

Abstract
It has been previously shown that spreading neuronal activation can generate a cortical spreading ischaemia (CSI) in rats. The purpose of the present study was to investigate whether vasodilators cause CSI to revert to a normal cortical spreading depression (CSD).
A KCl-induced CSD travelled from an open cranial window to a closed window where the cortex was superfused with physiological artificial cerebrospinal fluid (ACSF). At the closed window, recordings revealed a short-lasting negative slow potential shift accompanied by a variable, small and short initial hypoperfusion followed by hyperaemia and then oligaemia.
In contrast, spreading neuronal activation locally induced CSI at the closed window when ACSF contained a NO. synthase (NOS) inhibitor, NG-nitro-l-arginine, and an increased K+ concentration ([K+]ACSF). CSI was characterised by a sharp and prolonged initial cerebral blood flow decrease to 29 ± 11 % of the baseline and a prolonged negative potential shift.
Co-application of a NO. donor, S-nitroso-N-acetylpenicillamine, and NOS inhibitor with high [K+]ACSF re-established a short-lasting negative potential shift and spreading hyperaemia typical of CSD. Similarly, the NO.-independent vasodilator papaverine caused CSI to revert to a pattern characteristic of CSD.
In acute rat brain slices, NOS inhibition and high [K+]ACSF did not prolong the negative slow potential shift compared to that induced by high [K+]ACSF alone.
The data indicate that the delayed recovery of the slow potential was caused by vasoconstriction during application of high [K+]ACSF and a NOS inhibitor in vivo. This supports the possibility of a vicious circle: spreading neuronal activation induces vasoconstriction, and vasoconstriction prevents repolarisation during CSI. Speculatively, this pathogenetic process could be involved in migraine-induced stroke.
The coupling between neuronal activity and cerebral blood flow (CBF) is a fundamental process which underpins all cerebral functions (for review see Villringer & Dirnagl, 1995). The underlying mechanisms and how they might become disrupted in cerebrovascular disease are still far from being completely understood. Cortical spreading depression (CSD) represents a specific phenomenon associated with neuronal activation and secondary changes of CBF (for review see Lauritzen, 1994). CSD is characterised as a depolarisation wave in the cerebral cortex propagating at a rate of approximately 3 mm min−1. The wave is induced by factors interfering with neuronal and astroglial function, such as glutamate and K+. Large changes in the ionic homoeostasis and release of excitatory amino acids are associated with CSD. Inter- and extracellular communication between neurones and astrocytes are likely to be involved in the propagation of CSD under physiological conditions (Nedergaard et al. 1995; Basarsky et al. 1998; Kunkler & Kraig, 1998). Interactions between neurones and astrocytes during neuronal activation are also assumed at the level of neuronal nutrition (neurometabolic coupling) (Pellerin & Magistretti, 1994). Hence, both neurones and astrocytes probably collaborate in the process of matching the increased metabolic demand with CBF during spreading depression.
In the brains of rodents, CBF may slightly decrease during the onset of CSD but this initial vasoconstriction is variable and short. CSD then induces a brief cortical spreading hyperaemia, followed by a longer-lasting cortical spreading oligaemia (for review see Lauritzen, 1994). Adequate tissue oxygenation during CSD is reflected by the lack of histopathological changes (Nedergaard & Hansen, 1988), except for microglial activation (Caggiano & Kraig, 1996).
The physiological mechanism of the CBF response to CSD is not well understood. Much attention has been focused on the role of the potent vasodilator NO. (Goadsby et al. 1992; Duckrow, 1993; Zhang et al. 1994; Wahl et al. 1994; Fabricius et al. 1995; Wolf et al. 1996; Colonna et al. 1997; Dreier et al. 1998). A conceivable mechanism linking CSD to NO. is the coupling of NO. production to changes of intracellular [Ca2+]. Intracellular [Ca2+] rises during CSD in neurones (Kunkler & Kraig, 1998). This stimulates the neuronal NO. synthase (NOS) (Moncada et al. 1991). Endothelial NOS could be indirectly activated by acetylcholine or monoamines released during CSD. Indeed, an increase of NO. was directly measured at the initial phase of CSD (Read & Parsons, 2000). Hence, the vasodilator NO. may mask an initial vasoconstriction. Accordingly, Duckrow (1993) found that NOS inhibition augmented the initial CBF decrease which may depend on the rise of the extracellular K+ concentration ([K+]o), a strong vasoconstrictive stimulus (Hansen et al. 1980; McCulloch et al. 1982).
The findings by Duckrow (1993) were confirmed in some (Fabricius et al. 1995; Dreier et al. 1998) but not all (Zhang et al. 1994; Wolf et al. 1996) studies in rats. The reasons for these discrepancies remain unclear. However, a substantial augmentation and prolongation of the initial vasoconstriction was achieved by applying either a NOS inhibitor or the NO. scavenger oxyhaemoglobin in the presence of increased subarachnoid K+ yielding a cortical spreading ischaemia (CSI) (Dreier et al. 1998). CSI was similarly obtained by combined administration of oxyhaemoglobin, physiological K+ and low glucose in the subarachnoid space (Dreier et al. 2000). In these experiments, histopathological analysis revealed that CSIs produce widespread cortical infarction (Dreier et al. 2000). Thus, experimental evidence was provided that spreading neuronal activation can generate significant ischaemia if the coupling between neuronal metabolism and blood flow is inverted (Dreier et al. 1998, 2000).
