fig. 1) but were not affected by HOE 140 (MAP: -12 ± 2 vs. -9 ± 3 mmHg; HR: -23 ± 4 vs. -25 ± 5 beats min−1 before and after HOE 140, respectively). In both groups, LBK did not affect baseline haemodynamic parameters.
Figure 1.
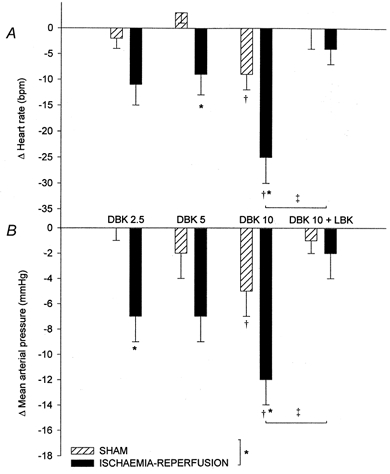
Variation of heart rate (A, beats min−1) and mean arterial blood pressure (B) (means ±s.e.m.) after i.a. injection of des-Arg9-bradykinin at 2.5 μg kg−1 (DBK 2.5), 5 μg kg−1 (DBK 5) and 10 μg kg−1 (DBK 10) and DBK 10, 10 min after [Leu8]-des-Arg9-bradykinin (10 μg kg−1 min−1) (DBK 10 + LBK) in the sham (n = 6%) and ischaemia-reperfusion (n = 5%) groups. *P < 0.05 vs. sham group, †P < 0.05 vs. control value, ‡P < 0.05 vs. corresponding value.
In vitro measurements
Isolated hearts
Baseline left ventricular developed pressure was lower in the ischaemia-reperfusion group (73 ± 7 mmHg) than in the sham group (102 ± 10 mmHg, P < 0.05). Heart rate was similar in the two experimental groups (162 ± 2 beats min−1 in the ischaemia-reperfusion group vs. 167 ± 9 beats min−1 in the sham group).
Perfusion with DBK caused a concentration-dependent decrease in perfusion pressure (Fig. 2) that was statistically significant in the ischaemia-reperfusion but not in the sham group. LBK tended to reverse the effect of DBK but this was not significant. The responses to acetylcholine and sodium nitroprusside were the same in both groups (-15 ± 6 vs. -17 ± 6 mmHg and -22 ± 4 vs. -23 ± 4 mmHg in ischaemia-reperfusion and sham groups, respectively). DBK did not significantly affect heart rate or LVDP.
Figure 2.
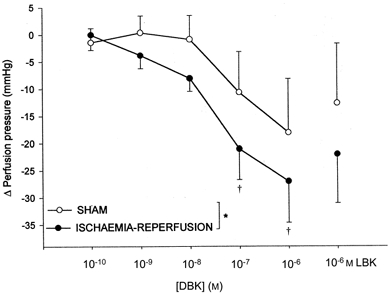
Cumulative decrease in perfusion pressure (means ±s.e.m.) of isolated hearts with increasing concentrations of des-Arg9-bradykinin (DBK; 10−10 to 10−6m), and the effect of DBK (10−6m) after [Leu8]-des-Arg9-bradykinin (LBK; 10−6m) in the sham (n = 5%) and ischaemia-reperfusion (n = 5%) groups. *P < 0.05 vs. sham group, †P < 0.05 vs. control value.
Isolated aorta, carotid and mesenteric arteries
In both groups, DBK induced a dose-dependent constriction of intact and de-endothelialised aorta that was statistically greater in de-endothelialised rings. LBK significantly inhibited this effect in de-endothelialised rings only (Fig. 3). There was no statistical difference between sham and ischaemia-reperfusion groups. DBK caused a dose-dependent relaxation of intact carotid arteries in the ischaemia-reperfusion but not in the sham group and this was significantly inhibited by LBK (Fig. 4). In both groups, DBK did not affect the isometric force of de-endothelialised carotid arteries.
Figure 3.
