

Abstract
The region of alternative splicing in the KCNQ2 potassium channel gene was determined by RNase protection analysis of KCNQ2 mRNA transcripts.
Systematic analysis of KCNQ2 alternative splice variant expression in rat superior cervical ganglia revealed multiple variant isoforms.
One class of KCNQ2 splice variants, those that contained exon 15a, was found to have significantly different kinetics to those of the other isoforms. These transcripts encoded channel subunits that, when co-expressed with the KCNQ3 subunit, activated and deactivated approximately 2.5 times more slowly than other isoforms. Deletion of exon 15a in these isoforms produced a reversion to the faster kinetics.
Comparison of the kinetic properties of the cloned channel splice variants with those of the native M-current suggests that alternative splicing of the KCNQ2 gene may contribute to the variation in M-current kinetics seen in vivo.
The KCNQ2 and KCNQ3 potassium channel genes encode subunits that co-assemble to form heteromeric channels that underlie the M-current in sympathetic neurons and most probably also contribute to M-currents in central neurons (Wang et al. 1998). The M-current is a slowly activating and deactivating current that plays a critical role in controlling neuronal excitability (Brown, 1988; Yamada et al. 1989; Wang & McKinnon, 1995). In accordance with this property, mutations in either gene can produce benign familial neonatal epilepsy (Singh et al. 1998; Biervert et al. 1998; Charlier et al. 1998).
The KCNQ2 gene has a complex genomic structure (Nakamura et al. 1998; Biervert & Steinlein, 1999) and can produce multiple differentially spliced transcripts (Nakamura et al. 1998; Tinel et al. 1998). To date, the functional properties of channels encoded by alternatively spliced transcripts produced from the KCNQ2 gene have not been studied systematically. For this reason, we have attempted to determine the complete diversity of KCNQ2 mRNA transcripts expressed in rat sympathetic ganglia and have examined the functional properties of the channels encoded by these alternative splice variants. We chose the rat superior cervical ganglia (SCG) because neurons in these ganglia are most commonly used for studies of M-channel function and the properties of the M-channel are best described in this tissue.
METHODS
Isolation of cDNA clones
Two studies on the structure of the KCNQ2 gene in mouse and human have produced results that are in general agreement, except for the use of different naming conventions (Nakamura et al. 1998; Biervert & Steinlein, 1999). In this paper we use the exon/intron nomenclature of Biervert & Steinlein (1999).
Rat KCNQ2 cDNA was initially identified by performing PCR amplification of partial cDNA clones from rat brain and SCG cDNA using the following degenerate primers directed against two regions, one approximately 15 amino acids upstream from the S3 region and the other close to the pore region:
forward: TGG GCN GCN GGN TGY TGY TG
reverse: RTA CCA DAT RCA NGC NAG CCA RTG.
We determined an initial sequence encompassing the entire open reading frame of the KCNQ2 gene by performing several rounds of 5′ and 3′ RACE PCR using initial anchor oligonucleotides complementary to the partial cDNA clone and SCG cDNA as a template for amplification. Once cDNAs were obtained that extended beyond both the 5′ and 3′ ends of the open reading frame, oligonucleotides complementary to non-coding regions at either end of the coding sequence were designed. Full-length clones were obtained using Expand Long Template PCR (Boehringer Mannheim, Indianapolis, IN, USA) and were sequenced in their entirety.
Mutation of cDNA clones
The Form B deletion mutation was made by inverse PCR of a full-length Form B clone using Vent DNA polymerase (NEB). PCR products were treated with DpnI to remove the original template, then purified and re-ligated. The mutated clone was sequenced in its entirety.
RNase protection assay
The procedures for the preparation of RNA from rat sympathetic ganglia (SCG) and brain and for the RNase protection assay were identical to those described previously (Dixon & McKinnon, 1996). Rats were killed by exposure to a rising concentration of carbon dioxide, followed by decapitation. RNA expression was quantified directly from dried RNase protection gels using a PhosphorImager (Molecular Dynamics, Sunnyvale, CA, USA).
Cell culture and transfection
CHO cells
The procedure for Chinese hamster ovary (CHO) hm1 cell culture and transfection has been described previously (Selyanko et al. 1999). Briefly, CHO hm1 cells (immortalized cell line), expressing the human M1 receptor (Mullaney et al. 1993), were grown at 37°C and 5 % CO2 in alpha-modified Eagle's medium (MEM; Gibco BRL) supplemented with 10 % fetal calf serum, 1 % L-glutamine and 1 % penicillin-streptomycin. Cells plated in 35 mm plastic dishes were transfected 1-2 days after plating using ‘LipofectAmine Plus’ (Gibco BRL). Plasmids containing KCNQ and CD8 cDNAs, driven by the CMV promoter, were co-transfected in a channel to marker ratio of 10:1. Equal amounts of KCNQ2 and KCNQ3 cDNA were used. CD8 cDNA was kindly provided by Dr B. Seed. Cells expressing channels were identified by adding CD8-binding Dynabeads (Dynal A/S) prior to recording.
SCG neurons
Cultured SCG neurons were prepared as described previously (Owen et al. 1990) from 14-day-old Sprague-Dawley rats, and used at 1-2 days. The rats were killed by exposure to a rising concentration of carbon dioxide, followed by decapitation, according to the Animals (Scientific Procedures) Act 1986.
