

Abstract
Exposure of hippocampal neurones to glutamate at toxic levels is associated with a profound collapse of mitochondrial potential and deregulation of calcium homeostasis. We have explored the contributions of reactive oxygen species (ROS) to these events, considered to represent the first steps in the progression to cell death.
Digital imaging techniques were used to monitor changes in cytosolic Ca2+ concentration ([Ca2+]c; fura-2FF) and mitochondrial potential (Δψm; rhodamine 123); rates of ROS generation were assessed using hydroethidium (HEt); and membrane currents were measured with the whole-cell configuration of the patch clamp technique.
Inhibitors of lipid peroxidation (trolox plus ascorbate) and scavengers of superoxide or hydrogen peroxide (manganese(III) tetrakis(4-benzoic acid) porphyrin (MnTBAP) and TEMPO plus catalase), had only minimal impact on the mitochondrial depolarisation and the sustained increase in [Ca2+]c during and following a 10 min exposure to glutamate.
The antioxidants completely suppressed ROS generated by xanthine with xanthine oxidase. No significant increase in ROS production was detected with HEt during a 10 min glutamate exposure.
A combination of antioxidants (TEMPO, catalase, trolox and ascorbate) delayed but did not prevent the glutamate-induced mitochondrial depolarisation and the secondary [Ca2+]c rise. However, this was attributable to a transient inhibition of the NMDA current by the antioxidants.
Despite their inability to attenuate the glutamate-induced collapse of Δψm and destabilisation of [Ca2+]c homeostasis, the antioxidants conferred significant protection in assays of cell viability at 24 h after a 10 min excitotoxic challenge. The data obtained suggest that antioxidants exert their protective effect against glutamate-induced neuronal death through steps downstream of a sustained increase in [Ca2+]c associated with the collapse of Δψm.
The accumulation of glutamate in the extracellular space in the CNS plays a major part in extending the cell death following a period of anoxia or ischaemia beyond the immediate ischaemic focus. This glutamate toxicity has been clearly attributed to a massive influx of Ca2+ through NMDA and non-NMDA channels and a sustained increase in [Ca2+]c, which initiates the exitotoxic processes culminating in a delayed neuronal death (see review by Choi & Rothman, 1990). It has become almost dogma that free-radical species (reactive oxygen species or ROS) produced in neurones during a toxic glutamate challenge play a central role in these processes. This view has emerged as a result of experiments involving a number of different experimental approaches. Thus, increased superoxide production has been detected using spin traps and electron paramagnetic resonance (Lafon-Cazal et al. 1993; Dugan et al. 1995), while the neuro-protective effects of antioxidants have been demonstrated repeatedly (Dykens et al. 1987; Monyer et al. 1990; Patel et al. 1996; Ciani et al. 1996; Dugan et al. 1997; Carriedo et al. 1998). A substantial body of work has also involved the use of the fluorescence indicators of superoxide or hydroxyl radicals: hydroethidine, dihydro-rhodamine 123 and dichlorodihydrofluorescein (Dugan et al. 1995; Reynolds & Hastings, 1995; Bindokas et al. 1996; Perez Velazquez et al. 1997; Sengpiel et al. 1998). It has then been argued that ROS impair plasma membrane ionic transport systems, including ion channels, ion pumps and ion exchangers (for review, see Kourie, 1998), and so may be responsible for the impaired ionic homeostasis that seems to precede ATP depletion. Furthermore, in isolated mitochondria, ROS and high intramitochondrial [Ca2+] may act together to trigger the opening of the mitochondrial permeability transition pore (mPTP) (Zoratti & Szabo, 1995; Ankarcrona et al. 1996; Crompton, 1999), perhaps accounting for the profound loss of mitochondrial membrane potential (Δψm) seen in some models of excitotoxicity.
In experiments with cerebellar granule cells (Khodorov et al. 1996a) and hippocampal neurones (Vergun et al. 1999), we have demonstrated a striking correlation between glutamate-induced deterioration of [Ca2+]c homeostasis and the collapse of Δψm. Thus in the majority of hippocampal neurones maintained in culture for more than ∼11 days (> 11 days in vitro - DIV), exposure to glutamate for 10 min caused a profound mitochondrial depolarisation associated with a secondary increase of [Ca2+]c followed by a sustained high [Ca2+]c plateau that remained despite washout of the glutamate. We have shown that the production of NO is strongly implicated in generating these responses (Keelan et al. 1999), but the possibility of an additional contribution by other free radical species, perhaps superoxide, which could combine with NO to form peroxynitrite, remains.
In order to explore further the role of ROS in this response, we have studied the impact of an array of different antioxidants on the cellular response to glutamate. These have included: MnTBAP (a superoxide dismutase mimic and hydrogen peroxide (H2O2) scavenger), TEMPO (a cell-permeable nitroxide spin trap), catalase (a scavenger of H2O2), and the analogue of vitamin E trolox, with ascorbate (which prevents lipid peroxidation). Previously, a mixture of these antioxidants consisting of TEMPO, catalase, trolox and ascorbate has been effectively employed to prevent mPTP opening by ROS released during illumination of cultured cortical atrocytes loaded with tetramethylrhodamine ethyl ester (Leyssens et al. 1995; Jacobson & Duchen, 1998; Duchen, 2000a). We fully expected that the antioxidants would also prevent or at least greatly diminish the mitochondrial depolarisation and corresponding neuronal Ca2+ overload following a toxic glutamate challenge. Much to our surprise, we have found that neither the inhibition of lipid peroxidation, nor the reduction of intracellular accumulation of superoxide (O2−) and H2O2 - confirmed using fluorescence techniques - could abolish the glutamate-induced collapse of Δψm or the deterioration of [Ca2+]c homeostasis, although both manipulations had a significantly protective effect against the toxic glutamate exposure in terms of cell viability and survival. Therefore, remarkably, it seems that antioxidants can effectively decrease the glutamate-induced neurotoxicity even without a concurrent protection of neurones against a severe destabilisation of [Ca2+]c homeostasis and mitochondrial dysfunction. We also found that a profound delay in mitochondrial depolarisation caused by a combination of antioxidants was attributable to the inhibition of NMDA-induced currents, suggesting that great care must be taken in the interpretation of experiments using these agents.
METHODS
Tissue culture
A culture of mixed hippocampal neurones and glial cells was prepared as described previously (Vergun et al. 1999). In brief, rat pups 2-4 days post-partum were killed by cervical dislocation and hippocampi were dissected and placed in ice-cold Gey’s salt solution (Life Technologies, UK), containing 20 μg ml−1 gentamycin. The tissue was minced then placed in Ca2+- and Mg2+-free Hanks’ buffered saline solution (Life Technologies), containing 0.1 % trypsin for 15 min at 36°C, after which the trypsin was inactivated by washing with normal Hanks’ saline. Cells were dissociated by trituration, plated on poly-d-lysine-coated coverslips and cultured in Minimal Essential Medium with Earle’s salts (MEM; Life Technologies), containing 8 % horse serum (Life Technologies). Cytosine arabinoside (5 μm) was added for 24 h on the second day in vitro. Cultures were maintained at 36°C in a humidified atmosphere of 5 % CO2 and 95 % air. Cultures were fed twice a week with fresh medium. Experiments were carried out on 11-15 DIV cultures.
Imaging studies
For [Ca2+]c measurements, cells were incubated for 30 min at room temperature with 5 μm fura-2FF and 0.005 % Pluronic in a Hepes-buffered salt solution (referred to below as recording saline, RS) composed of (mm): 156 NaCl, 3 KCl, 2 MgSO4, 1.25 KH2PO4, 2 CaCl2, 10 glucose and 10 Hepes, pH adjusted to 7.35 with NaOH. For simultaneous measurement of [Ca2+]c and Δψm, rhodamine 123 (Rh123; 10 μg ml−1) was added to the medium during the last 15 min of the fura-2FF incubation period. The cells were then washed with RS and placed on an epifluorescence inverted microscope (Nikon diaphot) equipped with a ×20 fluorite objective (NA 0.7). [Ca2+]c and Δψm were monitored in single cells using excitation light provided by a xenon arc lamp, the beam passing sequentially through 10 nm bandpass filters centred at 340, 380 and 490 nm housed in a computer-controlled stepping filter wheel (Cairn Research, Sittingbourne, Kent). Sequential excitation of cells at 340 and 380 nm allowed ratiometric measurements of fura-2FF fluorescence, while excitation at 490 nm allowed measurement of Rh123 fluorescence (see Keelan et al. 1999). Data were acquired every 15 s, and cells were protected from photo-toxicity by interposing a shutter in the light path between exposures. Emitted fluorescence light was reflected through a 515 nm long-pass filter to a frame transfer cooled CCD camera with an active sensor of 800 pixels × 600 pixels (Digital Pixel Ltd, UK), and digitised to 12 bits. All imaging data were collected and analysed using Kinetic Imaging software (Liverpool, UK). The fura-2FF data have not been calibrated in terms of [Ca2+]c because of the uncertainty arising from the use of different calibration techniques. Accumulation of Rh123 in polarised mitochondria quenches the fluorescence signal; in response to mitochondrial depolarisation the dye redistributes throughout the cell and fluorescence emission is dequenched; an increase in Rh123 signal therefore indicates mitochondrial depolarisation (Duchen & Biscoe, 1992).
HEt, also known as dihydroethidium, was used to assay O2− production. HEt was prepared as a 10 μg ml−1 stock in dry, N2-sparged dimethylsulfoxide (DMSO) and stored under N2 at -20°C. Working stocks were prepared from this solution by diluting in dry DMSO to 1 μg ml−1 and were also kept under N2. HEt was present at a concentration of 0.5-3.2 μm in all solutions during these experiments, and no preincubation was used, in order to limit the intracellular accumulation of the oxidised product. Ethidium fluorescence was excited at 490 nm, emitted fluorescence was measured at > 515 nm, and data were acquired every 10-15 s.
