

Abstract
We have investigated the effects of changing intracellular pH on intracellular free calcium concentration ([Ca2+]i) in voltage-clamped neurones of the snail Helix aspersa. Intracellular pH (pHi) was measured using the fluorescent dye 8-hydroxypyrene-1,3,6-trisulphonic acid (HPTS) and changed using weak acids and weak bases. Changes in [Ca2+]i were recorded using either fura-2 or calcium-sensitive microelectrodes.
Acidification of the neurones with 5 mM or 20 mM propionate (∼0.2 or 0.3 pH units acidification, respectively) caused a small reduction in resting [Ca2+]i of 5 ± 2 nM (n = 4) and 7 ± 16 nM (n = 4), respectively. The removal of the 20 mM propionate after ∼40 min superfusion resulted in an alkalinization of ∼0.35 pH units and an accompanying rise in resting [Ca2+]i of 31 ± 9 nM (n = 4, P < 0.05). The removal of 5 mM propionate did not significantly affect [Ca2+]i.
Alkalinizations of ∼0.2-0.4 pH units of Helix neurones induced by superfusion with 3 mM concentrations of the weak bases trimethylamine (TMA), ammonium chloride (NH4Cl) and procaine were accompanied by significant (P < 0.05) increases in resting [Ca2+]i of 42 ± 4 nM (n = 26), 30 ± 7 nM (n = 5) and 36 ± 4 nM (n = 3), respectively. The effect of TMA (0.5-6 mM) on [Ca2+]i was dose dependent with an increase in [Ca2+]i during pHi increases of less than 0.1 pH units (0.5 mM TMA).
Superfusion of neurones with zero calcium (1 mM EGTA) Ringer solution inhibited depolarization-induced calcium increases but not the calcium increase produced by the first exposure to TMA (3 mM). In the prolonged absence of extracellular calcium (∼50 min) TMA-induced calcium rises were decreased by 64 ± 10% compared to those seen in the presence of external calcium (P < 0.05).
The calcium rise induced by TMA (3 mM) was reduced by 60 ± 5% following a 10 min period of superfusion with caffeine (10 mM) to deplete the endoplasmic reticulum (ER) stores of calcium (P < 0.05).
Cyclopiazonic acid (10-30 μM CPA), an inhibitor of the ER calcium pump, inhibited the calcium rise produced by TMA (3 mM) and NH4Cl (3 mM) by 61 ± 4% compared to controls (P < 0.05).
These data are consistent with physiological intracellular alkaline shifts stimulating release of calcium, or inhibiting re-uptake of calcium by an intracellular store. The calcium increase was much reduced following application of caffeine, treatment with CPA or prolonged removal of external calcium. Hence the ER was likely to be the source of mobilized calcium.
Changes in intracellular pH (pHi) and extracellular pH (pHo) are associated with electrical activity of neurones (Ahmed & Connor, 1980; Chesler & Kaila, 1992; Schwiening et al. 1993; Trapp et al. 1996). Such changes in pH are likely to have important modulatory effects due to the high pH sensitivity of cellular proteins such as enzymes, ion channels and ion transporters (Busa, 1986; Deitmer & Rose, 1996). In particular these pH shifts may have significant effects on calcium regulation in nerve cells. Although changes in pHi (ΔpHi) have been associated with changes in intracellular calcium levels in a number of cell types the mechanisms underlying ΔpHi-induced calcium transients remains unclear. Increases in neuronal free calcium concentration ([Ca2+]i) occur as a consequence of electrical activity, some resulting from calcium influx through voltage-gated channels. These increases in [Ca2+]i can be modulated by several different processes. These are extrusion from the cell via the calcium-hydrogen pump (plasma membrane calcium-ATPase; PMCA), release from and/or uptake into intracellular stores and cytosolic calcium buffering. Increases in [Ca2+]i can cause acidifications in neurones due to increased activity of the PMCA following a calcium load (Schwiening et al. 1993; Trapp et al. 1996). On the other hand, alkalinizations can result from the activation of outwardly rectifying proton channels (Thomas & Meech, 1982). Changes in pHi can also result from neurotransmitter-dependent pathways (Amos et al. 1996; Ballanyi & Kaila, 1998).
There have been only a few studies that investigated the effects of experimentally induced pHi changes on resting calcium levels in neurones (Dickens et al. 1989; OuYang et al. 1994). These studies showed that relatively large experimentally imposed shifts in pHi (0.3-0.9 pH units) can modulate neuronal calcium regulation. However, the sensitivity of [Ca2+]i to smaller, more physiological changes in neuronal pH has received little attention. In addition the specific sites of calcium regulation that may be modulated by pHi shifts remain unclear. In this study we investigate the effects of relatively small pHi changes on resting calcium levels in the model neuronal system of the snail Helix aspersa.
We have found that intracellular alkalinization of snail neurones by as little as 0.1 pH units produces a significant [Ca2+]i increase. This increase appears to be due to calcium mobilization from internal stores. Preliminary data from this study have been presented at a meeting of the Physiological Society (Willoughby & Schwiening, 1999).
METHODS
General
All experiments were carried out on snail neurones in isolated suboesophageal ganglia. Cells were voltage-clamped using two microelectrodes and held at a potential of -60 mV. Changes in pHi were induced by superfusion of a weak base or a weak acid. pHi was measured using the dye 8-hydroxypyrene-1,3,6-trisulphonic acid (HPTS). Cytosolic calcium was measured using fura-2 or Ca2+-sensitive microelectrodes.
Snail brain preparation
Common garden snails, Helix aspersa, were collected locally and kept aestivating until killed by rapid removal of the brain. The suboesophageal ganglia were isolated as described by Kennedy & Thomas (1995) and superfused with normal Hepes-buffered snail Ringer solution. The inner connective sheath was then carefully torn to expose the nerve cell bodies. The cells were left to settle for 20-30 min. All experiments were performed at room temperature (18-24°C).
Solutions
Hepes-buffered snail Ringer solution contained (mm): NaCl 80, KCl 4, CaCl2 7, MgCl2 5, Hepes 20 titrated with NaOH to pH 7.5. Calcium-free snail Ringer solution contained (mm): NaCl 80, KCl 4, MgCl2 12, Hepes 20 and EGTA 1 titrated with NaOH to pH 7.5. The high potassium saline used for the calibration of the pH-sensitive fluorescent dye (HPTS) contained (mm): NaCl 5, KCl 77.5, MgCl2 1, Hepes 10 and was titrated with KOH. Cyclopiazonic acid was kept frozen as a 50 mm stock solution in dimethyl sulphoxide (DMSO) and diluted in snail Ringer solution on the day of use (final DMSO concentration < 0.1%). HPTS and the calcium sensor cocktail ingredients (see Kennedy & Thomas, 1995, for details) were purchased from Fluka (Dorset, UK). Fura-2 (pentapotassium salt) was purchased from Molecular Probes (Eugene, OR, USA).
