

Abstract
Betamethasone has been used extensively to accelerate fetal lung maturation, yet little is known of its effects on neuronal morphogenesis in the developing fetus. Microtubule-associated proteins (MAPs) are a diverse family of cytoskeletal proteins that are important for brain development and the maintenance of neuroarchitecture.
Vehicle (n = 7) or betamethasone (10 μg h−1, n = 7) was infused I.V. to fetal sheep over 48 h beginning at 0.87 of gestation (128 days of gestation), producing fetal plasma betamethasone concentrations resembling those to which the human fetus is exposed during antenatal glucocorticoid therapy.
Paraffin sections of the left hemisphere were stained with monoclonal antibodies against MAP1B and the MAP2 isoforms MAP2a,b,c and MAP2a,b. The level of the juvenile isoform MAP2c was determined by comparison of the two MAP2 immunostainings.
We were able to detect MAP1B and MAP2 immunoreactivity (IR) in the fetal sheep brain. MAP2c was the major MAP2, constituting 90.2 % of the total MAP2. Betamethasone exposure diminished MAP1B IR in the frontal cortex and caudate putamen (P < 0.05) but not in the hippocampus. A decrease of MAP2 IR was found in the frontal cortex, hippocampus and caudate putamen (P < 0.05). Loss of MAP2 IR was mainly due to the loss of MAP2c IR. Haematoxylin–eosin staining did not demonstrate irreversible neuronal damage.
Regional cerebral blood flow determined using coloured microspheres was significantly decreased by 28 % in the frontal cortex and by 36 % in the caudate putamen but not in the hippocampus 24 h after the onset of betamethasone exposure (P < 0.05). The loss of MAP1B and MAP2a,b,c IR showed a significant correlation to the cerebral blood flow decrease only in the frontal cortex (P < 0.05). These data suggest that mechanisms other than metabolic insufficiency caused by the decreased cerebral blood flow may contribute to the loss of MAPs.
The results suggest that clinical doses of betamethasone may have acute effects on cytoskeletal proteins in the fetal brain.
Cortisol is essential for normal maturation of the central nervous system (Meyer, 1985; De Kloet et al. 1988). However, increased exposure to glucocorticoids both in vitro and in vivo induces acute neurotoxic effects (McEwen et al. 1995) and apoptosis (Hassan et al. 1996). Glucocorticoids are known to increase the susceptibility of the hippocampus to metabolic insults (Sapolsky, 1994). Neurotoxic effects are induced by activation of type II glucocorticoid receptors in rats (Hassan et al. 1996). The type II receptor-specific synthetic glucocorticoids betamethasone and dexamethasone have both been used extensively in perinatal medicine to accelerate fetal lung maturation in fetuses of pregnant women in premature labour (Ballard & Ballard, 1995).
Unfortunately, there is little information on the effects of glucocorticoids on neuronal morphogenesis in the developing fetus. It has been shown that antenatal dexamethasone treatment causes degeneration and depletion of the hippocampal pyramidal and dentate granular neurons associated with dendrite degeneration in the CA3 region in non-human primates (Uno et al. 1994). Decreased neurogenesis (De Kloet et al. 1988), gliagenesis (Howard & Benjamins, 1975) and myelinisation (Howard & Benjamins, 1975; De Kloet et al. 1988) have been demonstrated in the developing rat brain.
Alterations of cytoskeletal proteins such as the microtubule-associated proteins (MAPs) are known to occur as early intracellular structural events in response to traumatic (Folkerts et al. 1998), seizure-related (Ballough et al. 1995) or ischaemic brain injuries in adult (Matesic & Lin, 1994; Schwab et al. 1998) and neonatal rodent brains (Malinak & Silverstein, 1996; Ota et al. 1997) as well as to exposure of neurotoxic substances (Nassogne et al. 1995; Noraberg & Zimmer, 1998; Bywood & Johnson, 2000). MAPs are a diverse family of cytoskeletal proteins apparently occurring in all vertebrates including man (Viereck et al. 1988; Arnold & Trojanowski, 1996). They perform important functions related to normal neuronal integrity, through the maintenance of nerve cell shape and intracellular transport (Bershadsky & Vasiliev, 1989), and to the regulation of neuronal morphogenesis (Tucker, 1990; Johnson & Jope, 1992). MAP1B and MAP2 are present throughout the developing nervous system (Tucker, 1990; Johnson & Jope, 1992). They are found in human embryos from 9 weeks of gestation (Arnold & Trojanowski, 1996). MAP2 exists in low molecular weight (LMW) and high molecular weight (HMW) isoforms that occur specifically in neurons (Tucker, 1990). The LMW isoform MAP2c as well as MAP1B appear in early embryogenesis during neuronal differentiation in rats and continue to be expressed at high levels until the end of axon and dendrite outgrowth (Riederer & Matus, 1985; Przyborski & Cambray Deakin, 1995). Then MAP2c is replaced by the HMW isoform MAP2a (Riederer & Matus, 1985; Tucker et al. 1988b). MAP2b is the second HMW isoform of MAP2. It first appears at a similar time to MAP2c during early development in rats but is present throughout life (Burgoyne & Cumming, 1984; Riederer & Matus, 1985). The temporary correlation of the disappearance of MAP1B and replacement of MAP2c by the HMW isoform MAP2a with synaptogenesis led to the assumption that the juvenile forms MAP1B and MAP2c are involved in the plasticity during the process of neuronal outgrowth (Tucker et al. 1988b;Johnson & Jope, 1992). The HMW isoforms MAP2a and MAP2b contribute to the assembly and stability of microtubules (Matus, 1990; Tucker, 1990). MAP2a and MAP2b are selectively associated to neuronal cell bodies and dendrites (Tucker et al. 1988b) whereas MAP2c is also present in axons (Meichsner et al. 1993; Albala et al. 1995). MAP1B occurs in dendrites and axons (Tucker et al. 1988b).