All experiments were performed at room temperature unless otherwise indicated. All drugs were applied by two changes of bath solution in order to ensure that exchange was complete, and in most experiments the NMDA antagonist MK801 (15 μm) was added to the washout solution after exposure to glutamate to limit the effects of endogenous glutamate release in the post-glutamate period (see Vergun et al. 1999). The antioxidants used had no effect at all on the fluorescence of fura-2FF or Rh123 when measured in a cuvette in solution.
Patch clamp measurements of ionic currents
The electrophysiological experiments were carried out on pyramidal neurones, acutely isolated from the CA-1 region of rat hippocampus, using the ‘vibrodissociation technique’ (Vorobjev, 1991). The experiments were begun after 3 h of incubation of the hippocampal slices in a medium containing (mm): 124 NaCl, 3 KCl, 1.4 CaCl2, 2 MgCl2, 10 glucose and 26 NaHCO3. The solution was bubbled with carbogen and maintained at 32°C. During the whole period of isolation and current recording, nerve cells were washed with a Mg2+-free solution (mm): 140 NaCl, 5 KCl, 2 CaCl2, 15 glucose and 10 Hepes, pH 7.3. Fast replacement of solutions was achieved using a simple perfusion system (Vorobjev et al. 1996). The time constant of the solution exchange, measured by the method of sodium concentration jumps (Vyklicky et al. 1990; Chen & Lipton, 1997), was 20-30 ms. The currents were recorded at 18°C in the whole-cell configuration using micropipettes made from Pyrex tubes and filled with an ‘intracellular’ solution (mm): 140 CsF, 4 NaCl and 10 Hepes, pH 7.2. The resistance of the filled micropipettes was 3-7 MΩ. The analog current signals were digitised at a frequency of 1 kHz.
Neurotoxicity experiments
Toxic exposure to glutamate (100 μm) was performed at room temperature for 10 min in Mg2+-free RS containing 10 μm glycine. Control cells were exposed to an exchange of the normal RS. After 10 min, the cells were washed twice to remove the glutamate, and the culture medium was replaced. The cells were returned to the incubator for 24 h. Mixtures of trolox and ascorbate or TEMPO and catalase were added for 45 min before this toxic exposure to the culture medium (36°C), during exposure to RS, and for 24 h after glutamate exposure, if it was required for the experimental protocol. Preincubation of the cells with MnTBAP for 30 min was performed at room temperature in RS; in this case the control cultures were exposed to RS at room temperature for the same period of time. Neurotoxicity was measured using propidium iodide (PI, 20 μm), which is normally excluded from viable cells but exhibits a red fluorescence in cells following a loss of membrane integrity, while Hoechst 33342 (4.5 μm), which binds to chromatin in intact cells, was used to count viable cells. Using phase contrast optics, a bright field image allows easy identification of neurones, which look quite different to the flatter glial component and also lie in a different focal plane, as they grow above the glial layer. The phase contrast image was used to identify the neurones and then their nuclei were counted - Hoechst 33342 stains all nuclei blue and the number of nuclei that also stain red with PI indicates the proportion of dead cells. We did not see fragmented nuclei, and it was easy to identify one nucleus per neuronal soma counted under phase contrast. A total number of 600-800 neurones were counted in 20-25 fields of each coverslip. Each experiment was repeated three times using separate cultures.
Chemicals
Fura-2FF was obtained from Teflabs (Austin, TX, USA). Rh123, PI and HEt were obtained from Molecular Probes. MnTBAP was obtained from Calbiochem (UK); a stock solution (20 mm) was prepared in 75 mm NaOH. Trolox was obtained from Aldrich; a stock solution (75 mm) was prepared in 75 % ethanol. Xanthine oxidase was obtained from Boehringer Mannheim Biochemical. Carbonyl cyanide p-trifluoro-methoxyphenyl hydrazone (FCCP) and all other chemicals were from Sigma.
Statistics
Statistical analysis was performed with the aid of the scientific and technical graphics computer program Microcal Origin 4.1 (Northampton, MA, USA). All the data are presented as means ±s.e.m., and comparisons were done by ANOVA, with P < 0.05 taken as significant, using the Dunn post-test.
RESULTS
changes in [ca2+]c and Δψm in response to glutamate treatment
Figure 1A illustrates an example of simultaneous measurement of fura-2FF ([Ca2+]c) and Rh123 (Δψm) signals in a field of 16 neurones before and after exposure of cells to glutamate for 10 min. In most cells, the application of 100 μm glutamate (in Mg2+-free, 10 μm glycine-containing solution) produced an initial [Ca2+]c peak followed by a secondary rise in [Ca2+]c to a high plateau value. In these cells the initial [Ca2+]c peak was followed by a relatively small mitochondrial depolarisation whereas the delayed, secondary increase of [Ca2+]c revealed with the low affinity [Ca2+]c indicator (see Vergun et al. 1999; Keelan et al. 1999) was associated with a delayed, secondary and profound collapse of Δψm. The secondary mitochondrial depolarisation coincided in time with the secondary [Ca2+]c increase. In all experiments, the amplitude of the glutamate-induced mitochondrial depolarisation was compared with the response to the mitochondrial uncoupler FCCP (1 μm), which dissipates Δψm completely. All data using Rh123 are presented normalised between zero, representing the resting Rh123 fluorescence, and unity, representing the maximal increase in fluorescence in response to FCCP. The magnitude of the glutamate-induced collapse of Δψm was similar to that caused by FCCP, indicating almost complete dissipation of Δψm in response to toxic glutamate exposure (and see Vergun et al. 1999).
Figure 1. Effect of trolox and ascorbate on glutamate-induced changes in Δψm and [Ca2+]c.
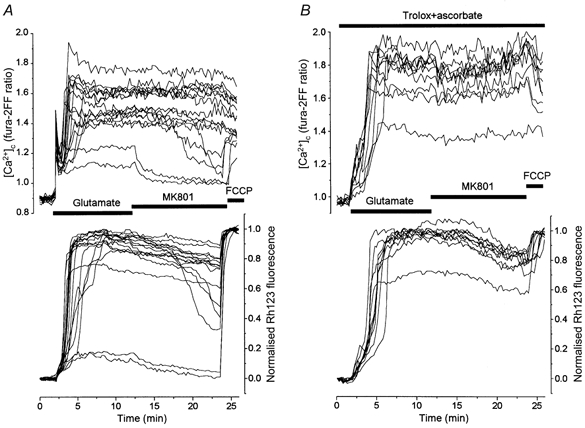
Changes in [Ca2+]c and Δψm in hippocampal neurones at 13 DIV during and after glutamate (100 μm, in Mg2+-free, 10 μm glycine-containing solution) exposure are shown. An increase in Rh123 fluorescence reflects mitochondrial depolarisation. The Rh123 fluorescence signal (F) is normalised between zero, representing the resting Rh123 fluorescence, and unity, representing the maximal increase in fluorescence (Fmax) in response to 1 μm FCCP. The NMDA channel blocker MK801 (15 μm) was added to the washout solution to exclude the effect of endogenous excitatory amino acid release (Vergun et al. 1999) on the post-glutamate [Ca2+]c recovery. A, [Ca2+]c and Δψm dynamics in a control experiment. B, the dynamics of [Ca2+]c and Δψm in neurones of a sister culture preincubated for 40 min with a mixture of trolox (750 μm) and ascorbate (1 mm) at room temperature; the antioxidants were present in all solutions during experiments.
In the majority of neurones, neither Δψm nor [Ca2+]c showed significant recovery after the termination of glutamate exposure. In a small proportion of neurones (2 of 16 shown in Fig. 1A), glutamate exposure caused only a small monophasic mitochondrial depolarisation, which recovered after the removal of glutamate from the medium associated with recovery of [Ca2+]c to baseline values. Qualitatively similar results were obtained in eight analogous experiments (n= 121 neurones). Within the temporal resolution of the measurements, the time taken for the onset of the secondary mitochondrial depolarisation and the secondary [Ca2+]c increase was identical, at 3.9 ± 2.0 min (n= 103 neurones). The peak amplitude of the glutamate-induced mitochondrial depolarisation amounted to 0.86 ± 0.14 (n= 103 neurones) of the amplitude of FCCP-induced mitochondrial depolarisation. As responses varied somewhat between cultures, matched cultures were used for all experiments involving antioxidant treatments. A more detailed quantitative description of the relationships between the glutamate-induced mitochondrial and [Ca2+]c responses has been given previously (Vergun et al. 1999).
effect of trolox and ascorbate on glutamate-induced changes in [ca2+]c and Δψm
It has been demonstrated by many groups that blockade of lipid peroxidation by different antioxidants may attenuate excitotoxic neuronal injury (Monyer et al. 1990; Nakao et al. 1996; Ciani et al. 1996; Carriedo et al. 1998). In order to investigate the relationship between this protective effect and the glutamate-induced loss of Δψm and deregulation of [Ca2+]c homeostasis, we examined the effect of agents that limit lipid peroxidation - ascorbate and trolox, an analogue of vitamin E - on the glutamate-induced changes in [Ca2+]c and Δψm. The neurones were treated with a mixture of 750 μm trolox and 1 mm ascorbate for 40 min at room temperature, after which glutamate was added to the antioxidant-containing solution for 10 min. These concentrations have been shown in our hands to be effective at limiting mitochondrial depolarisation by an imposed oxidant stress in other models (Leyssens et al. 1995; Jacobson & Duchen, 1998) and at protecting mitochondria from oxidative damage in other models of oxidative stress (e.g. see Brookes et al. 1998).