Perforated-patch whole-cell recording
All recordings were made at room temperature (20-22°C). Recordings from CHO cells were made 1-4 days after transfection. Cells were bath perfused with the following solution (mM): 144 NaCl, 2.5 KCl, 2 CaCl2, 0.5 MgCl2, 5 Hepes, 10 glucose; pH 7.4 with Tris base. Pipettes were filled with ‘internal’ solution containing (mM): 80 potassium acetate, 30 KCl, 40 Hepes, 3 MgCl2, 3 EGTA, 1 CaCl2; pH 7.4 with KOH. The junction potential was small (4 mV; Selyanko et al. 1995) so no correction was made. Amphotericin B was used to perforate the patch (Rae et al. 1991). Pipette resistance was 2-3 MΩ and the series resistance was compensated (60-90 %).
Data were acquired and analysed using pCLAMP software (Axon Instruments, Foster City, CA, USA). Currents were recorded using an Axopatch 200A (or 200) patch-clamp amplifier, filtered at 1 kHz and digitized at 4-8 kHz. Activation curves were fitted by the Boltzmann equation:
where I is the tail current recorded at -70 mV following a step to the membrane potential (Vm), I(50) is the current following a step to +50 mV, V0.5 is the membrane potential at which I is equal to 0.5I(50) and k is the slope factor. Current size and its inhibition by a muscarinic stimulant were measured from the amplitude of the deactivation tail recorded at -50 mV, measured from extrapolated exponential fits. The program Origin (Microcal Software Inc.) was used for fitting steady-state activation curves and for creating the figures.
Expression in Xenopus oocytes
All experimental procedures were subject to the Animal Welfare Assurance of the State University of New York (no. A3011-01). Full-length cRNA transcripts were synthesized in vitro. Oocytes were obtained from mature female Xenopus laevis using established procedures (Colman, 1984). Frogs were anaesthetized in ice water containing a 0.1 % solution of tricaine, and were humanely killed after the final collection of oocytes. Defolliculation was performed by incubation at room temperature for 2 h in 2 mg ml−1 collagenase (Type VIII, Sigma, St Louis, MO, USA) in Ca2+-free OR2 oocyte medium (see below) with gentle agitation. Oocytes were stored in OR3 solution (50 % L-15 medium (Gibco BRL), 1 mM glutamine, 15 mM Na-Hepes (pH 7.6), 0.1 mg ml−1 gentamicin) at 18°C. Oocytes were injected with 50 nl of cRNA (∼0.3 ng nl−1) using a microdispenser and a micropipette with a tip diameter of 10-20 μm. Injected oocytes were incubated at 18°C for 24-48 h prior to analysis.
Oocytes were voltage clamped using a two-microelectrode voltage clamp. Intracellular electrodes filled with 3 M KCl with resistances of 0.5-3 MΩ were used. The standard extracellular recording solution (OR2) contained (mM): 80 NaCl, 5 KCl, 1.8 CaCl2, 1 MgCl2 and 5 Na-Hepes (pH 7.6). Data collection and analysis were performed using pCLAMP software.
Data are shown as means ±s.e.m.; n, number of cells. P < 0.05 was considered to be statistically significant (t test).
RESULTS
Determination of the region of alternative splicing in KCNQ2 transcripts
During preliminary studies, a number of independent KCNQ2 cDNA clones were isolated from cDNA prepared from rat SCG mRNA. Most of these clones had unique splicing patterns suggesting that the rat KCNQ2 gene expressed multiple alternatively spliced mRNAs in the SCG. To determine which regions of the KCNQ2 transcript were subject to alternative splicing, RNase protection assays were performed using a series of probes encompassing the entire coding region of the gene (Fig. 1). It was found that alternative splicing of the KCNQ2 transcript was restricted to a region that encoded the first half of the presumptive intracellular C-terminus of the channel. A probe encompassing exons 8-15 was digested into multiple bands whereas probes to other regions of the transcript gave only single protection bands (Fig. 1). Essentially the same results were found for the total brain samples as for the SCG samples.
Figure 1. RNase protection analysis of KCNQ2 transcripts expressed in the SCG and brain of rat.

A diagrammatic representation of the KCNQ2 gene is shown in the upper panel with the exon boundary lengths marked to scale. Exon structure is based on mouse and human genomic structures (Nakamura et al. 1998; Biervert & Steinlein, 1999) and the nomenclature corresponds to that of Biervert & Steinlein (1999). Exon 15a corresponds to exon 14 in the mouse genomic structure (Nakamura et al. 1998). The location of the probes used in the RNase protection assays (P1-P6) is shown at the top. The positions of the six transmembrane domains (S1-S6) and the pore region (P) are shown below. In the lower panel, the RNase protection assays for probes P1-P5 are shown. In each assay the first lane corresponds to the probe (Px), the second lane to the tRNA sample (t), the third lane to the SCG sample (S) and the fourth lane to the brain sample (B). For probes P1, P2, P3 and P5 a single band was protected. In contrast, multiple protected bands were found using the P4 probe with no single band predominating. Probe P6 did not cross an exon boundary and gave a single band in RNase protection assays (see Fig. 9).