Microelectrodes
Microelectrodes for recording membrane potential (Em) and clamp current (Ic) were pulled from filamented borosilicate glass (1.5 mm diameter, Clark Electromedical Instruments, Reading, UK) and had a resistance of 7-15 MΩ when filled with 1 m KCl.
pHi measurements
The methods for pHi measurements using HPTS have been described in detail previously (Willoughby et al. 1998). In brief, a well-exposed neurone (80-250 μm in diameter) was selected and impaled with two microelectrodes. The first microelectrode was filled with 1 m KCl and 10 mm HPTS for voltage recording and pressure injection of HPTS. A second electrode, containing 1 m KCl, was used for current injection during voltage clamping. The neurone was illuminated with alternating 440 nm and 380 nm filtered light from a xenon arc lamp using a 200 μm diameter quartz optical fibre. A second quartz fibre was used to collect fluorescence > 510 nm which was detected by a photomultiplier tube (Thomas & Schwiening, 1992). The signal was calibrated in vitro as described previously (Willoughby et al. 1998).
Fluorimetric calcium measurements
The methods for calcium measurements with fura-2 were as described previously (Thomas & Schwiening, 1992; Kennedy & Thomas, 1995). In brief, a well exposed neurone was impaled with two microelectrodes. The first microelectrode was filled with 1 m KCl and 1 mm fura-2 for voltage recording and pressure injection of the calcium indicator. A second electrode, containing 1 m KCl, was used to voltage clamp the cell. The neurone was illuminated with alternating 340 nm, 360 nm and 380 nm filtered light from a xenon arc lamp using the quartz optical fibre method described above. The signal was calibrated in vitro using standard calibration solutions (Molecular Probes).
Calcium-sensitive microelectrodes
These were prepared as described by Kennedy & Thomas (1995) except that 1.2 mm diameter unfilamented aluminosilicate glass tubing was used. The calcium-sensitive electrodes had a tip diameter of ∼2 μm and resistance of 10-50 GΩ. Electrodes were tested prior to use by changing the superfusion of the bath from normal snail Ringer solution to a calcium-free Ringer solution to which 1 mm EGTA had been added (pCa > 7). Electrodes showing a response of less than 150 mV to this solution change were rejected. Ca2+-sensitive microelectrodes were calibrated using a Nernstian response of 28 mV per tenfold change in calcium (Kennedy & Thomas, 1996). The electrodes showed no change in potential when an intracellular pCa 7 calibration solution (Kennedy & Thomas, 1996) at pH 7.4 was changed to either pH 6.9 or pH 7.7 indicating that the electrodes were insensitive to pH over the range used in these experiments. This is in agreement with Ammann et al. (1987) who showed that the Ca2+-sensitive cocktail (ETH 129), used in this study, is insensitive to pH changes in this range.
Data collection and plotting
All data were collected at 4 Hz using a CED 1401 analog-to-digital converter (Cambridge Electronic Design, Cambridge, UK) following appropriate filtering. Calcium and pH data presented in figures were averaged to produce a final data rate of ∼24 data points per millimetre (600 d.p.i), corresponding to a frequency of between ∼0.13 Hz for data in Fig. 5 and ∼2 Hz for data in Fig. 8. Membrane potential and current data were plotted at the same data rate, but the minimum and maximum values within the period were plotted. This was done to retain the peak size of the depolarizations and currents. Calibrated HPTS data are shown as pH values where increasing proton concentrations are indicated as upward deflections. Calcium data are plotted as nanomolar, rather than pCa, values for several reasons. Firstly, most researchers are more comfortable with nanomolar calcium plots. Secondly, calcium buffering is constant over the physiological range with respect to molar concentrations rather than pCa changes (Schwiening & Thomas, 1996). Thirdly, fura-2 fluorescence ratios are almost linearly proportional to nanomolar values in the low physiological range. However, it should be noted that the calcium-sensitive microelectrodes produce a voltage linearly proportional to pCa. Since the nanomolar calcium plots may obscure the close relationship between pH and pCa we have included, for comparison, pCa scales.
Figure 5. The alkalinization-induced calcium increase is not dependent upon calcium entry.
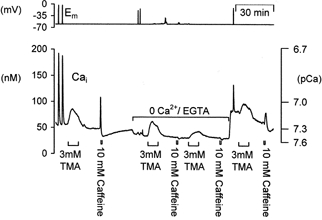
A typical sample experiment showing the effects of external calcium removal on the response to alkalinization in a voltage-clamped snail neurone. The effects of depolarization (20 steps to 0 mV for 100 ms at 5 Hz), trimethylamine (3 mm) and caffeine (10 mm) on cytosolic calcium were compared in the presence and absence of extracellular calcium. Zero calcium snail Ringer solution contained 1 mm EGTA. All trimethylamine applications were for ∼8 min, caffeine applications were for 1 min.
Figure 8.
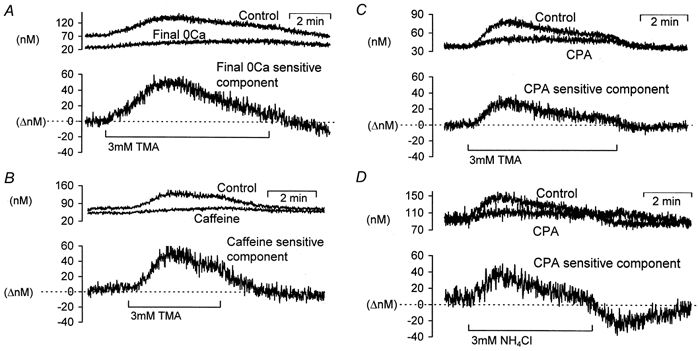
The extracellular calcium and CPA-dependent components of the calcium rises following exposure to a weak base
The upper two traces in each panel are recordings of [Ca2+]i during weak base exposure under control and test conditions. The lower trace in each panel is the difference between the two upper traces and represents the inhibited component. A, data taken from Fig. 5, showing the effect of following prolonged (∼50 min) external calcium removal on the TMA-induced calcium rise. B, data taken from Fig. 6 showing the caffeine-sensitive component of the TMA-induced calcium rise. C, the CPA-sensitive component of the TMA-induced calcium rise, data taken from Fig. 7A. D, the CPA-sensitive component of the NH4Cl-induced calcium changes (representative of 3 similar experiments).