A representative experiment is illustrated in Fig. 1B. Exposure of matched control cells to glutamate for 10 min produced the usual biphasic changes in Δψm associated with a concurrent increase in [Ca2+]c in most of the cells. As shown in Fig. 1B, treatment of the cells with trolox and ascorbate had no significant impact on either the secondary [Ca2+]c increase or the loss of mitochondrial potential. Responses were variable between cultures, but the mean percentage of neurones exhibiting a small reversible mitochondrial depolarisation in response to glutamate alone was 11 % (n= 53 neurones) and was 10 % in the cells treated with glutamate in the presence of the antioxidants (n= 64 neurones). Similarly, the mean of the maximum fura-2FF ratio was not significantly altered by the antioxidant treatment, being 1.73 ± 0.03 (n= 58) compared with the control value of 1.86 ± 0.05 (n= 47). The maximum of the glutamate-induced change in Rh123 signal was also not significantly affected by the antioxidants, having a value of 0.92 ± 0.01 (n= 47) in the control cells and 0.84 ± 0.02 (n= 58) in the treated cells (P > 0.05). The time from the beginning of the glutamate application to the maximum of both mitochondrial depolarisation and [Ca2+]c in treated neurones was significantly (P < 0.001) longer than in control cells: 3.79 ± 0.17 min in treated cells (n= 58) and 2.52 ± 0.29 min in control (n= 47). Thus, trolox and ascorbate slightly delayed the development of both the secondary increase of [Ca2+]c and mitochondrial depolarisation, but did not prevent glutamate-induced mitochondrial dysfunction or deterioration of [Ca2+]c homeostasis. The basis for this delay will be discussed below, but is probably attributable to direct effects of the antioxidants on the NMDA receptor (see below).
effect of mntbap, tempo and catalase on glutamate-induced changes in [ca2+]c and Δψm
In order to investigate the role of O2− and H2O2 in the collapse of Δψm and the deterioration of [Ca2+]c homeostasis produced by glutamate we used TEMPO. TEMPO is a cell-permeable nitroxide that acts to catalyse O2− dismutation (Samuni et al. 1988; Krishna et al. 1992) and it has been shown to protect cells from oxidative damage in some model systems (Mitchell et al. 1990; Grinberg & Samuni, 1994). To avoid the toxic action of H2O2 generated by O2− dismutation, TEMPO (500 μm) was used in combination with catalase (250 U ml−1), a scavenger of H2O2. The cells were incubated with TEMPO and catalase for 45 min at room temperature and were then exposed to glutamate in the presence of these antioxidants. Figure 2B shows the changes in [Ca2+]c and Δψm in response to glutamate in the presence of TEMPO plus catalase. The maximum mitochondrial depolarisation in neurones treated with TEMPO and catalase was slightly smaller than in control (0.83 ± 0.02, n= 32, in TEMPO-catalase-treated neurones; 0.90 ± 0.01, n= 41, in control). The time from the beginning of the glutamate application to the maximum of both [Ca2+]c and mitochondrial depolarisation was not significantly different in TEMPO plus catalase (3.46 ± 0.36, n= 32) compared with control (3.88 ± 0.45, n= 41).
Figure 2. Effect of O2− and H2O2 scavengers on glutamate-induced changes in Δψm and [Ca2+]c.
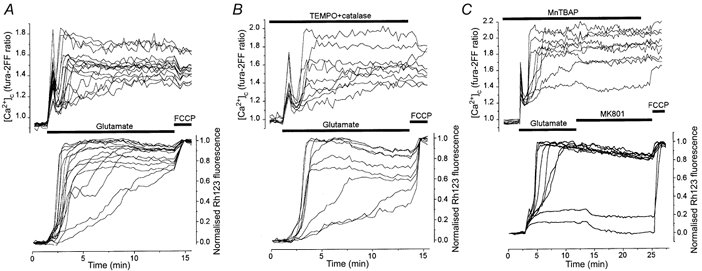
Simultaneous measurements of [Ca2+]c and Δψm in 14 DIV control neurones (A) and in sister neurones preincubated with TEMPO (500 μm) and catalase (250 U ml−1) mixture (B) or with 200 μm MnTBAP (C).
In addition to TEMPO and catalase, we also tested the action of MnTBAP, another scavenger of ROS. MnTBAP is a cell-permeable low molecular weight superoxide dismutase mimetic, which has been shown to protect several cellular systems against intracellularly generated O2− (Faulkner et al. 1994; Day et al. 1995). MnTBAP has also been shown to scavenge H2O2 and protect endothelial cells against H2O2-mediated injury (Day et al. 1997). This compound was protective against NMDA- and kainate-induced cell death in rat cortical cultures (Patel et al. 1996) and greatly reduced glutamate-induced delayed Ca2+ deregulation in rat cerebellar granule cells (Castilho et al. 1999). In the present experiments, cells were pretreated with 200 μm MnTBAP for 30-35 min at room temperature and MnTBAP was present in all solutions during the experiments. An example of such an experiment is presented in Fig. 2C. MnTBAP did not significantly change the maximal amplitude or the time from the beginning of glutamate application to the maximum mitochondrial depolarisation (0.86 ± 0.01 and 2.55 ± 0.49 min in control, n= 32 neurones; 0.96 ± 0.01 and 2.47 ± 0.29 min in MnTBAP, n= 56 neurones; 5 experiments). The second phase of the [Ca2+]c rise was also not apparently affected by the scavenger.
Measurements of ROS generation with HEt
In order to ensure that the antioxidant treatments were truly effective at scavenging superoxide, and to further examine the rate of ROS generation in response to glutamate, we used dihydroethidium. Dihydroethidium, also known as HEt, is oxidised by O2− to the fluorescent product ethidium. An increase in ethidium fluorescence is therefore considered to be a selective detector of O2− hyperproduction (Bindokas et al. 1996). However, as reported by Rottenberg (1984) and by Budd et al. (1997), ethidium is a lipophilic cation and accumulates into mitochondria where, at higher concentrations, its fluorescence is quenched. Mitochondrial depolarisation may therefore cause an increase in fluorescence that is independent of O2− production, in much the same way that Rh123 fluorescence increases with mitochondrial depolarisation (see Rottenberg, 1984). This ambiguity was avoided by using the lowest possible concentration of HEt compatible with an acceptable signal/noise ratio, and avoiding a preincubation period, which might allow time for oxidation of HEt and its accumulation into mitochondria. In our preparation, depolarisation of mitochondria with FCCP (1 μm) increased the ethidium fluorescence even when HEt was used at a concentration of 1 μm. It is necessary to emphasise that one should not expect to see an increase in O2− production following application of a mitochondrial uncoupler (e.g. see Budd et al. 1997). When the cells were loaded with a smaller (0.5 μm) concentration of HEt, which was not sensitive to FCCP, we could detect no significant increase in the rate of HEt oxidation in response to glutamate (data not shown, but see Fig. 3A). We wondered whether perhaps O2− might be scavenged by NO to form peroxynitrite, and so examined the response again in the presence of the inhibitor of nitric oxide synthase (NOS) N-nitro-l-arginine methyl ester (L-NAME, 1 mm), but still we were unable to detect any significant increase in the rate of HEt oxidation in response to glutamate (see Fig. 3A) - indeed, the data were well fitted by a straight line with a correlation coefficient of 0.997 throughout the control period and with no discernable deviation after exposure to glutamate.
Figure 3. Measurements of ROS generation in hippocampal neurones with HEt.
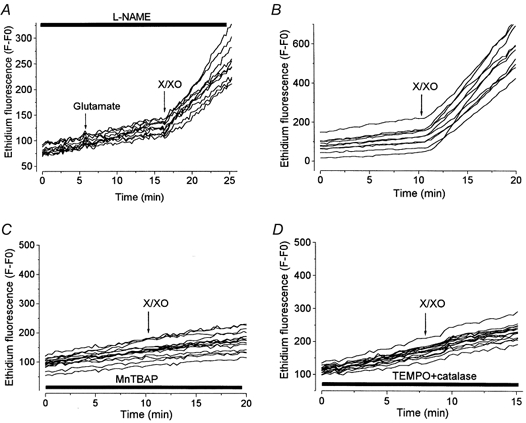
For all records, HEt was present throughout all experimental solutions, at 1 μm in A and at 3.2 μm in B-D. Xanthine (250 μm) and xanthine oxidase (50 mU ml−1) (X/XO) were used to induce O2− production in each case. In A, neurones were pretreated for 20 min with 1 mml-NAME, which was also present throughout as indicated by the bar. Application of glutamate had no effect on the HEt signal. In the experiment illustrated in C, cells were treated with 200 μm MnTBAP for 30 min before recording. The record shown in D comes from cells pretreated with 500 μm TEMPO and 250 U ml−1 catalase for 40 min.
To provide a positive control for the efficiency of HEt as a detection system and then to confirm the efficiency of the antioxidant systems, we used xanthine with xanthine oxidase (X/XO) as an O2−-generating system. Figure 3A and B shows the increase in ethidium fluorescence in the neurones following the application of 250 μm xanthine with 50 mU ml−1 xanthine oxidase. The slope of a linear regression line fitted to the HEt signals (all fitted with correlation coefficients > 0.99) showed an increase of ∼5-fold with X/XO (a mean of 5.1 times, s.e.m. of 0.66 in a sample of 10 cells). This superproduction of O2− was fully abolished by pretreatment of the cells with 200 μm MnTBAP for 30 min (Fig. 3C), demonstrating that MnTBAP can penetrate the cells and remove O2− effectively in this model. In order to confirm the efficiency of TEMPO as an O2−-scavenging system, we used X/XO-induced O2− generation similarly with HEt. Figure 3D shows that the mixture of TEMPO plus catalase under these conditions completely blocked the rise in the ethidium fluorescence induced by X/XO.