In the present study, CSI was induced by topical superfusion of ACSF containing a NOS inhibitor and increased [K+]ACSF in rats. We tested whether the NO. donor S-nitroso-N-acetylpenicillamine (SNAP) or the NO.-independent vasodilator papaverine re-established a short-lasting CSD with hyperaemia. We also investigated the effects of NOS inhibition and high [K+]ACSF on CSD in brain slices in which vascular effects are excluded.
METHODS
Animal preparation, experimental protocol, CBF measurement and electrophysiology in vivo
All animal experiments were approved by the Landesamt für Arbeitsschutz, Gesundheitsschutz und technische Sicherheit Berlin (G 0346/98). Male Wistar rats (n = 30; 250-350 g) were anaesthetised with 100 mg kg−1 thiopental-sodium intraperitoneally (i.p.) (Trapanal, BYK Pharmaceuticals, Konstanz, Germany), tracheotomised, and artificially ventilated (Effenberger Rodent Respirator, Effenberger Med.-Techn. Gerätebau, Pfaffing/Attel, Germany). The left femoral artery and vein were cannulated, and continuous saline solution (1 ml h−1) was infused. Body temperature was maintained at 38.0 ± 0.5°C using a heating pad. Systemic arterial pressure (RFT Biomonitor, Zwönitz, Germany) and endexpiratory PCO2 (Heyer CO2 Monitor EGM I, Bad Ems, Germany) were monitored. Arterial PO2 (Pa,O2), Pa,CO2 and pH were serially measured using a Compact 1 blood gas analyser (AVL Medizintechnik GmbH, Bad Homburg, Germany). Since the rats were not paralysed, the adequacy of the level of anaesthesia was assessed by testing motor responses to tail pinch. In addition, changes of blood pressure in response to tail pinch were used to control anaesthesia. Further thiopental doses (25 mg kg−1i.p.) were applied when necessary.
Parietally, a craniotomy was performed using a saline-cooled drill as previously reported (Dreier et al. 1998). The dura mater was removed. The craniotomy site was covered with a piece of glass cut from a coverslip so that a closed cranial window resulted. An inflow and outflow tube allowed superfusion of the brain cortex with ACSF at the closed window (Fig. 1). The composition of the ACSF was (mmol l−1): Ca2+ 1.5; Mg2+ 1.2; HCO3− 24.5; Cl− 135; glucose 3.7; urea 6.7. The K+ concentrations (3, 20 and 35 mmol l−1) determined the Na+ concentrations (152, 135 and 120 mmol l−1). The ACSF was equilibrated with a gas mixture comprising 6.6 % O2, 5.9 % CO2 and 87.5 % N2 leading to a PO2 of 125.9 ± 18.3 mmHg, a PCO2 of 38.1 ± 5.2 mmHg and a pH of 7.39 ± 0.05. CBF was continuously monitored by one or two laser-Doppler flow probes (Perimed AB, Järfälla, Sweden). The steady (direct current; DC) potential was measured by a silver chloride wire with an agar bridge inserted into the space between the cortex and the coverslip. The electrode was connected to a differential amplifier (Jens Meyer, Munich, Germany). Systemic arterial pressure, CBF, DC shift and endexpiratory PCO2 were continuously recorded using a PC and a chart recorder (DASH IV, Astro-Med, Inc, West Warwick, RI, USA).
Figure 1. Experimental set-up.

Aa, experimental set-up of groups 1-3. CSD was elicited at an open window (ow) using a drop of KCl. From the open window, the neuronal activation spread to a closed window (cw). The closed window was covered by a piece of glass cut from a coverslip. An inflow (ACSFin) and outflow (ACSFout) tube allowed the superfusion of the cortex with artificial cerebrospinal fluid (ACSF) at the closed window. CBF was measured by one or two laser-Doppler flow probes (LDF 1, LDF 2) at the closed window. The direct current (DC) shift was recorded using a silver chloride electrode. Ab, higher magnification of the windows in Aa. In response to the second cortical spreading depression (CSD) which started from the open window, a cortical spreading ischaemia (CSI) was recorded at the closed window while ACSF contained baseline [K+]ACSF at 20 mmol l−1 and l-NNA. B, experimental set-up of group 5. ACSF containing l-NNA and baseline [K+]ACSF at 35 mmol l−1 induced a CSI at the closed window (cw). From the closed window, the neuronal activation spread to an open frontal window (ow) over naive cortical tissue. At the open window, the spreading neuronal activation induced a cortical spreading depression (CSD). CBF was measured by a laser-Doppler flow probe (LDF 1) at the closed window. At the open window, CBF (LDF 2) and the DC potential were recorded.