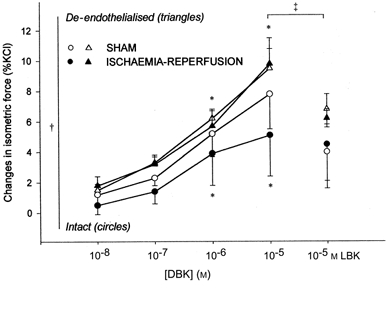
Changes in the isometric force of aortic rings (means ±s.e.m., percentage of maximal KCl contraction) after des-Arg9-bradykinin (DBK; 10−8 to 10−5m), and [Leu8]-des-Arg9-bradykinin (LBK; 10−5m) in the sham (n = 6) and ischaemia-reperfusion (n = 7) groups. *P < 0.05 vs. control tension for pooled values from both groups, †P < 0.05 intact vs. de-endothelialised vessels; ‡P < 0.05 LBK vs. DBK (10−5m). Comparison between intact (circles) and de-endothelialised (triangles) rings.
Figure 4.
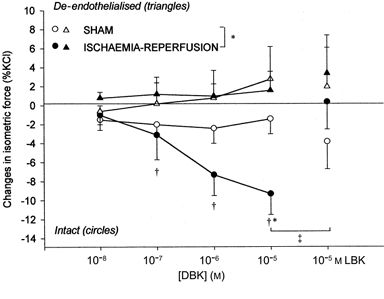
Changes in isometric force of carotid rings (means ±s.e.m., percentage of maximal KCl contraction) after des-Arg9-bradykinin (DBK; 10−8 to 10−5m), and [Leu8]-des-Arg9 bradykinin (LBK; 10−5m) in the sham (n = 6) and ischaemia-reperfusion (n = 7) groups. *P < 0.05 vs. sham group, †P < 0.05 vs. control tension. ‡P < 0.05 vs. corresponding value. Comparison between intact (circles) and de-endothelialised (triangles) rings.
Finally, DBK caused a dose-dependent relaxation of mesenteric arteries in the ischaemia-reperfusion but not in the sham group (Fig. 5). This effect was significantly antagonised by LBK.
Figure 5.
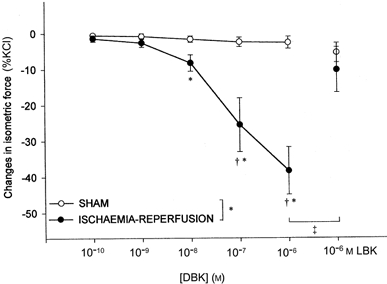
Changes in isometric force of intact mesenteric arteries (means ±s.e.m., percentage of maximal KCl contraction) after des-Arg9-bradykinin (DBK; 10−10 to 10−6m) and [Leu8]-des-Arg9-bradykinin (LBK; 10−6m), in the sham (n = 5%) and ischaemia-reperfusion (n = 6) groups. *P < 0.05 vs. sham group, †P < 0.05 vs. control tension, ‡P < 0.05 vs. corresponding value.
Myocardial ischaemia-reperfusion did not affect the response of any type of artery to acetylcholine or sodium nitroprusside.
Immunolocalisation of cardiovascular B1-receptors
Western blot analysis allowed us to verify that our antibody was specifically bound to the B1-receptor protein. Indeed, there was only one band on the autoradiogram and it corresponded to a 58 kDa protein. This band was prominent on lines corresponding to the heart, aorta, carotid and mesenteric arteries from ischaemia-reperfusion and LPS-pre-treated animals. Figure 6 shows a characteristic Western blot obtained with cardiac tissue isolated from the ischaemic zone; this is also representative of all vascular tissues.
Figure 6.
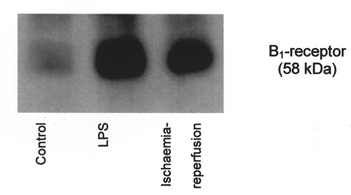
Western blot analysis for B1-receptors in tissue extracted from the ischaemic zone.
The immunohistology assay showed fluorescence in the endothelium and adventitia of all arteries studied and some fluorescence in the aortic media; this corresponded to the presence of B1-receptors in these territories after myocardial ischaemia-reperfusion (Fig. 7).