Thus, scavenging of O2− and H2O2 by MnTBAP and TEMPO plus catalase at concentrations that are able to remove a much greater oxidant load (generated by X/XO) than that seen with glutamate, nevertheless failed to influence the progression of the collapse of Δψm or the secondary rise in [Ca2+]c seen in response to exposure to glutamate. It is important also to emphasise here that we have not been able to identify any significant increase in HEt signal in response to glutamate under conditions in which FCCP did not cause an increase in HEt signal.
Effects of combined antioxidants (TEMPO, catalase, trolox and ascorbate) on glutamate-induced neuronal responses
Changes in [Ca2+]c and Δψm
As treatment of cells with antioxidants that prevented either the accumulation of ROS (MnTBAP, TEMPO and catalase) or lipid peroxidation (trolox and ascorbate) proved to be ineffective at preventing the glutamate-induced collapse of Δψm and [Ca2+]c overload, we decided to examine the effect of the full combination of antioxidants (TEMPO, catalase, trolox and ascorbate). Pretreatment of cells with this mixture of antioxidants for 40-50 min substantially changed the pattern of both mitochondrial and [Ca2+]c responses elicited by exposure to glutamate. Figure 4B illustrates a representative experiment, compared as usual to the response of a matched control culture (Fig. 4A). The cells were first preincubated for 40 min with the combined antioxidants, and were then treated with glutamate in the presence of the antioxidants, which also remained following glutamate washout. This caused a considerable reduction of the amplitude of the initial [Ca2+]c increase and of the rate of the secondary [Ca2+]c increase. Of special interest is that glutamate removal failed to stop this [Ca2+]c elevation and that despite the presence of antioxidants, [Ca2+]c still reached and stayed at a high plateau level. Mitochondrial depolarisation in the antioxidant-treated cells acquired a clearly biphasic time course. The initial small mitochondrial depolarisation appeared to remain at a low plateau level and then abruptly developed into the second phase of a profound depolarisation. The duration of the time delay between the beginning of glutamate application and the onset of the second phase of mitochondrial depolarisation varied among neurones as shown in Fig. 4B in the range 4.4-16.5 min, thus exceeding in some cases the duration of glutamate exposure. Despite such a delay, the mitochondria eventually depolarised fully and the signal remained practically unchanged despite glutamate washout. Qualitatively similar results were obtained in 10 analogous experiments. The mean time to the maximum of mitochondrial depolarisation in the antioxidant-treated cells was 10.16 ± 3.8 min (n= 126 neurones) as compared with 3.96 ± 2.9 min (n= 103) in control neurones. The maximal amplitude of the mitochondrial depolarisation in the presence of antioxidants was slightly, but significantly, smaller (0.74 ± 0.15, n= 94) than that in the control neurones (0.86 ± 0.14, n= 103), P < 0.01.
Figure 4. Effects of an antioxidant mixture on glutamate-induced changes in [Ca2+]c and Δψm.
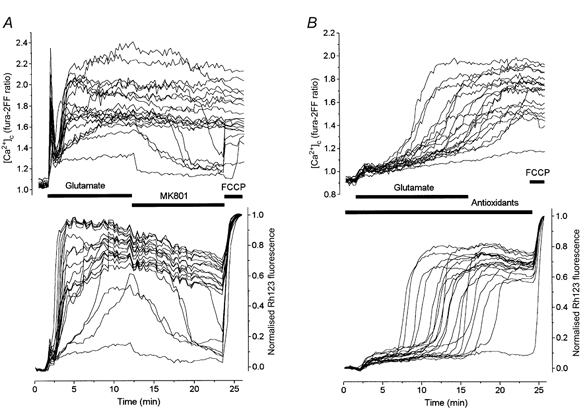
Neurones at 14 DIV were co-loaded with fura-2FF and Rh123 as shown above. A, control response to glutamate. B, response of cells exposed to a mixture of antioxidants containing TEMPO (500 μm), catalase (250 U ml−1), trolox (750 μm) and ascorbate (1 mm). These cells were incubated with the antioxidants for 40 min before glutamate application.
Further studies, however, revealed that pre-treatment and equilibration of cells with antioxidants was not a prerequisite for the manifestation of their inhibitory effect on the glutamate-induced changes in Δψm and [Ca2+]c. Figure 5 illustrates a representative experiment in which the mixture of the antioxidants was applied soon after the beginning of the glutamate treatment. Here exposure of cells to glutamate induced the usual biphasic increase in [Ca2+]c and accompanying mitochondrial depolarisation. Addition of antioxidants to the glutamate-containing solution then both acutely decreased the [Ca2+]c signal and induced the recovery of Δψm. This effect was only seen with the mixture of all antioxidants and not when the agents were used individually. The effect of the antioxidant mixture was reversible, and after washout of the antioxidants, [Ca2+]c again rose progressively and the mitochondria gradually depolarised.
Figure 5. A comparison of the inhibitory effect of antioxidants and the NMDA receptor antagonist AP-5 on the glutamate-induced changes in [Ca2+]c and Δψm.
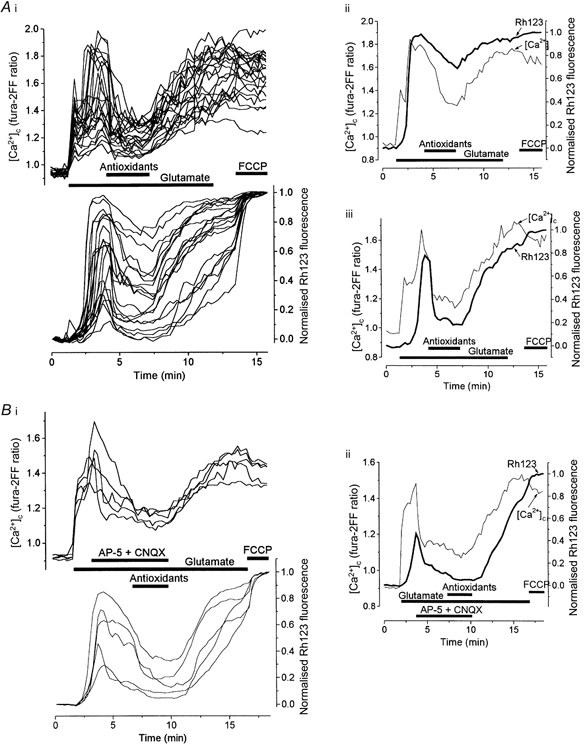
[Ca2+]c and Δψm were measured simultaneously in hippocampal neurones at 14 DIV (A) and at 12 DIV (B) during exposure to 100 μm glutamate. A, the full mixture of antioxidants (TEMPO, catalase, trolox and ascorbate) was added to the glutamate-containing solution 3 min after the beginning of the glutamate challenge. A family of records from all neurones in one field is presented in i, while superimposed traces reflecting the changes in [Ca2+]c and Δψm from two representative cells are shown in ii and iii. B, 500 μm AP-5 plus 75 μm CNQX were applied to the cells 2.5 min after the onset of glutamate exposure after which the perfusate was changed to the mixture of antioxidants as indicated. Changes in [Ca2+]c and Δψm are shown in five individual neurones in i, while records of [Ca2+]c and Δψm are superimposed for one representative cell in ii. This experiment clearly shows that at the beginning of a glutamate challenge the [Ca2+]c increase is readily reversible and that its decrease, caused either by the antioxidants or by AP-5, is followed by a more or less pronounced mitochondrial repolarisation. This evidently indicates that the mitochondrial depolarisation in this period is strongly dependent on sustained Ca2+ influx through glutamate receptor channels.
In contrast to the mixture of antioxidants, its ingredients, TEMPO, catalase, trolox and ascorbate, as well as the mixture of TEMPO, catalase and trolox without ascorbate when applied during glutamate exposure separately or in pairs caused only very small if any changes in [Ca2+]c and mitochondrial responses (not illustrated).
These observations raised the question whether the effects of antioxidants on the glutamate-induced changes in [Ca2+]c and Δψm really result from the interaction of these compounds with ROS or whether they are instead due to blockade of glutamate receptor channels. To this end we first of all compared the inhibitory effect of the antioxidants with that produced by the NMDA and non-NMDA receptor antagonists AP-5 (2-amino-5-phosphonopentanoic acid, 500 μm) and CNQX (6-cyano-7-nitroquinoxaline-2,3-dione, 75 μm) (Fig. 5B). Application of NMDA antagonists both decreased [Ca2+]c and restored Δψm, and addition of antioxidants caused a further reduction, which varied among the cells. Washout of the antioxidants led to a renewed rise in [Ca2+]c and mitochondrial depolarisation.
The suppression of the initial [Ca2+]c peak, the surprisingly immediate inhibitory effects of the combined antioxidants and the similarity between the effects of glutamate receptor antagonists and the mixture of antioxidants on the glutamate responses led us to wonder whether the antioxidants may act through some direct action on Ca2+ influx through the glutamate receptor-gated channels rather than through their action on ROS production. To test this supposition we carried out a series of direct measurements of the effects of antioxidants on NMDA-gated ionic currents induced either with their natural agonist aspartate or with glutamate.
Effects of antioxidants on ionic currents in NMDA channels
Whole-cell ionic currents in acutely isolated rat hippocampal neurones were elicited by fast application of 100 μm glutamate or 100 μm aspartate in Mg2+-free solution containing 3 μm glycine. Aspartate was used as it is the natural agonist at the NMDA receptor, with a higher affinity for NMDA receptors (Kd= 7.5 μm) than synthetic NMDA (Kd= 16 μm; Patneau & Mayer, 1990). At the holding membrane potential, Vh=−100 mV, glutamate and aspartate induced an inward current, which, after an initial fast rise, decreased to a plateau level, IC.