To determine the effects of the NO. donor SNAP (RBI/Sigma, Deisenhofen, Germany) and the NO.-independent vasodilator papaverine (Sigma Chemicals, Deisenhofen, Germany) on the CSI, 18 experiments were performed using the experimental set-up shown in Fig. 1Aa (groups 1-3). An open cranial window was laterally implanted over the temporal bone to later elicit a CSD there by application of a droplet of KCl (150 mmol l−1) followed by washout with physiological ACSF after approximately 30 s. The dura mater was cut at this open window. From the open window, CSD spread to the parietal closed window where CBF and the DC potential were measured. To begin with, CSD was triggered under two different conditions in each animal: superfusion of the cortex at the closed window with (1) physiological ACSF and (2) ACSF containing the NOS inhibitor NG-nitro-l-arginine (l-NNA) (Sigma) at 1 mmol l−1 and a [K+] ([K+]ACSF) of 20 mmol l−1. The first CSD which was triggered at the open cranial window induced a CSD at the closed cranial window while the cortex was superfused with physiological ACSF. The second CSD (1.5 h after the first CSD), starting from the open window, induced a CSI instead of CSD at the closed window when ACSF contained l-NNA and high [K+]ACSF. This second event is illustrated in Fig. 1Ab. After the second CSD, superfusion with l-NNA and increased [K+]ACSF were continued and the animals were assigned to three different groups. In group 1, SNAP was co-applied with l-NNA and high [K+]ACSF. In group 2, papaverine was co-applied instead of SNAP. Both vasodilators were administrated at a saturating concentration of 100 μmol l−1. No vasodilator was added in group 3. A third CSD (1 h after the second CSD) was triggered under the respective condition in each animal.
The experimental set-up in Fig. 1Ba was used to confirm the effect of SNAP on the CSI. To begin with, in six animals, only a closed cranial window was implanted. [K+]ACSF was increased at the closed window from 3 to 35 mmol l−1. From 35 mmol l−1, it was further increased by steps of 10 mmol l−1 until CSDs occurred (group 4). In another six animals, ACSF containing l-NNA and [K+]ACSF at 35 mmol l−1 was superfused (group 5). Under this condition, CSIs instead of CSDs spontaneously occurred at the closed window. A second open cranial window with intact dura mater was frontally implanted in these animals (Fig. 1Ba). There, CBF was also measured, and the DC potential was recorded using a calomel electrode. When a CSI occurred at the parietal closed window, a CSD arrived at the frontal open window with delay. This arrangement is illustrated in Fig. 1Bb. After several CSI→CSD cycles, SNAP was co-administrated with l-NNA and high [K+]ACSF at the closed window to test its effect on the CSI.
Brain slices and K+-selective microelectrodes
The experiments were performed using six slices from six different male Wistar rats (150-200 g). Combined entorhinal cortical- hippocampal slices were prepared as previously reported (Dreier & Heinemann, 1991). The brain was removed and washed in cold (5-8 °C) ACSF after decapitation under deep ether anaesthesia. Near horizontal slices (400 μm) were cut using a vibratome (752 M vibroslice, Campden Instruments Ltd, Loughborough, UK). The slices corresponded to the temporal area of plates 99-108 in the atlas of Paxinos & Watson (1986). They included ventral hippocampal formation, entorhinal cortex and neocortex.
Slices were transferred into an interface recording chamber and continuously perfused with prewarmed (35-36°C), carbogenated ACSF containing (mmol l−1): Ca2+ 2; Mg2+ 2; HCO3− 26; Cl− 133; glucose 10; SO22- 2; H2PO4− 1.25 (pH 7.4). The K+ concentrations (5 and 20 mmol l−1) determined the Na+ concentrations (151.25 and 136.25 mmol l−1). A warmed, humified 95 % O2 and 5 % CO2 gas mixture was directed over the surface of the slices which, according to polarographical measurements using platinum-iridium microelectrodes, results in a PO2 of approximately 22 mmHg at a depth of 100 μm where our recordings were performed (Bingmann & Kolde, 1982). This PO2 level corresponds to that in cortical tissue in vivo in the rat of 28.4 ± 3.6 mmHg recorded with the same method (Back et al. 1994). The recordings were started after > 1 h of slice equilibration in the ACSF. Glass-insulated bipolar platinum wire stimulation electrodes were placed into the Schaffer collaterals and a microelectrode measuring the extracellular field potential into the pyramidal layer of area CA1 of the hippocampus to test slice viability. Slices were accepted when they responded to a paired pulse at 50 ms interstimulus interval with single population spikes of more than 3 mV amplitude displaying frequency potentiation. Then, a K+-selective microelectrode was positioned in the neocortex. The K+-selective/reference microelectrode was manufactured and tested as previously reported (Lux & Neher, 1973). CSD was triggered by applying a droplet of KCl (200 mmol l−1) into the perfusion medium at a distance of approximately 300 μm near layer I of the medial entorhinal cortex using a KCl-filled pipette under five different conditions: (1) standard ACSF, (2) ACSF containing baseline [K+]ACSF at 20 mmol l−1, (3) baseline [K+]ACSF at 20 mmol l−1+l-NNA at 1 mmol l−1, (4) baseline [K+]ACSF at 20 mmol l−1+l-NNA at 1 mmol l−1+ SNAP at 100 μmol l−1 and (5) after wash with physiological ACSF. At the neocortical recording site (area Te2 or Te3), a significant slow increase of the extracellular K+ concentration ([K+]o) preceding the sharp rise related to CSD was not detected. This indicated that the KCl droplet did not reach this area by passive diffusion.