Figure 7.
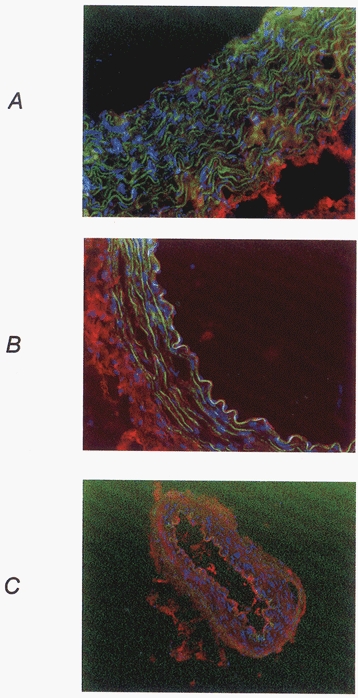
Immunohistological fluorescence photograph of aorta (A), carotid (B) and mesenteric (C) arteries from an animal submitted to a myocardial ischaemia-reperfusion sequence. Red, B1-receptors; blue, nuclei; green, autofluorescence of elastin.
DISCUSSION
The objective of this work was to evaluate whether myocardial ischaemia-reperfusion was able to induce a de novo synthesis of B1-receptors in the rabbit. The presence of B1-receptors was evaluated pharmacologically by assessing the effects of des-Arg9-bradykinin, the classical B1-receptor agonist (Marceau et al. 1998) and by immunolocalisation of the B1-receptor protein. Our results clearly demonstrate that a myocardial ischaemia- reperfusion sequence can induce cardiac as well as vascular B1-receptor expression. This confirms the recent study of Tschöpe et al. (2000) that demonstrates an increase in cardiac RNAs coding for B1-receptors after myocardial infarction. In addition, we have shown that myocardial ischaemia-reperfusion could induce B1-receptors at the cardiac level but also at a distance in systemic vessels and that these receptors were functionally active.
In vivo, after myocardial ischaemia-reperfusion, des-Arg9-bradykinin induced a bradycardia and a decrease in the mean arterial pressure. These effects were due to B1-receptors since they were antagonised by [Leu8]-des-Arg9-bradykinin and unaffected by HOE 140. The small response to DBK observed in the sham group could be explained by a weak inflammatory response to combined anaesthesia and surgical stress. However, since it is not significantly antagonised by LBK, we cannot exclude a B1-receptor-independent effect of DBK injection. The observed hypotension could be due to a decrease in vascular resistance as well as in cardiac output. Since no effect on heart rate was seen in isolated hearts, this suggests that the bradycardia seen in vivo could be due to a central effect of des-Arg9-bradykinin.
Since [Leu8]-des-Arg9-bradykinin had no effect on baseline arterial pressure or heart rate, B1-receptors do not appear to play an important role in the control of haemodynamic parameters after myocardial ischaemia- reperfusion. However, this effect was measured 3 h after reperfusion rather than at the beginning of reperfusion when kinin outflow from the ischaemic zone is maximal (Lamontagne et al. 1995). Moreover, in the isolated rat heart model, des-Arg9-bradykinin has been shown to be released from the coronary circulation after myocardial ischaemia-reperfusion only when ACE is blocked (Lamontagne et al. 1995). Indeed, inhibition of the most important degradation pathway of bradykinin could lead to an increased activity of the carboxypeptidase responsible for the synthesis of des-Arg9 fragments (Marceau, 1995). This suggests that, in patients treated with ACE inhibitors, the presence of a B1-agonist along with B1-receptors could lead to B1-receptor-dependent haemodynamic responses after myocardial ischaemia- reperfusion.