RESULTS
Effects of weak acid exposure on intracellular pH and calcium
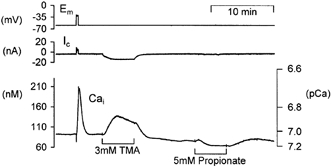
To investigate the effects of cytosolic acidification on intracellular calcium concentration ([Ca2+]i) we applied the weak acid propionate to voltage-clamped snail neurones (Szatkowski & Thomas, 1989). The membrane potential was held at -60 mV to minimize calcium entry through voltage-gated calcium channels. The neurones were also superfused with Hepes-buffered nominally HCO3/CO2-free Ringer solution to reduce the rate of pHi regulation. Figure 1 illustrates typical pHi and [Ca2+]i changes seen during 5 mm and 20 mm propionate application. The neurones were pressure injected with HPTS to measure pH, or fura-2 to measure [Ca2+]i. Mean ±s.e.m. values for these experiments are shown in Table 1. A peak cytosolic acidification was seen within the first 5 min of 5 mm and 20 mm propionate addition. During the 40 min superfusion with propionate pHi returned towards normal. When the propionate was removed cytosolic alkalinization was seen. In parallel experiments we found that resting [Ca2+]i fell slightly when 5 mm or 20 mm propionate was applied. In one of four experiments a small increase, rather than a fall, in resting [Ca2+]i was seen following 20 mm propionate application. The subsequent removal of 5 mm propionate resulted in a small, non-significant [Ca2+]i rise whilst removal of 20 mm propionate caused a rise in [Ca2+]i of 31 ± 9 nm(n = 4, P < 0.05). These results show that cytosolic acidification of ∼0.2-0.3 pH units, at constant membrane potential, often causes a small fall in [Ca2+]i. However, the removal of 20 mm propionate causes a cytosolic alkalinization of ∼0.35 pH units and an accompanying rise in [Ca2+]i. To investigate whether this rise in [Ca2+]i was caused by the rise in pHi we imposed further intracellular alkalinizations with a range of weak bases. The bases were chosen for their ability to produce different patterns of pHi changes.
Figure 1. Effects of weak acid application and removal on pHi and [Ca2+]i in voltage-clamped snail neurones.
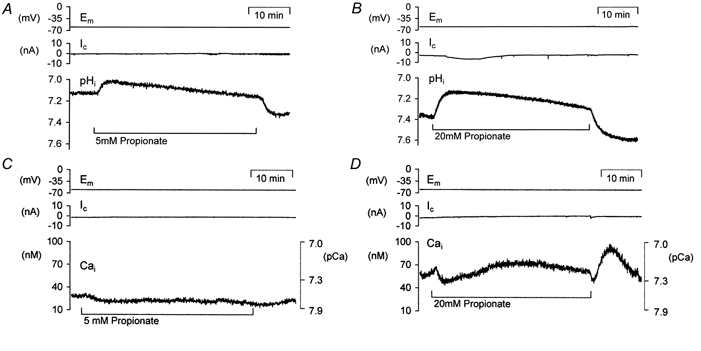
A and B, records of voltage-clamp potential (Em), clamp current (Ic) and pHi (using HPTS) during and after prolonged exposure to different concentrations of the weak acid propionate. C and D show parallel recordings of [Ca2+]i (using fura-2) during and after exposure to the same concentrations of propionate. Calcium data are plotted as nanomolar concentrations and secondary axes showing equivalent pCa values have been included (see Methods).
Table 1.
The effects of weak acid addition and removal on intracellular pH and cytosolic calcium levels
| 5 mM propionate addition | 5 mM propionate removal | 20 mM propionate addition | 20 mM propionate removal | |
|---|---|---|---|---|
| ΔpHi (pH units) | −0.18 ± 0.04 ** (6) | 0.25 ± 0.05 * (3) | −0.31 ± 0.04 ** (3) | 0.34 ± 0.08* (3) |
| Δ[Ca2+]i (nM) | −5 ± 2 (4) | 5 ± 3 (4) | −7 ± 16 (4) | 31 ± 9 † (4) |
All data are shown as means ±s.e.m.
P < 0.05
P < 0.01 compared to resting pHi values
P < 0.05 compared to resting [Ca2+]i; n values are presented in parentheses.
Intracellular pH and calcium changes imposed by weak bases
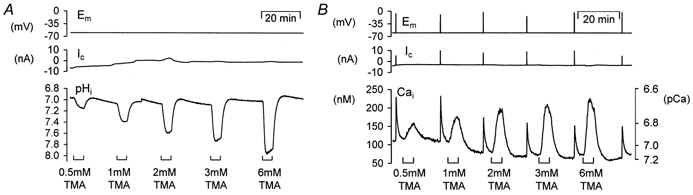
Cytosolic alkalinizations were imposed by superfusion of the isolated ganglia preparations with the weak bases trimethylamine (TMA), ammonium chloride (NH4Cl) and procaine. An example of the alkalinizations recorded in voltage-clamped snail neurones injected with HPTS during the superfusion of the three bases is shown in Fig. 2A. The neurone was held at -60 mV and had a resting pHi of ∼6.93. Each weak base was applied at 3 mm for 5-8 min. TMA and procaine produced similar alkalinizations of between ∼0.3 and 0.4 pH units (3 mm TMA, 0.38 ± 0.04 pH units, n = 7; 3 mm procaine 0.29 ± 0.04 pH units, n = 4). When they were removed pHi returned towards control values. The response seen with NH4Cl was different. The neurone alkalinized by only ∼0.2 pH units on superfusion with 3 mm NH4Cl (mean values 0.19 ± 0.01 pH units, n = 3) but on removal it showed an acidification of 0.4 pH units which overshot control values. These pHi shifts were expected due to the permeability of cells (Boron & De Weer, 1976). When NH4Cl is applied the NH3-induced intracellular alkalinization is reduced by the simultaneous influx of . On NH4Cl removal the NH3 efflux occurs with little concurrent flux.
Figure 2. Effect of three weak bases on pHi and [Ca2+]i of snail neurones.
A, the snail neurone was voltage clamped at -60 mV and pressure injected with HPTS (approximate final intracellular concentration 200 μm). Intracellular alkalinizations were induced by superfusing with 3 mm trimethylamine (TMA), 3 mm ammonium chloride (NH4Cl) and 3 mm procaine for ∼5 min. B, another snail neurone was held at -60 mV and pressure injected with fura-2 (approximate final concentration 50 μm). Superfusion with 3 mm trimethylamine (TMA), 3 mm ammonium chloride (NH4Cl) and 3 mm procaine for ∼5 min all resulted in a transient rise in [Ca2+]i. The neurone was periodically depolarized (burst of 10 steps to 0 mV for 100 ms at 5 Hz) to maintain store calcium levels.
In parallel experiments the effects of TMA, NH4Cl and procaine on resting calcium levels were investigated. Resting cytosolic calcium was 39 ± 4 nm(n = 26). Figure 2B shows that superfusion of all three of the weak bases in turn at concentrations of 3 mm produced increases in resting calcium levels that peaked within 2-3 min of the superfusion period and slowly began to return towards baseline levels. Upon removal of the weak bases calcium levels returned rapidly back to, or just below in the case of NH4Cl removal, resting calcium levels. Neurones were depolarized between weak base applications to ensure there was no progressive depletion of an intracellular calcium store. Analysis of experiments in which the fura-2 signal had been calibrated show average rises in calcium levels of 42 ± 4 nm during 3 mm TMA (n = 26), 30 ± 7 nm during 3 mm NH4Cl (n = 5) and 36 ± 4 nm during 3 mm procaine (n = 3) superfusion. These experiments support the hypothesis that it is the pHi changes, and not the addition of exogenous membrane-permeant buffers that cause changes in [Ca2+]i. However, it is possible that these are not real changes in [Ca2+]i and that they are due to the pH sensitivity of fura-2. We tested this directly by measuring [Ca2+]i with calcium-sensitive microelectrodes that are insensitive to pH changes over the physiological range (see Methods).
pH effects on [Ca2+]i recorded with ion-selective microelectrodes
Figure 3 shows recordings of membrane potential, clamp current and [Ca2+]i made in a voltage-clamped Helix neurone impaled with a calcium-sensitive microelectrode. The cell was first depolarized to -30 mV for 20 s which resulted in a clear calcium transient. The cell was then exposed to first 3 mm TMA and then 5 mm propionate to induce an intracellular alkalinization and then an intracellular acidification. Calibration of the calcium-sensitive microelectrode (see Methods for details) reveals a resting calcium level of ∼100 nm. Superfusion of 3 mm TMA for 5 min produced a ∼50 nm rise in [Ca2+]i which decayed slowly. A 5 mm propionate application induced a small fall in calcium levels and on removal of the propionate there was a small overshoot in [Ca2+]i. Similar findings were recorded in three separate experiments. It therefore seemed unlikely that the effects of pHi shifts on [Ca2+]i were simply an artefact due to the pH sensitivity of fura-2. To investigate this phenomenon further we tested the effect of varying concentrations of TMA on pHi and pCai.
