

Abstract
Permeation of a range of hydrophobic anions through the rat skeletal muscle chloride channel, rClC-1, expressed in Sf-9 (a Spodoptera frugiperda insect cell line) cells has been studied using the whole-cell patch-clamp technique.
Bi-ionic reversal potentials measured with external application of foreign anions gave the following permeability sequence: Cl− (1) ≫ benzoate (0·15) ≫ hexanoate (0·12) ≫ butyrate (0·09) ≫ propionate (0·047) x∼ formate (0·046). Anions with larger hydrophobic moieties were more permeant, which suggested that ClC-1 selectivity for hydrophobic anions is dominated by their interaction with a hydrophobic region in the external mouth of the pore.
All anions studied when applied from outside show an apparently paradoxical voltage-dependent block of inward currents; this voltage-dependent block could be qualitatively described by a discrete-state permeation model with two binding sites and three barriers.
Effects of the external anions with aliphatic side-chains on the apparent open probability (Po) suggested that they are unable to gate the channel, but can modulate ClC-1 gating, probably, by changing Cl− affinity to the gating site.
Effects of internal application of benzoate, hexanoate or propionate mimicked those of increasing internal pH, and similarly depended on the channel protonation from the external side. Results for internal benzoate support the concept of a negatively charged cytoplasmic particle being involved in the ClC-1 gating mechanism sensitive to the internal pH.
Substitution of the permeating ion is widely used in ion channel research for investigating such properties of a channel pore as its size, the nature of the residues lining the pore, number of binding sites and their distribution along the conducting pathway (Bormann et al. 1987; Halm & Frizzell, 1992; Hille, 1992; Linsdell et al. 1997a,b). In the case of ClC-1, the mammalian skeletal muscle chloride channel, ion substitution has been particularly informative as, in addition to characterising the pore properties, it has provided information about the gating mechanism of this channel (Rychkov et al. 1996, 1998; Fahlke et al. 1997a).
Early work on frog muscle with extensive testing of foreign anions (Woodbury & Miles, 1973) revealed the importance of hydrophobic interactions in the Cl− channel pore. Anions were separated into two groups, chloride like and benzoate like. For Cl−and chloride-like anions, which included Br−, and I−, membrane conductance decreased at low pH while for benzoate-like anions (benzoate, propionate, acetate and formate) conductance increased. This was explained in terms of an essential imidazole group involved in the binding of permeant anions and the presence of a hydrophobic region near the selectivity centre which can reduce the height of the energy barrier for the anions with larger hydrophobic moieties. This original work has been supported by two recent studies on ClC-1 using heterologous expression systems and an extensive range of foreign anions, which have shown that ClC-1 has a multi-ion pore with at least two binding sites and that binding to the outer site follows the lyotropic series suggesting the presence of a hydrophobic region adjacent to the anion-attracting group (Fahlke et al. 1997; Rychkov et al. 1998). Both of these studies have also shown that permeation and gating in ClC-1 are functionally linked. While the experimental results and some conclusions of these two studies are quite similar, the proposed explanation on the nature of the functional linkage between permeation and gating in ClC-1 are fundamentally different. Fahlke et al. (1995, 1996, 1997) consider that ClC-1 has a voltage sensor intrinsic to the channel protein that can be modulated by the permeating anion. In contrast, the explanation presented by Rychkov et al. (1998) is based on the idea that the permeating anion itself acts as a gating charge in ClC-1.
Our previous studies indicate that the interaction between permeation and gating of ClC-1 is quite complex. While some large hydrophobic anions such as anthracene-9- carboxylic acid (9-AC) bind to the site and occlude the pore, with no effect on gating (Astill et al. 1996), others, such as 2-(4-chlorophenoxy)propionic acid (CPP) modulate the affinity of the gating site to Cl− without occluding the pore (Aromataris et al. 1999).
In the present work we have investigated the role of the hydrophobic region within the ion-conduction pathway of ClC-1 in permeation and gating by using a homologous series of anions of aliphatic carboxylic acids and benzoate, an anion of an aromatic carboxylic acid, which we refer to collectively as hydrophobic anions. The results of this study provide further evidence that Cl− and some other anions can act as a gating charge in the ClC-1 channel and confirm the importance of the hydrophobic binding site in the pore in determining ClC-1 selectivity. In addition these studies have enabled us to make predictions about the location and height of energy barriers involved in anion permeation and provided a new insight into the complex mechanism of ClC-1 gating.
METHODS
Electrophysiology
Whole-cell patch-clamping was performed on Sf-9 cells infected with the recombinant virus BVDA6.3 carrying ClC-1 cDNA, using an Axopatch 200A and associated standard equipment as described previously (Astill et al. 1996). The usual bath solution contained (mM): 170 NaCl, 2 MgSO4, 2 calcium gluconate and 10 Hepes adjusted to pH 7.5 with NaOH. Pentobarbitone (0.5 mM) was added to the external solution routinely to block native anion channels in Sf-9 cells (Birnir et al. 1992). Lower concentrations of Cl− in the bath solution were achieved by equimolar substitution of 5, 10, 25, 50, 75, 90, 95 and 100 % of external Cl− by a particular anion. One of the anions in this study, octanoate, could not be used at concentrations higher than 45 mM because it produced cell damage; moreover it precipitated with divalent cations, so Ca2+ and Mg2+ had to be replaced with Na+ in the presence of octanoate. Removal of divalent cations from the external solution does not itself alter the properties of ClC-1 (authors’ unpublished observation).
Electrodes were pulled from borosilicate glass and had a resistance of 1-3 MΩ when filled with a normal internal solution containing (mM): 40 KCl, 120 potassium glutamate, 10 EGTA-Na and 10 Hepes adjusted to pH 7.2 with NaOH. Electrode solutions of higher and lower pH were prepared using Tris, Hepes or Mes buffers (10 mM) and titrating with glutamic acid or NaOH as appropriate. When sodium salts of different foreign anions were used in the internal solution, the concentration of potassium glutamate was reduced accordingly. Liquid junction potentials between the bath and electrode solutions were estimated using JPCalc (Barry, 1994) and corrected where specified.
Sodium salts of formate, propionate and benzoate were supplied by AJAX Chemicals (Sydney, Australia); butyrate, hexanoate and octanoate were purchased from Sigma (St Louis, MO, USA).
Data analysis
To obtain permeability ratios, membrane potentials for at least five different concentrations of each foreign anion were measured in current clamp mode. Shifts in potential (relative to control Cl− solution) were plotted against the mole fraction of the anion and fitted with a Goldman-Hodgkin-Katz (GHK) equation of the form:
| (1) |
where Y is the shift in membrane potential, Mf (mole fraction) is the concentration of foreign anion, X−, as a proportion of the total anion concentration (Mf=[X−]/([X−]+[Cl−])), and A is the permeability ratio for this anion relative to Cl− (PX/PCl).
Outward conductance was estimated as the chord conductance in the region of the I-V plot immediately to the right of the reversal potential. A constant difference from the reversal potential of +10 mV was used for each anion.
To approximate the time course of inward current deactivation raw current data points were fitted to an equation of the form:
| (2) |
where A1 and A2 represent the amplitude of the fast and slow exponential components, τ1 and τ2 are their time constants, respectively, C represents the amplitude of the steady-state component and t is time. Peak current (Imax) is current at time zero (eqn (2)).
To estimate the apparent Kd at different membrane potentials, peak current amplitudes were plotted against the concentration of a particular foreign anion and fitted with sigmoidal functions of variable slope using a four-parameter logistic equation.