Isolation of alternatively spliced KCNQ2 cDNA clones containing an in-frame exon 16
The cDNA clones that were isolated fell into three classes: those that contained an in-frame exon 16, those that contained an out-of-frame exon 16 and those that terminated prematurely to exon 16. Since only the clones containing an in-frame exon 16 expressed functional channels (see below), we examined in detail the splicing forms of the transcripts containing exon 16.
To determine the diversity and relative abundance of the splice isoforms expressed in SCG a total of 41 independent cDNA clones encompassing the region subject to differential splicing were obtained by PCR amplification and then sequenced. The isolated clones fell into two broad classes. The majority of clones (35/41) had a reading frame that extended through most of exon 16, which contains the stop codon terminating the main open reading frame. A minority of clones (6/41) had a reading frame that terminated early in exon 16 due to an alternative splice junction in exon 14 that resulted in a shift in the reading frame for exons 15 and 16. These two classes will be discussed separately since the presence of an in-frame exon 16 is necessary to encode a functional channel (see below).
For the 35 clones encoding an in-frame exon 16, five different sites of potential alternative splicing were found. This implies that there are 32 potential combinations of alternative exons and splice junctions. Of these 32 possible combinations only nine combinations were isolated (Fig. 2). There was, therefore, a non-random distribution of alternative exons and splice junctions. Although no isoform predominated, some forms were clearly more abundant than other potential isoforms (Fig. 2C). In particular, forms containing the alternative 5′ splice site in exon 13 were favoured as were, to a slightly lesser extent, those containing exon 8 and the alternative 5′ splice site of exon 13.
Figure 2. Alternatively spliced forms of rat KCNQ2 transcripts containing an in-frame exon 16 expressed in SCG.

A, expanded view of the KCNQ2 genomic structure corresponding to the region of alternative splicing. Exons that are alternatively spliced or have alternative splice junctions are shown shaded. Exons 12 and 13 have alternative splice junctions. Exon lengths are shown to scale but intron lengths are shortened. B, comparison of two composite KCNQ2 deduced amino acid sequences, one containing all the known inserts and one lacking all inserts. Corresponding exon boundaries are marked above the sequence. C, table showing the distribution of alternative (alt) exons or splice junctions in the different isoforms and the frequency of isolation.
Functional expression of KCNQ2 cDNA clones containing an in-frame exon 16
To determine whether alternatively spliced transcripts encoded KCNQ2 subunits with different functional properties we expressed four of the KCNQ2 variants (Forms A, B, C and E) in combination with the KCNQ3 subunit. All four isoforms could be expressed at relatively high levels in CHO cells or Xenopus oocytes and there was no significant difference in the peak current produced by the four variants.
The steady-state activation properties of the four isoforms were similar (Fig. 3 and Table 1). There were, however, clear differences in the activation and deactivation kinetics of the different isoforms (Figs 4A and 5A). As with native M-currents (Selyanko & Brown, 1999), the time courses of both activation and deactivation were best fitted using two exponentials. The two time constants for deactivation (τ1, τ2) for channels containing Form B were approximately 2.5 times slower than those for channels containing the other three splice variants (Table 2; P < 0.01vs. all other splice variants). The activation kinetics for channels containing Form B were also slower (Table 3). A mixture of cDNAs for both Forms B and C of KCNQ2 were also co-expressed with cDNA for KCNQ3. The resultant currents retained biexponential activation and deactivation kinetics, with deactivation time constants intermediate between those of Form B and Form C expressed individually with KCNQ3, though the time constant for the faster component (τ1, 101 ms) more closely approached that for the ‘slow’ variant Form B than those for the other variants (Table 2).
Figure 3. Activation of KCNQ2+KCNQ3 channels expressed in CHO cells.