Data analysis
Data were analysed by comparing relative changes of CBF and absolute changes of the DC potential and [K+]o. CBF changes were calculated in relation to baseline at the onset of the experiment (= 100 %). The calculated CBF and DC parameters are explained in Fig. 2 by means of an original recording of a CSI. All data in the text and figures are given as mean values ± standard deviation. Statistical comparisons were performed using either Student's t test or analysis of variance either completely randomised or with repeated measures followed by Bonferroni’s procedure. P < 0.05 was accepted as significant.
Figure 2. Calculation of the CBF and DC parameters.

The calculated parameters of the CBF and DC response to CSD are exemplified using a picture of a CSI. 1, baseline CBF level; 2, CBF level as a percentage of baseline immediately before the CSD was triggered; 3, the lowest CBF level as a percentage of baseline during the initial hypoperfusion; 4, the highest CBF level as a percentage of baseline during the hyperaemia; 5, the duration of the initial hypoperfusion in minutes; 6, the duration of the hyperaemia in minutes; 7, duration of the negative DC shift in minutes; 8, amplitude of the initial hypoperfusion as a percentage (= CBF level before CSD minus the lowest CBF level during the initial hypoperfusion); 9, amplitude of the negative DC shift. These parameters were used, for example, in Figs 4-6.
RESULTS
All systemic variables remained within normal limits throughout the preparation and experiment in all experimental groups.
In response to a CSD, a short negative DC shift associated with hyperaemia was recorded when physiological ACSF was superfused
In 18 animals (groups 1-3), CSD was triggered at an open cranial window by KCl. From there, it spread to a closed cranial window where the cortex was superfused with physiological ACSF. At the closed window, CSD induced a variable, small initial hypoperfusion. On average, CBF non-significantly decreased from 107 ± 17 % to 95 ± 15 % compared to baseline (= 100 %). The hypoperfusion lasted for 0.7 ± 0.6 min. Then, CBF increased to 293 ± 102 % for 1.6 ± 0.5 min followed by a longer-lasting decrease to 85 ± 16 %. The subarachnoid DC potential showed a negative shift of -2.6 ± 1.1 mV which lasted for 1.7 ± 0.5 min. Thus, the CBF and DC parameters were characteristic of a normal CSD (Fig. 3Aa, Ba and Ca). Occasionally, more than one CSD occurred in response to the droplet of KCl. This seemed not to influence the further course of the experiment.
Figure 3. SNAP and papaverine caused the CSI to revert to a short CSD associated with hyperaemia.
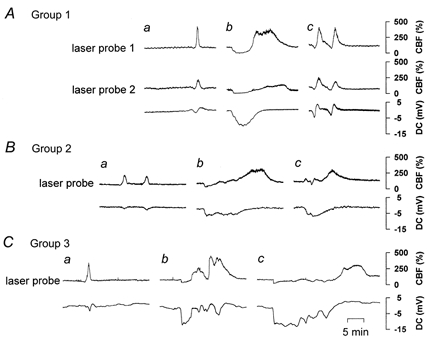
CBF and DC recordings of 3 out of 18 animals in which CSDs were distantly triggered from the closed window. Aa, Ba and Ca, when CSD arrived at the closed window, a short and small initial hypoperfusion variably preceded a spreading hyperaemia under physiological ACSF. The negative DC shift was short lasting. Ab, Bb and Cb, in response to CSD, a sharp and prolonged initial hypoperfusion was observed under ACSF containing l-NNA at 1 mmol l−1 and [K+]ACSF at 20 mmol l−1 (CSI). It was accompanied by a prolonged negative DC shift. Ac, co-application of SNAP at 100 μmol l−1 caused CSI to revert to a short initial hypoperfusion preceding spreading hyperaemia associated with a short-lasting negative DC shift (n = 6). Bc, co-application of papaverine at 100 μmol l−1 less potently caused CSI to revert (n = 6). Cc, a second CSI occurred in response to CSD if no vasodilator was co-applied (n = 6).