Peak tail currents (Imax, eqn (2)) for voltage steps to -100 mV after test pulses in the range -160 to +100 mV were used to produce apparent open probability (Po) curves by fitting with a Boltzmann distribution of the form:
| (3) |
where Po(∞) is an offset, V is the membrane potential, V½ is the potential at which Po(V) = (1 +Po(∞))/2, and k is the slope factor. The slope factor k was used to calculate the apparent gating valence (Z = 25.4/k).
The simplest permeation model which could be fitted to our data required two ion binding sites and three barriers. This model was used for qualitative examination of channel permeation properties in mixtures of chloride and hexanoate (for a detailed description of such models, see Anderson & Koeppe, 1992). For simplicity, no direct interaction, other than the single-file constraint, between ions in the pore was assumed. The electrical distances of the wells and barriers of the model were assumed to be identical for Cl− and hexanoate (Hex−). As a result, the model is described by a total of 15 parameters (electrical distances between barriers and wells, together with barrier heights and well depths). The possible occupation states of the model are:
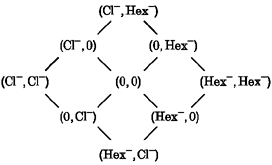
The absolute rate constants were calculated using the ‘frequency factor’kT/h, where k is the Boltzmann constant, T is absolute temperature and h is Planck's constant, even though other choices could have been made (Anderson & Koeppe, 1992). Within certain limits, the choice of the frequency factor does not change the qualitative predictions of such a model. The measured macroscopic I-V relationships were scaled to the current of a single channel by assuming a chord conductance of 1 pS at -160 mV in the absence of hexanoate (Pusch et al. 1994; Saviane et al. 1999). The fit was performed using the least-squares criterion and the Simplex algorithm.
RESULTS
Permeation and block of ClC-1 by hydrophobic anions
All anions studied in the present work were measurably permeant as assessed by the shifts of the membrane potential in the presence of different concentrations of each anion. Values obtained for all concentrations could be reliably fitted (r2 ≫ 0.97) with the modified GHK equation (eqn (1), Fig. 1A) which gave the following relative permeability ratios: Cl− (1) ≫ benzoate (0.15 ± 0.01) ≫ hexanoate (0.12 ± 0.01) ≫ butyrate (0.09 ± 0.01) ≫ propionate (0.047 ± 0.004) ∼ formate (0.046 ± 0.008). This permeability sequence correlated with the sequence of the octanol/water partition coefficients (logP) of the corresponding carboxylic acids (Leo et al. 1971; Fig. 1A, inset). The relative permeability of octanoate could not be determined because of the cell damage produced by the higher concentrations of this anion.
Figure 1. Effect of extracellular hydrophobic anions on conductance and permeability of rClC-1 expressed in Sf-9 cells.
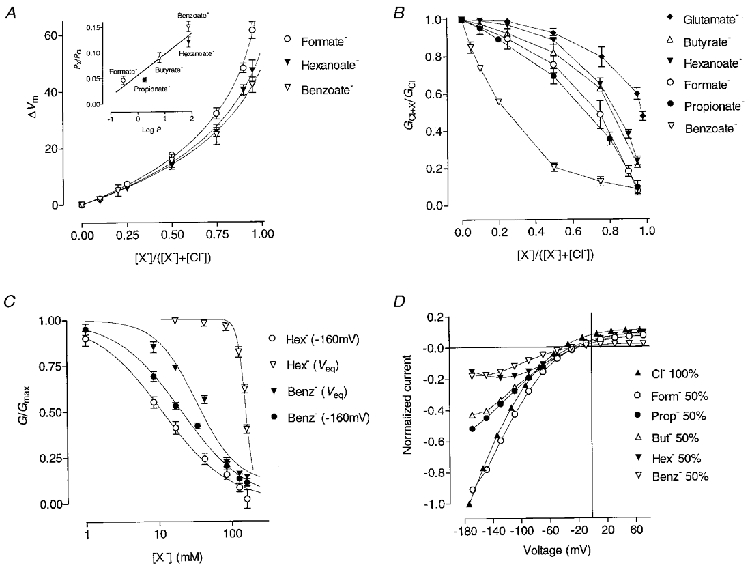
A, dependence of the membrane potential shift (ΔVm) on the mole fraction, [X−]/([X−]+[Cl−]), of foreign anions. Lines represent the fits of the GHK equation (see Methods, eqn (1)). Inset shows the dependence of relative permeability of hydrophobic anions (PX/PCl) on the octanol/water partition coefficient (logP). Experimental points are fitted by linear regression (r2∼ 0.9, P < 0.02). B, dependence of outward relative conductance GX + Cl/GCl, on mole fraction of the foreign anion. C, representative dose-response curves of the block of ClC-1 inward conductance at -160 mV and of outward conductance near the equilibrium potential (Veq) (see Methods) by benzoate (Benz−) and hexanoate (Hex−). D, I-V plots of the instantaneous peak currents through ClC-1 when 50 % of external Cl− was replaced with different anions (Form−, formate; Prop−, propionate; But−, butyrate; Hex−, hexanoate; Benz−, benzoate).
Similar to some other permeant and impermeant anions (Fahlke et al. 1997; Rychkov et al. 1998), foreign anions of this group also block ClC-1 to varying extents. Reduction in the currents in the presence of the foreign anions could result from occlusion of the pore or from changes in open probability. To estimate the degree of block free from contamination of possible effects on Po we relied on the fact that the apparent Po of ClC-1 is close to saturation at voltages positive to the Cl− equilibrium potential. The outward conductance and the peak inward currents after a positive prepulse are thus less affected by the changes in the Po (Rychkov et al. 1996). Outward conductance was strongly decreased by benzoate while anions with aliphatic carbon chains were much less effective (Fig. 1B). To minimise the effect of the reduction of Cl− concentration and obtain the apparent Kd values for the block, conductance (G) in the presence of each concentration of an anion was normalised to the conductance measured in the presence of the same concentration of glutamate (Gglut), which cannot pass through the pore or affect the gating (Rychkov et al. 1998). These values of G/Gglut were plotted against the log of the corresponding anion concentration and fitted with sigmoidal dose-response curves (Fig. 1C). The very steep dose- response curve for hexanoate probably reflects the fact that at very high concentrations of foreign anions effects of the reduction of the Cl− concentration can not be avoided. The potency of the block followed the sequence (with Kd in parentheses, n = 3-7): benzoate (55 ± 3 mM) ≫ propionate (124 ± 5 mM) ≫ formate (140 ± 5 mM) ≫ butyrate (156 ± 4 mM) ∼ hexanoate (158 ± 3 mM).
Surprisingly, instantaneous current-voltage relationships presented in Fig. 1D show that external application of all anions produced a voltage-dependent block (representative dose-response curves are shown in Fig. 1C) of the peak inward current (outward movement of Cl−). Compared with the outward conductance, the blocking potency sequence was reversed, except for benzoate: larger anions had a larger effect. Unlike voltage-dependent block by the anions investigated earlier (Fahlke et al. 1997; Rychkov et al. 1998) block by the external hydrophobic anions increased at more negative potentials, making a simple open channel block an unlikely explanation.
