

Abstract
We used whole-cell patch clamp to investigate steady-state activation of ATP-sensitive K+ channels (KATP) of rat arterial smooth muscle by protein kinase A (PKA) and the pathway by which angiotensin II (Ang II) inhibits these channels.
Rp-cAMPS, an inhibitor of PKA, did not affect KATP currents activated by pinacidil when the intracellular solution contained 0.1 mM ATP. However, when ATP was increased to 1.0 mM, inhibition of PKA reduced KATP current, while the phosphatase inhibitor calyculin A caused a small increase in current.
Ang II (100 nM) inhibited KATP current activated by the K+ channel opener pinacidil. The degree of inhibition was greater with 1.0 mM than with 0.1 mM intracellular ATP. The effect of Ang II was abolished by the AT1 receptor antagonist losartan.
The inhibition of KATP currents by Ang II was abolished by a combination of PKA inhibitor peptide 5-24 (5 μM) and PKC inhibitor peptide 19-27 (100 μM), while either alone caused only partial block of the effect.
In the presence of PKA inhibitor peptide, the inhibitory effect of Ang II was unaffected by the PKC inhibitor Gö 6976, which is selective for Ca2+-dependent isoforms of PKC, but was abolished by a selective peptide inhibitor of the translocation of the ε isoform of PKC.
Our results indicate that KATP channels are activated by steady-state phosphorylation by PKA at normal intracellular ATP levels, and that Ang II inhibits the channels both through activation of PKCε and inhibition of PKA.
Vasoconstrictors cause an increase in intracellular Ca2+ concentration of vascular smooth muscle, and therefore in contractile force, by increasing the influx of extracellular Ca2+ through voltage-dependent L-type Ca2+channels and by releasing Ca2+ from intracellular stores (Somlyo, 1985; Nelson et al. 1990). The increase in Ca2+ influx may be caused both by a direct activation of Ca2+-permeable channels and through membrane depolarization and so an increase in the open probability of voltage-dependent Ca2+ channels (Mulvany et al. 1982; Nelson et al. 1988). Several vasoconstrictors have also been shown to inhibit K+ channels, an effect that should contribute to their depolarizing action (Beech, 1997). For example, the potent physiological vasoconstrictor Angiotensin II (Ang II) has effects both on cation channels and K+ channels of smooth muscle. Ang II activates Ca2+ channels and non-selective cation channels (Ohya & Sperelakis, 1991; Hughes & Bolton, 1995), while it inhibits voltage-activated, Ca2+-activated and ATP-sensitive K+ (KATP) channels (Toro et al. 1990; Miyoshi & Nakaya, 1991; Minami et al. 1995; Clément-Chomienne et al. 1996; Kubo et al. 1997).
Vasoconstrictor inhibition of K+ channels has been shown to occur through protein kinase C (PKC) (Beech, 1997; Quayle et al. 1997), and a number of vasoconstrictors, including Ang II, inhibit KATP channels by activating PKC (Bonev & Nelson, 1996; Kubo et al. 1997). However, in addition to activation of PKC (through phospholipase C and diacylglycerol), Ang II receptors can cause inhibition of cyclic AMP-dependent protein kinase (PKA) through inhibition of adenylyl cyclase and reduction in cyclic AMP (Anand-Srivastava, 1983; Griendling et al. 1986; Nishizuka, 1992; Unger et al. 1996). Since PKA is known to activate KATP channels (Quayle et al. 1997), it is possible that Ang II could inhibit the channels by reducing their activation by PKA as well as directly through PKC.
In the present study we have therefore investigated the components of the signalling pathways between Ang II and the KATP channel, and the possible involvement of PKA. Previous work on the inhibition of KATP channels by vasoconstrictors, including our own, has used a low intracellular [ATP] of 0.1 mM to increase KATP channel activity, and under these conditions PKA does not appear to be involved (Bonev & Nelson, 1996; Kubo et al. 1997). Here we show that when the intracellular [ATP] is raised to 1 mM, steady-state KATP channel activation by PKA is observed. Under these conditions, Ang II, acting through the AT1 receptor, can reduce KATP channel activity both by inhibition of PKA to reduce this steady-state activation, and through activation of PKC. The PKC-dependent component of theaction of Ang II is abolished by a peptide inhibitor of the translocation of PKCɛ, suggesting that this isoform of PKC mediates the effect. Our results suggest that steady-state activation of KATP channels by PKA may be important in setting their background activity, and that a reduction in this PKA activation by inhibition of adenylyl cyclase may form an additional pathway for the actions of some vasodilators to their action through PKC.
METHODS
Dissociation of arterial smooth muscle cells
Single smooth muscle cells were isolated enzymatically from small branches of the rat mesenteric artery. Male adult Wistar rats were rendered unconscious by exposure to a rising concentration of CO2, and killed by exsanguination. The care of the animals conformed to the requirements of the Animals (Scientific Procedures) Act 1986. Mesenteric arteries were removed and cleaned of connective tissue in ice-cold solution containing (mM): 137 NaCl, 5.6 KCl, 0.42 Na2HPO4, 0.44 NaH2PO4, 1 MgCl2, 2 CaCl2, 10 Hepes and 10 glucose, adjusted with NaOH to pH 7.4. Second and third-order branches of the arteries were then transferred to the same solution except that CaCl2 was reduced to 0.1 mM (low calcium solution) for 10 min, and warmed to 35°C. Arteries were digested for 30-35 min in low calcium solution containing (mg ml−1): 0.9 albumin, 1.4 papain and 0.9 dithioerythritol; and then for 16-20 min in low calcium solution containing (mg ml−1): 0.9 albumin, 1.4 collagenase type F (Sigma) and 0.9 hyaluronidase type I-S (Sigma). Arteries were then transferred to low calcium solution containing albumin (1.0 mg ml−1). Single smooth muscle cells were obtained by trituration with a wide-bore pipette, stored at 4°C, and used on the day of preparation. The cells had membrane capacitances of 12.9 ± 0.5 pF (n = 69).
Solutions and chemicals
The intracellular solution for conventional whole-cell recordings of KATP current contained (mM): 110 KCl, 30 KOH, 10 Hepes, 10 EGTA, 1 MgCl2, 1 CaCl2, 0.1 or 1.0 Na2ATP, 0.1 ADP and 0.5 GTP; adjusted to pH 7.2. The free [Ca2+] calculated using the program Maxchelator (http//www.stanford.edu/≈cpatton/maxc.html) was 20 nM. The 6 mM K+ extracellular solution contained (mM): 134 NaCl, 6 KCl, 1 MgCl2, 0.1 CaCl2, 10 Hepes and 10 glucose; adjusted to pH 7.4. To separate KATP currents, we recorded at -60 mV to minimise activation of voltage-dependent K+ channels, and raised extracellular [K+] to 140 mM to give a substantial inward driving force for K+, as we have done previously (Kubo et al. 1997). The 140 mM K+ extracellular solution contained (mM): 140 KCl, 1 MgCl2, 0.1 CaCl2, 10 Hepes and 10 glucose; pH 7.4. The external Ca2+ was lowered to 0.1 mM to reduce Ca2+ influx. The external solution was changed by continuous perfusion of the experimental chamber (volume 0.4 ml); the dead time for the new solution to reach the bath was about 30 s and complete exchange took about 2 min. Pinacidil, angiotensin II, glibenclamide and 1, 2-dioctanoyl-rac-glycerol (DiC8) were purchased from Sigma. Gö 6976, PKCɛ translocation inhibitor peptide, myristoylated protein kinase C inhibitor 19-27, Rp-cAMPS (the Rp isomer of adenosine 3′, 5′-cyclic monophosphorothioate triethylammonium salt), protein kinase A inhibitor peptide 5-24, and calyculin A were obtained from Calbiochem (UK). Losartan was kindly provided by Dr A. Stanley, Department of Medicine, University of Leicester. Pinacidil, glibenclamide, DiC8 and calyculin A were dissolved in dimethylsulphoxide (DMSO) solution. The final concentration of DMSO was less than 0.2 %.