The aim of the present study was to investigate the effects of betamethasone on the neuronal cytoskeleton in fetal sheep brain. The dose of betamethasone used was chosen to produce plasma concentrations that are similar to those to which the human preterm fetus is exposed during antenatal administration of betamethasone to women in premature labour (Derks et al. 1997). We sought to evaluate effects that indicate an acceleration of brain maturation and induction of neuronal dysfunction. Loss of MAPs is an early gross morphological indicator of neurodegeneration in rats (Bywood & Johnson, 2000). In order to determine whether any loss of MAP immunoreactivity (IR) is associated with neuronal death we determined neuronal viability with haematoxylin- eosin staining. To determine whether a change of MAP IR is the result of betamethasone-induced cerebral hypoperfusion, shown in a previous study in fetal sheep (Schwab et al. 2000a), rather than a direct neurotoxic effect we correlated changes of MAP1B and MAP2a,b,c IR to the cerebral blood flow (CBF) values from the same brain regions.
METHODS
Surgical procedure
All procedures were approved by the Cornell University Animal Use and Care Committee and were performed in facilities approved by the American Association for the Accreditation of Laboratory Animal Care and the animal welfare commission of the Thuringian state government for animal research. Surgery was performed on 14 Rambouillet-Colombia ewes bred on a single occasion and of known gestational age using techniques previously described in detail (Nathanielsz et al. 1980). Briefly, ewes were instrumented at 120 ± 1 dGA (days of gestation) with catheters inserted into the carotid artery to obtain blood samples and into the jugular vein for post-operative antibiotic administration. The fetuses were instrumented with polyvinyl catheters (Tygon, Norton Performance Plastics; 0.1 mm i.d., 0.18 mm o.d.) inserted into the axillary artery, and the jugular and pedal vein as well as in the amniotic cavity for control of physiological variables and CBF measurement using coloured microspheres (Schwab et al. 2000a). After surgery the ewes were returned to a metabolism cage and allowed free access to food and water. All ewes received a daily dose of 1 g ampicillin (AMP-Equine; SmithKline Beecham, West Chester, PA, USA) i.v. and 1 g ampicillin into the amniotic cavity for 5 days. Phenylbutazone (0.5 g twice daily) was administered orally (Equiphene paste, Luitpold Pharmaceuticals, Shirley, NY, USA) for 3 days to provide post-operative analgesia. All catheters were maintained patent via a continuous infusion of heparin at 12.5 i.u. ml−1 in 0.9 % NaCl solution delivered at 0.5 ml h−1.
Experimental protocol
Daily fetal and maternal arterial blood samples were taken at 09.00 h throughout the study for measurement of blood gases and pH using a blood gas analyser (ABL600, Radiometer, Copenhagen, Denmark; measurements corrected to 39°C). The haemoglobin concentration and oxygen saturation were measured photometrically (Hemoximeter OSM2, Radiometer). At 128 dGA (0.87 of gestation), vehicle (isotonic saline, n = 7) or betamethasone infusion (n = 7, Celestone Soluspan, Schering, Kenilworth, NJ, USA) at a rate of 10 μg h−1 was started into the fetal jugular vein and maintained over the next 48 h. This dose produces a fetal plasma betamethasone concentration of about 10 ng ml−1 (Derks et al. 1997). As described in detail elsewhere (Schwab et al. 2000a), coloured microspheres were used to measure regional CBF during quiet sleep in five of the seven fetuses reported here. CBF measurements were made before and 24 and 48 h after the onset of vehicle or betamethasone infusion. The animals used for CBF data comprised a part of the group of animals used in another study that focused solely on CBF (Schwab et al. 2000a).
At the end of 48 h of infusion, ewes were anaesthetised with 4 % halothane after an i.v. injection of 0.3-0.4 g ketamine and fetuses were delivered by Caesarean section. Fetuses were killed by exsanguination while under halothane anaesthesia. Fetal brains were perfused via the left carotid artery with heparinised physiological saline followed by 500 ml phosphate-buffered solution containing 3 % paraformaldehyde and 0.5 % glutaraldehyde for 15 min. Ewes were killed by i.v. injection of pentobarbital sodium solution at a dose of 1 ml kg−1 (Fatal-Plus, Vortech Pharmaceuticals, Dearborn, MI, USA).
Histological processing
Whole fetal brains were removed and postfixed overnight in 3 % phosphate-buffered paraformaldehyde and 0.5 % glutaraldehyde. Hemispheres were divided and one randomly chosen hemisphere was used for microsphere processing. The other hemisphere was put into 1.5 % paraformaldehyde and 0.25 % glutaraldehyde for prolonged postfixation for up to 1 week. Hemispheres were cut into slices of about 7 mm thickness and embedded in paraffin. Adjacent 6 μm frontal slices were stained with monoclonal mouse antibodies against MAP1B, MAP2a,b,c and MAP2a,b (Sigma). In preliminary experiments we found immunopositive staining with the antibodies used demonstrating MAP1B-, MAP2a,b,c- and MAP2a,b-like IR in brain tissue of fetal sheep at 130 dGA. Immunolabelling was performed using the ABC technique (Vectastain Elite kit, Vector Labs, Burlingame, CA, USA). Tissue sections were pre-incubated with 1.5 % normal horse serum (Vector), followed by the primary antibody (1:500, overnight at 4°C). Slices were incubated with a biotinylated anti-mouse IgG secondary antibody (Vector, 1:200) for 1 h, followed by a preformed avidin-horseradish peroxidase complex (Vectastain Elite ABC-Reagent, Vector). Immunostaining was developed using diaminobenzidine (Sigma). To block endogenous peroxidase activity, slices were treated with 0.3 % hydrogen peroxide. For negative controls the primary antibody was replaced by normal mouse serum. Additionally, adjacent tissue slices were stained with haematoxylin- eosin to identify irreversibly damaged neurons.
Quantitative image analysis
Tissue areas of positive immunoreactivity in regions of interest were estimated within the frontal neocortex (cortical layer III), the hippocampus (CA1 region) and the dorsolateral caudate putamen by an individual (I. A.-S.) who was blinded to the experimental protocol. Slices of x190 magnification were digitised using a 3 CCD colour video camera (Sony, MC3215). MAP1B, MAP2a,b and MAP2a,b,c immunopositive areas were quantified using an image analysis program (Scion Image 1.62, NIH, USA). Differences in the areas stained by both MAP2 antibodies in adjacent slices correspond to the area immunoreactive for MAP2c (Tucker et al. 1988a).