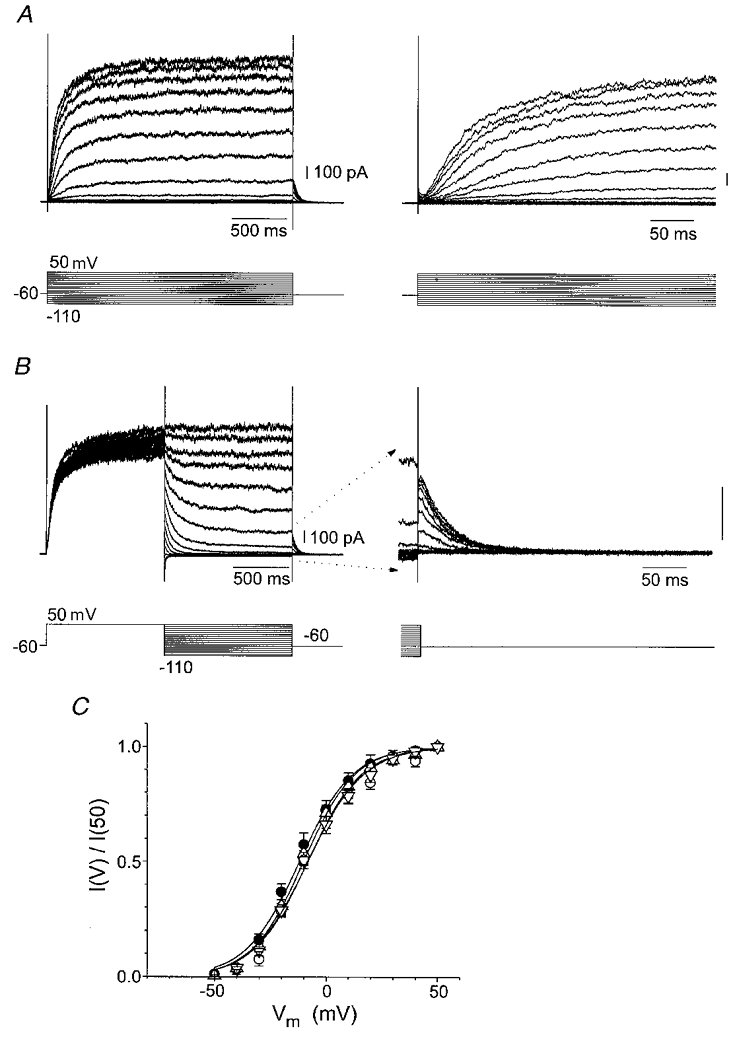
A, KCNQ2 (Form E)+KCNQ3 currents recorded in response to prolonged depolarizing voltage steps in a transfected CHO cell are shown at two different time scales. B, activation properties of channels recorded from the same transfected CHO cell were measured from deactivation tails recorded at -60 mV following steps over the range -110 to +50 mV. C, average steady-state activation curves for the KCNQ2 Forms A, B, C and E co-expressed with KCNQ3. For the parameters of activation see Table 1. ○, Form A; ○, Form B; ▵, Form C; ▿, Form E.
Table 1.
Steady-state activation properties of alternatively spliced KCNQ2 clones
| Splice variant | V0.5 (mV) | k (mV) | n |
|---|---|---|---|
| Form A | −11.9 ± 0.8 | 12.0 ± 0.7 | 5 |
| Form B | −8.2 ± 1.2 | 12.1 ± 1.0 | 5 |
| Form C | −10.1 ± 0.6 | 11.6 ± 0.6 | 4 |
| Form E | −8.1 ± 0.7 | 12.5 ± 0.7 | 5 |
Parameters were obtained by fitting activation curves like those shown in Fig. 3 with the Boltzmann equation. V0.5 and k are the halfactivation potential and the slope factor, respectively; n is the number of cells.
Figure 4. Deactivation kinetics of different KCNQ2 isoforms and the native M-current.

A, examples of KCNQ2 (Form C)+KCNQ3 and KCNQ2 (Form B)+KCNQ3 currents recorded from two different CHO cells in response to a deactivation voltage-clamp protocol: voltage steps over the range -20 to -100 mV in 10 mV increments from a holding potential of -20 mV. Deactivation of Form B currents was approximately 2.5 times slower than that for Form C. B, deactivation of the native M-current recorded from SCG neurons. Examples of cells with fast and slow deactivation kinetics are shown.
Figure 5. Kinetic analysis of KCNQ2+KCNQ3 channels expressed in CHO cells (A) and native M-currents in SCG neurons (B).
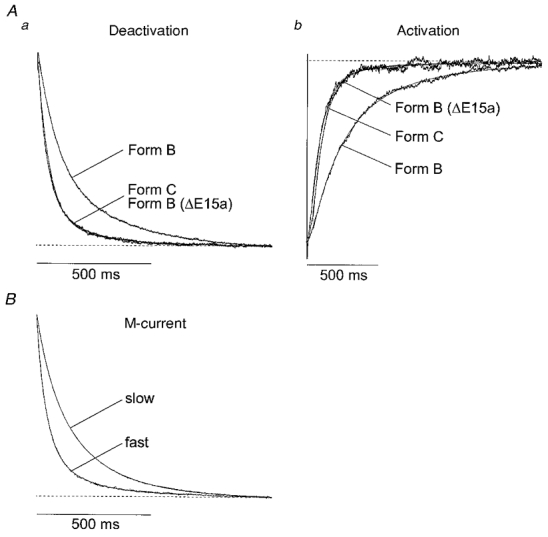
Normalized currents produced by stepping from -20 to -50 mV (Aa and B, deactivation) and from -50 to -20 mV (Ab, activation) are shown. Currents were fitted (smooth lines, superimposed) by the sums of two exponential curves. Values of the two time constants τ1 (fast) and τ2 (slow), with the relative magnitude of the slower component in parentheses, were: Aa (deactivation), 40 and 209 ms (11 %) for KCNQ2 Form C; 95 and 396 ms (37 %) for KCNQ2 Form B; and 47 and 168 ms (25 %) for KCNQ2 Form B(ΔE15a). Ab (activation): 92 and 226 ms (56 %) for KCNQ2 Form C; 240 and 4227 ms (37 %) for KCNQ2 Form B; and 81 and 797 ms (2 %) for Form B(ΔE15a). In B (M-current deactivation), τ1 and τ2, respectively, were 44 and 217 ms (12 %) for the fast M-current, and 99 and 492 ms (22 %) for the slow M-current.
Table 2.