Data recording and analysis
Whole-cell K+ currents were recorded from single smooth muscle cells in the conventional configuration of the patch-clamp technique (Hamill et al. 1981). Membrane currents were recorded, and voltage controlled, using an Axon interface and Axopatch 200 amplifier (Axon Instruments). Patch pipettes were made from thin-walled borosilicate glass (Clark Electromedical, Pangbourne, Berks, UK) using a pp-83 vertical puller (Narishige, Tokyo, Japan) and coated with sticky wax (Kemdent, Swindon, Wilts, UK) to reduce capacitance. Currents were filtered at 5 kHz. Electrode resistance before sealing was 3-5 MΩ, and after sealing was > 10 GΩ. In order to minimize activity of delayed rectifier K+ channels and large conductance Ca2+-activated potassium (BKCa) channels, experiments were performed at -60 mV and intracellular Ca2+ was buffered to a low level with EGTA.
Experiments were done at 20-25°C. Results are expressed as means ±s.e.m. Intergroup differences were analysed by analysis of variance followed by the Student-Newman-Keuls test and Student's t test as appropriate, and P < 0.05 was considered statistically significant.
RESULTS
Steady-state activation of KATP current by PKA
A number of vasodilators, including adenosine and calcitonin gene-related peptide, can activate KATP channels in arterial smooth muscle through stimulation of cyclic AMP-dependent protein kinase (PKA) (Quayle et al. 1994; Kleppisch & Nelson, 1995; Quayle et al. 1997; Wellman et al. 1998), but channel activation via PKA in the absence of vasodilators has not so far been reported. However, almost all work on whole-cell KATP channel currents in arterial cells has used low intracellular [ATP], usually 0.1 mM, which might lead to a correspondingly low rate of cAMP generation by adenylyl cyclase. We have therefore investigated the possibility that an increase in [ATP] towards more physiological levels (1.4-1.8 mM, Post & Jones, 1991) might reveal steady-state activation of KATP channels by PKA.
To test for such steady-state activation, we compared the effects of the inhibitor Rp-cAMPS, which inhibits PKA by binding to its regulatory subunit, on KATP current in cells with intracellular [ATP] of either 0.1 or 1.0 mM. Whole-cell current was recorded at a holding potential of -60 mV and extracellular K+ was increased to 140 mM so that K+ currents were inward at this potential. To increase the size of the KATP current and so our ability to detect any effect of inhibition of PKA, we used the KATP channel opener pinacidil (10 μM). Figure 1A shows the effect of Rp-cAMPS in a cell dialysed with a pipette solution containing 0.1 mM ATP. Raising the extracellular K+ concentration from 6 to 140 mM (arrow) caused a small increase in inward current, while pinacidil (10 μM) activated a much more substantial inward current, which was not affected by Rp-cAMPS (Fig. 1A and C). The current was subsequently inhibited by glibenclamide (10 μM), which selectively inhibits KATP channels at this concentration (Quayle et al. 1997). In cells dialysed with a solution containing 1.0 mM ATP, the size of the pinacidil-induced current was reduced compared with that seen with 0.1 mM ATP (-93 ± 9 pA, n = 12 cells vs. -210 ± 32 pA, n = 10, P < 0.01), as expected from the inhibitory effect of ATP that is characteristic of the channel. However, in contrast to its lack of effect with 0.1 mM ATP, Rp-cAMPS caused substantial inhibition of KATP current when intracellular ATP was 1.0 mM (Fig. 1B and C). In seven cells, 100 μM Rp-cAMPS decreased the current by 46.1 ± 2.9 %.
Figure 1. The reduction of KATP current by Rp-cAMPS is dependent on intracellular [ATP].
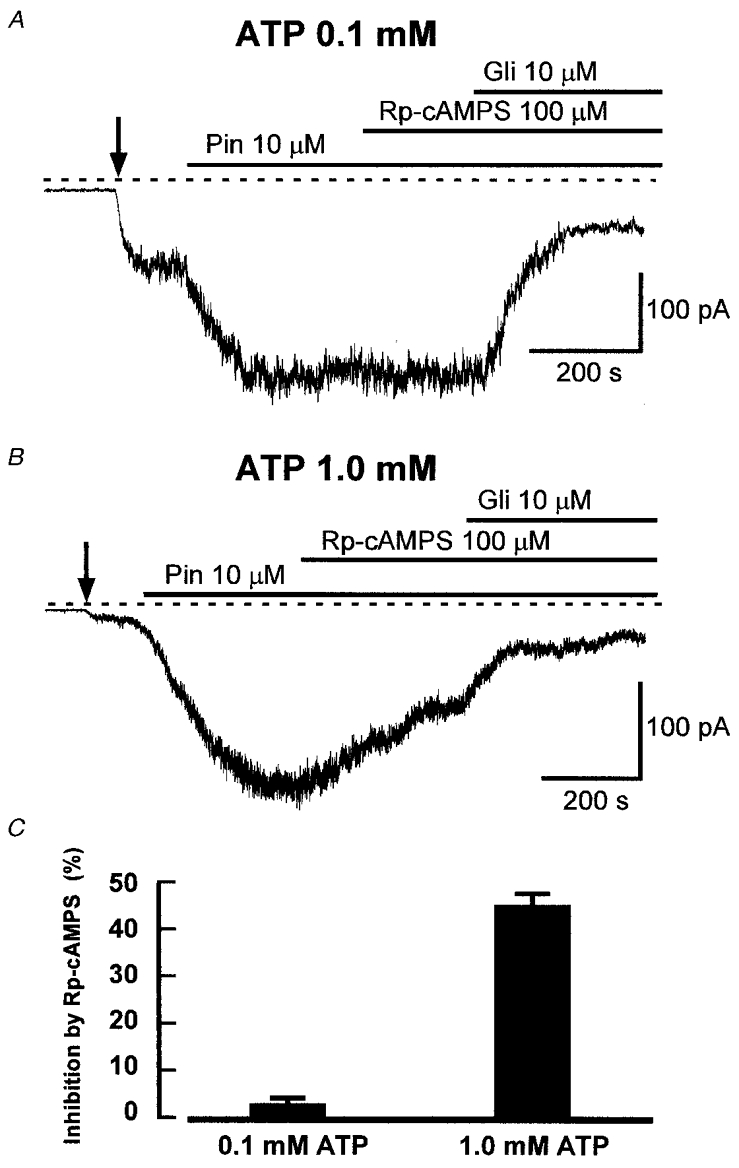
A, representative recording of whole-cell current from a cell held at -60 mV showing the lack of effect of Rp-cAMPS on glibenclamide-sensitive K+ current activated by pinacidil in a cell with 0.1 mM intracellular ATP. The dashed line indicates the zero current level. In all recordings, the cell was dialysed with a solution containing 140 mM K+, and extracellular K+ was changed from 6 to 140 mM as indicated by the vertical arrow in this and subsequent figures. Pinacidil (Pin), Rp-cAMPS and glibenclamide (Gli) were applied as indicated. B, current recording showing the effect of Rp-cAMPS with 1.0 mM intracellular ATP. Rp-cAMPS caused significant inhibition under these conditions, and glibenclamide inhibited the remaining current. C, mean (+s.e.m.) inhibition of KATP current by Rp-cAMPS in cells dialysed with solution containing ATP at either 0.1 or 1.0 mM. The bars show mean values +s.e.m. from 6 and 7 cells, respectively.