In response to CSD, CSI was recorded when ACSF was superfused containing NOS inhibitor and increased [K+]ACSF
Using the same animals (groups 1-3), a second CSD was elicited at the open window while ACSF was topically superfused containing the NOS inhibitor l-NNA at 1 mmol l−1 combined with a [K+]ACSF of 20 mmol l−1 at the closed window (Fig. 3Ab, Bb and Cb). Co-application of l-NNA and high [K+]ACSF decreased CBF to 80 ± 19 % at the closed window before the second CSD. In response to the CSD, a sharp initial hypoperfusion was observed to 29 ± 11 %, lasting for 4.2 ± 2.4 min. The hypoperfusion was followed by a CBF increase to 196 ± 85 % lasting for 10.7 ± 3.9 min. The negative DC shift reached -7.6 ± 3.2 mV, lasting for 9.7 ± 5.8 min. All CBF and DC parameters were significantly different from those under physiological ACSF (P < 0.01). The changes were typical of CSI (Dreier et al. 1998).
A NOá donor or papaverine restored the pattern of a short negative DC shift associated with spreading hyperaemia
Following the second CSD, the animals were assigned to three different groups containing six animals each. In all three groups, superfusion of l-NNA and high [K+]ACSF was continued. The NO. donor SNAP was co-applied at a concentration of 100 μmol l−1 in group 1, and the NO.-independent vasodilator papaverine at 100 μmol l−1 in group 2; in group 3, no vasodilator was added (control group). Figure 3Ac shows that SNAP caused the CSI to almost completely revert to a CSD associated with short-lasting negative DC shift and hyperaemia preceded by only a short initial hypoperfusion. The statistical analysis of the effect is illustrated in Fig. 4 (group 1). Papaverine also caused the CSI to revert to a CSD with short initial hypoperfusion, although less potently than SNAP (Fig. 3Bc). The effects of papaverine were also statistically significant as shown in Fig. 4 (group 2). Papaverine led to a significant recovery of the following parameters: CBF value during the initial hypoperfusion, duration of the hypoperfusion, CBF value during the hyperperfusion and the associated negative DC shift in comparison to values obtained for CSI. If no vasodilator was co-applied with l-NNA and high [K+]ACSF, the third CSD also induced a CSI at the closed window (Fig. 4, group 3). An example is given in Fig. 3Cc. In summary, both SNAP and papaverine caused the CSI to revert to a CSD associated with short-lasting DC potential and hyperaemia but SNAP was more effective than papaverine.
Figure 4. SNAP and papaverine caused the CSI to revert to a short CSD associated with hyperaemia (statistics).

□, physiological ACSF;  , 1 mmol l−1l-NNA + 20 mmol l−1[K+]ACSF;
, 1 mmol l−1l-NNA + 20 mmol l−1[K+]ACSF;  , 1 mmol l−1l-NNA + 20 mmol l−1[K+]ACSF+ 100 μmol l−1 papaverine; ▪, 1 mmol l−1l-NNA + 20 mmol l−1[K+]ACSF+ 100 μmol l−1 SNAP. The figure gives the results of the statistical analysis of groups 1-3 (compare Fig. 3). The calculated parameters are explained in Fig. 2. We statistically compared CSD in the presence of (1) physiological ACSF, (2) ACSF containing l-NNA and high baseline [K+]ACSF and (3) ACSF containing l-NNA, high baseline [K+]ACSF and vasodilator within each group using an analysis of variance with repeated measures followed by Bonferroni’s procedure. A star without bracket indicates that one condition was significantly different from both other conditions while a star with bracket means a significant difference between only two conditions (level of significance: P < 0.05). Regarding all parameters, SNAP seemed more effective than papaverine.
, 1 mmol l−1l-NNA + 20 mmol l−1[K+]ACSF+ 100 μmol l−1 papaverine; ▪, 1 mmol l−1l-NNA + 20 mmol l−1[K+]ACSF+ 100 μmol l−1 SNAP. The figure gives the results of the statistical analysis of groups 1-3 (compare Fig. 3). The calculated parameters are explained in Fig. 2. We statistically compared CSD in the presence of (1) physiological ACSF, (2) ACSF containing l-NNA and high baseline [K+]ACSF and (3) ACSF containing l-NNA, high baseline [K+]ACSF and vasodilator within each group using an analysis of variance with repeated measures followed by Bonferroni’s procedure. A star without bracket indicates that one condition was significantly different from both other conditions while a star with bracket means a significant difference between only two conditions (level of significance: P < 0.05). Regarding all parameters, SNAP seemed more effective than papaverine.