Deactivation kinetics of alternatively spliced KCNQ2 clones
| Splice variant | τ1 (ms) | τ2 (ms) | A2 (%) | n |
|---|---|---|---|---|
| Form A | 48 ± 4 | 208 ± 18 | 22 ± 4 | 14 |
| Form B | 125 ± 18 | 501 ± 70 | 37 ± 4 | 14 |
| Form C | 49 ± 5 | 188 ± 29 | 28 ± 5 | 9 |
| Form E | 53 ± 4 | 205 ± 23 | 28 ± 2 | 15 |
| Form B(ΔE15a) | 48 ± 3 | 212 ± 18 | 19 ± 4 | 10 |
| Forms B+C | 101 ± 15 | 309 ± 36 | 38 ± 5 | 8 |
Deactivation tails were recorded at −50 mV following a step from −20 mV (see Fig. 4). Tails were fitted with the sum of two exponentials, with fast and slow time constants τ1 and τ2, respectively. A2 is the elative contribution of the slow (τ2) component; n is the number of cells. Mean values for τ1 and τ2 for Form B ere significantly larger (P < 0.01) than the corresponding mean values for Forms A, C, E and B(ΔE15a).
Table 3.
Activation kinetics of alternatively spliced KCNQ2 clones
| Splice variant | τ1 (ms) | τ2 (ms) | A2 (%) |
|---|---|---|---|
| Form A | 126 ± 18 | 697 ± 104 | 17 ± 7 |
| Form B | 195 ± 24 | 1626 ± 365 | 25 ± 4 |
| Form C | 102 ± 15 | 571 ± 127 | 29 ± 9 |
| Form E | 129 ± 14 | 639 ± 109 | 17 ± 6 |
| Form B(ΔE15a) | 120 ± 13 | 671 ± 123 | 11 ± 2 |
Comparison with native M-currents recorded from SCG neurons
The different deactivation kinetics of the heterologously expressed KCNQ2+KCNQ3 currents have parallels with the properties of native M-currents recorded from SCG neurons (Fig. 4). M-currents in SCG neurons were recorded using identical recording conditions to those used for the CHO cells. The deactivation kinetics of the M-current in neurons showed significant variation from cell to cell (Fig. 4B). When measured in a sample of 41 SCG neurons, the time constants of the slow and fast components of the native current fell almost completely within the range of the time constants τ1 and τ2 for the alternatively spliced forms of KCNQ2+KCNQ3 channels (Figs 5 and 6), suggesting that alternative splicing of KCNQ2 transcripts might account for the variation in deactivation rates seen for the native current (see Discussion).
Figure 6. Comparison of the fast and slow time constants for deactivation of the native M-current with those of the KCNQ2 splice variants.
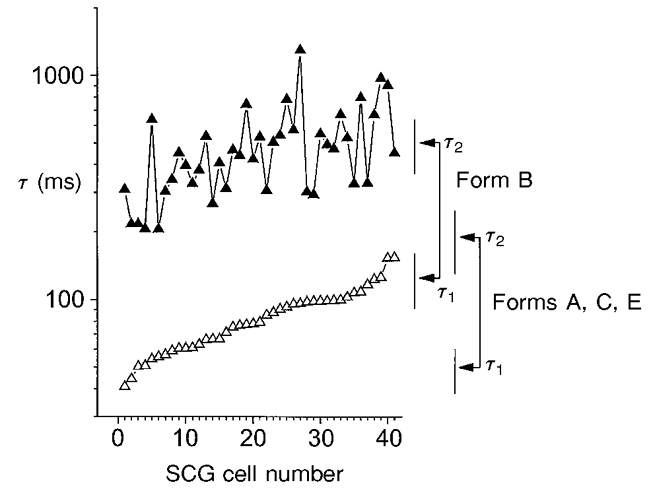
Fast (▵) and slow (▴) time constants for deactivation in 41 SCG neurons plotted vs. cell number. Pairs of data points were ordered so that the fast time constants increased from left to right. There is a correlation between the values of the fast time constant (τ1) and the slow time constant (τ2) (R = 0.52, P < 0.001). On the right, mean values (arrows) and 95 % confidence limits (vertical bars) of the corresponding experimentally observed fast and slow time constants of the different KCNQ2 splice variants are plotted (see Table 2; values for splice variants A, C and E are pooled).
Exon 15a accounts for the slow deactivation and activation kinetics
Based on the sequence differences between the four splice variants (Fig. 2), it seemed most likely that the difference in kinetics of Form B, compared with those of the other splice variants, was due to the presence in Form B of exon 15a. To test this possibility we constructed a Form B clone with a deletion of exon 15a (Form B (ΔE15a)). The deactivation-activation kinetics of this clone were essentially identical to those of Forms A, C and E (Fig. 5; Tables 2 and 3), confirming that the presence of exon 15a in Form B accounts for the slow deactivation-activation kinetics.
KCNQ2 variants are inhibited via activation of M1 muscarinic acetylcholine receptors
To test whether the splice variants differed in their sensitivity to muscarinic inhibition the channels were co-expressed with the human M1 muscarinic receptor. Currents produced by co-expression of KCNQ2 Forms A, B, C, and E with KCNQ3 in M1 receptor-transformed CHO cells were all inhibited by 10 μM of the muscarinic agonist oxotremorine methiodide (Oxo-M; n = 2, 4, 3, and 3, respectively; see Fig. 7 for examples). Oxo-M also inhibited both fast and slow M-currents in SCG neurons (Fig. 7).