Statistical analysis
Statistical comparison of the physiological variables and morphometric data between the vehicle- and betamethasone-treated groups was performed by Mann and Whitney's U test. Differences in CBF between baseline, 24 h and 48 h of infusion were tested for significance by Wilcoxon's sign rank test. Correlation between CBF data and the loss of MAP IR was computed by Pearson's test. Results are presented throughout as means ±s.e.m. Significance was assumed at a P value of < 0.05.
RESULTS
Fetal blood gases were within the physiological range during baseline and vehicle or betamethasone infusion. Betamethasone infusion had no effect on fetal blood gas values (Table 1).
Table 1.
Physiological parameters before and after vehicle or betamethasone infusion
| pH | PCO2 (mmHg) | PO2 (mmHg) | O2 (%) | Hb (g dl−1) | |
|---|---|---|---|---|---|
| Vehicle | |||||
| Baseline | 7.38 ± 0.02 | 48.73 ± 5.13 | 22.82 ± 3.01 | 59.37 ± 7.68 | 11.10 ± 2.58 |
| 48h infusion | 7.36 ± 0.04 | 50.25 ± 5.60 | 23.73 ± 4.19 | 56.20 ± 5.55 | 11.20 ± 1.04 |
| Betamethasone | |||||
| Baseline | 7.35 ± 0.02 | 50.33 ± 2.08 | 23.13 ± 3.56 | 62.57 ± 7.45 | 10.35 ± 1.22 |
| 48h infusion | 7.37 ± 0.03 | 49.25 ± 5.86 | 23.18 ± 3.84 | 60.43 ± 9.59 | 11.42 ± 1.98 |
Values are means ±s.e.m.; vehicle infusion, n = 7; betamethasone infusion, n = 7.
Brain tissue of fetal sheep at 130 dGA showed immunopositive staining with all of the monoclonal MAP1B, MAP2a,b,c and MAP2a,b antibodies. MAP1B and MAP2a,b,c IR was localised to neuronal processes and cell bodies (Figs 1 and 2). MAP2c was by far the most prominent MAP2, amounting to 90.2 % of all MAP2 isoforms investigated (Fig. 3 and 4).
Figure 1. Photomicrographs of MAP1B immunostaining (brown precipitate) of the frontal cerebral cortex (top), caudate putamen (middle) and hippocampus (bottom), counterstained with haematoxylin (blue), of a vehicle- (A, C, E) and a betamethasone-treated fetus (B, D, F) at 130 dGA.
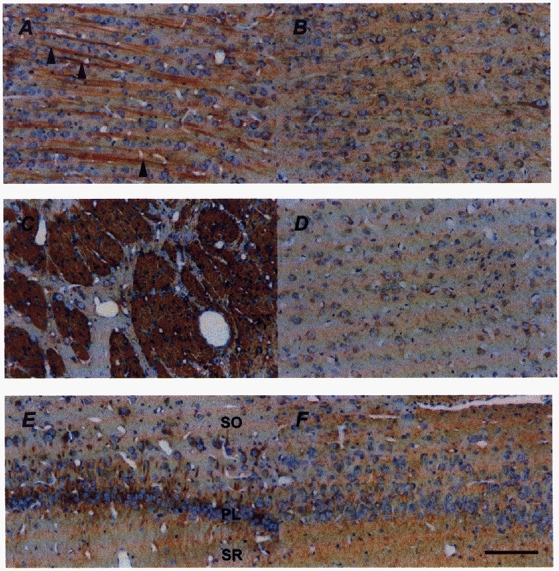
MAP1B IR is evident in cell bodies and processes of neurons and the neuropil. In the middle cortical layers (A) apical cortical dendrites (arrowheads) show their typical parallel orientation. The most dense MAP1B immunostaining was found in the caudate putamen (C). Loss of MAP1B IR in the frontal cortex (B) and caudate putamen (D) of the betamethasone-treated fetuses is clearly visible. Note the loss of clear demarcation of the neuronal processes in the stratum radiatum (SR) and stratum oriens (SO) of the hippocampus after betamethasone treatment (F). PL, pyramidal cell layer of the CA1 region. Nuclei counterstained with haematoxylin do not show irreversible neuronal damage. Scale bar, 100 μm.
Figure 2. Photomicrographs of MAP2a,b,c immunostaining (brown precipitate) of the frontal cerebral cortex (top), caudate putamen (middle) and hippocampus (bottom), counterstained with haematoxylin (blue), of a vehicle- (A, C, E) and a betamethasone-treated fetus (B, D, F) at 130 dGA.
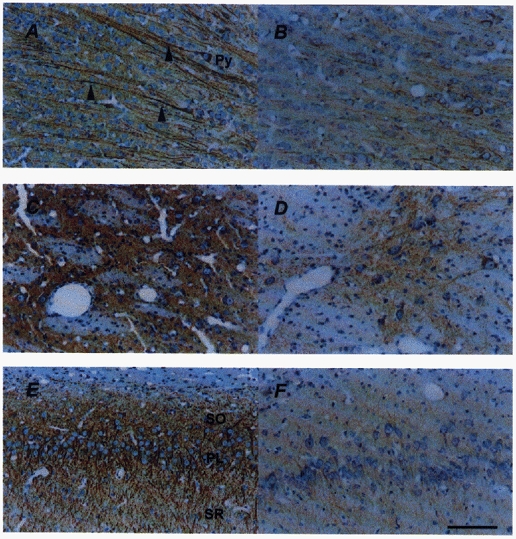
MAP2a,b,c IR is evident in neuronal perikarya and processes. In the cerebral cortex (A), the anatomical orientation of the apical dendrites (arrowheads) of pyramidal cells (Py) is clearly visible. In the caudate putamen (C), neuronal populations are recognised by MAP2a,b,c antibody in contrast to white fibre bundles. In the CA1 region of the hippocampus (E) the pyramidal cell layer (PL), stratum oriens (SO) and stratum radiatum (SR) are densely labelled by MAP2a,b,c. Loss of MAP2 IR was found in the frontal cortex (B), caudate putamen (D) and hippocampus (F) 48 h after the onset of betamethasone treatment. Nuclei counterstained with haematoxylin do not show irreversible neuronal damage. Scale bar, 100 μm.
Figure 3. Photomicrographs of MAP2a,b,c (A) and MAP2a,b (B) immunostaining (brown precipitate) in the frontal cerebral cortex counterstained with haematoxylin (blue).