The above finding is consistent with a steady-state level of KATP channel activation by PKA in the presence of 1.0 mM ATP. It is likely that the level of channel activity will be set by the balance between phosphorylation by PKA and dephosphorylation by protein phosphatases. To see whether we could induce additional channel activation by inhibiting dephosphorylation, we tested the effect of calyculin A, a cell-permeant inhibitor of protein phosphatases 1 (PP1) and 2A (PP2A) (Herzig & Neumann, 2000). Figure 2 shows the effect of calyculin A (50 nM). In 0.1 mM ATP, calyculin A was without effect on KATP current, either in the absence or presence of pinacidil, as expected if there was no steady-state phosphorylation of KATP channels at this low [ATP]. However, when intracellular ATP was increased to 1.0 mM, calyculin A caused a small but significant increase in KATP current either with or without pinacidil, as shown in Fig. 2C and D. These results are consistent with the effects of Rp-cAMPS and suggest that when intracellular ATP is 1.0 mM there is steady-state phosphorylation of KATP channels by PKA, and that inhibition of phosphatases causes a small increase in KATP current because of the enhancement of phosphorylation.
Figure 2. The effect of calyculin A on KATP current.
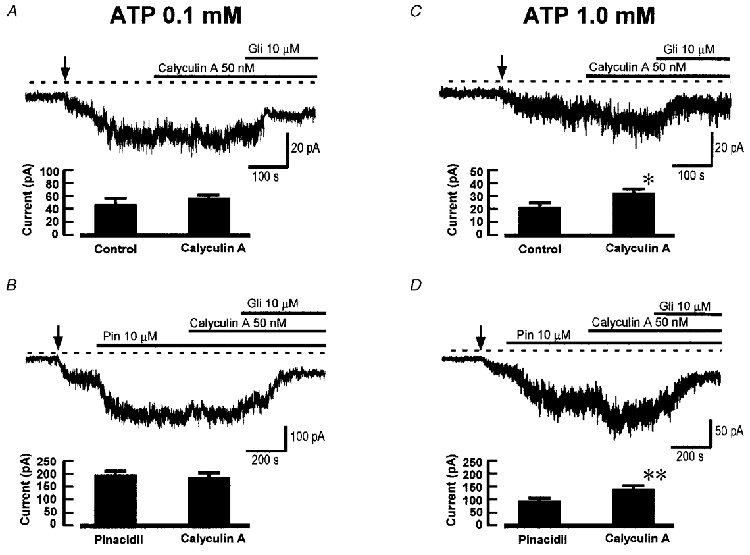
In each panel the traces show whole-cell recordings at -60 mV and the histograms show mean (+s.e.m.) results from 6 or 7 cells in each case. Calyculin A was used at 50 nM. A, lack of effect of calyculin A on background KATP current with 0.1 mM intracellular ATP. B, lack of effect of calyculin A on pinacidil-induced KATP current with 0.1 mM intracellular ATP. C, effect of calyculin A on background KATP current with 1 mM intracellular ATP; * results significantly different from control (P < 0.05, paired t test). D, effect of calyculin A on pinacidil-induced KATP current with 1 mM intracellular ATP; ** results significantly different from pinacidil only (P < 0.01).
Angiotensin II causes greater inhibition of KATP current with 1.0 mM than 0.1 mM ATP
We have shown previously that angiotensin II (Ang II) inhibits KATP currents of rat mesenteric arterial myocytes (Kubo et al. 1997). In those experiments we used an intracellular [ATP] of 0.1 mM, and found that the adenylyl cyclase-PKA system did not contribute to the inhibition, which appeared to occur solely via PKC (Kubo et al. 1997). Our present finding of steady-state channel activation by PKA when intracellular ATP is increased to 1.0 mM, however, raises the possibility that with this higher [ATP] Ang II might also be able to reduce channel activity by inhibition of PKA, since Ang II receptors can inhibit adenylyl cyclase (Anand-Srivastava, 1983; Unger et al. 1996). We have therefore examined the inhibition of KATP current by Ang II with either 0.1 or 1.0 mM intracellular [ATP].
Figure 3 shows that, with 0.1 mM ATP, Ang II (100 nM) reduced KATP current activated by 10 μM pinacidil (Fig. 3A), the mean inhibition being 41.4 ± 2.3 % (n = 10 cells, Fig. 3C), a value quite close to that of 36 %, which we measured previously for Ang II inhibition of KATP current activated by levcromakalim rather than pinacidil (Kubo et al. 1997). In cells with 1.0 mM ATP, Ang II caused a greater inhibition of KATP current (Fig. 3B): in 12 cells the mean inhibition was 72.1 ± 7.4 % (P < 0.01 compared with 0.1 mM ATP). This finding is consistent with Ang II having an additional mechanism of action in the higher [ATP]. Since an intracellular ATP concentration of 1.0 mM revealed the effect of PKA on KATP channels, we have used this concentration in our subsequent experiments designed to investigate the signal transduction pathways involved in channel inhibition by Ang II.
Figure 3. Angiotensin II inhibition of KATP current with 0.1 and 1.0 mM ATP.
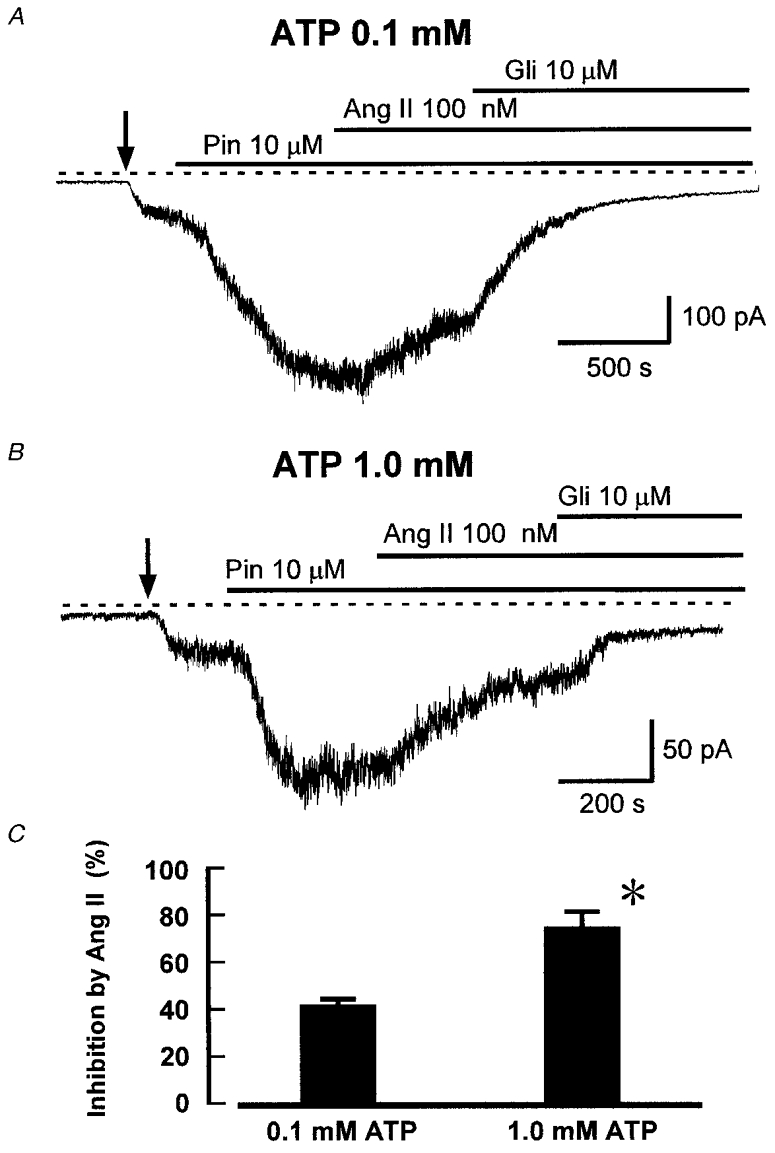
A, recording of whole-cell current at -60 mV from a cell dialysed with 0.1 mM ATP. Pinacidil (Pin), Ang II, and glibenclamide (Gli) were added as indicated. Ang II inhibited KATP current by 30 % in this cell. B, effect of Ang II in a cell under the same conditions as in A, except that intracellular ATP was 1.0 mM. C, mean inhibition (+s.e.m.) of KATP current by Ang II (100 nM) in cells with either 0.1 or 1.0 mM intracellular ATP (n = 10 and 12, respectively). *P < 0.01 compared with 0.1 mM ATP, Student's t test.