Figure 3. The effects of weak base and weak acid on resting calcium levels recorded with a calcium-selective microelectrode.
The neurone was held at -60 mV and depolarized once for 20 s. Trimethylamine (3 mm) and propionate (5 mm) were superfused sequentially for periods of ∼5 min.
Dose dependency of alkalinization-induced [Ca2+]i increases
A range of TMA concentrations (0.5-6 mm) was used to examine the sensitivity of the alkalinization-induced calcium increase. Figure 4A shows the effect of successive increasing TMA concentrations on pHi in a neurone held at -60 mV. TMA was superfused for 5 min periods with 20 min intervals between each application. Increasing concentrations of weak base produced progressively larger and faster alkalinizations. During superfusion with the higher TMA concentrations pHi showed some small recovery towards control. Figure 4B shows a parallel experiment where we measured the effects of identical TMA concentrations on resting calcium levels. The neurone was depolarized between each TMA application Collection of data from six experiments in total reveals a clear dose dependence with increasing concentrations of TMA producing larger and faster increases in [Ca2+]i. A TMA concentration of 0.5 mm induced an average intracellular alkalinization of less than 0.09 ± 0.01 pH units but was accompanied by a clear rise in [Ca2+]i of 19 ± 11 nm. The highest dose of TMA tested (6 mm) induced an alkalinization of 0.59 ± 0.17 pH units and a calcium rise of 70 ± 39 nm. One possible source of these calcium rises is enhanced influx across the plasma membrane. We tested this hypothesis by removing extracellular calcium.
Figure 4. Dose dependence of trimethylamine on intracellular alkalinizations and calcium rises.
A, a voltage-clamped snail neurone was superfused with a series of increasing concentrations of TMA whilst pHi was monitored with HPTS. B shows the calcium increases induced by superfusion with increasing concentrations of TMA as monitored by fura-2. All applications of weak base were for 5 min with at least 20 min between each application.
Removal of calcium from extracellular Ringer solution
Figure 5 shows a typical experiment in which we tested the effects of removing external calcium on the TMA-induced [Ca2+]i increase measured with fura-2. At the start of the experiment shown in Fig. 5 the neurone was depolarized twice, this resulted in large rises in [Ca2+]i which rapidly recovered to baseline. Superfusion with 3 mm TMA for 8 min produced a rise in calcium that returned to just below baseline upon TMA removal. A 30 s application of 10 mm caffeine also produced a large transient increase in [Ca2+]i followed by a calcium undershoot, presumably due to enhanced calcium-induced calcium release followed by store refilling (Orkand & Thomas, 1995). External calcium was then removed. This caused a small fall in baseline [Ca2+]i. Subsequent depolarization did not evoke a rise in cytosolic calcium. Under these conditions superfusion with 3 mm TMA produced a similar calcium rise to that seen in the presence of external calcium. Subsequent exposure to 10 mm caffeine, in zero calcium Ringer solution, failed to produce a rise in [Ca2+]i, whilst a second superfusion with 3 mm TMA produced only a small calcium rise. On replacement of external calcium there was a large increase in resting calcium levels. This was not seen in all experiments and may be a consequence of store depletion and/or calcium leakage across the plasma membrane. Further depolarizations and superfusion of 3 mm TMA produced similar [Ca2+]i increases to those seen in control conditions. Application of 10 mm caffeine for 30 s once again produced a calcium rise, followed by a calcium undershoot. Similar results were seen in five other experiments. In two other neurones repeated applications of 3 mm TMA in zero calcium Ringer solution produced progressively smaller calcium increases, consistent with a gradual emptying of intracellular stores. Analysis of data from all seven experiments with TMA applied in calcium-free Ringer solution show an increase in calcium levels of 31 ± 10 nm following short-term calcium removal (∼10 min) compared to an average 35 ± 7 nm increase in controls. As a result of long-term external calcium removal (∼50 min) significantly reduced the size of the TMA-induced calcium rise to just 14 ± 6 nm (P < 0.05). Since caffeine application, following TMA in zero calcium Ringer solution, did not induce a calcium rise it seems likely that some internal store was depleted of calcium. However, the lower resting calcium levels seen during superfusion with zero calcium Ringer solution might also explain this result due to a calcium-dependent reduction in calcium-induced calcium release. These data are not consistent with intracellular alkalinization stimulating calcium influx across the plasma membrane and they support the involvement of a depletable intracellular store. There are two known intracellular stores that could underlie this phenomenon of alkalinization-induced calcium rise: endoplasmic reticulum (ER) and mitochondria.
Effect of caffeine-induced store depletion on the alkalinization-induced calcium rise
The ER stores of Helix neurones can be depleted of calcium during prolonged application of 10 mm caffeine (Kennedy & Thomas, 1995; Collins & Thomas, 1998). As shown in Fig. 6 we investigated whether emptying the caffeine-sensitive calcium stores could influence the TMA-induced calcium rise seen in these neurones under voltage-clamp conditions. Superfusion of 3 mm TMA, under control conditions, produced a calcium rise similar to that described previously in this paper. A 10 min application of 10 mm caffeine produced an initial large calcium transient followed minutes later by a second transient that was greatly reduced in amplitude. When 3 mm TMA was applied immediately following the caffeine the calcium rise was less than half that seen previously. After a 30 min wash the size of the calcium rise seen during 3 mm TMA superfusion returned to that seen in control conditions. Similar results were seen in three out of four experiments. In one experiment prior application of 10 mm caffeine did not reduce the size of the TMA-induced calcium rise. Analysis of data from all four experiments reveals a significant decrease in the size of the weak base-induced calcium rise from 62 ± 10 nm in control conditions to 25 ± 3 nm immediately following a 10 min caffeine application (P < 0.05). This is equivalent to a 60 ± 5% reduction in the size of the alkalinization-induced calcium rise.
Figure 6. The effect of endoplasmic reticulum store depletion on the alkalinization-induced calcium rise.