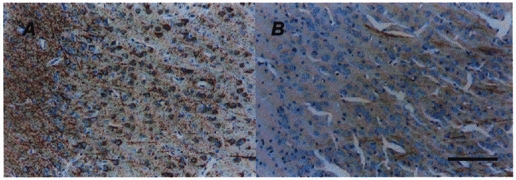
Note the small amount of immunoreactivity of MAP2a,b (B) in comparison to that of MAP2a,b,c (A). Scale bar, 100 μm.
Figure 4. Comparison of the acute effects of antenatal betamethasone (BETA) treatment on MAP2a,b,c and MAP2a,b IR in fetal sheep at 130 dGA in the frontal cerebral cortex.
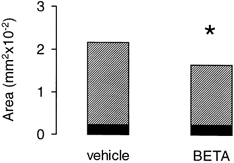
Filled bars, MAP2a,b; hatched bars, MAP2a,b,c; vehicle-treated fetuses, n = 7; betamethasone (BETA)-treated fetuses, n = 7; *P < 0.05. Difference between MAP2a,b,c and MAP2a,b IR gives the amount of MAP2c. Note the loss of MAP2a,b,c IR (P < 0.05) in contrast to MAP2a,b IR after betamethasone treatment.
Betamethasone-treated fetuses showed a 41.3 % loss of MAP1B IR in the frontal cortex and a 45.4 % loss in the caudate putamen (Figs 1 and 5, P < 0.05). In contrast, no change of MAP1B IR was noted in the hippocampus in comparison to the vehicle-treated fetuses (Fig. 5). However, neuronal processes lost their good demarcation in favour of a more diffuse immunolabelling of the neuropil (Fig. 1E and F). It seems the diffuse immunolabelling of the neuropil characterises a major step towards the loss of MAP since it was also found in the other brain regions showing a loss of MAP1B or MAP2 IR (Figs 1 and 2).
Figure 5. Acute effects of antenatal betamethasone (BETA) treatment on MAP1B IR in fetal sheep at 130 dGA.
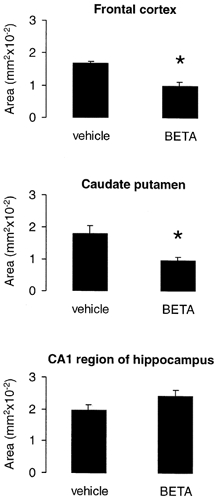
Vehicle-treated fetuses, n = 7; betamethasone-treated fetuses, n = 7; means ±s.e.m., *P < 0.05.
Betamethasone exposure resulted in a decrease of MAP2 IR in the frontal cortex by 24 %, in the caudate putamen by 45.5 % and in the hippocampus by 52.5 % (P < 0.05, Figs 2 and 6). Loss of MAP2 in the betamethasone-treated animals was mainly the result of a loss of MAP2c since MAP2a,b IR did not differ between the vehicle- and betamethasone-treated animals (Fig. 4).
Figure 6. Acute effects of antenatal betamethasone (BETA) treatment on MAP2a,b,c IR in fetal sheep at 130 dGA.
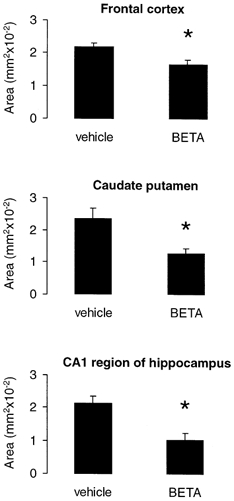
Vehicle-treated fetuses, n = 7; betamethasone-treated fetuses, n = 7; means ±s.e.m.; *P < 0.05.
Haematoxylin–eosin staining did not demonstrate irreversible neuronal damage in any of the brain regions investigated (Fig. 1 and 2).
CBF in the frontal cortex, caudate putamen and hippocampus did not change significantly during vehicle infusion. In betamethasone-treated fetuses, CBF was decreased by 34 % in the frontal cortex and by 43 % in the caudate putamen 24 h after the onset of infusion (P < 0.05). In the hippocampus, CBF decreased by 28 %. However, this decrease was not significant. Forty-eight hours after the onset of betamethasone infusion, no significant difference from baseline values could be proven. The loss of MAP1B and MAP2a,b,c IR showed a correlation to the CBF decrease 24 h after the onset of betamethasone infusion in the frontal cortex and hippocampus (Fig. 7). However, significance at the 5 % level was present only in the frontal cortex (Fig. 7). No correlation between the loss of MAP IR and CBF was found 48 h after the onset of betamethasone exposure.
Figure 7. Correlation of the area of MAP1B IR and MAP2a,b,c IR to the relative CBF 24 h after the onset of betamethasone exposure.
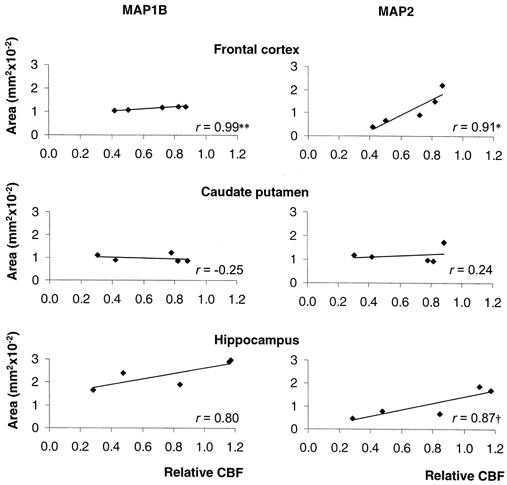
CBF is given relative to baseline. Vehicle-treated fetuses, n = 5; betamethasone-treated fetuses, n = 5. **P < 0.01, *P < 0.05, †P < 0.06.
DISCUSSION
We have been able to demonstrate that fetal exposure to betamethasone plasma concentrations similar to those to which the human fetus is exposed during antenatal glucocorticoid therapy leads to acute disturbances in the neuronal cytoskeleton in the fetal sheep brain at 130 dGA (0.87 of gestation). These disturbances were not accompanied by irreversible neuronal damage monitored with Haematoxylin–eosin staining. This is the first demonstration of MAP1B and of the HMW isoforms of MAP2 in comparison to the LMW isoform MAP2c in the ovine fetal brain. Rees et al. (1999) have detected MAP2 immunohistochemically in the fetal sheep brain cerebellum at 125 dGA without determination of the distinct isoforms of MAP2.