Figure 6 illustrates representative currents evoked by glutamate (100 μm, Fig. 6A) and aspartate (100 μm, Fig. 6B) co-applied either with one of the antioxidants used or with the mixture of all these compounds as indicated. Mean values of the steady-state current in the presence of antioxidants, IB, are plotted as a function of the control current value in Fig. 6C. TEMPO (500 μm), ascorbate (1 mm) and trolox (750 μm) each caused a moderate inhibition of both glutamate- and aspartate-induced currents. In the case of glutamate, the ratio IB/IC was 0.83 ± 0.02 for TEMPO, 0.72 ± 0.02 for ascorbate and 0.65 ± 0.05 for trolox (n= 4). In the case of aspartate, the IB/IC value was 0.75 ± 0.02 for TEMPO, 0.72 ± 0.02 for ascorbate and 0.66 ± 0.07 for trolox (n= 10). Catalase (250 U ml−1) potentiated glutamate-induced currents (IB/IC= 1.14 ± 0.02) and did not affect aspartate-induced currents (IB/IC= 0.97 ± 0.06; IB and IC were not significantly different, P > 0.5). Glutamate- and aspartate-induced currents were also differentially affected by the mixture of these drugs. Thus, this antioxidant cocktail inhibited only about 80 % of the glutamate-induced current (IB/IC= 0.22 ± 0.03), but almost completely blocked the current elicited by aspartate (IB/IC= 0.027 ± 0.006). Such a difference between the effects of antioxidants on currents induced by aspartate and by glutamate suggested that antioxidants blocked only, or mainly, the NMDA subtype of glutamate receptors. In this respect the effect of antioxidants resembled that of the classical NMDA channel blocker MK801, which at concentrations sufficient for a total blockade of NMDA currents (15 μm) reduced the 100 μm glutamate-induced inward currents by about 80 % (not illustrated).
Figure 6. Effect of antioxidants on whole-cell currents induced by glutamate and aspartate.
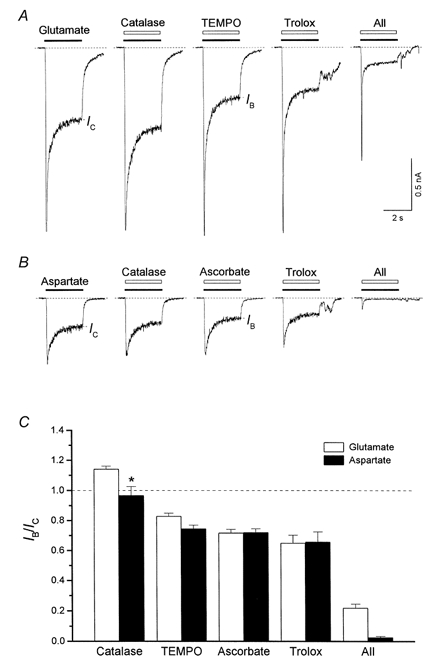
A, membrane current seen in response to application of glutamate (100 μm) and coapplication of glutamate with each of catalase (250 U ml−1), TEMPO (500 μm), trolox (750 μm), and a mixture of TEMPO, catalase, trolox and ascorbate (1 mm; All). IC and IB indicate the steady-state currents in the control and in the presence of antioxidants, respectively. B, responses to application of aspartate (100 μm) and aspartate coapplication with each of catalase (250 U ml−1), ascorbate (1 mm), trolox (750 μm), and a mixture of TEMPO, catalase, trolox and ascorbate. C, the mean values of IB/IC in each condition. The IB/IC value for catalase coapplication with aspartate did not differ significantly from 1 (IB and IC were not significantly different, *P > 0.05). In all other cases, the stationary current value in the presence of antioxidants, IB, differed significantly from the control current value, IC (P < 0.001).
Comparison of the effects of antioxidants and MK801 on [Ca2+]c and Δψm
The results of these electrophysiological experiments confirmed our supposition that antioxidants are able to block NMDA receptors (or channels) directly. Therefore one might expect that the changes in [Ca2+]c and Δψm induced by a prolonged co-application of glutamate with MK801 would be similar to those induced by glutamate in the presence of the antioxidants. However, this was not the case. In Fig. 7A a 20 min application of 100 μm glutamate during MK801 (15 μm) exposure induced only a small [Ca2+]c increase associated with a slight monophasic mitochondrial depolarisation. During the prolonged exposure to glutamate in the presence of MK801, none of the cells exhibited the secondary elevation of [Ca2+]c or the mitochondrial depolarisation typical of the effects of a combined glutamate and antioxidant treatment (e.g. Fig. 4B). These data ruled out the possibility that the secondary [Ca2+]c increase and mitochondrial depolarisation seen during glutamate exposure in the presence of antioxidants resulted from Ca2+ influx through Ca2+-permeable non-NMDA channels or from a reversal of Na+-Ca2+ exchange caused by excessive Na+ influx into the cell via these channels.
Figure 7. Comparison of the effects of antioxidants and MK801 on [Ca2+]c and Δψm.
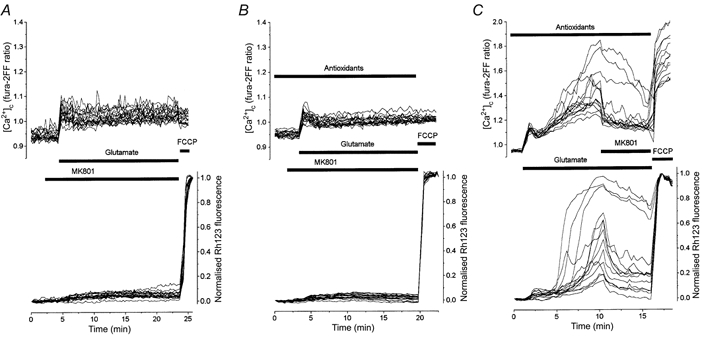
Glutamate was applied to neurones (at 13 DIV) 2.5 min after exposure to MK801 (A) or to the combined antioxidants plus MK801 (B). In B and C cells were treated with antioxidants (TEMPO, catalase, trolox and ascorbate at the concentrations given in Fig. 4) for 40 min before recording. C, MK801 was applied to the antioxidant-treated cells in the period when most neurones exhibited a secondary increase of [Ca2+]c and a loss of Δψm.
Another possibility was that the secondary delayed [Ca2+]c increase and mitochondrial depolarisation during the combined application of glutamate and antioxidants was caused by some toxic side-effect of the antioxidants leading either to a non-specific increase in ionic permeability of the neuronal membrane or to a deterioration of neuronal Ca2+ extrusion systems. To test this possibility we pre-incubated the cells with the combination of antioxidants and then co-applied them with glutamate and MK801. Figure 7B shows that when NMDA channels were blocked, no additional [Ca2+]c increase or mitochondrial depolarisation was seen in response to glutamate in the presence of the cocktail of antioxidants. All these data led us to suggest that the antioxidant-induced blockade of NMDA receptors, in contrast to that produced by MK801, dissipates gradually during a prolonged glutamate application and is replaced by a delayed activation resulting in a secondary increase in both [Ca2+]c and mitochondrial depolarisation. To test this hypothesis we carried out the experiment presented in Fig. 7C. Here MK801 was applied 8 min after the beginning of glutamate and antioxidant co-application, just in the period during which most neurones showed a mitochondrial depolarisation and a secondary increase in [Ca2+]c. Clearly, MK801 now effectively reversed this slow response, strongly indicating that it was generated through the activation of NMDA channels after their escape from the antioxidant-induced blockade.
It is important to note here that the effect of NMDA antagonists on [Ca2+]c and Δψm in the post-glutamate period depends critically on the timing of the application (and hence the apparent differences between the data in Figs 4 and 5) in relation to other intracellular events. Thus, much published data suggest that application of the glutamate receptor antagonist MK801 or removal of external Ca2+ fail to reverse the sustained post-glutamate increase of [Ca2+]c, elicited by toxic glutamate exposure (deErausquin et al. 1990; Khodorov et al. 1996a; Vergun et al. 1999). However, in a recent paper (Vergun et al. 1999) we showed that a sustained [Ca2+]c plateau that is insensitive to MK801 only appears in the post-glutamate period once Δψm has collapsed and has reached its steady state. If glutamate action was curtailed before the mitochondrial depolarisation was established, then both the increase in [Ca2+]c and the loss of Δψm were still reversible (see Fig. 8 in Vergun et al. 1999). Thus, in the data shown in Figs 5 and 7C of the present paper we are demonstrating analogous examples: antioxidants or MK801 were applied before the mitochondrial depolarisation and [Ca2+]c had reached their irreversible steady state.
Figure 8. Effect of antioxidants on neuronal viability.
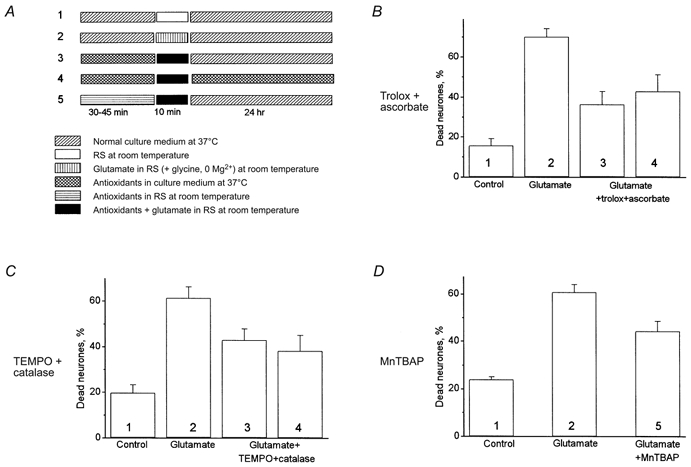
Propidium iodide fluorescence was used to detect dead neurones 24 h after treatment with glutamate (100 μm) for 10 min according to the protocols (1-5) shown in A. Dead cells were counted with respect to the total number of neurones present, identified by staining the nuclei with Hoechst 33342. Effects of each protocol on viability are shown as follows: B, effect of trolox (750 μm) and ascorbate (1 mm); C, TEMPO (500 μm) and catalase (250 U ml−1) for 45 min before glutamate treatment at 36°C; D, incubation with MnTBAP (200 μm) at room temperature for 30 min. Clearly, in each case a degree of neuroprotection by the antioxidants was evident.