Figure 7. Muscarinic inhibition of fast and slow KCNQ2+KCNQ3 channels expressed in CHO cells and fast and slow native M-currents in SCG neurons.

Currents recorded using the standard deactivation protocol (bottom trace) before, during and after washout of Oxo-M (10 μM).
Alternatively spliced KCNQ2 cDNA clones containing out-of-frame exons 15 and 16
In addition to the clones containing in-frame exons 15 and 16, six of the 41 clones contained an alternative splice junction in exon 14 leading to a reading frame shift in exons 15 and 16 (Fig. 8). This class of clones comprised 15 % of the total cDNA clones suggesting that they make up a significant fraction of the total KCNQ2 transcripts. This did not appear to be a cloning artifact, because RNase protection analysis with a probe specific to the alternative splice junction in exon 14 showed that transcripts with this splice junction made up approximately 13 % of the total KCNQ2 mRNA (data not shown). Curiously, however, these clones did not encode functional channels when expressed either alone or in combination with the KCNQ3 subunit in Xenopus oocytes. These clones also did not function as dominant negatives in Xenopus oocytes. Expression of a frame-shifted KCNQ2 subunit did not reduce peak currents when co-expressed in a 1:1:1 stoichiometry of KCNQ2 (frame-shift):KCNQ2 (Form A):KCNQ3, in comparison to control cells injected only with equimolar concentrations of KCNQ2 (Form A) and KCNQ3 mRNA (data not shown). The function of these frame-shifted subunits remains unknown.
Figure 8. KCNQ2 transcripts containing an alternative exon 14 splice junction.

A, expanded view of the KCNQ2 genomic structure indicating the location of the alternative exon 14 splice junction (hatched region) that results in a downstream reading frame shift. Exon lengths are shown to scale but intron lengths are shortened. B, deduced amino acid sequence of the KCNQ2 transcript containing the alternative exon 14 splice junction. Exon junctions are marked above the sequence. The asterisk indicates the stop codon.
These clones also had alternative splice patterns that were similar to those found for the full-length clones. The number of clones obtained was too small to determine whether the distribution of splice variants was affected by the presence of the alternative exon 14 splice site.
Isolation of truncated KCNQ2 cDNA clones
During initial cDNA cloning studies we isolated multiple cDNA clones that terminated before exon 16. Most of these clones contained the exons encoding the hydrophobic core of the channel but terminated soon after the core. Similar results have been described in mouse (Nakamura et al. 1998). These truncated clones were potentially of interest since they might encode subunits that function as dominant negatives or have some other novel function. Although these truncated clones represented a significant percentage of the total number of cDNA clones isolated in both rat and mouse it was uncertain whether mRNA transcripts encoding truncated channels were actually present at significant levels in vivo. Because a wide variety of different truncated cDNA clones were isolated it was not practical to directly test the abundance of each unique transcript using RNase protection assays. Instead, the relative abundance of the transcripts containing exon 16 (probe P6) was compared with the expression of transcripts containing the core region (probe P2) using an RNase protection assay (Fig. 9). Transcripts recognized by probe P6 were as abundant as those recognized by the P2 probe, which suggests that the vast majority of KCNQ2 transcripts contain exon 16. It is likely, therefore, that the multiple truncated isoforms of the channel described previously in mouse (Nakamura et al. 1998) and that we also found in rat are cloning artifacts, possibly due to partially spliced pre-mRNAs in the RNA used as a template for the cDNA clones.
Figure 9. RNase protection analysis of KCNQ2 expression in SCG RNA.

RNase protection analysis of KCNQ2 probes P6 and P2 using two independent SCG RNA samples. To allow comparison of the relative efficiency with which the two probes hybridized to KCNQ2 transcripts in SCG RNA both probes were also hybridized to in vitro transcribed KCNQ2 transcripts in parallel experiments (not shown). For quantification, the results were normalized based on the efficiency of probe hybridization to the in vitro transcripts. cyc indicates cyclophilin probe, used as an internal control.
DISCUSSION
In this paper we show that a large number of alternatively spliced KCNQ2 gene transcripts are expressed in the SCG. One class of transcripts, those containing exon 15a, encodes channels that have unique functional properties. When co-assembled with the KCNQ3 subunit these channels deactivate at significantly slower rates than do KCNQ2 subunits that lack this region. The presence of transcripts containing exon 15a can account, at least in part, for the heterogeneous deactivation kinetics seen for native M-currents recorded from SCG neurons.