Increased [K+]ACSF without NOS inhibitor induced a short-lasting CSD with sharp and short initial hypoperfusion at the closed window (group 4)
In these experiments, ACSF without l-NNA but with increasing [K+]ACSF was superfused (n = 6). [K+]ACSF at 35 mmol l−1 increased CBF to 162 ± 61 %. CBF and DC potential alterations indicating CSD were seen at 35 mmol l−1 in one animal, only at 45 mmol l−1 in three animals and only at 55 mmol l−1 in two animals. Interestingly, a sharp initial hypoperfusion was recorded in response to CSD (Fig. 5). This was similar to that known from NOS inhibition with normal [K+]ACSF (Duckrow, 1993; Fabricius et al. 1995; Dreier et al. 1998) but started from a higher level of CBF. On average, CBF decreased from 140 ± 59 % to 110 ± 41 %. The hypoperfusion lasted for 54 ± 78 s. Then, CBF shortly increased to 203 ± 44 % followed by a decrease to 108 ± 46 %. The hyperaemic reponse to CSD was significantly smaller in the presence of high baseline [K+]ACSF compared with that in the presence of physiological ACSF (first CSD in groups 1-3) (P < 0.01). The subarachnoid negative DC shift was -4.2 ± 1.5 mV lasting for 2.4 ± 0.7 min. Its amplitude was significantly larger than that in the presence of physiological ACSF (P < 0.05).
Figure 5. Cortical spreading depression triggered at the closed window by high baseline [K+]ACSF without l-NNA.

CSDs also originated at the closed window when ACSF contained increased concentrations of K+ without l-NNA. In B, simultaneous original recordings from the closed window are shown. Interestingly, the CBF response was characterised by a sharp but short-lasting initial hypoperfusion. This pattern is also known to occur under the condition of NOS inhibition and normal cerebrospinal K+ concentration (Duckrow et al. 1993). In A, the CBF parameters recorded at the closed window in the presence of high [K+]ACSF and l-NNA (group 4) () are compared with those in the presence of high [K+]ACSF without l-NNA (group 4) (□). Stars indicate a level of significance of at least P < 0.05. The meaning of the CBF parameters is explained in Fig. 2.
A CSI occurred at the parietal closed cranial window superfused with NOS inhibitor and high [K+]ACSF and induced a CSD at a second frontal window over naive cortical tissue (group 5)
CBF changes typical of CSI occurred at the closed window when ACSF containing l-NNA and [K+]ACSF at 35 mmol l−1 was superfused (n = 6) (Fig. 6Ba parietal laser probe). From the closed window, the neuronal activation spread to a frontal window with intact dura mater. At the naive cortical region of the frontal window, a short negative DC shift associated with hyperaemia characteristic of CSD was recorded with a delay (Fig. 6Ba frontal laser probe and DC recording).
Figure 6. CSI originating at the closed window was converted by SNAP into a CSD associated with short-lasting negative DC shift and hyperaemia.
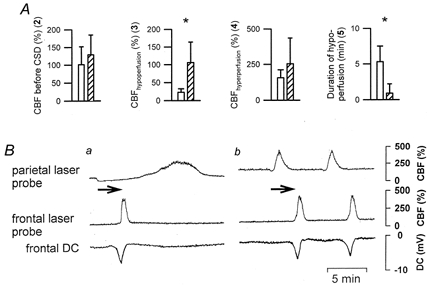
In these experiments, the spreading neuronal activation was not distantly triggered but originated at the closed window superfused with l-NNA and [K+]ACSF at 35 mmol l−1. From there, the neuronal activation spread to an open frontal window where CBF and the DC potential were recorded. In B simultaneous original recordings from the parietal and frontal window are shown. The arrow indicates the spreading of the event from the parietal to the frontal window. While CSI was observed at the closed parietal window, a DC potential and CBF pattern typical of CSD were measured at the naive cortical region of the frontal window (Ba). When SNAP was co-applied, an almost normal CBF response instead of CSI was also generated at the closed parietal window which spread to the open window (Bb). In A, the CBF parameters recorded at the closed window are compared before (□) and after co-application of SNAP (). Stars indicate a level of significance of at least P < 0.05. The meaning of the CBF parameters is explained in Fig. 2.
A NOá donor caused the CSI to revert to a short hyperaemia at the closed window (group 5)
In the same animals, after several CSI→CSD cycles, SNAP was co-applied with l-NNA and high [K+]ACSF at the parietal closed window. Under this condition, CSI reverted to an almost normal CBF response to CSD at the closed window (Fig. 6Bb). The CSD arrived at the frontal window with the same delay as before. No change in the CSD parameters was observed at the naive frontal region. A statistical comparison is given in Fig. 6A regarding the CBF parameters at the closed window before and after co-application of SNAP.
Combined NOS inhibition with high baseline [K+]ACSF did not prolong the DC potential in rat brain slices in comparison to high baseline [K+]ACSF alone
To determine the effects of high baseline [K+]ACSF, l-NNA or SNAP on the neuronal-astroglial network independent of vascular effects, six brain slice experiments were performed. CSD was induced by droplet application of KCl into the perfusate near layer I of the medial entorhinal cortex. The changes of the DC potential and [K+]o were recorded using a K+-sensitive microelectrode placed in neocortical areas Te2 or Te3. CSD showed a negative shift of -12.2 ± 7.0 mV lasting for 1.0 ± 0.2 min accompanied by a sharp increase of [K+]o from 5 to 62 ± 16 mmol l−1 under standard ACSF (Fig. 7Aa and Ba). When baseline [K+]ACSF was increased from 5 to 20 mmol l−1, the duration of both the negative DC shift and the rise of [K+]o was significantly prolonged in response to CSD. Neither the peak level of [K+]o nor the negative DC shift was significantly altered (Fig. 7Ab and Bb). l-NNA and SNAP, continuously applied via the ACSF, had no additional effects on [K+]o or the DC potential (Fig. 7Ac, Ad and Bc). The effect of high [K+]ACSF was reversible (Fig. 7Ae and Be).