Effects of extracellular application of hydrophobic anions on ClC-1 gating
Whole-cell Cl− currents through ClC-1 channels expressed in Sf-9 cells and other heterologous expression systems deactivate with a double exponential time course to a new steady-state level when the membrane potential is stepped to values negative to the Cl− reversal potential (Fig. 2A;Fahlke et al. 1996; Rychkov et al. 1996). Time constants and the relative amplitudes of the exponential components, as well as the amplitude of the steady-state component, depend on voltage, internal and external pH and the anions present in the internal and external solutions (Fahlke et al. 1996, 1997; Rychkov et al. 1996, 1998). Hydrophobic anions used in this study as Cl− substitutes also altered the kinetics of ClC-1 current deactivation at negative potentials (Fig. 2). The smallest of them, formate, significantly slowed down the rate of current deactivation (Fig. 2B). Analysis of time constants and relative amplitudes of the current components revealed that the change is mainly due to a decrease in the relative amplitude of the fast exponential component (A1) and an increase in the steady-state component (C) (Fig. 3A, D and E). In contrast, with Cl− substitution by propionate or butyrate, the amplitude of the slow exponential component (A2) became smaller and for butyrate the time constants of both exponential components also became faster (Figs 2C and D and 3B-E). With a further increase in the size of the hydrophobic attachment to the carboxyl group (hexanoate, benzoate and octanoate) only one exponential component plus a steady state component could be reliably extracted from the 100 ms currents (Figs 2E-G and 3D and E). The reason for disappearance of the fast exponential component could be that it became too fast and/or too small to be reliably separated from the capacitive currents; or that the time constants of both exponentials became too close to each other at potentials negative to -100 mV to be resolved as separate components. Time constants of the single exponentials extracted from the inward currents in the presence of hexanoate, benzoate or octanoate became larger at less negative potentials but their voltage dependence was less pronounced than that of the slower time constant of the control current (Fig. 3E).
Figure 2. Whole-cell currents in Sf-9 cells expressing rClC-1 with partial replacement of external Cl− with different anions.
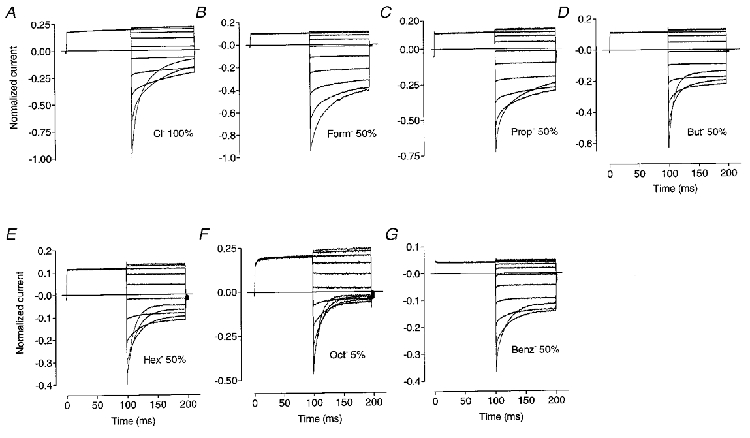
Currents were activated by the following voltage protocol: holding potential was -30 mV; prepulse to +40 mV was followed by steps which ranged from -120 to +80 mV in 20 mV increments. Since experiments were performed on different cells, currents were normalised to the peak inward current in control solution at -120 mV. The amplitude of peak inward currents ranged between -5 and -15 nA in the different cells.
Figure 3. Effect of the external application of foreign anions on the parameters of inward current deactivation.
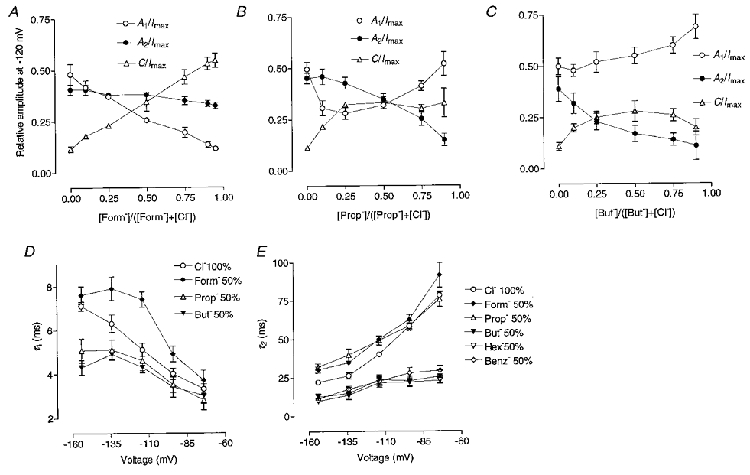
Relative amplitudes of the inward current components at -120 mV are plotted against the mole fraction of formate (A), propionate (B), and butyrate (C). D and E, voltage dependence of the fast (D) and slow (E) time constants at 50 % Cl− replacement by different anions. Relative amplitudes (A1, A2 and C) and time constants (τ1 and τ2) were determined using equation: I(t) =A1exp(-t/τ1) +A2exp(-t/τ2) +C, as described in Methods.
Changes in ClC-1 current kinetics produced by formate, propionate, butyrate, hexanoate and octanoate followed a clear pattern: the larger the hydrophobic side chain the faster the current deactivation and the smaller the non-deactivating component of the inward current. Octanoate, the largest anion tested, was the most potent; at just 5 % Cl− substitution it changed current kinetics as much as 50 % Cl− substitution by hexanoate. Benzoate, although being larger in size, could be placed between propionate and butyrate, judging by the effects it produced on kinetics (Fig. 2).
The open probability of ClC-1 increases with the Cl− concentration in the external solution similarly to the open probability of the fast single protopore gate of the homologous ClC-0 chloride channel from Torpedo (Pusch et al. 1995; Chen & Miller, 1996; Rychkov et al. 1996). If Cl− is replaced with an impermeant anion like glutamate, the V½ of a Boltzmann curve shifts to more positive potentials; meanwhile, in the presence of other anions, like I−, it shifts to more negative potentials (Rychkov et al. 1996, 1998). Two anions of the group investigated in this study, formate and benzoate, shifted apparent Po curves to slightly more negative potentials, effectively substituting for Cl− as ClC-1 openers; all other anions showed a complex mole fraction effect on the apparent Po of ClC-1 (Fig. 4A and B). In the presence of low concentrations of propionate and, to a lesser extent, butyrate, V½ was shifted to more negative potentials and after reaching a minimum, it was shifted progressively to more positive potentials. Dependence of V½ on concentration for these anions appeared as an anomalous mole fraction effect, similar to that reported for ClC-0 in mixtures of Cl− and (Pusch et al. 1995). In ClC-0 it was accompanied by an anomalous mole fraction effect of on macroscopic conductance as well. In the present study anions that showed an anomalous mole fraction effect on the open probability of the ClC-1 channel did not show that effect on macroscopic conductance (Fig. 1B); neither was it apparent in the relative permeability ratios.
Figure 4. Effect of the external foreign anions on the apparent Po.