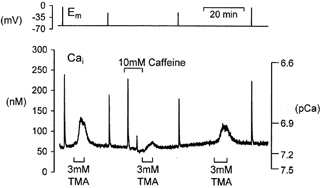
The neurone was pressure injected with fura-2 and held under voltage clamp. Depolarization (20 steps to 0 mV for 100 ms at 5 Hz) or 3 mm TMA application induced a transient rise in calcium. Caffeine, 10 mm, was applied for 10 min. Immediately following caffeine application the size of the TMA-induced calcium rise was reduced. A third application of TMA, after 30 min superfusion in normal Ringer solution, produced a larger calcium rise.
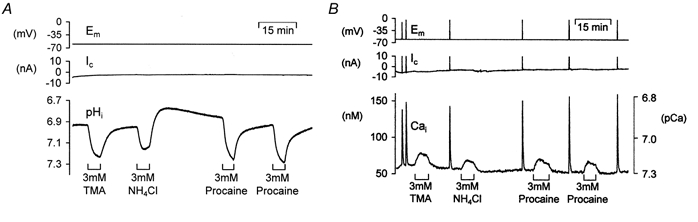
Effect of ER calcium-ATPase inhibition on the alkalinization-induced calcium rise
To further test the hypothesis that alkalinization might interfere with store-dependent calcium handling we investigated the effects of TMA following treatment with cyclopiazonic acid (CPA) to deplete the ER calcium stores. Figure 7A shows an example of an experiment where we compared the TMA-induced [Ca2+]i increase, assessed with fura-2, prior to and after superfusion with CPA. At the start of the experiment the neurone was exposed to 3 mm TMA, depolarized and then exposed to 10 mm caffeine. All of these treatments produced rises in calcium. Superfusion of the neurone with 10 μm CPA for 15 min produced a slow rise in cytosolic calcium levels that partially recovered. This rise is believed to be due to a gradual release of calcium from the stores and inhibition of calcium re-uptake into the ER via the Ca2+-ATPase (SERCA). CPA was then removed. Subsequent treatment with caffeine produced no calcium increase; however, the depolarization-induced calcium rise still occurred. When we applied 3 mm TMA a much smaller rise in calcium was seen when compared to control. Similar results were seen in six other experiments investigating the effects of CPA on calcium increases induced by 3 mm TMA or NH4Cl. Following ER store depletion with CPA the mean alkalinization-induced [Ca2+]i increase was significantly reduced in size from 41 ± 10 nm in control conditions to just 15 ± 4 nm following CPA treatment (P < 0.01). This is equivalent to a 61 ± 4% reduction in the size of the alkalinization-induced calcium rise.
Figure 7. The effect of the endoplasmic reticulum calcium pump (SERCA) inhibitor CPA on alkalinization-induced calcium rise assessed with either fura-2 or a calcium-sensitive microelectrode.
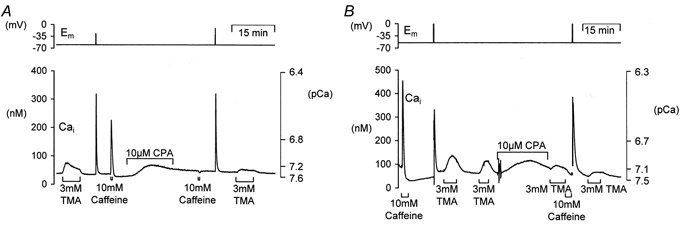
A, the neurone was pressure injected with fura-2 and held under voltage clamp. TMA application induced a transient rise in calcium. Depolarization (20 steps to 0 mV for 100 ms at 5 Hz) and caffeine exposure (10 mm for 30 s) caused larger rises in calcium. Following exposure to 10 μm CPA the caffeine-induced rise in calcium was abolished, but the depolarization-induced calcium transient was unaffected. Following CPA superfusion TMA produced a smaller and more sustained calcium rise. B, recording of [Ca2+]i made with a Ca2+-sensitive microelectrode showing control caffeine, depolarization and TMA-induced calcium transients. CPA superfusion abolished the caffeine-induced calcium rise and attenuated the TMA-induced calcium rise.
To ensure that these findings were not due to either an effect of CPA on fura-2, accumulation of fura-2 by an intracellular store, or spatially averaged calcium signals of widely differing magnitudes we performed similar experiments to that shown in Fig. 7A but using calcium-sensitive microelectrodes. Figure 7B shows an example of such an experiment. It can be seen that the changes in [Ca2+]i measured by the microelectrode at a point were qualitatively very similar to those reported by the whole-cell fura-2 signal. CPA produced a very similar inhibition of the calcium rise, induced by TMA, as that reported by the dye. However, there are several notable differences between the signals recorded by the dye and the electrodes. Typically the calcium-sensitive microelectrodes report somewhat higher calcium values than the fluorescent dye fura-2. They also record larger TMA-induced calcium rises and larger falls in calcium following removal of caffeine (see Discussion).
Temporal aspects of the TMA-induced calcium rise
In Figs 5, 6 and 7 we have shown the effect of external calcium removal, prior caffeine application and CPA application, respectively, on the alkalinization-induced calcium rise. None of these manoeuvres completely inhibited these rises. A comparison of the control alkalinization-induced calcium rises and those following either calcium removal, caffeine or CPA applications are shown in Fig. 8. We have also plotted the difference traces to obtain the inhibited components. Figure 8A shows the component that is sensitive to prolonged removal of external calcium (∼50 min in zero Ca2+ Ringer solution). Figure 8B shows the component of the calcium rise inhibited by prolonged application of 10 mm caffeine immediately prior to TMA superfusion. Figure 8C and D shows the CPA-inhibited component of the TMA- and NH4Cl-induced calcium rises, respectively. It can be seen from Fig. 8 that depletion of store calcium by either prolonged external calcium removal, caffeine or CPA addition produce similar inhibited components. This store-dependent component of the alkalinization-induced calcium rise reaches a peak increase of 28 ± 5 nm above baseline (n = 14) after weak base addition and declines within ∼5 min to baseline. The store-independent component is sustained and can account for the 18 ± 2 nm calcium increase (n = 14). This component is unlikely to be a simple pH effect on fura-2 since it is also seen with calcium-sensitive microelectrodes (Fig. 7B).
DISCUSSION
Alkalinization induces calcium increases
In this study we demonstrate that changes in intracellular pH can have marked effects on neuronal [Ca2+]i. The initial addition of weak acid produced an intracellular acidification and a small but non-significant decrease in calcium levels. Removal of 20 mm propionate after a prolonged period of superfusion produced a clear increase in resting [Ca2+]i. Similar calcium rises were seen when we superfused voltage-clamped neurones with the weak bases TMA, NH4Cl and procaine suggesting that cytosolic alkalinization was responsible for inducing the [Ca2+]i increases. These rises were relatively transient throughout the period of weak base superfusion with a peak increase seen 2-3 min into the superfusion period. Although the peak calcium rise imposed by superfusion with 3 mm TMA was 42 ± 4 nm this (relatively small absolute rise in calcium) is equivalent to a doubling of resting [Ca2+]i (39 ± 4 nm in Helix neurones, n = 26). Experimentally imposed cytosolic alkalinization has been associated with increases in resting calcium levels in a number of cell types including parotid cells (Snowdowne et al. 1992), acinar cells (Yodozawa et al. 1997), pituitary cells (Shorte et al. 1991), epithelial cells (Danthuluri et al. 1990; Wiegmann et al. 1993; Benning et al. 1996; Nitschke et al. 1996) and smooth muscle cells (Siskind et al. 1989; Batlle et al. 1993; Austin et al. 1996). In contrast Kaila & Voipio (1990) have shown in single crayfish muscle fibres that resting cytosolic calcium and tension were decreased by intracellular alkalosis. More specifically, a few neuronal studies have shown alkalinization to be associated with increases in cytosolic calcium levels (axons: Abercrombie & Gammeltoft, 1987; cultured neurones: Dickens et al. 1989; OuYang et al. 1994; microglial cells: Minelli et al. 2000). Acidic pHi has been associated with a decrease in cytosolic calcium in other cell types including parotid cells (Snowdowne et al. 1992) and smooth muscle cells (Austin et al. 1996). However, other studies have associated cellular acidification with a calcium increase in crayfish muscle fibres (Kaila & Voipio, 1990) and smooth muscle (Batlle et al. 1993).