Figure 7. NOS inhibition and high baseline [K+]ACSF did not prolong the DC potential in rat brain slices in comparison to high baseline [K+]ACSF alone.

A, statistical comparison of 6 slices. Stars indicate a level of significance of P < 0.05. B, example of original traces of the extracellular K+ concentration ([K+]o) and the DC potential in response to CSD. Aa and Ba, CSD under physiological ACSF. Ab and Bb, CSD under baseline [K+]ACSF at 20 mmol l−1. The repolarisation of the neuronal network was significantly prolonged (= increased duration of the negative DC shift). Ac and Bc, CSD under baseline [K+]ACSF at 20 mmol l−1 and l-NNA at 1 mmol l−1. NOS inhibition had no additional effect on the repolarisation. Ad, CSD under [K+]ACSF at 20 mmol l−1, l-NNA at 1 mmol l−1 and SNAP at 100 μmol l−1. SNAP had no additional effect. Ae and Be, complete recovery after wash with physiological ACSF.
DISCUSSION
CSI was produced by NOS inhibition and increased subarachnoid K+in vivo. In one series, CSD was triggered by KCl at an open cranial window. CSD spread from there to a closed cranial window where CBF and the DC potential were measured. At this second cranial window, the cortex was superfused with ACSF containing the NOS inhibitor and [K+]ACSF at 20 mmol l−1 so that the normal CBF response to CSD was locally transformed into a CSI. When a vasodilator was co-applied, CSI reverted to a short-lasting CSD with normal spreading hyperaemia. The NO. donor was more effective than papaverine.
In another series, the sequence was reversed. CSI originated at the closed cranial window where the cortex was superfused with ACSF containing NOS inhibitor and [K+]ACSF at 35 mmol l−1. With some delay, the spreading neuronal activation arrived at a second cranial window with intact dura mater. A normal, short-lasting CSD with hyperaemia was observed at this naive cortical region. After several of these CSI→CSD cycles, a NO. donor was co-applied with NOS inhibitor and high [K+]ACSF at the closed cranial window. This locally converted the CSI into a normal hyperaemia. The neuronal activation spread to the second window with the same delay as before. The CBF response was unaltered there.
In addition, without NOS inhibitor [K+]ACSF was increased above the threshold to induce CSD at the closed window. The CBF response to CSD was not normal but was characterised by a sharp and short initial hypoperfusion. This was similar to that reported from NOS inhibition with physiological [K+]ACSF (Duckrow, 1993; Fabricius et al. 1995; Dreier et al. 1998). The data indicate that the rise of baseline [K+]ACSF directly altered the vascular reactivity to CSD.
The K+ threshold inducing CSD was determined to be 10-20 mmol l−1 higher than that under co-application of the NOS inhibitor. This may be due to inhibition of the NMDA receptor by NO. (Lipton et al. 1993) or the higher CBF level promoting glial buffering (Paulson & Newman, 1987).
In brain slices, baseline [K+]ACSF at 20 mmol l−1 significantly prolonged the repolarisation after CSD compared to baseline [K+]ACSF at 3 mmol l−1. It has been demonstrated that a rise of baseline [K+]ACSF down-regulated sodium pump activity in cultured cortical astrocytes (Hajek et al. 1996). Whether the neuronal sodium pump is also down-regulated has not been studied yet. The Na+-K+-ATPase is critically involved in the process of repolarisation after CSD (Rosenthal & Somjen, 1973; Mayevsky et al. 1974). Hence, down-regulation of its activity might well explain the delay of repolarisation by increased baseline [K+]ACSF. Neither co-application of the NOS inhibitor nor co-application of the NO. donor with high baseline [K+]ACSF had an additional impact on the repolarisation process after CSD in the slice in contrast to the in vivo experiments. This suggests that the delay of repolarisation related to the NOS inhibitor and the accelaration by the NO. donor in vivo were secondary to their vascular actions. Neither increased baseline [K+]ACSF nor the NO.-related compounds significantly altered other CSD-related parameters such as the peak level of [K+]o or the negative DC shift.
A vicious circle of vasoconstriction induced by spreading neuronal depolarisation, and inhibition of neuronal repolarisation by vasoconstriction
The data unequivocally demonstrate that NO. is involved in the CBF response to CSD. This action is unmasked by, for example, a sustained increase in [K+]ACSF in the subarachnoid space.