Angiotensin acts through an AT1 receptor
Ang II receptors can be divided into AT1 and AT2 subtypes (Unger et al. 1996). The AT1 receptor is responsible for most of the known effects of Ang II, including the major part of its vasoconstrictor effect, is coupled to phospholipase C through Gq/11 G proteins (Griendling et al. 1986; Nishizuka, 1992) and is also negatively coupled to adenylyl cyclase (Anand-Srivastava, 1983; Unger et al. 1996) through Gi/o. In order to investigate the type of receptor involved in KATP channel inhibition, we tested the effect of Ang II in the presence of the AT1-selective antagonist losartan. Losartan binds to the AT1 receptor with affinities in the range 10-200 nM, shows > 30 000-fold selectivity for the AT1 over the AT2 receptor, and is without effect on other receptors at concentrations < 10 μM (Chiu et al. 1990; Timmermans et al. 1993). We used losartan at 1 μM, a concentration at which it is highly selective and has been shown to block both phosphoinositide hydrolysis and inhibition of adenylyl cyclase in response to Ang II (Timmermans et al. 1993). Figure 4 shows that the inhibitory effect of Ang II on KATP current was almost abolished (reduced to 2.3 ± 1.3 %) after 10 min pretreatment with losartan. As expected, subsequent inhibition of PKA with Rp-cAMPS (100 μM, Fig. 4A) or activation of PKC with DiC8 (10 μM, Fig. 4B) still caused equivalent reductions in KATP current to those seen in the absence of losartan (Fig. 4C).
Figure 4. Ang II is ineffective in the presence of losartan.
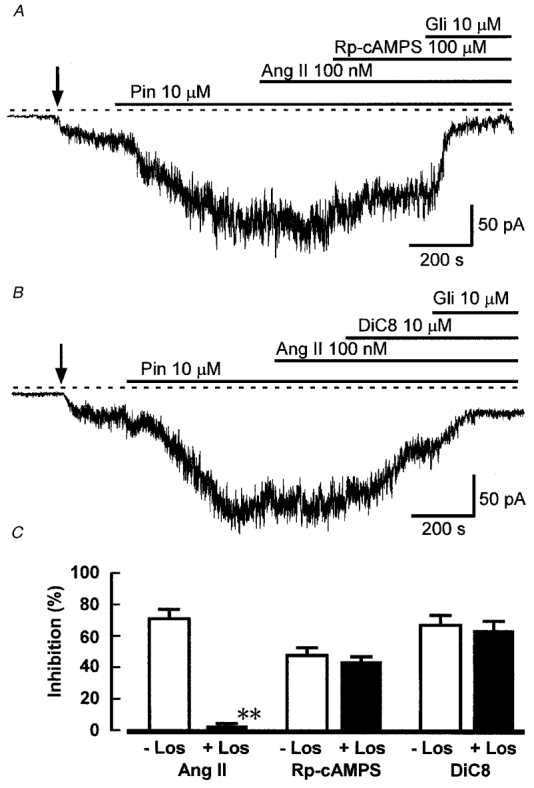
A and B, the traces show whole-cell currents recorded at -60 mV from cells pretreated with 1 μM losartan, and with 1 mM intracellular ATP. Ang II did not inhibit KATP current under these conditions, while subsequent application of Rp-cAMPS (A) or DiC8 (B) was still effective. C, mean inhibition (+s.e.m.) of KATP current by Ang II (100 nM), Rp-cAMPS (100 μM) and DiC8 (10 μM) in cells without (open bars) and with pretreatment with 1 μM losartan (Los) (filled bars). The cells were dialysed with solution containing 1 mM ATP. The number of cells in each group was: Ang II, 12, 12; Rp-cAMPS, 7, 6; DiC8, 6, 6. **P < 0.001 compared with control (without losartan), Student's t test.
The combination of PKC and PKA inhibitor peptides blocks inhibition by Ang II
We propose above that Ang II reduces KATP current both by activation of PKC and inhibition of PKA. To test this hypothesis, and to assess the relative contributions of PKC and PKA to the effect of Ang II, we used the selective and potent inhibitor peptides for PKA and PKC, PKA inhibitor peptide 5-24 (PKA-IP) and PKC inhibitor peptide 19-27 (PKC-IP). The selectivity of PKA-IP 5-24 has been reviewed by Kemp et al. (1988) and it is highly selective for PKA over other protein kinases at 5 μM. PKC-IP 19-27 is a selective pseudosubstrate inhibitor of PKC, giving 98 % inhibition at 100 μM (Eichholtz et al. 1993). As a positive control for PKA-IP in our cells, and to confirm the lack of effect of PKC-IP on PKA, we tested the effect of these inhibitors on the activation of KATP current by dibutyryl cAMP (db-cAMP). PKA-IP (5 μM) was applied intracellularly by inclusion in the patch pipette solution, and PKC-IP (100 μM) was applied extracellularly in its myristoylated cell-permeant form. Figure 5 shows that 0.5 mM db-cAMP caused activation of KATP current (Fig. 5A and D) and that, as expected, this effect was abolished in the presence of PKA-IP (Fig. 5B). However, in the presence of PKC-IP, the db-cAMP-activated current was not significantly different from its control value in the absence of the inhibitor peptide (Fig. 5C and D), confirming that PKC-IP did not affect PKA at this concentration.
Figure 5. Effects of inhibitor peptides PKA-IP and PKC-IP on KATP current activation by dibutyryl cAMP.