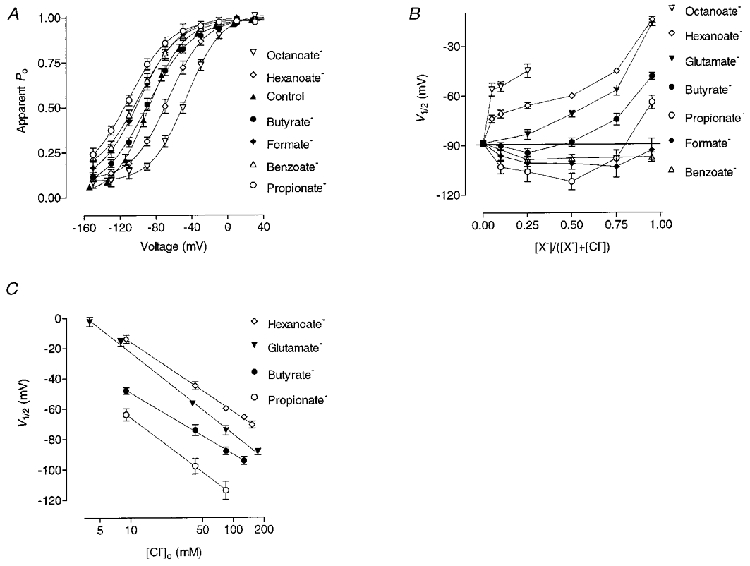
A, apparent Po as a function of membrane potential in control condition and 25 % of Cl− replacement by different anions. B, dependence of V½ on mole fraction of the foreign anions. C, dependence of V½ on log of Cl− concentration with different anions as Cl− substitutes. The slope of the lines are -45.9 ± 1.0 mV (hexanoate), -51.2 ± 0.6 mV (propionate), -40.5 ± 1.0 mV (butyrate), -54.1 ± 1.3 mV (glutamate) per decade of external Cl− concentration change.
Hexanoate and octanoate at concentrations below 10 % shifted V½ sharply to more positive potentials but further increase of the concentration of these anions resulted in a much less steep change of V½ (Fig. 4B). The dependence of V½ on the log of external Cl− concentration when Cl− is replaced with glutamate was linear in the whole range of concentrations between 174 and 4 mM (Fig. 4C). The slope of the line indicates that the open probability of the ClC-1 channel shifts to the right by 54.1 ± 1.3 mV per 10-fold decrease in Cl− concentration. For the other anions shown in the plot the relationship between V½ and Cl− concentration became linear only at a higher percentage of Cl− replacement (Fig. 4B). The slopes of the lines, though, are very similar to that for glutamate, indicating that within this range the change in Po is largely due to the reduction of Cl− concentration.
Effects of intracellular benzoate on ClC-1 gating
It has been reported previously that the effects of a foreign anion on permeation and block of the ClC-1 channel pore were different depending on whether it was applied internally or externally (Fahlke et al. 1997; Rychkov et al. 1998). Preliminary results with internal application of benzoate revealed a strong dependence of inward current deactivation on the presence of this anion in the pipette solution, and it was suggested that binding of anions to a site accessible from the cytoplasmic side may be one of the mechanisms modulating the gating of this channel (Rychkov et al. 1996). In the next series of experiments we, therefore, studied in more detail the effects of hydrophobic anions applied from the cytoplasmic side of the channel. It was found that propionate, benzoate and hexanoate acted very similarly on the properties of ClC-1, so the results for only one of them, benzoate, are shown.
Although the effects of benzoate were similar at any internal pH between 5.5 and 7.2, currents presented in Fig. 5A are recorded at an internal pH of 5.5 to have a better picture of the action of this anion on the kinetics of the inward current deactivation. At low internal pH the time constants of both exponential components of the inward current are slower and the non-deactivating component is larger; at pHi 5.5 the inward current recorded at -120 mV deactivates by only about 40 % during a 100 ms step (Fig. 5A). Addition of benzoate to the internal solution resulted in a faster and more complete deactivation of the inward currents (Fig. 5A); at the same time, the apparent Po curves were shifted to more positive potentials. The voltage of half-maximal open probability of ClC-1 was shifted by ∼60 mV per 10-fold increase of benzoate concentration. No significant difference in the slope was found at different internal pH levels (Fig. 5B). Analysis of the current kinetics revealed that the time constants of both exponential components become faster (Fig. 5C and D), and that the relative contribution of each component of the current is significantly changed (Fig. 6). These effects were concentration related. While the relative amplitude of the slow exponential component appears to have a complex dependence on benzoate (Fig. 6B), this may be an artefact, as with an internal concentration of 15 mM benzoate, the fast time constant is decreased below 2 ms which makes it difficult to record it faithfully, and underestimation of its contribution would overestimate the relative contribution of the second component. At higher concentrations of benzoate in the internal solution (30-60 mM) the open-channel block became apparent, as the amplitude of the inward current was reduced during the first few minutes of intracellular perfusion (not shown).
Figure 5. Dependence of ClC-1 gating on the concentration of internal benzoate.
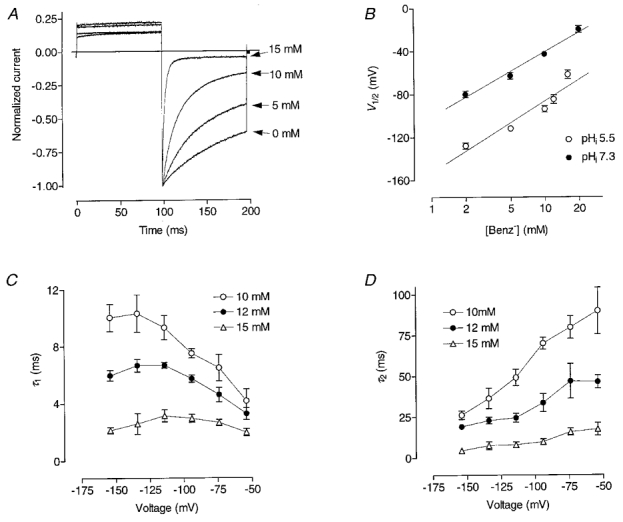
A, ClC-1 currents recorded from different cells in response to -120 mV steps after a prepulse to +40 mV and normalised to the peak current amplitude. B, dependence of V½ on the internal benzoate concentration. The slope of the lines are 66.5 ± 7.8 mV (pHi 5.5) and 60.1 ± 4.5 mV (pHi 7.2) per decade of internal benzoate change. C and D, voltage dependence of the fast (C) and slow (D) time constants at different concentrations of benzoate in the pipette solution. Internal pH was 5.5; the benzoate concentration in the pipette solution is indicated next to the keys in A, C and D.
Figure 6. Effect of internal benzoate on the relative amplitudes of the inward current components.
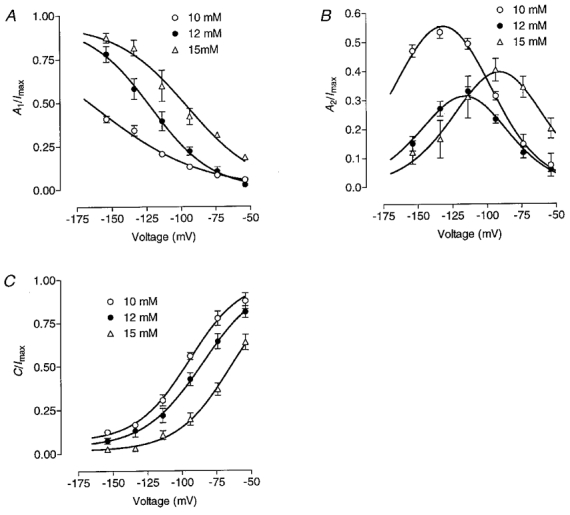
Voltage dependence of the relative amplitudes of the fast exponential (A1), slow exponential (A2) and non-deactivating (C) components (see Methods, eqn (2)) of the inward current at different internal benzoate concentrations are presented in panels A, B and C, respectively. Internal pH was 5.5; the benzoate concentration in the pipette solution is indicated next to the keys. Continuous lines represent fits of the fast and steady-state components by Boltzmann distributions B1 and B2 and of the slow component by 1 - (B1+B2) in each case.
Almost identical changes in the parameters describing ClC-1 current kinetics can be achieved by raising internal pH above 5.5. Similarly to increasing internal benzoate, raising internal pH leads to a faster and more complete deactivation of the inward current (Fig. 7A, C and D) and to the shift of the apparent Po curves (Fig. 7B). Relative amplitudes of the inward current components are also affected in a very similar way (Fig. 8).