Two exponential components were present in the activation and deactivation time courses of both native M-currents (see also Marrion et al. 1992; Selyanko & Brown, 1999) and KCNQ2+KCNQ3 currents. This is not itself a consequence of the heteromeric nature of channels because currents generated by homomeric KCNQ2 channels also showed biexponential kinetics (A. A. Selyanko, unpublished observations); instead, it reflects some fundamental property of channel gating, since biexponential kinetics can be predicted from the voltage dependence of the open and closed time distributions of single native M-channels (Selyanko & Brown, 1999). Nevertheless, the results obtained in the present experiments do suggest that subunit structure does affect M-current kinetics overall, without disturbing the basic gating properties, and that the natural variation in kinetics from one cell to another could well result from variable contributions of the ‘fast’ and ‘slow’ KCNQ2 splice variants to the make-up of the native channels. Thus, the variation in the deactivation kinetics of native M-currents (which is quite considerable – up to about 3-fold for the faster time constant τ1; Fig. 6) is almost completely encompassed by the deactivation rates of the cloned channels.
It might, of course, be expected that more than two exponential components should be present in the deactivation time course of the native M-current, due to the presence of heteromeric channels containing both slow and fast forms of KCNQ2 (and perhaps also due to the variable expression of homomers containing only slow or fast subunits). This might well be the case, and indeed the intermediate values for most of the time constants found in the native current support this possibility. In practice, however, no significant improvement to the fits could be obtained using more than two exponential functions. Further, expression of a mixture of ‘fast’ (C) and ‘slow’ (B) KCNQ2 splice variants with KCNQ3 yielded currents that also retained biexponential kinetics. The proportion of native neurons showing ‘slow’ kinetics within the range encompassed by the ‘slow’ splice variants containing exon 15a was rather larger than that suggested by the relative abundance of these splice variants as predicted from the number of clones. However, the selection of individual cells for electrophysiological study was unlikely to have been strictly random, and may bear little relation to the amounts of RNA in whole ganglia. Further, when equal amounts of cDNA for ‘fast’ and ‘slow’ (B and C) KCNQ2 splice variants were co-expressed with KCNQ3, the kinetics of the resultant current more closely approached those of the ‘slow’ splice variant than the ‘fast’ splice variant, suggesting that the ‘slow’ splice variant might exert a disproportionate effect on current kinetics.
The functional significance of the variation in channel kinetics is not understood. Slow M-current deactivation will, in principle, make a neuron less excitable for a longer period following activation of the M-current. It is thought that there are functionally distinct groups of neurons within the SCG (Janig & McLachlan, 1992) so expression of the slow currents might correlate with one subgroup of cells. There is currently no way of testing this possibility, however, because functional subtypes of sympathetic neurons cannot be identified anatomically.
Exon 15a occurs in a region that is well conserved among all KCNQ channels. This region is thought to be involved in subunit recognition and assembly (Schmitt et al. 2000). The results presented in this paper suggest that this region is also important in determining deactivation and activation properties. A naturally occurring mutation in this region of the KCNQ1 channel (R555C) produces a change in the rate of deactivation and a shift in the steady-state activation properties of the channel (Chouabe et al. 1997). One simple explanation that could account for these results is that the segment encoded by exon 15a affects the ease with which subunits change conformation between the open and closed states by modifying how they interact with each other.
The other sites of alternative splicing occur in less well-conserved regions except for the exon 12 alternative 5′ splice junction, which occurs in a region of partial conservation for all the KCNQ genes except KCNQ1. The function of these other regions is not well established and the KCNQ2 splice variants did not vary significantly in the functional properties that were tested other than deactivation-activation kinetics. They were all expressed with similar efficiency, had similar steady-state activation properties and were similarly susceptible to inhibition by the muscarinic agonist Oxo-M when co-expressed with the human M1 muscarinic receptor. It should be noted that homomeric KCNQ1, 2, 3 and 4 channels expressed in these cells can all be inhibited by stimulating M1 receptors (Selyanko et al. 2000), even though their C-tails vary considerably.
Thus, variation in KCNQ2 splicing appears to be an important source of the variation in M-channel kinetics seen in vivo. However, we cannot exclude other possible causes of variation, such as post-translational modification of the channel subunits or the variable expression of other KCNQ genes in these neurons.
Acknowledgments
This work was supported by grants NS-29755, HL-2895816 and HL-61269 from the National Institutes of Health and programme grant G7909913 from the UK Medical Research Council. We would like to thank Steven Sattler and Fe C. Abogadie for expert technical assistance.