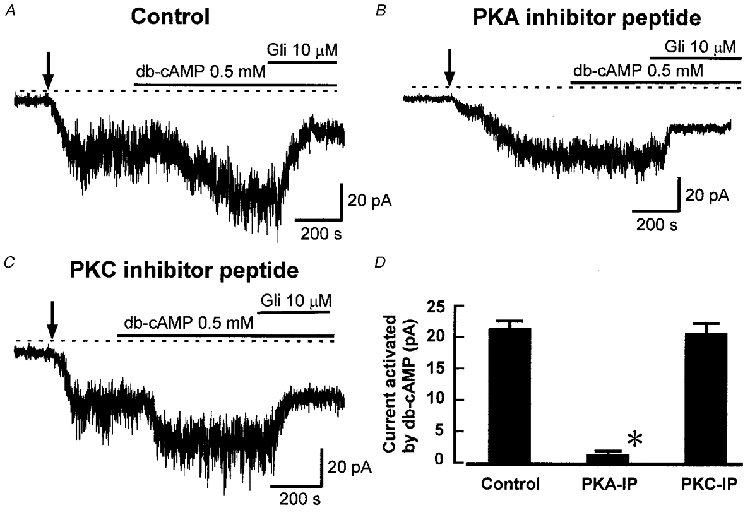
A, recording showing the activation of KATP current by db-cAMP (0.5 mM). B, recording showing that db-cAMP was ineffective in a cell dialysed with PKA-IP 5-24 (5 μM). C, recording showing that PKC-IP 19-27 (100 μM) did not block activation of KATP current by db-cAMP. D, mean activation (+s.e.m.) of KATP current by db-cAMP (0.5 mM) in control cells and in the presence of either PKA-IP 5-24 or PKC-IP 19-27 (n = 5 in each case). *P < 0.05 compared with control, Student's t test.
To examine the roles of PKA and PKC in the inhibition of KATP channels by Ang II, we pretreated smooth muscle cells with either PKC-IP (100 μM) or PKA-IP (5 μM), or both, applied as described above. In the absence of Ang II, PKC-IP did not affect the current induced by pinacidil (mean current -87 ± 9 pA, n = 6). However, as expected if there is steady-state activation of current by PKA, PKA-IP did reduce pinacidil activated current to a mean of -67 ± 3 pA (n = 6, P < 0.05 compared with control).
Figure 6 shows the effect of PKA and PKC inhibitor peptides on the action of Ang II. PKA-IP decreased the inhibition of KATP current by Ang II from its control value of 72.1 ± 7.4 % (Fig. 3) to 44.3 ± 6.4 % (n = 6, Fig. 6A and D). PKC-IP decreased the inhibition by Ang II to 46.1 ± 7.2 % (n = 6, Fig. 6B and D). Thus inhibition of either PKA or PKC significantly reduced the action of Ang II, but neither blocked its effect completely. However, when we applied both inhibitor peptides together, the effect of Ang II on KATP current was abolished, as shown in Fig. 6C and D.
Figure 6. Inhibitor peptides for PKC and PKA prevent inhibition by Ang II.
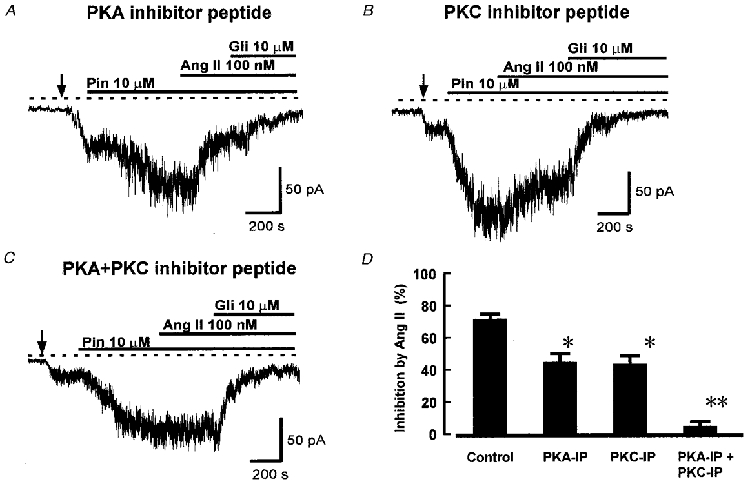
The traces show recordings of whole-cell current at -60 mV in cells with 1 mM intracellular ATP. A, effect of PKA inhibitor peptide 5-24. The cell was dialysed with pipette solution containing 5 μM PKA inhibitor for 10 min before the application of Ang II. Pinacidil (Pin), Ang II and glibenclamide (Gli) were added as indicated. B, effect of PKC inhibitor peptide 19-27. Myristoylated PKC-IP (100 μM) was applied in the extracellular solution for 10 min before the application of Ang II. C, effect of application of both PKA-IP and PKC-IP, applied as in A and B above. Ang II did not inhibit KATP current under these conditions. D, mean (+s.e.m.) inhibition of KATP current by 100 nM Ang II under control conditions and in the presence of PKA-IP (5 μM), PKC-IP (100 μM), and both peptides applied together (n = 12, 6, 6 and 7 cells, respectively). *P < 0.05, **P < 0.001 compared with control, ANOVA followed by Student-Newman-Keuls test.
Ang II acts through the PKCɛ isoform
The PKC isoform that is involved in KATP channel inhibition by Ang II, or other vasoconstrictors, has not been determined, and the PKC inhibitor peptide used above inhibits all PKC isoforms. Vascular smooth muscle has been shown to express the Ca2+-dependent PKC isoforms α and β and the Ca2+-independent isoforms ɛ and ζ (Dixon et al. 1994; Lee & Severson 1994; Clément-Chomienne et al. 1996). To investigate the involvement of different PKC isoforms in the action of Ang II, we tested the effects of isoform-selective PKC inhibitors. In all these experiments the intracellular solution contained PKA inhibitor peptide (5 μM) to block effects mediated through PKA. First, we tested the effect of Ang II on KATP current in the presence of Gö 6976, which selectively inhibits the Ca2+-dependent PKC isoforms α and β at nanomolar concentrations, while having no effect on PKCδ, ɛ and ζ at micromolar concentrations (Martiny-Baron et al. 1993). The action of Ang II was not affected by Gö 6976 (1 μM), since Ang II still inhibited KATP current in its presence (Fig. 7A), and the degree of this inhibition was not significantly different from that seen in the presence of PKA inhibitor peptide alone (Fig. 7C). This finding suggests that a Ca2+-dependent PKC isoform is not involved, and the ζ isoform also seems an unlikely candidate, since it is not activated by diacylglycerol (DAG) or its analogues (Nishizuka, 1992; Lee & Severson, 1994). We therefore tested the effect of the specific octapeptide inhibitor of PKCɛ translocation EAVSLKPT (40 μM, Johnson et al. 1996). This peptide, in combination with PKA-IP, abolished the effect of Ang II on KATP current (Fig. 7B and C), suggesting that the PKC component of the action of Ang II occurs through the ɛ isoform. As a negative control, we used the scrambled version of the PKCɛ translocation inhibitor peptide, LSETKPAV (40 μM), which was without effect on the inhibition of KATP current by Ang II (Fig. 7C).
Figure 7. The effects of isoform-selective protein kinase C inhibitors on the inhibition of KATP current by Ang II.