Effects of antioxidants on neuronal viability
In a further series of experiments we studied the ability of antioxidants to protect neurones against delayed glutamate-induced neuronal death. Experiments were performed using two different protocols as indicated in the scheme shown in Fig. 8A. (i) Cultures were treated with antioxidants for 30 min at room temperature (MnTBAP) or for 45 min at 36°C (trolox plus ascorbate or TEMPO plus catalase) and then glutamate (100 μm, in Mg2+-free, 10 μm glycine-containing solution) was coapplied with antioxidants for 10 min at room temperature. The cells were then washed and replaced into the normal culture medium for 24 h at 36°C (columns 3 and 5 in Fig. 8B–D). (ii) As for i, but antioxidants were also present for 24 h in the culture medium after application of glutamate (column 4 in Fig. 8B–D).
Treatment of neurones with the mixture of trolox and ascorbate had a considerable protective effect against glutamate toxicity (Fig. 8B). Thus, the exposure of cells to glutamate for just 10 min produced 70 ± 4.1 % (n= 3 experiments) neuronal death measured 24 h later. Trolox with ascorbate reduced this percentage to 36.3 ± 6.5 % (n= 3) when they were present only before and during glutamate application (column 3) and to 42.8 ± 8.3 % (n= 3) when the antioxidants remained during the 24 h after glutamate treatment (column 4). There was no significant difference between the percentage of dead cells in these two experimental protocols (P > 0.05). Similar results were obtained in the experiment with TEMPO plus catalase (Fig. 8C). TEMPO and catalase reduced glutamate-induced neuronal death from 62.4 ± 4.9 to 43 ± 4.9 and 38.2 ± 6.8 % in the TEMPO plus catalase-treated cells. Again, the percentage of dead neurones in experiments in which the cells were incubated during the 24 h post-glutamate period in the normal medium (column 3) did not differ significantly from that seen in experiments in which the antioxidants remained over the 24 h incubation period (column 4, P > 0.05, n= 3). A 30 min pre-incubation of the cells with MnTBAP reduced the percentage of dead neurones from 60.7 ± 3.3 to 44.2 ± 4.2 % (n= 3, difference significant at P < 0.05).
Thus, both groups of antioxidants, those limiting lipid peroxidation and those scavenging O2− and H2O2, were able to improve survival and protect cells against glutamate neurotoxicity. The presence of antioxidants in the post-glutamate period did not increase this protective effect. Taking into account the strong blockade of ionic currents through NMDA channels by the full mixture of these antioxidants produced in electrophysiological experiments (see Fig. 6), we did not examine the effect of the mixture of these antioxidants on cell viability following a toxic glutamate challenge.
DISCUSSION
It has become dogma that glutamate toxicity results from the combined effects of mitochondrial calcium overload and increased generation of reactive oxygen species (ROS). We and others have shown previously that application of glutamate under conditions that lead to cell death is associated with stereotypical changes in [Ca2+]c and in Δψm (White & Reynolds, 1996; Schinder et al. 1996; Khodorov et al. 1996a; Vergun et al. 1999), although details of the response do vary between preparations. At the outset of these experiments we fully expected that effective antioxidant treatments would limit both the collapse of Δψm and the secondary increase in [Ca2+]c that we had attributed to the combined action of oxidative stress and mitochondrial calcium overload. These expectations were based on the following logic. The [Ca2+]c plateau in the post-glutamate period is associated with a failure of Ca2+ extrusion (Khodorov et al. 1993, 1996b). It is also established that ROS can impair cellular Ca2+ transport systems (for review, see Kourie, 1998) including the plasma membrane Ca2+ ATPase (Rohn et al. 1996). Similarly, the profound collapse of Δψm associated with glutamate toxicity has been assumed by many to result from opening of the mitochondrial permeability transition pore (mPTP) as a result of the combined action of ROS and mitochondrial Ca2+ overload (for a review on the mPTP, see Crompton, 1999; and Duchen, 2000b). Much to our surprise, neither scavengers of superoxide or peroxide radicals (MnTBAP or TEMPO plus catalase) nor inhibitors of lipid peroxidation (trolox plus ascorbate) had any significant impact on either the [Ca2+]c or the mitochondrial responses, despite an unequivocal demonstration that the scavengers were effective at removing much larger loads of ROS generated by xanthine with xanthine oxidase.
Indeed, we were initially encouraged to find that a more complex mixture of antioxidants did cause a profound delay in the collapse of Δψm, but were troubled that this also caused a significant suppression of the initial rise in [Ca2+]c. These observations led directly to the demonstration that most of the antioxidants used in a variety of combinations caused a substantial suppression of the NMDA-induced current.
These observations together dictate a re-evaluation of the evidence for a role of ROS in these early events of glutamate toxicity. Scrutiny of the literature shows that by far the bulk of the evidence arises from two experimental approaches - use of fluorescent indicators of ROS generation, such as HEt, dihydrorhodamine and dicarboxyfluorescein (Dugan et al. 1995; Reynolds & Hastings, 1995; Bindokas et al. 1996; Perez Velazquez et al. 1997; Sengpiel et al. 1998), or study of the effects of antioxidants on viability (Dykens et al. 1987; Monyer et al. 1990; Patel et al. 1996; Ciani et al. 1996; Dugan et al. 1997; Carriedo et al. 1998). Only two published studies (Lafon-Cazal, 1993; Dugan et al. 1995) used electron paramagnetic resonance techniques to demonstrate increased ROS generation in response to glutamate. Use of the fluorescent indicators is fraught with difficulty. HEt is oxidised to ethidium. This is a lipophilic cation and, as such, will accumulate in mitochondria where the fluorescence signal is quenched (see Rottenberg, 1984; Budd, 1997). Thus, if mitochondria also depolarise this will increase the fluorescence signal irrespective of any ROS generation. It is presumably this process that is responsible for descriptions of increased ROS generation in response to mitochondrial uncouplers (Bindokas et al. 1996; Sengpiel et al. 1998), whereas uncouplers do not increase ROS generation in other model systems (e.g. see Budd et al. 1997). Similarly, dihydro-rhodamine is oxidised to Rh123 which will then accumulate in mitochondria and dequench exactly as we have seen with Rh123 in the present paper. Carboxyfluorescein in its various forms is extremely sensitive to pH (Reynolds & Hastings, 1995), and as glutamate toxicity is associated with a profound intracellular acidosis, these signals may be extremely hard to interpret. In the present study, we used HEt at the lowest concentration consistent with a reasonable signal/noise ratio, and without any preincubation period to limit spontaneous breakdown and intramitochondrial accumulation. Under these conditions, we could detect no significant increase in signal with FCCP or glutamate, suggesting that we were able to avoid the artefactual increase in signal associated with changing Δψm, and also that ROS, if produced at all, were generated in response to glutamate only at very low levels. The demonstration that treatments with antioxidants that could completely suppress the signal generated by xanthine with xanthine oxidase, which gave much greater loads of ROS than glutamate, nevertheless had no significant impact at all on the changes in [Ca2+]c or Δψm throws into doubt any significant role of superoxide generation in the initial phases of glutamate toxicity, at least in this model. It is particularly interesting here to compare our data with those of Castilho et al. (1999), who found that, while HEt reported an increased rate of ROS generation in cerebellar granule cells in response to glutamate, this was only seen some 30-40 min after glutamate application.
One might reasonably assume that if mitochondria undergo a major depolarisation, the cells will proceed to die. This may result either from ATP depletion, the collapse of the energy supply and necrotic cell death, or through the release of cytochrome c and a progression to apoptotic cell death. Therefore, again, we were surprised to find that despite the failure to pre-empt the secondary changes in [Ca2+]c or Δψm, antioxidant treatment was able to limit the progression to cell death. This means that the loss of Δψm must be reversible - sustained mitochondrial depolarisation over 24 h is almost inevitably lethal. These longer term changes in cell pathophysiology are not readily amenable to study using imaging techniques, but clearly need to be addressed.
At a glance, the failure of antioxidants to suppress the secondary [Ca2+]c increase while limiting cell death also seems to be at variance with the assumption of a causal link between the glutamate-induced alteration of [Ca2+]c homeostasis and delayed neuronal death. For example, Manev et al. (1989) found that in cerebellar granule cells a removal of external Ca2+ (with 1 mm EGTA) just after the termination of a 15 min glutamate (50 μm) exposure dramatically reduced the delayed neuronal degeneration. Using the same model of neuronal culture, deErausquin et al. (1990) showed that the protracted rise in [Ca2+]c in a single neurone is correlated with (r= 0.87, P > 0.01), and consequently predictive of, the time of appearance of neuronal death. Limbrick et al. (1995) established a strong correlation (r= 0.973) between the prolonged increases in [Ca2+]c induced by glutamate and a delayed death of cultured hippocampal neurones. Kiedrowski (1998) has shown that cultured cerebellar cells that failed to restore Δψm following glutamate withdrawal also failed to restore low [Ca2+]c, and later died. Meanwhile, in our experiments a decrease in cell death caused by antioxidants was apparently independent of any improvement of [Ca2+]c homeostasis in the short term. It seems likely that this disagreement might be resolved if the causal link between deregulation of [Ca2+]c homeostasis and delayed neuronal death is indeed mediated by ROS, but that ROS represent a downstream mechanism rather than an early step in the progression to cell death. Surprisingly, we found that the protective effects of the antioxidants did not depend on the timing of application in relation to the glutamate treatment or post-glutamate period (see Fig. 8), suggesting that the switch to the progression to cell death is in fact set in the period of toxic glutamate exposure.