Almost all prenatal in vivo studies of MAP distribution available at the present time have been undertaken in animals in which brain development is predominantly postnatal, such as rodents or chickens. In contrast, the sheep brain develops mainly prenatally (Dobbing & Sands, 1979). The prominent occurrence of the juvenile MAP1B and MAP2c at the stage of development we have studied suggests that neuronal outgrowth and synaptic maturation (Tucker et al. 1988b;Johnson & Jope, 1992) have not been completed in the fetal sheep brain by 0.87 gestation. This conclusion is supported by functional studies which demonstrate continued maturation of electrocortical brain activity at this stage of gestation as shown by linear (Szeto et al. 1985) and non-linear measurements (Schmidt et al. 1998). In humans, the adult pattern of neuronal cytoskeletal protein expression is attained by the second postnatal year (Arnold & Trojanowski, 1996).
Loss of MAP1B and MAP2 paralleled the CBF decrease in the same brain regions 24 h after the onset of betamethasone infusion. In a previous study, glucocorticoid exposure to the same dose of betamethasone as used in the present study also induced transient significant reductions of regional CBF in several other brain regions (Schwab et al. 2000a). The exact mechanisms responsible for the loss of MAPs during ischaemia are unknown. Loss of MAP IR may reflect conformational changes, diminished protein expression or degradation of the protein. The favourite hypothesis is that elevation of Ca2+ leads to proteolytic degradation by calcium-activated proteases such as calpain (Matesic & Lin, 1994; Pettigrew et al. 1996). Fischer et al. (1991) have shown that MAP1B is more resistant to degradation by calpain than MAP2. This difference in susceptibility may well contribute to the absence of a significant alteration of hippocampal MAP1B demonstrated in our study.
Although both CBF and MAP1B and MAP2 IR decreased during betamethasone exposure, a significant correlation was found only in the frontal cerebral cortex. Neuronal damage due to cerebral hypoperfusion depends on the extent and duration of the decrease in CBF. Loss of MAP caused by cerebral hypoperfusion does not reach its maximal level for at least 24 h after the insult as shown in both adult (Pettigrew et al. 1996) and neonatal (Malinak & Silverstein, 1996) rats. Thus, any relationship between decreased blood flow and loss of MAPs is likely to be related to the period of the most pronounced CBF decrease. However, that correlation does not necessarily prove a causal connection. Rather, the absence of a correlation in the caudate putamen in spite of a fall in CBF and a loss of MAP1B and MAP2 similar to that in the frontal cortex suggests that mechanisms other than the decrease in CBF contribute to the loss of MAP1B and MAP2. Glucocorticoid neurotoxicity may be responsible for the disturbance of the neuronal cytoskeleton. Synthetic glucocorticoids induce dendrite degeneration in the CA3 region in non-human primates (Uno et al. 1994) and dendritic loss is consistent with disruption of MAP2 IR in rats (Bywood & Johnson, 2000).
Evidence suggests that the effects of glucocorticoids depend on the receptor type that they occupy. Activation of type I or mineralocorticoid receptors is likely to produce neuroprotective effects (Hassan et al. 1996). Neurotoxic and apoptosis-inducing effects (Packan & Sapolsky, 1990; Hassan et al. 1996) as well as catabolic effects (Horner et al. 1990) are known to be type II or glucocorticoid receptor mediated. Saturation of type I receptors occurs at much lower steroid concentrations than saturation of type II receptors because type I receptors have an approximately tenfold higher affinity for cortisol as shown in rats (Reul & de Kloet, 1985). Consequently, plasma concentrations of cortisol in the saturation range of type I receptors have neuroprotective effects. Cortisol at higher concentrations typical for stressed individuals (50-60 ng ml−1; McEwen et al. 1987) and type II-specific synthetic glucocorticoids such as betamethasone or dexamethasone activate type II receptors and induce neurotoxic effects (Hassan et al. 1996). At the gestational age examined in this study, high affinity type II receptors are present in the fetal sheep brain (Rose et al. 1985; Yang et al. 1990). Moreover, the biological potency of synthetic glucocorticoids resulting from type II receptor binding is higher in comparison to cortisol (Yang et al. 1990). Therefore, external administration of betamethasone as a specific type II receptor ligand may sufficiently change the ratio of activated type I and type II receptors resulting in neurotoxic effects in the ovine fetal brain.
Studies in rats demonstrate that type II receptors occur in most brain regions including the cerebral cortex, hippocampus and caudate putamen (Ahima et al. 1991). We found alterations of MAP2 IR in all three of these regions and altered MAP1B IR in two of the three regions. The highest density of type I receptors, however, was found in limbic regions, i.e. in hippocampal neurons and in the hypothalamus (Ahima et al. 1991). The distribution pattern of ‘neuroprotective’ type I receptors might modulate the ‘neurotoxic’ effects of betamethasone via type II receptors in the hippocampus since endogenous cortisol production is not completely blocked by the dose of betamethasone used here (Derks et al. 1997). Differential activation of the two receptor types could prevent the hippocampus from the loss of MAP1B IR.
In spite of a lack of clear evidence that the breakdown of microtubules contributes to neuronal death it has been shown that the loss of MAP2 correlates with neuronal degeneration after brain injury in rodents (Matesic & Lin, 1994; Hicks et al. 1995). Alteration of the cytoskeleton is a major step in the initiation of apoptosis in several cell types (Tsukidate et al. 1993; Bonfoco et al. 1995). However, when recovery of protein synthesis occurs during the early post-ischaemic phase, the number of dendritic microtubules increases (Furuta et al. 1993). It has been suggested that the post-ischaemic increase of MAP2c expression represents a plastic response in neurons that will survive ischaemic injury (Saito et al. 1995). Further experiments utilising extended post-treatment periods are necessary to confirm whether the glucocorticoid-induced alteration in MAPs represents a state of transition towards irreversible neuronal damage or represents a reversible state of functional disturbance.