Effects of pHi on the fura-2 signal
In our study the majority of intracellular calcium measurements were obtained using the fluorescent indicator fura-2. However, previous work by Martinez-Zaguilan et al (1996) has shown that pH can affect the calcium sensitivity of fura-2. In particular the dissociation constant (Kd) of fura-2 decreases with increasing pH. Hence, two approaches were taken to ascertain whether a pH-sensitive shift in the Kd for fura-2 could account for the apparent increases in calcium that were seen during superfusion with weak base in this study.
Firstly, we calculated the size of possible rises in the fura-2 ratio signal (340/380 nm) that might be accounted for by a pH-sensitive shift in the Kd for fura-2. Based on the study by Martinez-Zaguilan et al. (1996) we calculated that an alkalinization of 0.4 pH units would produce a change in the Kd value from 145 nm at pH 7.2 to 128 nm at pH 7.6. This is equivalent to an apparent increase in calibrated calcium levels of around 13%. From a resting calcium level of 39 nm (estimated for this study) alkalinization with 3 mm TMA could produce an apparent calcium rise of 5 nm due to a shift in the fura-2 Kd value alone. This could only account for a small proportion of the alkalinization-induced [Ca2+]i rise (42 nm).
Secondly, we performed parallel experiments using Ca2+-sensitive microelectrodes to record [Ca2+]i. The microelectrodes are insensitive to changes in pH over the range investigated (see Methods). Our experiments illustrate a very clear calcium increase during weak base superfusion and support evidence for alkalinization-induced calcium increases obtained using the fluorescent probe fura-2. Alvarez-Leefmans et al. (1981) used Ca2+-sensitive microelectrodes to record the effects of CO2-induced acidifications in unclamped Helix aspersa neurones. They saw either a fall or no change in resting calcium levels. In Fig. 3 we have presented data showing a small fall in resting calcium during superfusion with the weak acid propionate (5 mm). This would suggest that the fall in calcium recorded during propionate superfusion in Fig. 1 of this paper was not simply due to a shift in the Kd value for fura-2 during changes in pHi (Kd= 161 nm at pH 7.0, equivalent to ∼10% fall in calculated calcium levels). Similarly, Alvarez-Leefmans and colleagues never saw a rise in calcium during periods of intracellular acidification.
Comparison of the dye and microelectrode calcium measurements
Figure 7 illustrates two common differences between the calibrated fura-2 and calcium-sensitive microelectrode measurements of [Ca2+]i. The calcium-sensitive microelectrodes usually report higher resting calcium levels and larger calcium undershoots following caffeine application. One possible explanation for the high resting calcium levels is a leak of calcium into the cell near the tip of the calcium-sensitive microelectrode (Kennedy & Thomas, 1996). This elevated calcium signal may be due to damage by the microelectrode or it could be a micro-domain with elevated calcium due to release of stored calcium or influx of calcium from outside. Collins & Thomas (1998) showed that using fura-2, the size of the caffeine-induced undershoot was proportional to the resting calcium level.
Sensitivity of [Ca2+]i to pHi changes
We were able to illustrate that the size of the alkalinization-induced calcium rise was dependent upon the concentration of weak base (and consequently the size of the cytosolic alkalinization). Analyses of the TMA dose-response data showed a clear increase in resting [Ca2+]i during an alkalinization of less than 0.1 pH units. This high sensitivity of [Ca2+]i to pHi implies that pHi shifts seen during normal neuronal activity might influence [Ca2+]i also. In Helix neurones we have found that a burst of depolarizations to 0 mV (50 times for 100 ms at 5 Hz) produced an acidification of 0.06 pH units (Willoughby et al. 1999). We propose that following a burst of neuronal activity the recovery from acidification (i.e. an alkalinization) could promote the release of calcium from the ER stores. Such a release of calcium may be necessary for re-setting the calcium integrating function of the ER, or have a signalling function. Equally the acidification during electrical activity may hinder the release of calcium from the ER, which may have a protective role. This work has important implications for calcium homeostasis in all neurones where activity-induced pHi shifts are seen (Ahmed & Connor, 1980; Schwiening et al. 1993; Trapp et al. 1996; Ballanyi & Kaila, 1998).
Source of the calcium rise
We considered two main sources for the [Ca2+]i increase seen during neuronal alkalinization. These were an increase in calcium entry across the plasma membrane or calcium release from the ER. We have shown that alkalinization-induced calcium increases in Helix neurones were still present following removal of external calcium, with a significant reduction in the size of the calcium rise after long periods in zero calcium Ringer solution (∼50 min). These results suggested that the calcium was coming from a source within the neurones. Dickens et al. (1989) similarly found that 30 mm NH4Cl produced a transient calcium rise in ‘low calcium (∼10−7m)’ conditions. On the other hand in rat primary cultured neurones OuYang et al. (1994) suggested that alkalinization with NH4Cl increased calcium influx since in zero external calcium they recorded only a decrease in [Ca2+]i.
Our studies with zero external calcium indicated that the ER was the probable source of the calcium increase. Under zero external calcium conditions the ER was unable to refill following calcium release. Consequently the full TMA-induced [Ca2+]i rise was only evident during the first period of superfusion with the weak base. Subsequent alkalinizations produced smaller calcium increases and the response to caffeine was also lost indicating depletion of the ER calcium stores. In other experiments, in the presence of external calcium, we used prolonged applications of 10 mm caffeine to deplete the internal stores of calcium. A significant reduction in the size of the TMA-induced calcium rise was seen in these experiments indicating that the ER could be the source of the calcium rise. We obtained further evidence that alkalinization-induced [Ca2+]i increases were dependent upon the ER by comparing the effects of weak bases before and after treatment with CPA (10-30 μm). The SERCA inhibitor significantly reduced the calcium rise induced by TMA and NH4Cl by ∼60% on average when compared to control conditions.