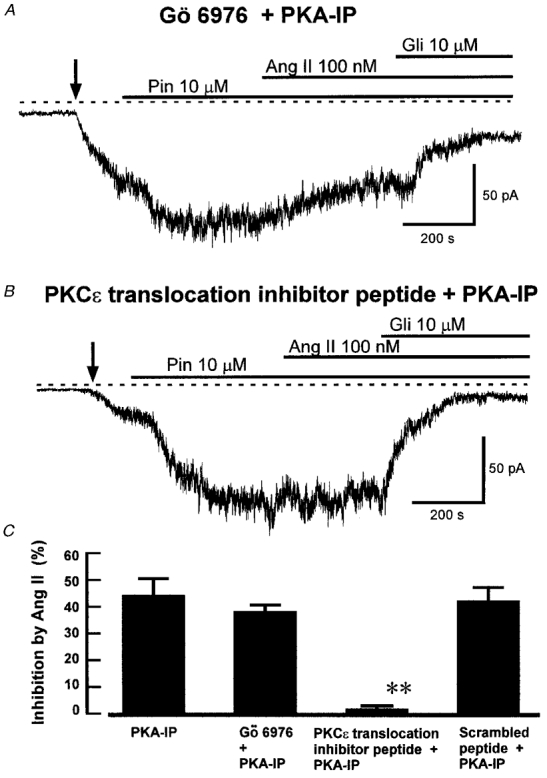
The traces show recordings of whole-cell current at -60 mV in cells with 1 mM ATP and 5 μM PKA-IP in the intracellular solution. A, effect of Gö 6976. The cell was pretreated for 10 min with 1 μM Gö 6976. The recording was made in the continued presence of Gö 6976, and pinacidil (Pin), Ang II and glibenclamide (Gli) were added as indicated. Ang II still inhibited KATP current under these conditions. B, effect of PKCɛ translocation inhibitor peptide. The cell was dialysed with 40 μM of the peptide. Pinacidil, Ang II and glibenclamide were added as indicated. Ang II did not inhibit KATP current under these conditions. C, mean (+s.e.m.) inhibition of KATP current by Ang II in PKA-IP alone (from Fig. 6D, for comparison), and in the presence of Gö 6976, PKCɛ translocation inhibitor peptide, and scrambled PKCɛ translocation inhibitor peptide, with PKA-IP in each case (n = 6, 7 and 5 cells, respectively). **P < 0.001 compared with PKA-IP alone, ANOVA followed by Student-Newman-Keuls test.
DISCUSSION
The results presented in this paper provide the first evidence for steady-state activation of arterial KATP channels through protein kinase A, and show that angiotensin II, at a physiological concentration, causes inhibition of these channels by acting at the AT1 receptor to cause both inhibition of PKA and activation of PKCɛ.
Steady-state KATP channel activation through PKA
A number of vasodilators, including adenosine and calcitonin gene-related peptide, can activate KATP channels in arterial smooth muscle through stimulation of cyclic AMP-dependent protein kinase (PKA) (see Quayle et al. 1997 for review). Our results suggest that PKA causes steady-state activation of KATP current in the absence of exogenous agonists for receptors coupled to adenylyl cyclase, providing that we raise intracellular ATP to 1.0 mM.
Under these conditions, inhibition of PKA with either Rp-cAMPS or PKA inhibitor peptide 5-24 caused a reduction in the steady-state KATP current activated by pinacidil. Furthermore, inhibition of protein phosphatases 1 and 2A with calyculin A led to a small but significant increase in current either in the absence or presence of pinacidil. These effects of Rp-cAMPS, PKA inhibitor peptide (Kubo et al. 1997), and calyculin A were not seen if intracellular [ATP] was only 0.1 mM. These results suggest that the lower ATP concentration may reduce the production of cAMP by adenylyl cyclase, while 1.0 mM ATP provides sufficient cAMP to lead to steady-state activation of PKA and so channel activity. Since previous whole-cell studies of arterial KATP channel activation by vasodilators and inhibition by vasoconstrictors, including our own, have used low (0.1 mM) intracellular ATP in order to reduce channel inhibition by ATP and so increase current size, the steady-state activation by PKA would have been obscured (Quayle et al. 1994; Bonev & Nelson, 1996; Kubo et al. 1997; Wellman et al. 1998). Our present findings suggest that PKA may make an important contribution to regulation of the channels. Furthermore, reduction of PKA activity by inhibition of the steady-state activity of adenylyl cyclase may provide another pathway for vasoconstrictor action on the channel.
Pathways for channel inhibition by angiotensin II
Ang II modulates several different ion channels in vascular smooth muscle cells, and various signalling pathways have been implicated in these effects. The inhibition of delayed rectifier K+ channels occurs through activation of PKC (Clément-Chomienne et al. 1996), but Ang II inhibits BKCa channels by a PKC-independent mechanism in cells cultured from pig coronary arteries (Minami et al. 1995). The mechanism of activation of non-selective cation channels in rabbit ear artery cells is unclear, but is likely to be independent of IP3 (Hughes & Bolton, 1995).
We have previously shown that Ang II inhibits arterial KATP channels through activation of PKC, but we found no evidence for an effect through inhibition of PKA (Kubo et al. 1997). Those experiments used 0.1 mM intracellular ATP, and we show in the present paper that raising ATP towards physiological levels (to 1.0 mM) reveals an important component of the action of Ang II that occurs through inhibition of PKA. Under these conditions inhibition of either PKC with PKC inhibitor peptide 19-27, or PKA with PKA inhibitor peptide 5-24, reduced the inhibitory effect of Ang II on KATP current, but did not abolish it. In order to completely block the effects of Ang II, it was necessary to apply both PKC and PKA inhibitor peptides simultaneously. These results suggest that Ang II inhibits KATP channels both by activation of PKC and by inhibition of PKA, as summarized in Fig. 8. Under our experimental conditions the PKC and PKA pathways appear to contribute about equally to channel inhibition by Ang II.
Figure 8. The proposed pathways for angiotensin II inhibition of KATP channels.
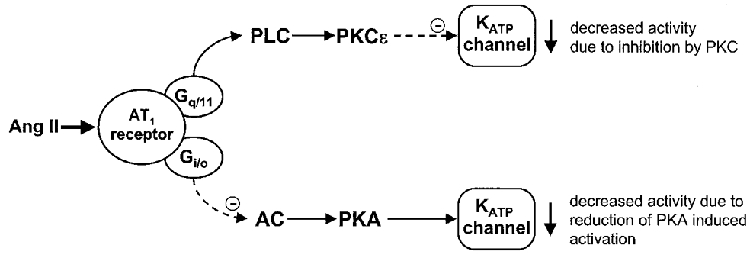
Binding of Ang II to an AT1 receptor is likely to act through Gq/11 to activate phospholipase C (PLC), and through Gi/o to inhibit adenylyl cyclase (AC). PLC will generate diacylglycerol, which activates PKCɛ to inhibit the KATP channel. Inhibition of adenylyl cyclase will decrease the level of cyclic AMP and so reduce activity of PKA, reducing steady-state phosphorylation of KATP by this kinase, and so reducing channel activity.
Several other vasoconstrictors have been reported to inhibit arterial KATP channels. Phenylephrine (α1-adrenoceptor), neuropeptide Y, serotonin (5HT2), and histamine (H1) all act through PKC (Bonev & Nelson, 1996), although the low ATP used (0.1 mM) could have obscured any action through PKA. Vasopressin, endothelin and α2D-adrenoceptors also cause channel inhibition (Miyoshi et al. 1992; Wakatsuki et al. 1992; Tateishi & Faber, 1995). The receptors for most of these agonists are coupled primarily to Gq/11, suggesting that their main effect may be through activation of phospholipase C and so protein kinase C. However, neuropeptide Y and α2D-adrenoceptors both couple to Gi/o, so that it is likely that inhibition of PKA may play a role in their actions, as we have shown for Ang II.
Ang II can bind to at least two high-affinity receptors, designated AT1 and AT2 receptors (Unger et al. 1996). The AT1 receptor is selectively blocked by losartan, and we found that losartan abolished the inhibitory action of Ang II on KATP current. Since it appears that the inhibition of KATP current involves both activation of PKC and inhibition of PKA, the involvement of an AT1 receptor is in agreement with the known positive coupling of this receptor to phospholipase C (Griendling et al. 1986; Nishizuka, 1992) and its negative coupling to adenylyl cyclase (Anand-Srivastava, 1983; Unger et al. 1996).