It has been suggested that calpain-mediated proteolysis plays a role in the regulation of MAP levels during brain development (Fischer et al. 1991; Johnson et al. 1991). Calcium-activated protein kinases and phosphatases change the phosphorylation state of MAP1B and MAP2. This change in state is important for regulating the dynamics of the neuronal cytoskeleton and neuronal plasticity (reviewed in Sanchez et al. 2000). The antibodies used in the present study, however, have not been characterised for their ability to recognise distinct phosphorylation patterns. Changes in MAP2 levels or phosphorylation state accompany synaptic modifications and play a critical role in cognitive processing and neural network learning (Johnson & Jope, 1992; Dayhoff et al. 1994; Sanchez et al. 2000). Thus, the immunocytochemical changes described here suggest a disturbed neuronal function that may provide a partial explanation for the alterations of electrocortical brain function in fetal sheep that we have demonstrated using the same dose of betamethasone (Schwab et al. 2000b). In human fetuses, acute changes in fetal behaviour, for example in fetal body movements, fetal breathing movements and heart rate during maternal betamethasone treatment, also suggest altered fetal brain function (Mulder et al. 1997; Senat et al. 1998).
In conclusion, juvenile forms of MAPs are present in the fetal sheep brain at 0.87 of gestation, suggesting that neuronal outgrowth and synaptogenesis have not been completed at that age (Tucker, 1990). Using MAPs as sensitive markers of early intracellular structural events in response to metabolic and neurotoxic insults, we were able to detect alterations of the neuronal cytoskeleton caused by betamethasone treatment at fetal exposure levels that occur clinically. This effect does not seem to be due solely to metabolic insufficiency caused by the decrease in CBF. Neurotoxic effects of betamethasone very probably contribute to the alteration of MAPs. The disturbance of MAPs, which are important regulator proteins of synaptic plasticity and neuronal function, coincided with the altered behavioural and electrocortical function shown in human and sheep fetuses during glucocorticoid treatment.
Acknowledgments
We are grateful to Dr Xiu-Ying Ding for skillful surgical assistance, Dr Cun Li for her advice on brain preparation, Claudia Hiepe for histological processing and Karen Moore for the help with the manuscript. This work was supported by the Deutsche Akademie der Naturforscher Leopoldina, Boehringer Ingelheim Fonds and NIH grant HD 21840.
References
- Ahima RS, Krozowski Z, Harlan RE. Type I corticosteroid receptor-like immunoreactivity in the rat CNS: distribution and regulation by corticosteroids. Journal of Comparative Neurology. 1991;313:522–538. doi: 10.1002/cne.903130312. [DOI] [PubMed] [Google Scholar]
- Albala JS, Kress Y, Liu WK, Weidenheim K, Yen SH, Shafit Zagardo B. Human microtubule-associated protein-2c localizes to dendrites and axons in fetal spinal motor neurons. Journal of Neurochemistry. 1995;64:2480–2490. doi: 10.1046/j.1471-4159.1995.64062480.x. [DOI] [PubMed] [Google Scholar]
- Arnold SE, Trojanowski JQ. Human fetal hippocampal development: II. The neuronal cytoskeleton. Journal of Comparative Neurology. 1996;367:293–307. doi: 10.1002/(SICI)1096-9861(19960401)367:2<293::AID-CNE10>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Ballard PL, Ballard RA. Scientific basis and therapeutic regimens for use of antenatal glucocorticoids. American Journal of Obstetrics and Gynecology. 1995;173:254–262. doi: 10.1016/0002-9378(95)90210-4. [DOI] [PubMed] [Google Scholar]
- Ballough GP, Martin LJ, Cann FJ, Graham JS, Smith CD, Kling CE, Forster JS, Phann S, Filbert MG. Microtubule-associated protein 2 (MAP-2): a sensitive marker of seizure-related brain damage. Journal of Neuroscience Methods. 1995;61:23–32. doi: 10.1016/0165-0270(95)00019-q. [DOI] [PubMed] [Google Scholar]
- Bershadsky AD, Vasiliev JM. Cytoskeleton. New York: Plenum Press; 1989. [Google Scholar]
- Bonfoco E, Ceccatelli S, Manzo L, Nicotera P. Colchicine induces apoptosis in cerebellar granule cells. Experimental Cell Research. 1995;218:189–200. doi: 10.1006/excr.1995.1147. [DOI] [PubMed] [Google Scholar]
- Burgoyne RD, Cumming R. Ontogeny of microtubule-associated protein 2 in rat cerebellum: differential expression of the doublet polypeptides. Neuroscience. 1984;11:156–167. doi: 10.1016/0306-4522(84)90220-3. [DOI] [PubMed] [Google Scholar]
- Bywood PT, Johnson SM. Dendrite loss is a characteristic early indicator of toxin-induced neurodegeneration in rat midbrain slices. Experimental Neurology. 2000;161:306–316. doi: 10.1006/exnr.1999.7259. [DOI] [PubMed] [Google Scholar]
- Dayhoff J, Hameroff S, Lahoz Beltra R, Swenberg CE. Cytoskeletal involvement in neuronal learning: a review. European Biophysical Journal. 1994;23:79–93. doi: 10.1007/BF00208862. [DOI] [PubMed] [Google Scholar]
- De Kloet ER, Rosenfeld P, Van Eekelen JA, Sutanto W, Levine S. Stress, glucocorticoids and development. Progress in Brain Research. 1988;73:101–120. doi: 10.1016/S0079-6123(08)60500-2. [DOI] [PubMed] [Google Scholar]
- Derks JB, Giussani DA, Jenkins SL, Wentworth RA, Visser GH, Padbury JF, Nathanielsz PW. A comparative study of cardiovascular, endocrine and behavioural effects of betamethasone and dexamethasone administration to fetal sheep. Journal of Physiology. 1997;499:217–226. doi: 10.1113/jphysiol.1997.sp021922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbing J, Sands J. Comparative aspects of the brain growth spurt. Early Human Development. 1979;3:79–83. doi: 10.1016/0378-3782(79)90022-7. [DOI] [PubMed] [Google Scholar]