In vitro, the isolated aorta, the classic model for the study of B1-receptors (Regoli et al. 1977), did not show a B1-receptor induction by myocardial ischaemia- reperfusion. Isolated aortic rings from both groups responded to des-Arg9-bradykinin by a constriction. This could also be explained either by the previously mentioned weak inflammatory response and/or by an in vitro induction by incubation, since the response to des-Arg9-bradykinin was evaluated approximately 1.5 h after isolation. Indeed, although a 1 h incubation time is not sufficient to induce aortic B1-receptors in control animals (Bouthillier et al. 1987), after surgical stress this delay could be reduced. De-endothelialised aortic rings responded more intensely than intact ones to des-Arg9-bradykinin. This suggests that vasodilatory endothelial B1-receptors in the aorta could counteract the smooth muscle-induced vasoconstriction. B1-receptors leading to smooth muscle-dependent vasoconstriction were first characterised in isolated aortic strips (Regoli et al. 1977), which do not permit the study of endothelial function. Thereafter, the use of aortic rings showed that the effect of des-Arg9-bradykinin on the intact aortic wall was a vasoconstriction (Levesque et al. 1995). To our knowledge, this is the first study using de-endothelialised and intact aortic rings to evaluate the effect of des-Arg9-bradykinin on the aortic endothelium. Our results suggest that it induces an endothelial vasodilatation that is masked by a more pronounced media-induced vasoconstriction. This is confirmed by the immunolocalisation of B1-receptors on both endothelium and media in the aorta. A strong fluorescence was also seen in the adventitia of all the vessels studied. Specific experiments will be needed to determine the role of these adventitial B1-receptors.
Three hours after myocardial ischaemia-reperfusion but not after sham operation, we observed a dose-dependent and endothelium-dependent vasodilatation in response to des-Arg9-bradykinin on isolated carotid rings. Indeed, immunohistochemical staining showed the presence of B1-receptors in the endothelium of carotid arteries with little fluorescence in the media. Our results are in accordance with those of Pruneau & Bélichard (1993), who reported that, after induction of B1-receptors by 5 h in vitro incubation, isolated rabbit carotid arteries respond to des-Arg9-bradykinin by a vasodilatation. These authors have also shown that acutely denuded carotid arteries do not respond to des-Arg9-bradykinin (Pruneau et al. 1994). The mechanisms involved in the B1-receptor-dependent relaxation are known to implicate NO (Pruneau & Bélichard, 1993) and prostaglandin (de Blois et al. 1987) synthesis.
The effect of des-Arg9-bradykinin on large arteries cannot explain the decrease in arterial pressure observed in vivo. We thus tested its effect on small resistance arteries such as the coronary bed of the isolated heart and isolated mesenteric arteries. After myocardial ischaemia- reperfusion, isolated hearts responded to des-Arg9- bradykinin by a decrease in perfusion pressure demonstrating the presence of B1-receptors at the site of ischaemia. Sham-operated hearts tended to respond to des-Arg9-bradykinin, although not significantly. This suggests that, in the heart, a small proportion of B1-receptors could have been induced by inflammation brought about by surgical stress, in particular the silk thread passed around the coronary artery, as well as by the isolated perfusion protocol, which can be considered as an in vitro incubation. The inflammation related to the surgical protocol could also explain the tendency of sham animals to respond to des-Arg9-bradykinin.
After myocardial ischaemia-reperfusion, small mesenteric arteries responded to des-Arg9-bradykinin by an almost complete relaxation. The immunohistochemical study showed a preferential distribution of B1-receptors at the endothelial level in mesenteric arteries after myocardial ischaemia-reperfusion. The fact that B1-receptors are present in systemic vessels is an interesting finding since a local stress (myocardial ischaemia) was able to induce B1-receptors at a distance and not only locally as previously shown (Tschöpe et al. 2000). Thus, the induction of endothelial B1-receptors on resistance arteries could (along with the bradycardia) explain the hypotension observed in vivo. After myocardial ischaemia-reperfusion we did not observe a decreased endothelial function in the vessels studied. This is in agreement with other authors. Indeed, no endothelial dysfunction is seen in the aorta, femoral and mesenteric arteries following myocardial ischaemia (Brandes et al. 1998). However, ischaemia-reperfusion is associated with an endothelial dysfunction in vessels submitted to ischaemia, as reported for coronary arteries isolated distally from the occlusion site (Richard et al. 1994), or for isolated hearts submitted to a global low-flow ischaemia (Bouchard et al. 1998). Our results are in accordance with this since no decreased response to acetylcholine was seen in vessels not submitted to ischaemia. In the heart, we did not observe an endothelial dysfunction since it was evaluated globally and not only in the previously ischaemic zone.