We have also sought to establish the isoform of PKC that is involved in the PKC component of the effect of Ang II. Activation of PKC is generally associated with translocation to particular subcellular sites, and it is suggested that translocation may involve binding of PKC to anchoring proteins specific to particular PKC isoforms (Mochly-Rosen et al. 1991; Mochly-Rosen, 1995). These anchoring proteins are known as RACKs (receptors for activated C-kinase) and the functional specificity of each PKC isoform may be determined by the localization of the RACK specific for that isoform (Mochly-Rosen, 1995; Nadimet al. 1995). Several isoforms of PKC are expressed in vascular smooth muscle, including α, β, ɛ and ζ (Dixon et al. 1994; Lee & Severson, 1994). Of these the Ca2+-independent isoform PKCɛ seemed a likely candidate for involvement in KATP channel inhibition by Ang II (Kubo et al. 1997), since it has been shown to be translocated in response to Ang II in pulmonary arterial smooth muscle (Minamiet al. 1995). Our present results provide strong evidence that Ang II acts through PKCɛ to inhibit KATP current, since the effect was abolished by an octapeptide, EAVSLKPT, which inhibits the translocation of PKCɛ (Johnson et al. 1996), while the selective inhibitor of Ca2+-dependent PKC isoforms Gö 6976 was without effect.
The mechanisms by which PKA activates and PKCɛ inhibits arterial KATP channels remain to be determined. The most economical hypothesis is that the kinases phosphorylate the channel proteins directly to alter their open probability, and both the Kir6 family and sulphonylurea receptor subunits that form KATP channels have consensus sequences for phosphorylation by PKA and PKC (Aguilar-Bryan et al. 1998). The subunit combination that forms KATP channels of smooth muscle is not definitively established, but Béguin et al. (1999) have recently shown that recombinant human β-cell KATP channels (Kir6.2/SUR1) can be phosphorylated by PKA on both subunits, and have identified the sites at which this occurs. Furthermore, Kir6.2 can be phosphorylated at its PKA site in response to Gs-coupled receptor stimulation, and phosphorylation increases channel activity. It seems likely that PKA activates KATP in arterial smooth muscle in an essentially similar manner.
KATP channel inhibition and vasoconstriction
Vasoconstrictors activate Ca2+ and other cation channels in smooth muscle that cause depolarization and Ca2+ entry. Concurrent inhibition of K+ channels should enhance depolarization and so contribute to vasoconstriction. There is evidence that KATP channels are active in the steady state and contribute to resting blood flow in a number of vascular beds, including the mesenteric and coronary circulations (e.g. Gardiner et al. 1996; see Quayle et al. 1997, for review). Thus KATP channel inhibition would be expected to contribute to the effects of vasoconstrictors in reducing blood flow. So far there have been few functional studies of the role of KATP channels in vasoconstriction, though Tateishi & Faber (1995) have provided evidence that vasoconstrictor activation of α2D-adrenoceptors can inhibit KATP channels in intact skeletal muscle arterioles. There is a clear need for future functional studies of the role of KATP channel inhibition in vasoconstriction by Ang II and other endogenous vasoconstrictors.
Acknowledgments
We thank Diane Everitt for skilled technical assistance and The Wellcome Trust and the British Heart Foundation for support.
References
- Aguilar-Bryan L, Clement JP, Gonzalez G, Kunjilwar K, Babenko A, Bryan J. Towards understanding the assembly and structure of KATP channels. Physiological Reviews. 1998;78:227–245. doi: 10.1152/physrev.1998.78.1.227. [DOI] [PubMed] [Google Scholar]
- Anand-Srivastava MB. Angiotensin II receptors negatively coupled to adenylate cyclase in rat aorta. Biochemical and Biophysical Research Communications. 1983;117:420–427. doi: 10.1016/0006-291x(83)91217-2. [DOI] [PubMed] [Google Scholar]
- Beech DJ. Actions of neurotransmitters and other messengers on Ca2+ channels and K+ channels in smooth muscle cells. Pharmacology and Therapeutics. 1997;73:91–119. doi: 10.1016/s0163-7258(97)87271-3. [DOI] [PubMed] [Google Scholar]
- Béguin P, Nagashima K, Nishimura M, Gonoi T, Seino S. PKA-mediated phosphorylation of the human KATP channel: separate roles of Kir6.2 and SUR1 subunit phosphorylation. EMBO Journal. 1999;18:4722–4732. doi: 10.1093/emboj/18.17.4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonev AD, Nelson MT. Vasoconstrictors inhibit ATP-sensitive K+ channels in arterial smooth muscle through protein kinase C. Journal of General Physiology. 1996;108:315–323. doi: 10.1085/jgp.108.4.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu AT, McCall DE, Ardecky RJ, Duncia JV, Nguyen TT, Timmermans PBMWM. Angiotensin II receptor subtypes and their selective nonpeptide ligands. Receptor. 1990;1:33–40. [PubMed] [Google Scholar]
- Clément-Chomienne O, Walsh MP, Cole WC. Angiotensin II activation of protein kinase C decreases delayed rectifier K+ current in rabbit vascular myocytes. The Journal of Physiology. 1996;495:689–700. doi: 10.1113/jphysiol.1996.sp021626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon BS, Sharma RV, Dickerson T, Fortune J. Bradykinin and angiotensin II: Activation of protein kinase C in arterial smooth muscle. American Journal of Physiology. 1994;266:C1406–1420. doi: 10.1152/ajpcell.1994.266.5.C1406. [DOI] [PubMed] [Google Scholar]
- Eichholtz T, DeBont DBA, DeWidt J, Liskamp RMJ, Ploegh HL. A myristoylated pseudosubstrate peptide, a novel protein-kinase-C inhibitor. Journal of Biological Chemistry. 1993;268:1982–1986. [PubMed] [Google Scholar]
- Gardiner SM, Kemp PA, March JE, Fallgren B, Bennett T. Effects of glibenclamide on the regional haemodynamic actions of α-trinositol and its influence on responses to vasodilators in conscious rats. British Journal of Pharmacology. 1996;117:507–515. doi: 10.1111/j.1476-5381.1996.tb15219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griendling KK, Rittenhouse SE, Brock TA, Ekstein LS, Gimbrone MA, Jr, Alexander RH. Sustained diacylglycerol formation from inositol phospholipids in angiotensin II-stimulated vascular smooth muscle cells. Journal of Biological Chemistry. 1986;261:5901–5906. [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Herzig S, Neumann J. Effects of serine/threonine protein phosphatases on ion channels in excitable membranes. Physiological Reviews. 2000;80:173–210. doi: 10.1152/physrev.2000.80.1.173. [DOI] [PubMed] [Google Scholar]
- Hughes AD, Bolton TB. Action of angiotensin II, 5-hydroxytryptamine and adenosine triphosphate on ionic currents in single ear artery cells of the rabbit. British Journal of Pharmacology. 1995;116:2148–2154. doi: 10.1111/j.1476-5381.1995.tb16424.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JA, Gray MO, Chen CH, Mochly-Rosen D. A protein kinase C translocation inhibitor as an isozyme-selective antagonist of cardiac function. Journal of Biological Chemistry. 1996;271:24962–24966. doi: 10.1074/jbc.271.40.24962. [DOI] [PubMed] [Google Scholar]