Figure 7. Dependence of ClC-1 gating on the internal pH.
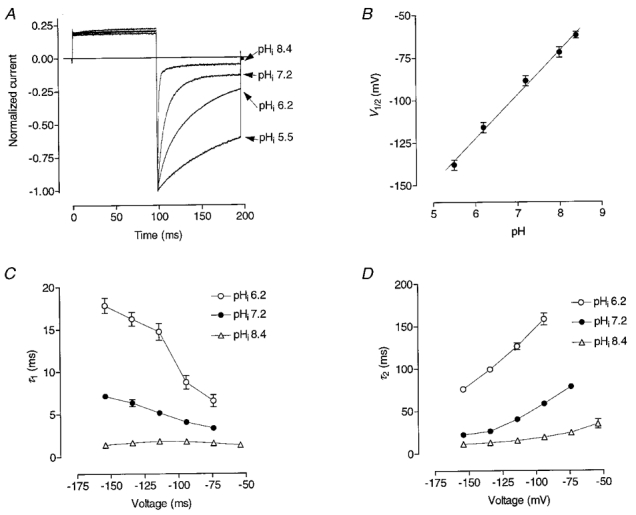
A, ClC-1 currents recorded from different cells in response to -120 mV steps after a prepulse to +40 mV and normalised to the peak current amplitude. B, dependence of V½ on pHi. The slope of the line is 25.9 ± 1.8 mV per unit of the internal pH change. C and D, voltage dependence of the fast (C) and slow (D) time constants at different pH of the pipette solution.
Figure 8. Effect of internal pH on the relative amplitudes of the inward current components.
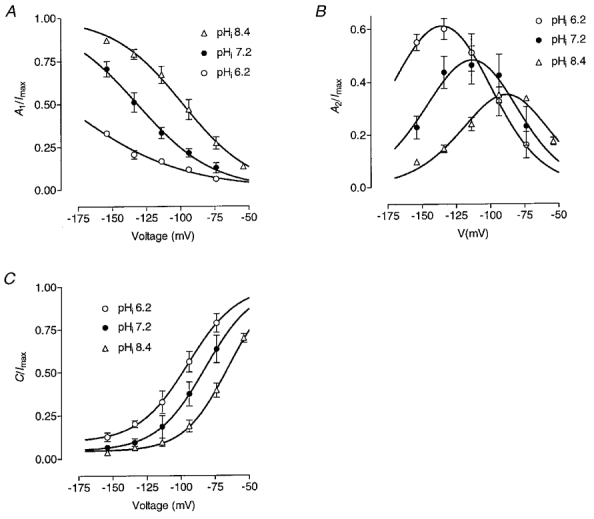
Voltage dependence of the relative amplitudes of the fast exponential (A1), slow exponential (A2) and non-deactivating (C) components (see Methods, eqn (2)) of the inward current at different internal pH are presented in panels A, B and C, respectively. Continuous lines represent fits of the fast and steady-state components by Boltzmann distributions B1 and B2 and of the slow component by 1 - (B1+B2) in each case.
It has been shown previously that protonation of the channel from the external side prevents ClC-1 fast deactivation gating (Rychkov et al. 1996; Fahlke et al. 1996), leaving a large non-deactivating inward current component. Changing internal pH in this condition does not have any effect on the inward current (Rychkov et al. 1996). In view of the high level of similarity between the effects of increasing internal benzoate and of raising internal pH, we further explored this relationship under the condition of low external pH. With external pH 7.4 and internal pH 5.5, the addition of 12 mM benzoate to the inside of the cell produced rapidly deactivating currents almost indistinguishable from the control (compare Figs 2A and 9A). When the external pH was lowered to 5.5, benzoate could not induce deactivation (Fig. 9B) and currents appeared the same as those seen with these values of pHo and no benzoate inside (see Fig. 4 in Rychkov et al. 1996). Thus an increased rate of deactivation of ClC-1 can be produced by elevating internal pH or benzoate concentration only when the channel is not protonated from the external side. It appears, therefore, that internal benzoate does not simply induce an open pore block.
Figure 9. The dependence of ClC-1 currents on the external pH in the presence of 12 mM benzoate in the internal solution.
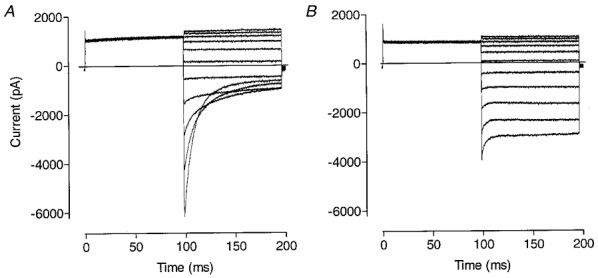
Currents were recorded in response to the same voltage protocol as in Fig. 2 with the external pH of 7.5 (A), and 5.5 (B). The internal pH in these experiments was 5.5.
DISCUSSION
Permeability sequence
Partitioning of an ion between water and the channel pore generally depends on the balance between the free energy of hydration and the energy of electrostatic interaction of the ion with the protein structures within the channel pore. According to the Eisenman equilibrium theory, water-channel partitioning of an ion is dominated by the hydration energy if the electrostatic interaction with the channel is weak, and bigger ions, which are more easily dehydrated, are favoured. Ion binding in the channel pore follows, in this case, the so-called weak field-strength sequence (Eisenman, 1965; Eisenman & Horn, 1983). If smaller ions are preferred it means that ion binding is dominated by the energy differences of electrostatic interaction of different ions with the site rather than by their differences in the energy of dehydration; in this case ion binding follows the strong field-strength sequence. The permeability sequence for halides in ClC-1 (Cl− ≫ Br− ≫ I− ≫ F−) (Rychkov et al. 1998) corresponds, in terms of the Eisenman theory (Eisenman, 1965), to anion binding to a cationic site of a moderately strong field strength (Eisenman sequence 4). In contrast, the ability of foreign anions to block the ClC-1 channel pore (SCN− ≫ I− ≫ ≫ Br−) (Rychkov et al. 1998) follows the weak field-strength Eisenman sequence 1 or so called ‘lyotropic series’ (Dani et al. 1983). This is strikingly different from the other Cl− channels including cystic fibrosis transmembrane conductance regulator (CFTR), GABA receptor, glycine receptor and epithelial Cl− channels, where both the permeability sequence and binding follows the same lyotropic series (Smith et al. 1999). Surprisingly, the relative permeability for hydrophobic anions presented in this study (unlike the halides), was higher for the larger ions: benzoate ≫ hexanoate ≫ butyrate ≫ propionate ≫ formate, giving the impression that selectivity follows a weak field-strength sequence. The apparent inconsistency of this permeability sequence with a moderately strong field-strength selectivity centre proposed earlier (Rychkov et al. 1998) could be explained as follows. The organic anions tested here are asymmetric and the negative charge is associated with the carboxyl group (COO−) and is not distributed over the hydrophobic sidechain. Therefore, increasing the size of the hydrophobic component will hardly change the effective radius of the negative charge which interacts with the anion attracting groups in the channel pore. This fact is reflected in a strong deviation of the energy of hydration of polyatomic molecules from the Born model of a charged sphere in a polarisable medium; the free energy of hydration of formate is the same as that of propionate or even gluconate (Smith et al. 1999). As a result, the ‘electrostatic’ term in the energy of the transition between channel and water will be very similar for all anions in this study. The presence of a hydrophobic region, or in terms of the Born theory a region with a low dielectric constant, which could be a part of the outer binding site (Rychkov et al. 1998), would reduce the energy barrier and/or increase the well depth for anions with larger hydrophobic moieties. The permeability sequence for these anions would, therefore, coincide with an increase in the size of the hydrophobic sidechains, provided the size of the pore is large enough to accommodate the anion.