B1-receptors, which were first demonstrated to be induced by in vitro incubation (Regoli et al. 1977), are also promoted by various pro-inflammatory substances such as LPS, IL-1, IL-2, IL-8, adjuvant and bradykinin or by inflammatory pathological processes (immune complex arthritis, balloon catheter injury, septicaemia or UV irradiation) (Marceau, 1995). This is due to a de novo synthesis of the receptor since the induction is prevented by inhibitors of RNA synthesis (Marceau, 1995) and of protein synthesis or trafficking (Audet et al. 1994). IL-1 appears to play a primordial role since all substances responsible for B1-receptor induction in vivo or in vitro can induce IL-1 synthesis or release (Marceau, 1995). There is a strong body of evidence that inflammation occurs during myocardial ischaemia and reperfusion (Hansen, 1995). The role of cytokines in mediating inflammation in this situation seems to be particularly important since their expression is increased during ischaemia and early reperfusion, particularly in coronary vessels (Galea et al. 1996). Moreover, IL-1 is released by endothelial cells submitted to hypoxia-reoxygenation (Ala et al. 1992). The production of IL-1 in the vascular bed of ischaemic myocardium could participate in the induction of B1-receptors in coronary arteries and also, as demonstrated by our results, in systemic vessels. Finally, although the involvement of kinins in the induction of B1-receptors remains to be confirmed (Nwator et al. 1989; Marceau, 1995), their release upon myocardial ischaemia could also contribute to the induction observed in this study. Some studies have shown that B1-receptors present in the isolated heart model can decrease noradrenaline outflow during ischaemia (Chahine et al. 1993; Feng et al. 1997). Others report that they can mimic the effect of ischaemic preconditioning in protecting the coronary endothelium (Bouchard et al. 1998). Taken together with our results showing that myocardial ischaemia- reperfusion is able to promote induction of bradykinin B1-receptors, this indicates that under ischaemic conditions kinins could interact with B1-receptors and have an eventual protective role. Finally, a combined action of IL-1 and kinins has been shown to stimulate angiogenesis through the activation of B1-receptors (Hu et al. 1993). Thus, the implication of these receptors in the neovascularisation occurring after myocardial ischaemia- reperfusion should also be investigated and could have clinical relevance, especially in patients treated with ACE inhibitors. In conclusion, our study is the first to provide evidence that myocardial ischaemia-reperfusion induces a global induction of functional kinin B1-receptors in the endothelium.
References
- Abd Alla S, Buschko J, Quitterer U, Maidhof A, Haasemann M, Breipohl G, Knolle J, Müller-Esterl W. Structural features of the human bradykinin B2 receptor probed by agonists, antagonists, and anti-idiotypic antibodies. Journal of Biological Chemistry. 1993;268:17277–17285. [PubMed] [Google Scholar]
- Ala Y, Palluy O, Favero J, Bonne C, Modat G, Dornand J. Hypoxia/reoxygenation stimulates endothelial cells to promote interleukin-1 and interleukin-6 production. Effects of free radical scavengers. Agents Actions. 1992;37:134–139. doi: 10.1007/BF01987902. [DOI] [PubMed] [Google Scholar]
- Audet R, Petitclerc E, Drapeau G, Rioux F, Marceau F. Further analysis of the upregulation of bradykinin B1 receptors in isolated rabbit aorta by using metabolic inhibitors. European Journal of Pharmacology. 1994;271:551–555. doi: 10.1016/0014-2999(94)90819-2. [DOI] [PubMed] [Google Scholar]