- Fischer I, Romano Clarke G, Grynspan F. Calpain-mediated proteolysis of microtubule associated proteins MAP1B and MAP2 in developing brain. Neurochemistry Research. 1991;16:891–898. doi: 10.1007/BF00965538. [DOI] [PubMed] [Google Scholar]
- Folkerts MM, Berman RF, Muizelaar JP, Rafols JA. Disruption of MAP-2 immunostaining in rat hippocampus after traumatic brain injury. Journal of Neurotrauma. 1998;15:349–363. doi: 10.1089/neu.1998.15.349. [DOI] [PubMed] [Google Scholar]
- Furuta S, Ohta S, Hatakeyama T, Nakamura K, Sakaki S. Recovery of protein synthesis in tolerance-induced hippocampal CA1 neurons after transient forebrain ischemia. Acta Neuropathologica. 1993;86:329–336. doi: 10.1007/BF00369444. [DOI] [PubMed] [Google Scholar]
- Hassan AH, von Rosenstiel P, Patchev VK, Holsboer F, Almeida OF. Exacerbation of apoptosis in the dentate gyrus of the aged rat by dexamethasone and the protective role of corticosterone. Experimental Neurology. 1996;140:43–52. doi: 10.1006/exnr.1996.0113. [DOI] [PubMed] [Google Scholar]
- Hicks RR, Smith DH, McIntosh TK. Temporal response and effects of excitatory amino acid antagonism on microtubule-associated protein 2 immunoreactivity following experimental brain injury in rats. Brain Research. 1995;678:151–160. doi: 10.1016/0006-8993(95)00179-t. [DOI] [PubMed] [Google Scholar]
- Horner HC, Packan DR, Sapolsky RM. Glucocorticoids inhibit glucose transport in cultured hippocampal neurons and glia. Neuroendocrinology. 1990;52:57–64. doi: 10.1159/000125539. [DOI] [PubMed] [Google Scholar]
- Howard E, Benjamins JA. DNA, ganglioside and sulfatide in brains of rats given corticosterone in infancy, with an estimate of cell loss during development. Brain Research. 1975;92:73–87. doi: 10.1016/0006-8993(75)90528-4. [DOI] [PubMed] [Google Scholar]
- Johnson GV, Jope RS. The role of microtubule-associated protein 2 (MAP2) in neuronal growth, plasticity and degeneration. Journal of Neuroscience Research. 1992;33:505–512. doi: 10.1002/jnr.490330402. [DOI] [PubMed] [Google Scholar]
- Johnson GV, Litersky JM, Jope RS. Degradation of microtubule-associated protein 2 and brain spectrin by calpain: a comparative study. Journal of Neurochemistry. 1991;56:1630–1638. doi: 10.1111/j.1471-4159.1991.tb02061.x. [DOI] [PubMed] [Google Scholar]
- McEwen B, Chao H, Spencer R, Brinton R, Macisaac L, Harrelson A. Corticosteroid receptors in brain: relationship of receptors to effects in stress and aging. Annals of the New York Academy of Sciences. 1987;512:394–401. doi: 10.1111/j.1749-6632.1987.tb24975.x. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Albeck D, Cameron H, Chao HM, Gould E, Hastings N, Kuroda Y, Luine V, Magarinos AM, McKittrick CR. Stress and the brain: a paradoxical role for adrenal steroids. Vitamins and Hormones. 1995;51:371–402. doi: 10.1016/s0083-6729(08)61045-6. [DOI] [PubMed] [Google Scholar]
- Malinak C, Silverstein FS. Hypoxic-ischemic injury acutely disrupts microtubule-associated protein 2 immunostaining in neonatal rat brain. Biology of the Neonate. 1996;69:257–267. doi: 10.1159/000244319. [DOI] [PubMed] [Google Scholar]
- Matesic DF, Lin R. Microtubule-associated protein 2 as an early indicator of ischemia-induced neurodegeneration in the gerbil forebrain. Journal of Neurochemistry. 1994;63:1012–1020. doi: 10.1046/j.1471-4159.1994.63031012.x. [DOI] [PubMed] [Google Scholar]
- Matus A. Microtubule-associated proteins and the determination of neuronal form. Journal of Physiology Paris. 1990;84:134–137. [PubMed] [Google Scholar]
- Meichsner M, Doll T, Reddy D, Weisshaar B, Matus A. The low molecular weight form of microtubule-associated protein 2 is transported into both axons and dendrites. Neuroscience. 1993;54:873–880. doi: 10.1016/0306-4522(93)90581-y. [DOI] [PubMed] [Google Scholar]
- Meyer JS. Biochemical effects of corticosteroids on neural tissues. Physiological Reviews. 1985;65:946–1020. doi: 10.1152/physrev.1985.65.4.946. [DOI] [PubMed] [Google Scholar]
- Mulder EJ, Derks JB, Visser GH. Antenatal corticosteroid therapy and fetal behaviour: a randomised study of the effects of betamethasone and dexamethasone. British Journal of Obstetrics and Gynaecology. 1997;104:1239–1247. doi: 10.1111/j.1471-0528.1997.tb10969.x. [DOI] [PubMed] [Google Scholar]
- Nassogne MC, Evrard P, Courtoy PJ. Selective neuronal toxicity of cocaine in embryonic mouse brain cocultures. Proceedings of the National Academy of Sciences of the USA. 1995;92:11029–11033. doi: 10.1073/pnas.92.24.11029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathanielsz PW, Bailey A, Poore ER, Thorburn GD, Harding R. The relationship between myometrial activity and sleep state and breathing in fetal sheep throughout the last third of gestation. American Journal of Obstetrics and Gynecology. 1980;138:653–659. doi: 10.1016/0002-9378(80)90083-6. [DOI] [PubMed] [Google Scholar]
- Noraberg J, Zimmer J. Ethanol induces MAP2 changes in organotypic hippocampal slice cultures. NeuroReport. 1998;9:3177–3182. doi: 10.1097/00001756-199810050-00010. [DOI] [PubMed] [Google Scholar]
- Ota A, Ikeda T, Ikenoue T, Toshimori K. Sequence of neuronal responses assessed by immunohistochemistry in the newborn rat brain after hypoxia-ischemia. American Journal of Obstetrics and Gynecology. 1997;177:519–526. doi: 10.1016/s0002-9378(97)70139-x. [DOI] [PubMed] [Google Scholar]
- Packan DR, Sapolsky RM. Glucocorticoid endangerment of the hippocampus: tissue, steroid and receptor specificity. Neuroendocrinology. 1990;51:613–618. doi: 10.1159/000125400. [DOI] [PubMed] [Google Scholar]