Permeation model
Current-voltage plots of instantaneous inward currents through the open ClC-1 in the presence of the hydrophobic anions applied externally (Fig. 1B) are, in a simple view, consistent with the appearance of a fast potential-dependent block by an anion from the inside or cation from the outside. This seems to be paradoxical because all previous work on ClC-1 suggests that channel properties do not depend on cations, and our hydrophobic anions were applied from the outside not the inside. It might be argued that the apparent voltage-dependent block is really due to an inability to resolve the faster kinetics in the presence of anions such as benzoate and hexanoate. Inflexions, however, appear in the I-V curves for other hydrophobic anions as well, even formate (at -160 mV), where the kinetics are much slower and quite resolvable (Fig. 1D). To find out if a particular energy profile for hydrophobic anions inside the channel pore can account for these effects on conductance a discrete-state permeation model, as described in Methods, was fitted to the instantaneous I-V relationships obtained in the presence of various concentrations of hexanoate. Inclusion of two anion binding sites in such a model was a necessary and sufficient requirement to fit the experimental data. A further constraint on the model was that none of the energy barriers could be made extremely high for hexanoate since it was measurably permeant. Due to the large number of the parameters the data do not allow their unique determination for this model; its qualitative features, however, are likely to be valid (Miller, 1999). Indeed it describes the experimental data quite well and the corresponding ‘energy profile’ has the following features (Fig. 10): both binding sites are located closer to the extracellular side of the channel and the outer binding site is effectively located at the outside, i.e. there is no voltage drop from the outer bath medium to this site. The strong inward rectification of currents in pure chloride solutions derives from the fact that the outer binding site has a much higher affinity for chloride than the inner site. At positive voltages, the rate-limiting step is the jump from the outer to the inner binding site (effective barrier of almost 20kT), whereas the inner binding site is cleared completely of Cl− and the outer binding site is practically always occupied. The jump from the outer to the inner binding site has only a small voltage dependence (effective electrical distance, δ= 0.1), therefore the I-V curve is flat in that region and the current is small because of the large effective barrier. At negative voltages (below -50 mV) the rate-limiting step is the jump from the inner to the outer binding site. The activation energy is, however, much smaller (11.6kT) such that the inward current (outward movement of Cl−) is much larger. In addition, due to the asymmetry of the electrical distances, the jump from inside to outside has a considerable voltage dependence (δ= 0.25). Other qualitative features of this model can explain both the invariance of the outward currents and the voltage-dependent reduction of inward currents by small additions of hexanoate. At positive voltages any hexanoate bound inside the channel is ‘cleared’ to pass to the inside. The rather high outer energy barrier for hexanoate in combination with the voltage independence of hexanoate entry from the outside thus makes Cl− movement from outside to inside strongly favoured. At negative voltages instead, hexanoate is ‘trapped’ in the outer binding site, preventing the passage of Cl−, coming from the inside, out through the channel. At progressively more negative potentials binding of hexanoate at the outer site is not changed, but the ‘clearance’ rate of hexanoate to the inside becomes smaller and consequently the block becomes stronger.
Figure 10. Modelling the permeation properties of ClC-1.
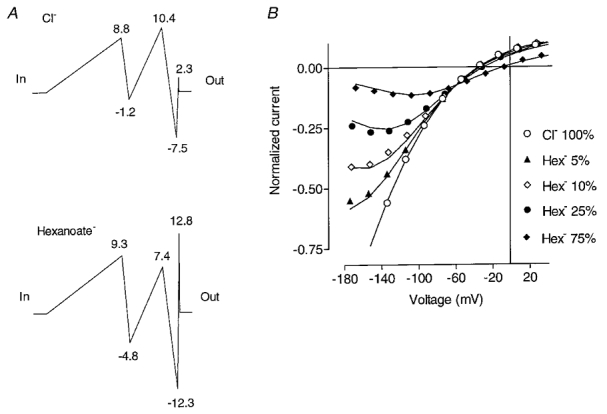
The energy profile for Cl− and hexanoate in the ClC-1 pore fitted assuming two binding sites (A), and I-V plots at different hexanoate concentrations (B), calculated by the proposed model. The energy levels relative to the external solution are presented on the charts in A, next to the corresponding barriers and wells in multiples of kT. The electrical distances for the transitions between the critical points of the energy profile, expressed as a fraction of total membrane potential drop, are as follows (in to out): 0.563; 0.058; 0.250; 0.110; 0.017; 0.002. Symbols in panel B represent the experimental data and lines represent the fits calculated from the model.
The different effects of various externally applied foreign anions on the conduction properties can be classified according to their respective affinities and barriers for the external superficial binding site: halides like bromide, and small less hydrophobic anions like formate do not bind strongly to the superficial site, and therefore their block of inward currents is relieved rather then exacerbated at more negative potentials. In contrast, more hydrophobic anions have a higher affinity for the superficial site, producing the above-described voltage-dependent block of inward currents.
Admittedly, this presents just one of many possible models that might be able to describe voltage-dependent block by hexanoate. Due to its simplifying assumptions (two binding sites, absence of interaction between ions in the pore, etc.) our model cannot explain all known properties of ClC-1 and should be considered only as a relatively simple starting point from which further theoretical consideration may be developed.
Effects of the external anions on gating
The voltage dependence of gating of the majority of ion channels can be attributed to the movement of an intrinsic part of the protein molecule known as the voltage sensor. However, in a number of cation channels, particularly in potassium inward rectifiers, voltage dependence of gating is comprised of the movements of an intrinsic voltage sensor and voltage-dependent binding of K+ ions inside the channel pore (Pennefather et al. 1992). In ClC-0, gating of the fast single protopore gate is rendered voltage dependent by a voltage-dependent movement of the permeating anion itself that serves as the source of the gating charge and no charge movement intrinsic to the protein seems to be involved (Pusch et al. 1995; Chen & Miller, 1996). The current models proposed to explain Cl− and voltage-dependent ‘fast’ gating in ClC-0 differ in details but there is a general agreement about the absence of an intrinsic voltage sensor (for review see Pusch, 1996). No such agreement is apparent for explaining voltage-dependent gating of the ClC-1 muscle chloride channel. While Rychkov et al. (1996, 1998) attributed most of the voltage dependence of ClC-1 to voltage-dependent movement of Cl− through the channel pore, Fahlke et al. (1995, 1996, 1997) proposed a gating model that includes a voltage sensor intrinsic to the ClC-1 channel protein. The former model is based on three main observations. First, ClC-1 has a high level of homology to ClC-0 and possibly a similar double-barrelled structure. Second, in extensive studies using site-directed mutagenesis, no structure in ClC-1 could be implicated as a voltage sensor so far. Third, the macroscopic open probability of ClC-1 is shifted by ∼54 mV to more positive potentials per 10-fold decrease in Cl− concentration, which makes it obvious that gating depends on Cl− binding in the channel pore, and which is very similar to the Cl− dependence of ClC-0. In light of these arguments in favour of a strong coupling between permeation and gating, we will base all interpretations of the effects of foreign anions on gating on the hypothesis that the channel is gated by the permeating anion.