- Baumgarten CR, Linz W, Kunkel G, Scholkens BA, Wiemer G. Ramiprilat increases bradykinin in outflow from isolated hearts of rat. British Journal of Pharmacology. 1993;108:293–295. doi: 10.1111/j.1476-5381.1993.tb12797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard J-F, Chouinard J, Lamontagne D. Role of kinins in the endothelial protective effect of ischaemic preconditioning. British Journal of Pharmacology. 1998;123:413–420. doi: 10.1038/sj.bjp.0701619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouthillier J, de Blois D, Marceau F. Studies on the induction of pharmacological responses to des-Arg9-bradykinin in vitro and in vivo. British Journal of Pharmacology. 1987;92:257–264. doi: 10.1111/j.1476-5381.1987.tb11319.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandes RP, Walles T, Koddenber G, Gwinner W, Mugge A. Endothelium-dependent vasodilatation in Sprague-Dawley rats with postinfarction hypertrophy: lack of endothelial dysfunction in vitro. Basic Research in Cardiology. 1998;93:463–469. doi: 10.1007/s003950050116. [DOI] [PubMed] [Google Scholar]
- Chahine R, Adam A, Yamaguchi N, Gaspo R, Regoli D, Nadeau R. Protective effects of bradykinin on the ischaemic heart: implication of the B1 receptor. British Journal of Pharmacology. 1993;108:318–322. doi: 10.1111/j.1476-5381.1993.tb12802.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Blois D, Bouthillier J, Marceau F. Effects of glucocorticoids, monokinines and growth factors on the spontaneously developing responses of the rabbit isolated aorta to des-Arg9-bradykinin. British Journal of Pharmacology. 1988;93:969–977. doi: 10.1111/j.1476-5381.1988.tb11487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Blois D, Marceau F. The ability of des-Arg9-bradykinin to relax rabbit isolated mesenteric arteries is acquired during in vitro incubation. European Journal of Pharmacology. 1987;142:141–144. doi: 10.1016/0014-2999(87)90664-9. [DOI] [PubMed] [Google Scholar]
- Entman ML, Michael L, Rossen RD, Dreyer WJ, Anderson DC, Taylor AA, Smith CW. Inflammation in the course of early myocardial ischemia. FASEB Journal. 1991;5:2529–2537. doi: 10.1096/fasebj.5.11.1868978. [DOI] [PubMed] [Google Scholar]
- Feng J, Yamaguchi N, Foucart S, Chahine R, Lamontagne D, Nadeau R. Transient ischemia inhibits nonexocytotic release of norepinephrine following sustained ischemia in rat heart: is bradykinin involved? Canadian Journal of Physiology and Pharmacology. 1997;75:665–670. doi: 10.1139/cjpp-75-6-665. [DOI] [PubMed] [Google Scholar]
- Galea J, Armstrong J, Gadsdon P, Holden H, Francis SE, Holt CM. Interleukin-1 beta in coronary arteries of patients with ischemic heart disease. Arteriosclerosis Thrombosis and Vascular Biology. 1996;16:1000–1006. doi: 10.1161/01.atv.16.8.1000. [DOI] [PubMed] [Google Scholar]
- Hansen PR. Role of neutrophils in myocardial ischemia and reperfusion. Circulation. 1995;91:1872–1885. doi: 10.1161/01.cir.91.6.1872. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Hamamoto H, Honda Y, Hirose M, Furukawa S, Kimura E. Changes in components of kinin system and hemodynamics in acute myocardial infarction. American Heart Journal. 1978;95:619–626. doi: 10.1016/0002-8703(78)90304-6. [DOI] [PubMed] [Google Scholar]
- Herskowitz A, Choi S, Ansari A, Wesselingh S. Cytokine mRNA expression in postischemic/reperfused myocardium. American Journal of Pathology. 1995;146:419–428. [PMC free article] [PubMed] [Google Scholar]