- Pettigrew LC, Holtz ML, Craddock SD, Minger SL, Hall N, Geddes JW. Microtubular proteolysis in focal cerebral ischemia. Journal of Cerebral Blood Flow and Metabolism. 1996;16:1189–1202. doi: 10.1097/00004647-199611000-00013. [DOI] [PubMed] [Google Scholar]
- Przyborski SA, Cambray Deakin MA. Developmental regulation of MAP2 variants during neuronal differentiation in vitro. Developmental Brain Research. 1995;89:187–201. doi: 10.1016/0165-3806(95)00117-v. [DOI] [PubMed] [Google Scholar]
- Rees S, Breen S, Loeliger M, McCrabb G, Harding R. Hypoxemia near mid-gestation has long-term effects on fetal brain development. Journal of Neuropathology and Experimental Neurology. 1999;58:932–945. doi: 10.1097/00005072-199909000-00004. [DOI] [PubMed] [Google Scholar]
- Reul JM, de Kloet ER. Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology. 1985;117:2505–2511. doi: 10.1210/endo-117-6-2505. [DOI] [PubMed] [Google Scholar]
- Riederer B, Matus A. Differential expression of distinct microtubule-associated proteins during brain development. Proceedings of the National Academy of Sciences of the USA. 1985;82:6006–6009. doi: 10.1073/pnas.82.17.6006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose JC, Kute TE, Winkler L. Glucocorticoid receptors in sheep brain tissues during development. American Journal of Physiology. 1985;249:E345–349. doi: 10.1152/ajpendo.1985.249.4.E345. [DOI] [PubMed] [Google Scholar]
- Saito N, Kawai K, Nowak TS., Jr Reexpression of developmentally regulated MAP2c mRNA after ischemia: colocalization with hsp72 mRNA in vulnerable neurons. Journal of Cerebral Blood Flow and Metabolism. 1995;15:205–215. doi: 10.1038/jcbfm.1995.26. [DOI] [PubMed] [Google Scholar]
- Sanchez C, Diaz-Nido J, Avila J. Phosphorylation of microtubule-associated protein 2 (MAP2) and its relevance for the regulation of the neuronal cytoskeleton function. Progress in Neurobiology. 2000;61:133–168. doi: 10.1016/s0301-0082(99)00046-5. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM. The physiological relevance of glucocorticoid endangerment of the hippocampus. Annals of the New York Academy of Sciences. 1994;746:294–304. doi: 10.1111/j.1749-6632.1994.tb39247.x. [DOI] [PubMed] [Google Scholar]
- Schmidt K, Schwab M, Kott M, Szeto HH. Nonlinear investigation of developmental changes of the ECoG activity in fetal sheep. Proceedings of the IEEE. 1998;20:2034–2037. [Google Scholar]
- Schwab M, Antonow-Schlorke I, Zwiener U, Bauer R. Brain-derived peptides reduce the size of cerebral infarction and loss of MAP2 immunoreactivity after focal ischemia in rats. Journal of Neural Transmission Supplement. 1998;53:299–311. doi: 10.1007/978-3-7091-6467-9_26. [DOI] [PubMed] [Google Scholar]
- Schwab M, Roedel M, Anwar MA, MüLLER T, Schubert H, Buchwalder LF, Walter B, Nathanielsz PW. Effects of betamethasone administration to the fetal sheep in late gestation on fetal cerebral blood flow. Journal of Physiology. 2000a;528:619–632. doi: 10.1111/j.1469-7793.2000.00619.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab M, Schmidt K, Roedel M, MÜller T, Schubert H, Anwar MA, Nathanielsz PW. Non-linear changes of electrocortical activity after antenatal betamethasone treatment in fetal sheep. Journal of Physiology. 2000b doi: 10.1111/j.1469-7793.2001.0535i.x. in the Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senat MV, Minoui S, Multon O, Fernandez H, Frydman R, Ville Y. Effects of dexamethasone and betamethasone on fetal heart rate variability in preterm labour: a randomised study. British Journal of Obstetrics and Gynaecology. 1998;105:749–755. doi: 10.1111/j.1471-0528.1998.tb10206.x. [DOI] [PubMed] [Google Scholar]
- Szeto H, Vo TD, Dwyer G, Dogramajian ME, Cox MJ, Senger G. The ontogeny of fetal lamb electrocortical activity: a power spectral analysis. American Journal of Obstetrics and Gynaecology. 1985;153:462–466. doi: 10.1016/0002-9378(85)90088-2. [DOI] [PubMed] [Google Scholar]
- Tsukidate K, Yamamoto K, Snyder JW, Farber JL. Microtubule antagonists activate programmed cell death (apoptosis) in cultured rat hepatocytes. American Journal of Pathology. 1993;143:918–925. [PMC free article] [PubMed] [Google Scholar]
- Tucker RP. The roles of microtubule-associated proteins in brain morphogenesis: a review. Brain Research Reviews. 1990;15:101–120. doi: 10.1016/0165-0173(90)90013-e. [DOI] [PubMed] [Google Scholar]
- Tucker RP, Binder LI, Matus AI. Differential localization of the high- and low-molecular weight variants of MAP2 in the developing retina. Developmental Brain Research. 1988a;38:313–318. doi: 10.1016/0165-3806(88)90059-4. [DOI] [PubMed] [Google Scholar]
- Tucker RP, Binder LI, Matus AI. Neuronal microtubule-associated proteins in the embryonic avian spinal cord. Journal of Comparative Neurology. 1988b;271:44–55. doi: 10.1002/cne.902710106. [DOI] [PubMed] [Google Scholar]
- Uno H, Eisele S, Sakai A, Shelton S, Baker E, Dejesus O, Holden J. Neurotoxicity of glucocorticoids in the primate brain. Hormones and Behavior. 1994;28:336–348. doi: 10.1006/hbeh.1994.1030. [DOI] [PubMed] [Google Scholar]
- Viereck C, Tucker RP, Binder LI, Matus A. Phylogenetic conservation of brain microtubule-associated proteins MAP2 and tau. Neuroscience. 1988;26:893–904. doi: 10.1016/0306-4522(88)90107-8. [DOI] [PubMed] [Google Scholar]
- Yang K, Jones SA, Challis JR. Changes in glucocorticoid receptor number in the hypothalamus and pituitary of the sheep fetus with gestational age and after adrenocorticotropin treatment. Endocrinology. 1990;126:11–17. doi: 10.1210/endo-126-1-11. [DOI] [PubMed] [Google Scholar]