If all of the [Ca2+]i increase was due to mobilization from the ER we would expect CPA, caffeine or prolonged periods of external calcium removal to completely abolish the alkalinization effect. However, this was not the case with ∼40% of the TMA response still remaining. The effects of changing pHi on the fura-2 Kd value can only account for 13% of the total calcium rise. This leaves 27% of the calcium increase that is of unknown source. It is possible that the ER is not completely depleted of calcium under the conditions used for these experiments. Alternatively, effects of pHi increases on the mitochondria may play a role as described for axonal preparations of Myxicola (Abercrombie & Gammeltoft, 1987). At present this cannot be ruled out for Helix neurones although the latter study revealed inhibition of mitochondrial calcium uptake in Myxicola axons at fairly high pH values (pH 8.3). Grinstein & Goetz (1985) suggest that alkalinization in rat lymphocytes could inhibit calcium extrusion via the plasma membrane calcium ATPase. It seems unlikely that this should be the case in Helix neurones as lowering cytosolic proton concentration would if anything tend to stimulate proton entry on the membrane pump in exchange for calcium extrusion (Schwiening et al. 1993).
Our findings showing alkalinization-induced calcium mobilization from the ER in neurones are comparable to studies in muscle and pancreatic cells. There is clear evidence that calcium handling by the sarcoplasmic reticulum (SR) is pHi sensitive (isolated SR vesicles: Meissner & Henderson, 1987; Meissner, 1994). Alkalinization has been shown to stimulate SR calcium release by increasing the opening of calcium release channels whilst store refilling was reduced (Dettbarn & Palade, 1991). Earlier work also showed alkaline pH to increase open probability of the ryanodine receptor from both the cytoplasmic and the luminal sides of the channel (Ma et al. 1988) with the cytoplasmic side of the channel being more sensitive to pH (Rousseau & Pinkos, 1990). In pancreatic and glial cells alkalinization has been shown to enhance activity of the inositol trisphosphate (IP3)-sensitive calcium release pathway (Yodozawa et al. 1997; Minelli et al. 2000). At present we have not established whether an intracellular alkalinization within Helix neurones can directly stimulate release of calcium from the ER due to increased opening of the ryanodine or IP3 receptors, or inhibit store re-filling. Nevertheless, the clear sensitivity of calcium levels to pH suggests that changes in pHi resulting from neuronal activity are likely to have significant effects on calcium mobilization. This could be particularly important in regions where transient local pHi shifts may be fairly large, for example just beneath the plasma membrane.
Acknowledgments
We would like to thank the MRC for financial support and local gardeners for collecting snails.
References
- Abercrombie RF, Gammeltoft K. High cytosolic pH inhibits Ca uptake by Myxicola axon mitochondria. American Journal of Physiology. 1987;252:C68–76. doi: 10.1152/ajpcell.1987.252.1.C68. [DOI] [PubMed] [Google Scholar]
- Ahmed Z, Connor JA. Intracellular pH changes induced by calcium influx during electrical activity in molluscan neurones. Journal of General Physiology. 1980;75:403–426. doi: 10.1085/jgp.75.4.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Leefmans FJ, Rink TJ, Tsien RY. Free calcium ions in neurones of Helix aspersa measured with ion-selective micro-electrodes. Journal of Physiology. 1981;315:531–548. doi: 10.1113/jphysiol.1981.sp013762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammann D, Bührer T, Schefer U, Müller M, Simon W. Intracellular neutral carrier-based Ca2+ microelectrode with subnanomolar detection limit. Pflügers Archiv. 1987;409:223–228. doi: 10.1007/BF00583469. [DOI] [PubMed] [Google Scholar]
- Amos BJ, Mathie A, Richards CD. Effect of metabotropic glutamate receptor activation on the intracellular pH and calcium of rat neurones and glia maintained in culture. Journal of Physiology. 1996;491.P:136P. [Google Scholar]
- Austin C, Dilly K, Eisner D, Wray S. Simultaneous measurement of intracellular pH, calcium, and tension in rat mesenteric vessels: Effects of extracellular pH. Biochemical and Biophysical Research Communications. 1996;222:537–540. doi: 10.1006/bbrc.1996.0779. [DOI] [PubMed] [Google Scholar]
- Ballanyi K, Kaila K. Activity-evoked changes in intracellular pH. In: Kaila K, Ransom BR, editors. pH and Brain Function. New York: Wiley-Liss; 1998. chap. 16. [Google Scholar]
- Batlle DC, Peces R, Lapointe MS, Ye M, Daugiras JT. Cytosolic free calcium regulation in response to acute changes in intracellular pH in vascular smooth muscle. American Journal of Physiology. 1993;264:C932–943. doi: 10.1152/ajpcell.1993.264.4.C932. [DOI] [PubMed] [Google Scholar]
- Benning N, Leipziger J, Greger R, Nitsche R. Effect of alkalinization of cytosolic pH by amines on intracellular Ca2+ activity in HT29 cells. Pflügers Archiv. 1996;432:126–133. doi: 10.1007/s004240050114. [DOI] [PubMed] [Google Scholar]
- Boron WF, Deweer P. Intracellular pH transients in squid giant axons caused by CO2, NH3, and metabolic inhibitors. Journal of General Physiology. 1976;67:91–112. doi: 10.1085/jgp.67.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busa WB. Mechanisms and consequences of pH-mediated cell regulation. Annual Review of Physiology. 1986;48:389–402. doi: 10.1146/annurev.ph.48.030186.002133. [DOI] [PubMed] [Google Scholar]
- Chesler M, Kaila K. Modulation of pH by neuronal activity. Trends in Neurosciences. 1992;15:396–402. doi: 10.1016/0166-2236(92)90191-a. [DOI] [PubMed] [Google Scholar]
- Collins RO, Thomas RC. Intracellular calcium, caffeine and store refilling in snail neurones. Journal of Physiology. 1998;511.P:42P. [Google Scholar]
- Danthuluri NR, Kim D, Brock TA. Intracellular alkalinization leads to Ca2+ mobilization from agonist sensitive pools in bovine aortic endothelial cells. Journal of Biological Chemistry. 1990;265:19071–19076. [PubMed] [Google Scholar]
- Deitmer JW, Rose CR. pH regulation and proton signaling by glial-cells. Progress in Neurobiology. 1996;48:73–103. doi: 10.1016/0301-0082(95)00039-9. [DOI] [PubMed] [Google Scholar]
- Dettbarn C, Palade P. Effects of alkaline pH on sarcoplasmic-reticulum Ca2+ release and Ca2+ uptake. Journal of Biological Chemistry. 1991;266:8993–9001. [PubMed] [Google Scholar]