- Hu DE, Fan TP. [Leu8]des-Arg9-bradykinin inhibits the angiogenic effect of bradykinin and interleukin-1 in rats. British Journal of Pharmacology. 1993;109:14–17. doi: 10.1111/j.1476-5381.1993.tb13525.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamontagne D, Nadeau R, Adam A. Effects of enalaprilat on bradykinin and des-Arg9-bradykinin release following reperfusion of the ischaemic rat heart. British Journal of Pharmacology. 1995;115:476–478. doi: 10.1111/j.1476-5381.1995.tb16357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levesque L, Larrivee J-F, Bachvarov DR, Rioux F, Drapeau G, Marceau F. Regulation of kinin-induced contraction and DNA synthesis by inflammatory cytokines in the smooth muscle of the rabbit aorta. British Journal of Pharmacology. 1995;116:1673–1679. doi: 10.1111/j.1476-5381.1995.tb16390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marceau F. Kinin B1 receptors: a review. Immunopharmacology. 1995;30:1–26. doi: 10.1016/0162-3109(95)00011-h. [DOI] [PubMed] [Google Scholar]
- Marceau F, Hess JF, Bachvarov DR. The B1-receptors for kinins. Pharmacological Reviews. 1998;50:357–386. [PubMed] [Google Scholar]
- Marceau F, Larrivee F, Saint-Jacques E, Bachvarov D. The kinin B1 receptor: an inducible G protein-coupled receptor. Canadian Journal of Physiology and Pharmacology. 1997;75:725–730. [PubMed] [Google Scholar]
- Matsuki T, Shoji T, Yoshida S, Kudoh Y, Motoe M, Inoue M, Nakata T, Hosoda S, Shimamoto K, Yellon D, Imura O. Sympathetically-induced myocardial ischemia causes the heart to release plasma kinin. Cardiovascular Research. 1987;21:428–432. doi: 10.1093/cvr/21.6.428. [DOI] [PubMed] [Google Scholar]
- Mulvany MJ, Halpern W. Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circulation Research. 1977;41:19–26. doi: 10.1161/01.res.41.1.19. [DOI] [PubMed] [Google Scholar]
- Nwator I, Whalley ET. Angiotensin converting enzyme inhibitors and expression of des-Arg9-bradykinin (kinin B1) receptors in vivo. European Journal of Pharmacology. 1989;160:125–132. doi: 10.1016/0014-2999(89)90661-4. [DOI] [PubMed] [Google Scholar]
- Pruneau D, Bélichard P. Induction of bradykinin B1 receptor-mediated relaxation in the isolated rabbit carotid artery. European Journal of Pharmacology. 1993;239:63–67. doi: 10.1016/0014-2999(93)90976-o. [DOI] [PubMed] [Google Scholar]
- Pruneau D, Luccarini JM, Bélichard P. Induction of kinin B1 receptor-dependent vasoconstriction following balloon catheter injury to the rabbit carotid artery. British Journal of Pharmacology. 1994;111:1029–1034. doi: 10.1111/j.1476-5381.1994.tb14847.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regoli D, Barabe J, Park WK. Receptors for bradykinin in rabbit aortae. Canadian Journal of Physiology and Pharmacology. 1977;55:855–867. doi: 10.1139/y77-115. [DOI] [PubMed] [Google Scholar]
- Regoli D, Marceau F, Lavigne J. Induction of B1-receptors for kinins in the rabbit by a bacterial lipopolysaccharide. European Journal of Pharmacology. 1981;71:105–115. doi: 10.1016/0014-2999(81)90391-5. [DOI] [PubMed] [Google Scholar]
- Richard V, Kaeffer N, Tron C, Thuillez C. Ischaemic preconditioning protects against coronary endothelial dysfunction induced by ischaemia and reperfusion. Circulation. 1994;89:1254–1261. doi: 10.1161/01.cir.89.3.1254. [DOI] [PubMed] [Google Scholar]
- Tschope C, Heringer-Walther S, Koch M, Spillmann F, Wendorf M, Leitner E, Schultheiss HP, Wakther T. Upregulation of bradykinin B1-receptor expression after myocardial infarction. British Journal of Pharmacology. 2000;129:1537–1538. doi: 10.1038/sj.bjp.0703239. [DOI] [PMC free article] [PubMed] [Google Scholar]