The bulk of the remaining evidence for a role of ROS in glutamate-induced cell death has come from the protective effects of a variety of antioxidants. A major unexpected finding in this paper was that the mixture of antioxidants consisting of TEMPO, catalase, trolox and ascorbate exerted a profound blocking action on ionic currents in NMDA channels of neuronal membrane. This blockade underlies a dramatic delay in the development of the mitochondrial depolarisation and a secondary increase in [Ca2+]c during a prolonged glutamate challenge (see Fig. 4). A comparison of the blockade of [Ca2+]c and mitochondrial responses induced by the antioxidant cocktail with that caused under analogous conditions by the classic blocker of NMDA channels MK801 (see Fig. 7) led us to conclude that the block of glutamate responses associated with the antioxidants, in contrast to that caused by MK801, is only transient. This would explain why addition of the antioxidant cocktail delays but does not prevent the secondary increase in [Ca2+]c and the collapse of Δψm in the post-glutamate period. The unexpected finding that the antioxidants along with their specific actions on ROS-related damage are also able to reduce the NMDA response may be relevant to the clinical usefulness of these agents in medical practice. However, it is also clear that the failure to recognise the blocking effects of antioxidants on NMDA receptors may be seriously misleading in the interpretation of experiments of this kind, and that the protective effects of these agents may not in fact be attributable to their specific actions as ROS scavengers.
The mechanism of the transient blockade of the NMDA response by the antioxidants is not clear. It seems likely that the antioxidants exert their blocking effect by the modulation of redox sites of the NMDA receptor (see for review Gozlan & Ben-Ari, 1995). NMDA receptors apparently exist in an equilibrium between at least two redox states: one fully oxidised state with moderate activity, and a fully reduced state, having higher efficacy. A number of endogenous compounds modulate or maintain this redox equilibrium. Therefore perhaps during a prolonged glutamate treatment the equilibrium between redox states of NMDA receptors alters, decreasing their affinity for the antioxidants, perhaps in response to the changing ionic balance induced by glutamate. Further investigations are required to clarify this problem. However, independently of its final solution, it is already quite clear that neither the intracellular accumulation of ROS, nor ROS-induced lipid peroxidation during the toxic glutamate exposure is a prerequisite for either the deregulation of [Ca2+]c homeostasis or a collapse of Δψm. The latter point needs to be emphasised, since it is impossible to be sure whether ROS play a role in the perturbation of [Ca2+]c homeostasis caused by glutamate in some other model systems. Indeed, according to Castilho et al. (1999) MnTBAP potently retards (by 40-60 min) the so-called delayed calcium deregulation (DCD) caused by a very prolonged treatment of young (7-9 DIV) cerebellar granule cells with glutamate for 60-100 min. Further studies are required to clarify the relationship between the secondary [Ca2+]c increase observed in our experiments at room temperature in hippocampal neurones during a glutamate challenge for 10 min and DCD developing in cerebellar granule cells at 36°C during a continuous glutamate exposure for up to 2 h.
At present, the only way that we can reconcile these different data is to suggest that the NO radical plays a more significant role than superoxide or peroxide in defining the collapse of Δψm, as we have shown that NO and calcium together seem to be required to precipitate the profound loss of Δψm seen in our model in response to glutamate (Keelan et al. 1999). This is also consistent with recent data from Sattler et al. (1999) which showed that close coupling of NOS to the NMDA receptor by the scaffolding protein PSD95 conferred the specificity of the NMDA-induced calcium rise and cell death in hippocampal neurones. ROS are also clearly involved in the final progression to cell death, as the antioxidants improved viability even at concentrations that did not inhibit NMDA receptor activity, but this role does not seem to include an influence on the early collapse of Δψm. Indeed, the failure of antioxidants to affect the collapse of Δψm undermines the logic that has sustained the suggestion for a role of the mPTP, and the underlying mechanism for the loss of Δψm remains to be fully established. It certainly seems clear that the basis for both the loss of calcium regulation and the loss of Δψm may not be as clearly understood as often implied and that work is still needed to identify the precise cellular mechanisms responsible for glutamate-induced cell death.
Acknowledgments
We thank the Royal Society, the Wellcome Trust and INTAS for financial support, and Ms D. Lilian Patterson for her help with tissue culture, without which the work would not be possible.
References
- Ankarcrona M, Dypbukt JM, Orrenius S, Nicotera P. Calcineurin and mitochondrial function in glutamate-induced neuronal cell death. FEBS Letters. 1996;394:321–324. doi: 10.1016/0014-5793(96)00959-3. [DOI] [PubMed] [Google Scholar]
- Bindokas VP, Jordan J, Lee CC, Miller RJ. Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. Journal of Neuroscience. 1996;16:1324–1336. doi: 10.1523/JNEUROSCI.16-04-01324.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes PS, Land JM, Clark JB, Heales SJ. Peroxynitrite and brain mitochondria: evidence for increased proton leak. Journal of Neurochemistry. 1998;70:2195–2202. doi: 10.1046/j.1471-4159.1998.70052195.x. [DOI] [PubMed] [Google Scholar]
- Budd SL, Castilho RF, Nicholls DG. Mitochondrial membrane potential and hydroethidine-monitored superoxide generation in cultured cerebellar granule cells. FEBS Letters. 1997;415:21–24. doi: 10.1016/s0014-5793(97)01088-0. [DOI] [PubMed] [Google Scholar]
- Castilho RF, Ward MW, Nicholls DG. Oxidative stress, mitochondrial function, and acute glutamate excitotoxicity in cultured cerebellar granule cells. Journal of Neurochemistry. 1999;72:1394–1401. doi: 10.1046/j.1471-4159.1999.721394.x. [DOI] [PubMed] [Google Scholar]
- Carriedo SG, Yin HZ, Sensi SL, Weiss JH. Rapid Ca2+ entry through Ca2+ permeable AMPA/kainate channels triggers marked intracellular Ca2+ rises and consequent oxygen radical production. Journal of Neuroscience. 1998;18:7727–7738. doi: 10.1523/JNEUROSCI.18-19-07727.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H-SV, Lipton SA. Mechanism of memantine block of NMDA-activated channels in rat retinal ganglion cells: uncompetitive antagonism. Journal of Physiology. 1997;499:27–46. doi: 10.1113/jphysiol.1997.sp021909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW, Rothman SM. The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annual Review of Neuroscience. 1990;13:171–182. doi: 10.1146/annurev.ne.13.030190.001131. [DOI] [PubMed] [Google Scholar]
- Ciani E, Groneng L, Voltattorni M, Rolseth V, Contestabile A, Paulsen RE. Inhibition of free radical production or free radical scavenging protects from the excitotoxic cell death mediated by glutamate in cultures of cerebellar granule neurons. Brain Research. 1996;728:1–6. [PubMed] [Google Scholar]
- Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochemical Journal. 1999;341:233–249. [PMC free article] [PubMed] [Google Scholar]
- Day BJ, Fridovich I, Crapo JD. Manganic porphyrins possess catalase activity and protect endothelial cells against hydrogen peroxide-mediated injury. Archives of Biochemistry and Biophysics. 1997;347:256–262. doi: 10.1006/abbi.1997.0341. [DOI] [PubMed] [Google Scholar]
- Day BJ, Shawen S, Liochev SI, Crapo JD. A metalloporphyrin superoxide dismutase mimetic protects against paraquat-induced endothelial cell injury, in vitro. Journal of Pharmacology and Experimental Therapeutics. 1995;275:1227–1232. [PubMed] [Google Scholar]
- deErausquin GA, Manev H, Guidotti A, Costa E, Brooker G. Gangliosides normalize distorted single-cell intracellular free Ca2+ dynamics after toxic doses of glutamate in cerebellar granule cells. Proceedings of the National Academy of Sciences of the USA. 1990;87:8017–8021. doi: 10.1073/pnas.87.20.8017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR. Contributions of mitochondria to animal physiology: from homeostatic sensor to calcium signalling and cell death. Journal of Physiology. 1999;516:1–17. doi: 10.1111/j.1469-7793.1999.001aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR. Mitochondria and Ca2+ in cell physiology and pathophysiology. Cell Calcium. 2000a;28:339–348. doi: 10.1054/ceca.2000.0170. [DOI] [PubMed] [Google Scholar]