- Kemp BE, Cheng H-C, Walsh DA. Peptide inhibitors of cAMP-dependent protein kinase. Methods in Enzymology. 1988;159:173–183. doi: 10.1016/0076-6879(88)59018-3. [DOI] [PubMed] [Google Scholar]
- Kleppisch T, Nelson MT. Adenosine activates ATP-sensitive potassium channels in arterial myocytes via A2 receptors and cAMP-dependent protein kinase. Proceedings of the National Academy of Sciences of the USA. 1996;92:12441–12445. doi: 10.1073/pnas.92.26.12441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo M, Quayle JM, Standen NB. Angiotensin II inhibition of ATP-sensitive K+ currents in rat arterial smooth muscle cells through protein kinase C. The Journal of Physiology. 1997;503:480–496. doi: 10.1111/j.1469-7793.1997.489bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee ML, Severson DL. Signal transduction in vascular smooth muscle: diacylglycerol second messengers and PKC action. American Journal of Physiology. 1994;267:C659–678. doi: 10.1152/ajpcell.1994.267.3.C659. [DOI] [PubMed] [Google Scholar]
- Martiny-Baron G, Kazanietz MG, Mischak H, Blumberg PM, Kochs G, Hug H, Marme D, Schatchtele C. Selective inhibition of protein-kinase-C isozymes by the indolocarbazole Gö 6976. Journal of Biological Chemistry. 1993;268:9194–9197. [PubMed] [Google Scholar]
- Minami K, Hirata Y, Tokumura A, Nakaya Y, Fukuzawa K. Protein kinase C-independent inhibition of the Ca2+-activated K+ channel by angiotensin II and endothelin-1. Biochemical Pharmacology. 1995;49:1051–1056. doi: 10.1016/0006-2952(95)98500-9. [DOI] [PubMed] [Google Scholar]
- Miyoshi Y, Nakaya Y. Angiotensin II blocks ATP-sensitive K+ channels in porcine coronary artery smooth muscle cells. Biochemical and Biophysical Research Communications. 1991;181:700–706. doi: 10.1016/0006-291x(91)91247-a. [DOI] [PubMed] [Google Scholar]
- Miyoshi Y, Nakaya Y, Wakatsuki T, Nakaya S, Fujino K, Saito K, Inoue I. Endothelin blocks ATP-sensitive K+-channels and depolarizes smooth muscle cells of porcine coronary artery. Circulation Research. 1992;70:612–616. doi: 10.1161/01.res.70.3.612. [DOI] [PubMed] [Google Scholar]
- Mochly-Rosen D. Localization of protein kinases by anchoring proteins: a theme in signal transduction. Science. 1995;268:247–251. doi: 10.1126/science.7716516. [DOI] [PubMed] [Google Scholar]
- Mochly-Rosen D, Khaner H, Lopez J. Identification of intracellular receptor proteins for activated protein kinase C. Proceedings of the National Academy of Sciences of the USA. 1991;88:3997–4000. doi: 10.1073/pnas.88.9.3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulvany MJ, Nilsson H, Flatman JA. Role of membrane potential in the response of rat small mesenteric arteries to exogenous noradrenaline stimulation. The Journal of Physiology. 1982;322:363–373. doi: 10.1113/jphysiol.1982.sp014418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadim HS, Damron DS, Murray PA. Differential subcellular translocation of protein kinase C isoforms in cultured canine pulmonary artery smooth muscle cells by angiotensin II. Circulation. 1995;92:1701. [Google Scholar]
- Nelson MT, Patlak JB, Worley JF, Standen NB. Calcium channels, potassium channels, and voltage-dependence of arterial smooth muscle tone. American Journal of Physiology. 1990;259:C3–18. doi: 10.1152/ajpcell.1990.259.1.C3. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Standen NB, Brayden JE, Worley JF. Noradrenaline contracts arteries by activating voltage-dependent calcium channels. Nature. 1988;336:382–385. doi: 10.1038/336382a0. [DOI] [PubMed] [Google Scholar]
- Nishizuka Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science. 1992;258:607–614. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- Ohya Y, Sperelakis N. Involvement of a GTP-binding protein in stimulating action of angiotensin II on calcium channels in vascular smooth muscle. Circulation Research. 1991;68:763–771. doi: 10.1161/01.res.68.3.763. [DOI] [PubMed] [Google Scholar]
- Post JM, Jones AW. Stimulation of arterial 42K efflux by ATP depletion and cromakalim is antagonized by glyburide. American Journal of Physiology. 1991;260:H848–854. doi: 10.1152/ajpheart.1991.260.3.H848. [DOI] [PubMed] [Google Scholar]
- Quayle JM, Bonev AD, Brayden JE, Nelson MT. Calcitonin gene-related peptide-activated ATP-sensitive K+ currents in rabbit arterial smooth muscle via protein kinase A. The Journal of Physiology. 1994;475:9–13. doi: 10.1113/jphysiol.1994.sp020045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quayle JM, Nelson MT, Standen NB. ATP-sensitive and inwardly rectifying potassium channels in smooth muscle. Physiological Reviews. 1997;77:1165–1232. doi: 10.1152/physrev.1997.77.4.1165. [DOI] [PubMed] [Google Scholar]
- Ron D, Luo J, Mochly-Rosen D. C2 region-derived peptides inhibit translocation and function of β protein kinase C in vivo. Journal of Biological Chemistry. 1995;269:21395–21398. doi: 10.1074/jbc.270.41.24180. [DOI] [PubMed] [Google Scholar]
- Somlyo AP. Excitation-contraction coupling and ultrastructure of smooth muscle. Circulation Research. 1985;57:497–507. doi: 10.1161/01.res.57.4.497. [DOI] [PubMed] [Google Scholar]
- Tateishi J, Faber JE. ATP-sensitive K+ channels mediate α-adrenergic receptor contraction of arteriolar smooth muscle and reversal of contraction by hypoxia. Circulation Research. 1995;76:53–63. doi: 10.1161/01.res.76.1.53. [DOI] [PubMed] [Google Scholar]
- Timmermans PBMWM, Wong PC, Chiu AT, Herblin WF, Benfield P, Carini DJ, Lee RJ, Wexler RR, Saye JAM, Smith RD. Angiotensin II receptors and angiotensin II antagonists. Pharmacological Reviews. 1993;45:205–251. [PubMed] [Google Scholar]
- Toro L, Amador M, Stefani E. ANG II inhibits calcium-activated potassium channels from coronary smooth muscle in lipid bilayers. American Journal of Physiology. 1990;258:H912–915. doi: 10.1152/ajpheart.1990.258.3.H912. [DOI] [PubMed] [Google Scholar]
- Unger T, Chung O, Csikos T, Culman J, Gallinat S, Gohlke P, Hohle S, Meffert S, Stoll M, Stroth U, Zhu YZ. Angiotensin receptors. Journal of Hypertension. 1996;14(suppl. 5):S95–103. [PubMed] [Google Scholar]
- Wakatsuki T, Nakaya Y, Inoue I. Vasopressin modulates K+-channel activities of cultured smooth muscle cells from porcine coronary artery. American Journal of Physiology. 1992;263:H491–496. doi: 10.1152/ajpheart.1992.263.2.H491. [DOI] [PubMed] [Google Scholar]
- Wellman GC, Quayle JM, Standen NB. ATP-sensitive K+ channel activation by calcitonin gene-related peptide and protein kinase A in pig coronary arterial smooth muscle. The Journal of Physiology. 1998;507:117–129. doi: 10.1111/j.1469-7793.1998.117bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]