The various effects of foreign anions on inward current kinetics and steady-state open probability can, in principle, be caused by several basic mechanisms. First, the relevant binding sites can have different affinities for the various anions; second, different anions bound to the ‘gating’ site might have a different ‘ability’ to effectively open the channel; third, foreign anions could prevent or favour the binding of Cl− to the ‘gating’ site either by binding there themselves or by binding to another site and thereby modulating the occupancy of the ‘gating’ site. In combination, these could make the dependence of the open probability on Cl− concentration quite complicated as happens in the presence of hydrophobic anions with aliphatic side-chains. At higher concentrations they all shift Po curves to the right by ∼40-52 mV per 10-fold decrease of Cl− concentration (Fig. 4), which indicates that their ability to open the channel is very low, and in fact, not significantly different from that of glutamate. However, the effects of these anions on the Po curve at lower concentrations are different and might be explained by the interference of these anions with Cl− binding to the regulatory gating site. The smaller propionate and butyrate anions appear to increase the affinity for Cl− at the gating site or increase the ability of bound Cl− ions to open the channel, while the larger hexanoate and octanoate anions have an opposite effect. At higher percentages of Cl− replacement all these effects are cancelled by the progressive lack of the channel opener.
Two other anions in this study, formate and benzoate, could replace Cl− at the gating site, which is indicated by only a small shift of the apparent Po curve as they increasingly replace Cl−. They both differ from the others by the absence of an aliphatic carbon chain in their structure; formate is missing a hydrophobic group, and benzoate is the only anion in this study with an aromatic ring. Presumably, the presence of an aliphatic hydrophobic group in propionate, butyrate, hexanoate and octanoate makes them ineffective in ClC-1 channel opening, probably due to some steric restriction at the gating site.
Single channel recording of ClC-1 revealed that the relaxation time constants of the fast and the slow gate closely match the time constants of the two exponential components of the inward currents (Saviane et al. 1999). Using this fact, significant reduction of the relative amplitude of the fast exponential component in the presence of formate can be interpreted as an increase of Po of the fast gate so current deactivation seen when 95 % of Cl− is replaced with this anion is mainly due to the slow gating. Changes of the amplitudes of the exponential components in the presence of other anions are not so straight forward, but it seems that both gating processes are affected by butyrate, benzoate and hexanoate.
Effects of the internal application of foreign anions
Gating of the ClC-1 channel depends not only on the external anions but also on the internal pH (Fahlke et al. 1996; Rychkov et al. 1996; Saviane et al. 1999). Lowering internal pH slows down the kinetics of current deactivation and shifts the voltage dependence of the apparent open probability to more negative potentials (Fig. 7); these changes are very similar to those caused by variations of the external Cl− concentration (Rychkov et al. 1996). Fahlke et al. (1996) postulated the presence in ClC-1 of a cytoplasmic blocking particle composed of titratable residues which would explain the dependence of current kinetics on the internal pH. It was also suggested that the particle is negatively charged and can interact with the channel pore. The concept of a negative cytoplasmic particle being involved in gating was supported by the results of Rychkov et al. (1996) who found that the addition of benzoate to the inside of the cell mimicked, at least superficially, the effect on gating of increasing pHi. This effect could be compatible with the gating particle at a physiological pHi being a soluble ion, OH−, rather than a negatively charged residue of the channel itself. No experimental data, so far, can decisively exclude either of the possibilities, but some indirect evidence argues against the fast gating involving a major conformational change of the channel protein. In ClC-0, in which fast gating shows a pH dependence similar to ClC-1 (Hanke & Miller, 1983), the temperature dependence of the fast gating (Q10∼2.2) is much lower than for a ‘ball-and-chain’ gating mechanism reported for other channels (Nobile et al. 1997). While Q10 is somewhat higher than the temperature dependence of a voltage-dependent block of an open pore, which is limited by diffusion (Q10∼1.5), it does not rule out a mechanism in which OH− binding from the internal side of the channel is accompanied by some conformational change of the channel protein (Pusch et al. 1997). It would be difficult to interpret similar data for ClC-1 because in this channel the fast gating cannot be separated from the slow gating as clearly as it can for ClC-0.
In the present study, we have analysed in detail the characteristics of the block produced by benzoate to determine if they are identical to those of fast gating of the channel. Increasing the internal benzoate concentration has very similar effects to increases in pHi on the amplitudes of all components of the inward current as well as on the time constants of both exponentials (Figs 5–8). In addition, both fast gating and block by benzoate are abolished by decreasing pHo (Fig. 9B, and Fig. 4A in Rychkov et al. 1996) with a pKa of ∼6. By contrast, the effect of pHi on V½ does not resemble what one would expect from titration of a charged group on a protein, the dependence is linear and does not have a clear pKa (Fig. 7B).
Regardless of the exact nature of the blocking particle, results shown in Fig. 9 strongly suggest that the mechanism of its action cannot be viewed as a simple voltage-dependent block of the open pore. Protonation of the channel from the extracellular side of the membrane abolishes channel deactivation at negative potentials at any internal pH and also abolishes the deactivation caused by internal benzoate. This additional positive charge somewhere in the channel would not be expected to reduce benzoate binding, and on the contrary, at low external pH, block by external benzoate is increased severalfold (authors’ unpublished observation). Therefore it seems logical to suggest that binding of benzoate from the inner side of the channel somehow modulates the affinity of the intrapore gating site for external Cl−. Similarly, modulation of that site by OH− or some intrinsic blocking particle binding at the internal mouth of the channel seems to be one of the most probable reasons for ClC-1 gating dependence on the internal pH.
In summary, we have shown for the first time that: (a) larger hydrophobic anions are more permeant through ClC-1 than smaller ones; (b) all anions studied, applied from outside, show a voltage-dependent block of inward current, intensified by more negative potentials; (c) a two-site three-barrier permeation model is sufficient to describe strong rectification of ClC-1 currents and potential-dependent block by hydrophobic anions; (d) anions with aliphatic side-chains applied externally at high concentrations shift apparent Po curves to more positive potentials similarly to glutamate; at lower concentrations their effect on Po depends on the size of the anion; (e) effects of the internal application of benzoate on inward current kinetics are very similar to that of increasing internal pH; and (f) effects of both, changing the internal benzoate concentration and varying internal pH, are abolished by channel protonation from the external side. The results of this study allowed us to make the following conclusions: (a) ClC-1 selectivity for hydrophobic anions is dominated by their interaction with a hydrophobic region in the external mouth of the pore; (b) anions with aliphatic side-chains constitute a separate group of measurably permeant anions which are unable to gate the channel, but which can modulate ClC-1 gating from the external side of the pore by changing the Cl− binding at the gating site, changing its accessibility or changing its gating efficacy; (c) benzoate applied internally can replace the cytoplasmic blocking particle which constitutes part of the gating mechanism sensitive to the internal pH; (d) the most probable mechanism of action of the cytoplasmic negatively charged particle, which could possibly be the OH− ion, is once again modulation of the apparent affinity of the gating site for Cl−.
Acknowledgments
We are grateful to Professor T. J. Jentsch and Dr K. Steinmeyer of the Centre for Molecular Neurobiology, Hamburg, for providing the rat ClC-1 clone. This work was supported by the Neuromuscular Research Foundation of the Muscular Dystrophy Association of South Australia, the Australian Research Council (to A.H.B. and M.L.R.) and Telethon Italy, Grant 1079, (to M.P.).