- Duchen MR. Mitochondria and calcium: from cell signalling to cell death. Journal of Physiolgy. 2000b;529:57–68. doi: 10.1111/j.1469-7793.2000.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR, Biscoe TJ. Relative mitochondrial membrane potential and [Ca2+]i in type I cells isolated from the rabbit carotid body. Journal of Physiology. 1992;450:33–61. doi: 10.1113/jphysiol.1992.sp019115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugan LL, Sensi SL, Canzoniero LM, Handran SD, Rothman SM, Lin TS, Goldberg MP, Choi DW. Mitochondrial production of reactive oxygen species in cortical neurons following exposure to N-methyl-D-aspartate. Journal of Neuroscience. 1995;15:6377–6388. doi: 10.1523/JNEUROSCI.15-10-06377.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugan LL, Turetsky DM, Du C, Lobner D, Wheeler M, Almli CR, Shen CK, Luh TY, Choi DW, Lin TS. Carboxyfullerenes as neuroprotective agents. Proceedings of the National Academy of Sciences of the USA. 1997;94:9434–9439. doi: 10.1073/pnas.94.17.9434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dykens JA, Stern A, Trenkner E. Mechanism of kainate toxicity to cerebellar neurons in vitro is analogous to reperfusion tissue injury. Journal of Neurochemistry. 1987;49:1222–1228. doi: 10.1111/j.1471-4159.1987.tb10014.x. [DOI] [PubMed] [Google Scholar]
- Faulkner KM, Liochev SI, Fridovich I. Stable Mn(III) porphyrins mimic superoxide dismutase in vitro and substitute for it in vivo. Journal of Biological Chemistry. 1994;269:23471–23476. [PubMed] [Google Scholar]
- Gozlan H, Ben-Ari Y. NMDA receptor redox sites: are they targets for selective neuronal protection? Trends in Pharmacological Sciences. 1995;16:368–374. doi: 10.1016/s0165-6147(00)89077-x. [DOI] [PubMed] [Google Scholar]
- Grinberg LN, Samuni A. Nitroxide stable radical prevents primaquine-induced lysis of red blood cell. Biochimica et Biophysica Acta. 1994;1201:284–288. doi: 10.1016/0304-4165(94)90052-3. [DOI] [PubMed] [Google Scholar]
- Jacobson D, Duchen M. Fluorescence imaging of the mitochondrial permeability transition in rat cortical astrocytes in culture. Journal of Physiology. 1998;506.P:75–76. P. [Google Scholar]
- Keelan J, Vergun O, Duchen MR. Excitotoxic mitochondrial depolarisation requires both calcium and nitric oxide in rat hippocampal neurons. Journal of Physiology. 1999;520:797–813. doi: 10.1111/j.1469-7793.1999.00797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiedrowski L. The difference between mechanisms of kainate and glutamate excitotoxicity in vitro: osmotic lesion versus mitochondrial depolarization. Restorative Neurology and Neuroscience. 1998;12:71–79. [PubMed] [Google Scholar]
- Khodorov B, Pinelis V, Golovina V, Fajuk D, Andreeva N, Uvarova T, Khaspekov L, Victorov I. On the origin of a sustained increase in cytosolic Ca2+ concentration after a toxic glutamate treatment of the nerve cell culture. FEBS Letters. 1993;324:271–273. doi: 10.1016/0014-5793(93)80132-e. [DOI] [PubMed] [Google Scholar]
- Khodorov B, Pinelis V, Vergun O, Storozhevykh T, Vinskaya N. Mitochondrial deenergization underlies neuronal calcium overload following a prolonged glutamate challenge. FEBS Letters. 1996a;397:230–234. doi: 10.1016/s0014-5793(96)01139-8. [DOI] [PubMed] [Google Scholar]
- Khodorov BI, Fayuk DA, Koshelev SG, Vergun OV, Pinelis VG, Vinskaya NP, Storozhevykh TP, Arsenyeva EN, Khaspekov LG, Lyzhin AP, Isaev N, Victorov IV, Dubinsky JM. Effect of a prolonged glutamate challenge on plasmalemmal calcium permeability in mammalian central neurones. Mn2+ as a tool to study calcium influx pathways. International Journal of Neuroscience. 1996b;88:215–241. doi: 10.3109/00207459609000616. [DOI] [PubMed] [Google Scholar]
- Kourie JI. Interaction of reactive oxygen species with ion transport mechanisms. American Journal of Physiology. 1998;275:C1–24. doi: 10.1152/ajpcell.1998.275.1.C1. [DOI] [PubMed] [Google Scholar]
- Krishna MC, Grahame DA, Samuni A, Mitchell JB, Russo A. Oxoammonium cation intermediate in the nitroxide-catalyzed dismutation of superoxide. Proceedings of the National Academy of Sciences of the USA. 1992;89:5537–5541. doi: 10.1073/pnas.89.12.5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafon-Cazal M, Pietri S, Culcasi M, Bockaert J. NMDA-dependent superoxide production and neurotoxicity. Nature. 1993;364:535–537. doi: 10.1038/364535a0. [DOI] [PubMed] [Google Scholar]
- Leyssens A, Anderson M, Craske M, Rastoghi R, Crompton M, Duchen MR. Transient depolarisations of mitochondria localised to discrete areas of single rat cardiomyocytes caused by free radicals and local calcium release: a model for free radical induced cell injury. Journal of Physiology. 1995;487.P:123. P. [Google Scholar]
- Limbrick DD, Jr, Churn SB, Sombati S, Delorenzo RJ. Inability to restore resting intracellular calcium levels as an early indicator of delayed neuronal cell death. Brain Research. 1995;690:145–156. doi: 10.1016/0006-8993(95)00552-2. [DOI] [PubMed] [Google Scholar]
- Manev H, Favaron M, Guidotti A, Costa E. Delayed increase of Ca2+ influx elicited by glutamate: role in neuronal death. Molecular Pharmacology. 1989;36:106–112. [PubMed] [Google Scholar]
- Mitchell JB, Samuni A, Krishna MC, Degraff WG, Ahn MS, Samuni U, Russo A. Biologically active metal-independent superoxide dismutase mimics. Biochemistry. 1990;29:2802–2807. doi: 10.1021/bi00463a024. [DOI] [PubMed] [Google Scholar]
- Monyer H, Hartley DM, Choi DW. 21-Aminosteroids attenuate excitotoxic neuronal injury in cortical cell cultures. Neuron. 1990;5:121–126. doi: 10.1016/0896-6273(90)90302-v. [DOI] [PubMed] [Google Scholar]
- Nakao N, Grasbon-Frodl EM, Widner H, Brundin P. Antioxidant treatment protects striatal neurons against excitotoxic insults. Neuroscience. 1996;73:185–200. doi: 10.1016/0306-4522(96)00034-6. [DOI] [PubMed] [Google Scholar]
- Patel M, Day BJ, Crapo JD, Fridovich I, McNamara JO. Requirement for superoxide in excitotoxic cell death. Neuron. 1996;16:345–355. doi: 10.1016/s0896-6273(00)80052-5. [DOI] [PubMed] [Google Scholar]
- Patneau DK, Mayer ML. Structure-activity relationships for amino acid transmitter candidates acting at N-methyl-D-aspartate and quisqualate receptors. Journal of Neuroscience. 1990;10:2385–2399. doi: 10.1523/JNEUROSCI.10-07-02385.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez Velazquez JL, Frantseva MV, Carlen PL. vitro ischemia promotes glutamate-mediated free radical generation and intracellular calcium accumulation in hippocampal pyramidal neurons. Journal of Neuroscience. 1997;17:9085–9094. doi: 10.1523/JNEUROSCI.17-23-09085.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds IJ, Hastings TG. Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. Journal of Neuroscience. 1995;15:3318–3327. doi: 10.1523/JNEUROSCI.15-05-03318.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohn TT, Hinds TR, Vincenzi FF. Inhibition of Ca2+-pump ATPase and the Na+/K+-pump ATPase by iron-generated free radicals. Protection by 6, 7-dimethyl-2,4-DI-1- pyrrolidinyl-7H-pyrrolo[2,3-d] pyrimidine sulfate (U-89843D), a potent, novel, antioxidant/free radical scavenger. Biochemical Pharmacology. 1996;51:471–476. doi: 10.1016/0006-2952(95)02222-8. [DOI] [PubMed] [Google Scholar]
- Rottenberg H. Membrane potential and surface potential in mitochondria: uptake and binding of lipophilic cations. Journal of Membrane Biology. 1984;81:127–138. doi: 10.1007/BF01868977. [DOI] [PubMed] [Google Scholar]
- Samuni A, Krishna CM, Riesz P, Finkelstein E, Russo A. A novel metal-free low molecular weight superoxide dismutase mimic. Journal of Biological Chemistry. 1988;263:17921–17924. [PubMed] [Google Scholar]
- Sattler R, Xiong Z, Lu WY, Hafner M, MacDonald JF, Tymianski M. Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science. 1999;284:1845–1848. doi: 10.1126/science.284.5421.1845. [DOI] [PubMed] [Google Scholar]
- Schinder AF, Olson EC, Spitzer NC, Montal M. Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. Journal of Neuroscience. 1996;16:6125–6133. doi: 10.1523/JNEUROSCI.16-19-06125.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengpiel B, Preis E, Krieglstein J, Prehn JH. NMDA-induced superoxide production and neurotoxicity in cultured rat hippocampal neurons: role of mitochondria. European Journal of Neuroscience. 1998;10:1903–1910. doi: 10.1046/j.1460-9568.1998.00202.x. [DOI] [PubMed] [Google Scholar]
- Vergun O, Keelan J, Khodorov BI, Duchen MR. Glutamate-induced mitochondrial depolarisation and perturbation of calcium homeostasis in cultured rat hippocampal neurones. Journal of Physiology. 1999;519:451–466. doi: 10.1111/j.1469-7793.1999.0451m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorobjev VS. Vibrodissociation of sliced mammalian nervous tissue. Journal of Neuroscience Methods. 1991;38:145–150. doi: 10.1016/0165-0270(91)90164-u. [DOI] [PubMed] [Google Scholar]
- Vorobjev VS, Sharonova IN, Haas HL. A simple perfusion system for patch-clamp studies. Journal of Neuroscience Methods. 1996;68:303–307. doi: 10.1016/0165-0270(96)00097-0. [DOI] [PubMed] [Google Scholar]
- Vyklicky L, Benveniste M, Mayer M. Modulation of NMDA receptor desensitization by glycine in mouse cultured hippocampal neurons. Journal of Physiology. 1990;428:313–331. doi: 10.1113/jphysiol.1990.sp018214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RJ, Reynolds IJ. Mitochondrial depolarization in glutamate-stimulated neurons: an early signal specific to excitotoxin exposure. Journal of Neuroscience. 1996;16:5688–5697. doi: 10.1523/JNEUROSCI.16-18-05688.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoratti M, Szabo I. The mitochondrial permeability transition. Biochimica et Biophysica Acta. 1995;1241:139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]