- Dickens CJ, Gillespie JI, Greenwell JR. Interactions between intracellular pH and calcium in single mouse neuroblastoma (N2A) and rat pheochromocytoma cells (P12) Quarterly Journal of Experimental Physiology. 1989;74:671–679. doi: 10.1113/expphysiol.1989.sp003319. [DOI] [PubMed] [Google Scholar]
- Grinstein S, Goetz JD. Control of free cytoplasmic calcium by intracellular pH in rat lymphocytes. Biochimica et Biophysica Acta. 1985;819:267–270. doi: 10.1016/0005-2736(85)90183-x. [DOI] [PubMed] [Google Scholar]
- Kaila K, Voipio J. Dependence of intracellular free calcium and tension on membrane potential and intracellular pH in single crayfish muscle fibres. Pflügers Archiv. 1990;416:501–511. doi: 10.1007/BF00382682. [DOI] [PubMed] [Google Scholar]
- Kennedy HJ, Thomas RC. Intracellular calcium and its sodium-independent regulation in voltage-clamped snail neurones. Journal of Physiology. 1995;484:533–548. doi: 10.1113/jphysiol.1995.sp020684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy HJ, Thomas RC. Effects of injecting calcium-buffer solutions on [Ca2+]i in voltage-clamped snail neurones. Biophysical Journal. 1996;70:2120–2130. doi: 10.1016/S0006-3495(96)79778-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Fill M, Knudson CM, Campbell KP, Coronado R. Ryanodine receptor of skeletal muscle is a gap junction type channel. Science. 1988;242:99–102. doi: 10.1126/science.2459777. [DOI] [PubMed] [Google Scholar]
- Martinez-Zaguilan R, Parnami G, Lynch RM. Selection of fluorescent ion indicators for simultaneous measurments of pH and Ca2+ Cell Calcium. 1996;19:337–349. doi: 10.1016/s0143-4160(96)90074-3. [DOI] [PubMed] [Google Scholar]
- Meissner G. Ryanodine receptor/Ca2+ release channels and their regulation by endogenous effectors. Annual Review of Physiology. 1994;56:485–508. doi: 10.1146/annurev.ph.56.030194.002413. [DOI] [PubMed] [Google Scholar]
- Meissner G, Henderson JS. Rapid calcium release from cardiac sarcoplasmic reticulum vesicles is dependent on Ca2+ and is modulated by Mg2+, adenine nucleotide, and calmodulin. Journal of Biological Chemistry. 1987;262:3065–3073. [PubMed] [Google Scholar]
- Minelli A, Lyons S, Nolte C, Verkhratsky A, Kettenmann H. Ammonium triggers calcium elevation in cultured mouse microglial cells by initiating Ca2+ release from thapsigargin-sensitive intracellular stores. Pflügers Archiv. 2000;439:370–377. doi: 10.1007/s004249900188. [DOI] [PubMed] [Google Scholar]
- Nitschke R, Riedel A, Ricken S, Leipziger J, Benning N, Fischer KG, Greger R. The effect of intracellular pH on cytosolic Ca2+ in HT29 cells. Pflügers Archiv. 1996;433:98–108. doi: 10.1007/s004240050254. [DOI] [PubMed] [Google Scholar]
- Orkand RK, Thomas RC. Effects of low doses of caffeine on [Ca2+]i in voltage-clamped snail (Helix aspersa) neurones. Journal of Physiology. 1995;489:19–28. doi: 10.1113/jphysiol.1995.sp021026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang YB, Mellergard P, Kristian T, Kristianova V, Siesjo BK. Influence of acid-base changes on the intracellular calcium concentration of neurons in primary culture. Experimental Brain Research. 1994;101:265–271. doi: 10.1007/BF00228746. [DOI] [PubMed] [Google Scholar]
- Rousseau E, Pinkos J. pH modulates conducting and gating behaviour of single calcium release channels. Pflügers Archiv. 1990;415:645–647. doi: 10.1007/BF02583520. [DOI] [PubMed] [Google Scholar]
- Schwiening CJ, Kennedy HJ, Thomas RC. Calcium-hydrogen exchange by the plasma membrane Ca-ATPase of voltage-clamped snail neurons. Proceedings of the Royal Society. 1993;B 253:285–289. doi: 10.1098/rspb.1993.0115. [DOI] [PubMed] [Google Scholar]
- Shorte SL, Collingridge GL, Randall AD, Chappel JB, Schofield JG. Ammonium-ions mobilize calcium from an internal pool which is insensitive to TRH and ionomycin in bovine anterior-pituitary cells. Cell Calcium. 1991;12:301–312. doi: 10.1016/0143-4160(91)90004-x. [DOI] [PubMed] [Google Scholar]
- Siskind MS, McCoy CE, Chobanian A, Schwartz JH. Regulation of intracellular calcium by cell pH in vascular smooth muscle cells. American Journal of Physiology. 1989;256:C234–240. doi: 10.1152/ajpcell.1989.256.2.C234. [DOI] [PubMed] [Google Scholar]
- Snowdowne KW, Way B, Thomas G, Chen HY, Cashman JR. pHi controls cytoplasmic calcium in rat parotid cells. Biochimica et Biophysica Acta. 1992;1108:145–152. doi: 10.1016/0005-2736(92)90019-i. [DOI] [PubMed] [Google Scholar]
- Szatkowski MS, Thomas RC. The intrinsic intracellular H+ buffering power of snail neurones. Journal of Physiology. 1989;409:89–101. doi: 10.1113/jphysiol.1989.sp017486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas RC, Meech RW. Hydrogen ion currents and intracellular pH in depolarised voltage-clamped snail neurones. Nature. 1982;299:826–828. doi: 10.1038/299826a0. [DOI] [PubMed] [Google Scholar]
- Thomas RC, Schwiening CJ. Optical fibre fluorescence system for intracellular Ca2+ measurement: one lamp and filter wheel supplies two set-ups. Journal of Physiology. 1992;452:153P. [Google Scholar]
- Trapp S, Luckermann M, Kaila K, Ballanyi K. Acidosis of hippocampal-neurons mediated by a plasmalemmal Ca2+/H+ pump. NeuroReport. 1996;7:2000–2004. doi: 10.1097/00001756-199608120-00029. [DOI] [PubMed] [Google Scholar]
- Wiegmann TB, Welling LW, Beatty DM, Howard DE, Vamos S, Morris SJ. Simultaneous imaging of intracellular [Ca2+] and pH in single MDCK and glomerular epithelial cells. American Journal of Physiology. 1993;265:C1184–1190. doi: 10.1152/ajpcell.1993.265.4.C1184. [DOI] [PubMed] [Google Scholar]
- Willoughby D, Thomas RC, Schwiening CJ. Comparison of simultaneous pH measurements made with 8-hydroxypyrene-1,3,6-trisulphonic acid (HPTS) and pH-sensitive microelectrodes in snail neurones. Pflügers Archiv. 1998;436:615–622. doi: 10.1007/s004240050679. [DOI] [PubMed] [Google Scholar]
- Willoughby D, Thomas RC, Schwiening CJ. A role for Na/H exchange in pH regulation in Helix neurones. Pflügers Archiv. 1999;438:741–749. doi: 10.1007/s004249900122. [DOI] [PubMed] [Google Scholar]
- Willoughby D, Schwiening CJ. The effects of intracellular pH on calcium handling in snail neurones. Journal of Physiology. 1999;515.P:124–125P. doi: 10.1111/j.1469-7793.2001.0405k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yodozawa S, Speake T, Elliott A. Intracellular alkalinization mobilizes calcium from agonist sensitive pools in rat lacrimal acinar cells. Journal of Physiology. 1997;499:601–611. doi: 10.1113/jphysiol.1997.sp021953. [DOI] [PMC free article] [PubMed] [Google Scholar]