References
- Biervert C, Schroeder BC, Kubisch C, Berkovic SF, Propping P, Jentsch TJ, Steinlein OK. A potassium channel mutation in neonatal epilepsy. Science. 1998;279:403–406. doi: 10.1126/science.279.5349.403. [DOI] [PubMed] [Google Scholar]
- Biervert C, Steinlein OK. Structural and mutational analysis of KCNQ2, the major gene locus for benign familial neonatal convulsions. Human Genetics. 1999;104:234–240. doi: 10.1007/pl00008713. [DOI] [PubMed] [Google Scholar]
- Brown DA. M currents. In: Narahashi T, editor. Ion Channels. New York: Plenum Press; 1988. pp. 55–94. [DOI] [PubMed] [Google Scholar]
- Charlier C, Singh NA, Ryan SG, Lewis TB, Reus BE, Leach RJ, Leppert M. A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nature Genetics. 1998;18:53–55. doi: 10.1038/ng0198-53. [DOI] [PubMed] [Google Scholar]
- Chouabe C, Neyroud N, Guicheney P, Lazdunski M, Romey G, Barhanin J. Properties of KvLQT1 K+ channel mutations in Romano-Ward and Jervell and Lange-Nielsen inherited cardiac arrhythmias. EMBO Journal. 1997;16:5472–5479. doi: 10.1093/emboj/16.17.5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colman A. Translation of eukaryotic messenger RNA in Xenopus oocytes. In: Hames BD, Higgins SJ, editors. Transcription and Translation. Oxford: IRL Press; 1984. pp. 271–302. [Google Scholar]
- Dixon JE, McKinnon D. Potassium channel mRNA expression in prevertebral and paravertebral sympathetic neurons. European Journal of Neuroscience. 1996;8:183–191. doi: 10.1111/j.1460-9568.1996.tb01179.x. [DOI] [PubMed] [Google Scholar]
- Janig W, McLachlan EM. Characteristics of function-specific pathways in the sympathetic nervous system. Trends in Neurosciences. 1992;15:475–481. doi: 10.1016/0166-2236(92)90092-m. [DOI] [PubMed] [Google Scholar]
- Marrion NV, Adams PR, Gruner W. Muliple kinetic states underlying macroscopic M-currents in bullfrog sympathetic neurons. Proceedings of the Royal Society. 1992;B248:207–214. doi: 10.1098/rspb.1992.0063. [DOI] [PubMed] [Google Scholar]
- Mullaney I, Dodd MW, Buckley N, Milligan G. Agonist activation of transfected human M1 muscarinic acetylcholine receptors in CHO cells results in downregulation of both the receptor and the α-subunit of the G-protein Gq. Biochemical Journal. 1993;289:125–131. doi: 10.1042/bj2890125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M, Watanabe H, Kubo Y, Yokoyama M, Matsumoto T, Sasai H, Nishi Y. KQT2, a new putative potassium channel family produced by alternative splicing. Isolation, genomic structure, and alternative splicing of the putative potassium channels. Receptors and Channels. 1998;5:255–271. [PubMed] [Google Scholar]
- Owen DG, Marsh SJ, Brown DA. M-current noise and putative M-channels in cultured rat sympathetic ganglion cells. The Journal of Physiology. 1990;431:269–290. doi: 10.1113/jphysiol.1990.sp018330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rae J, Cooper K, Gates P, Watsky M. Low access resistance perforated patch recordings using amphotericin B. Journal of Neuroscience Methods. 1991;37:15–26. doi: 10.1016/0165-0270(91)90017-t. [DOI] [PubMed] [Google Scholar]
- Schmitt N, Schwarz M, Peretz A, Abitbol I, Attali B, Pongs O. A recessive C-terminal Jervell and Lange-Nielsen mutation of the KCNQ1 channel impairs subunit assembly. EMBO Journal. 2000;19:332–340. doi: 10.1093/emboj/19.3.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selyanko AA, Brown DA. M-channel gating and simulation. Biophysical Journal. 1999;77:701–713. doi: 10.1016/S0006-3495(99)76925-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selyanko AA, Hadley JK, Wood IC, Abogadie FC, Delmas P, Buckley NJ, London B, Brown DA. Two types of K+ channel subunit, Erg1 and KCNQ2/3, contribute to the M-like current in a mammalian neuronal cell. Journal of Neuroscience. 1999;19:7742–7756. doi: 10.1523/JNEUROSCI.19-18-07742.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selyanko AA, Hadley JK, Wood IC, Abogadie FC, Jentsch TJ, Brown DA. Inhibition of KCNQ 1–4 potassium channels expressed in mammalian cells via M1 muscarinic acetylcholine receptors. The Journal of Physiology. 2000;522:349–355. doi: 10.1111/j.1469-7793.2000.t01-2-00349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selyanko AA, Robbins J, Brown DA. Putative M-type potassium channels in neuroblastoma-glioma hybrid cells: inhibition by muscarine and bradykinin. Receptors and Channels. 1995;3:147–159. [PubMed] [Google Scholar]
- Singh NA, Charlier C, Stauffer D, Dupont BR, Leach RJ, Melis R, Ronen GM, Bjerre I, Quattlebaum T, Murphy JV, McHarg ML, Gagnon D, Rosales TO, Peiffer A, Anderson VE, Leppert M. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nature Genetics. 1998;18:25–29. doi: 10.1038/ng0198-25. [DOI] [PubMed] [Google Scholar]
- Tinel N, Lauritzen I, Chouabe C, Lazdunski M, Borsotto M. The KCNQ2 potassium channel: splice variants, functional and developmental expression. Brain localization and comparison with KCNQ3. FEBS Letters. 1998;438:171–176. doi: 10.1016/s0014-5793(98)01296-4. [DOI] [PubMed] [Google Scholar]
- Wang H-S, McKinnon D. Potassium channel expression in prevertebral and paravertebral sympathetic neurones: control of firing properties. The Journal of Physiology. 1995;485:319–335. doi: 10.1113/jphysiol.1995.sp020732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H-S, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science. 1998;282:1890–1893. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- Yamada WM, Koch C, Adams PR. Multiple channels and calcium dynamics. In: Koch C, Segev I, editors. Methods in Neuronal Modeling. Cambridge MA USA: Bradford; 1989. pp. 97–133. [Google Scholar]