References
- Aromataris EC, Astill DStJ, Rychkov GY, Bryant SH, Bretag AH, Roberts ML. Modulation of the gating of ClC-1 by S-(-) 2-(4-chlorophenoxy) propionic acid. British Journal of Pharmacology. 1999;126:1375–1382. doi: 10.1038/sj.bjp.0702459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson OS, Koeppe RED. Molecular determinants of channel function. Physiological Reviews. 1992;72:S89–158. doi: 10.1152/physrev.1992.72.suppl_4.S89. [DOI] [PubMed] [Google Scholar]
- Astill DStJ, Rychkov G, Clarke JD, Hughes BP, Roberts ML, Bretag AH. Characteristics of skeletal muscle chloride channel ClC-1 and point mutant R304E expressed in Sf-9 insect cells. Biochimica et Biophysica Acta. 1996;1280:178–186. doi: 10.1016/0005-2736(95)00281-2. [DOI] [PubMed] [Google Scholar]
- Barry PH. JPCalc, a software package for calculating liquid junction potential corrections in patch-clamp, intracellular, epithelial and bilayer measurements and for correcting junction potential measurements. Journal of Neuroscience Methods. 1994;51:107–116. doi: 10.1016/0165-0270(94)90031-0. [DOI] [PubMed] [Google Scholar]
- Birnir B, Tierney ML, Howitt SM, Cox GB, Gage PW. A combination of human α1 and β1 subunits is required for formation of detectable GABA-activated chloride channels in Sf-9 cells. Proceedings of the Royal Society. 1992;B 250:307–312. doi: 10.1098/rspb.1992.0163. [DOI] [PubMed] [Google Scholar]
- Bormann J, Hamill OP, Sakmann Sakmann. Mechanism of anion permeation through channels gated by glycine and γ-aminobutyric acid in mouse cultured spinal neurones. The Journal of Physiology. 1987;385:243–286. doi: 10.1113/jphysiol.1987.sp016493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T-Y, Miller C. Nonequilibrium gating and voltage dependence of the ClC-0 Cl− channel. Journal of General Physiology. 1996;108:237–250. doi: 10.1085/jgp.108.4.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani JA, Sanchez JA, Hille Hille. Lyotropic anions: Na+ channel and Ca2+ electrode response. Journal of General Physiology. 1983;81:255–281. doi: 10.1085/jgp.81.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenman G. Some elementary factors involved in specific ion permeation. Proceedings of Physiological Sciences. 1965;4:489–506. [Google Scholar]
- Eisenman G, Horn R. Ionic selectivity revisited: the role of kinetic and equilibrium processes in ion permeation through channels. Journal of Membrane Biology. 1983;76:197–225. doi: 10.1007/BF01870364. [DOI] [PubMed] [Google Scholar]
- Fahlke C, Dürr C, George AL. Mechanism of ion permeation in skeletal muscle chloride channels. Journal of General Physiology. 1997;110:551–564. doi: 10.1085/jgp.110.5.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahlke C, Rosenbohm A, Mitrovic N, George AL, Rüdel R. Mechanism of voltage-dependent gating in skeletal muscle chloride channels. Biophysical Journal. 1996;71:695–706. doi: 10.1016/S0006-3495(96)79269-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahlke C, Rüdel R, Mitrovic N, Zhou M, George AL. An aspartic acid residue important for voltage-dependent gating of human muscle chloride channel. Neuron. 1995;15:463–472. doi: 10.1016/0896-6273(95)90050-0. [DOI] [PubMed] [Google Scholar]
- Halm DR, Frizzell RA. Anion permeation in an apical membrane chloride channel of a secretory epithelial cell. Journal of General Physiology. 1992;99:339–366. doi: 10.1085/jgp.99.3.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanke W, Miller C. Single chloride channel from Torpedo electroplax: activation by protons. Journal of General Physiology. 1983;82:25–45. doi: 10.1085/jgp.82.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. Ionic Channels of Excitable Membranes. 2. Sunderland, MA, USA: Sinauer Associates, Inc; 1992. pp. 348–368. [Google Scholar]
- Leo A, Hansch C, Elkins D. Partition coefficients and their uses. Chemical Reviews. 1971;71:525–616. [Google Scholar]
- Linsdell P, Tabcharani JA, Hanrahan JW. Multi-ion mechanism for ion permeation and block in the cystic fibrosis transmembrane conductance regulator chloride channel. Journal of General Physiology. 1997a;110:365–377. doi: 10.1085/jgp.110.4.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linsdell P, Tabcharani JA, Rommens JM, Hou Y-X, Chang X-B, Tsui L-C, Riordan JR, Hanrahan JW. Permeability of wild-type and mutant cystic fibrosis transmembrane conductance regulator chloride channels to polyatomic anions. Journal of General Physiology. 1997b;110:355–364. doi: 10.1085/jgp.110.4.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C. Ionic hopping defended. Journal of General Physiology. 1999;113:783–787. doi: 10.1085/jgp.113.6.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobile M, Olcese R, Toro L, Stefani E. Fast inactivation of Shaker K+ channels is highly temperature dependent. Experimental Brain Research. 1997;114:138–142. doi: 10.1007/pl00005613. [DOI] [PubMed] [Google Scholar]
- Pennefather P, Oliva C, Mulrine N. Origin of the potassium and voltage dependence of the cardiac inwardly rectifying K-current (IK1) Biophysical Journal. 1992;61:448–462. doi: 10.1016/S0006-3495(92)81850-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch M. Knocking on channel's door: the permeating chloride ion acts as the gating charge in ClC-0. Journal of General Physiology. 1996;108:233–236. doi: 10.1085/jgp.108.4.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch M, Jordt SE, Stein V, Jentsch TJ. Chloride dependence of hyperpolarization-activated chloride channel gates. The Journal of Physiology. 1999;515:341–353. doi: 10.1111/j.1469-7793.1999.341ac.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch M, Ludewig U, Rehfeldt A, Jentsch TJ. Gating of the voltage-dependent chloride channel ClC-0 by the permeant anion. Nature. 1995;373:527–531. doi: 10.1038/373527a0. [DOI] [PubMed] [Google Scholar]
- Pusch M, Steinmeyer K, Jentsch TJ. Low single channel conductance of the major skeletal muscle chloride channel, ClC-1. Biophysical Journal. 1994;66:149–152. doi: 10.1016/S0006-3495(94)80753-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rychkov GY, Pusch M, Astill DStJ, Roberts ML, Jentsch TJ, Bretag AH. Concentration and pH dependence of skeletal muscle chloride channel ClC-1. The Journal of Physiology. 1996;497:423–435. doi: 10.1113/jphysiol.1996.sp021778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rychkov GY, Pusch M, Roberts ML, Jentsch TJ, Bretag AH. Permeation and block of the skeletal muscle chloride channel, ClC-1, by foreign anions. Journal of General Physiology. 1998;111:653–665. doi: 10.1085/jgp.111.5.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saviane C, Conti F, Pusch M. The muscular chloride channel ClC-1 has a double barrelled appearance that is differentially affected in dominant and recessive myotonia. Journal of General Physiology. 1999;113:457–468. doi: 10.1085/jgp.113.3.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SS, Steinle ED, Meyerhoff ME, Dawson DC. Cystic fibrosis transmembrane conductance regulator: physical basis for lyotropic anion selectivity patterns. Journal of General Physiology. 1999;114:799–817. doi: 10.1085/jgp.114.6.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodbury JW, Miles PR. Anion conductance of frog muscle membranes: one channel, two kinds of pH dependence. Journal of General Physiology. 1973;62:324–353. doi: 10.1085/jgp.62.3.324. [DOI] [PMC free article] [PubMed] [Google Scholar]