Intracortical [K+]o acutely rises to a level of 60 mmol l−1 in response to CSD even if baseline [K+]ACSF is physiological (Hansen et al. 1980). Therefore, it is surprising that, additionally, an increase of baseline [K+]ACSF to 20 mmol l−1 is necessary to produce CSI. However, the effect of the increase of baseline [K+]ACSF may be related to the above-mentioned down-regulation of sodium pump activity leading to a delay of repolarisation after CSD. Thus, it was demonstrated earlier that CSI was also generated by the combination of NOS inhibition with the sodium pump inhibitor ouabain instead of high baseline [K+]ACSF (Dreier et al. 1997).
During CSD, the release of K+ into the extracellular space is related to the depolarisation of neurones and astrocytes. As mentioned above, [K+]o acutely rises to a level of approximately 60 mmol l−1 during CSD. At extracellular concentrations above 20 mmol l−1, K+ acts as a strong vasoconstrictor (McCulloch et al. 1982). This implies that the release of a strong vasoconstrictor is directly linked to the depolarisation of the neuronal-astroglial network. On the other hand, elevated [K+]o is known to release NO. (Dickie et al. 1990), which antagonises the vasoconstrictive effect of K+ (Minato et al. 1985; Nishiye et al. 1989). Thus, under otherwise normal conditions, the CSD-induced rise of [K+]o only induces an initial vasoconstriction when NO. is absent (Duckrow, 1993). If, in addition to a state of low NO., the repolarisation of the neuronal-astroglial network is partially inhibited by a decreased sodium pump activity, K+-induced vasoconstriction will be prolonged. If the vasoconstrictive phase is prolonged, the oxygen and glucose reserves of neurones and astrocytes will be finally exhausted. This, in turn, will produce a secondary, vascular-dependent inhibition of the neuronal-astroglial repolarisation which maintains vasoconstriction, and so on.
Hence, in vivo, two different mechanisms might inhibit the network repolarisation under the condition of low NO. and high baseline [K+]ACSF: an initial direct neuronal-astroglial and a secondary indirect vascular mechanism. The direct mechanism is due to the sustained increase of [K+]ACSF probably down-regulating the activity of the Na+-K+-ATPase (Hajek et al. 1996) and thereby inhibiting repolarisation. This mechanism could explain why elevated baseline [K+]ACSF prolonged the negative DC shift in response to CSD in the brain slices. The vascular mechanism is likely to be related to a combined effect of NOS inhibition and the rise of baseline [K+]ACSF. The resulting vasoconstriction indirectly inhibits the ATP-dependent sodium pump through oxygen/glucose deprivation. Consistently, an increased delay of repolarisation was only observed in vivo but not in vitro by combined NOS inhibition and high baseline [K+]ACSF compared to high baseline [K+]ACSF alone. Also in support of a vascular component is the finding that vasodilators caused the CSI (characterised by a prolonged negative DC shift) to revert to a CSD with shorter negative DC shift and hyperaemia. This was demonstrated for a NO. donor and the NO.-independent vasodilator papaverine in this study and, earlier on, also for the NO.-independent vasodilator nimodipine (Dreier et al. 1998). Nimodipine was similarly effective to the NO. donor.
Clinical implications
All recent population- and hospital-based epidemiological studies demonstrate an increased risk of migraine patients developing ischaemic stroke (Buring et al. 1995; Tzourio et al. 1995; Carolei et al. 1996; Merikangas et al. 1997). The risk was significantly higher for migraineurs with aura compared to those without (Tzourio et al. 1995; Carolei et al. 1996). There are different types of migraine-related stroke. The diagnosis of so called migraine-induced stroke has been based on several operational diagnostic criteria: (a) the patient has previously fulfilled criteria for migraine with aura; (b) the present attack is typical of previous attacks, but neurological deficits are not completely reversible within 7 days and/or neuroimaging demonstrates ischaemic infarction in the relevant area; and (c) other causes of infarction are ruled out by appropriate investigations (Headache Classification Committee, 1988). The pathophysiological correlate of the migrainous aura is thought to originate in the neuronal-astroglial network of the brain cortex leading to secondary vascular changes (Leão & Morison, 1945; Moskowitz & MacFarlane, 1993; Lauritzen, 1994). Thus, a slowly spreading wave of cortical depression (cortical spreading depression) has been proposed to induce spreading oligaemia of the cerebral cortex during the migrainous aura (Lauritzen, 1994; Woods et al. 1994). It remains enigmatic why focal oligaemia progresses to migraine-induced infarction in a small fraction of patients. Clinical data have suggested that research in migraine-induced stroke should rather focus on the neuronal metabolism-
blood flow cascade which may lead to irreversible cellular necrosis than on cardiac or arterial lesions (Bogousslavsky et al. 1988). In the present study, mechanisms were investigated which might be related to such an inversion of the physiological coupling between the spreading depolarisation of the neuronal-astroglial network and CBF.
Naturally, it remains speculative whether CSI is pathogenetically involved in cerebrovascular diseases. The combination of a local NOS inhibitor and high baseline [K+]ACSF is an artificial condition. However, several other combinations are also known to produce CSI. So far, all combinations consist of either a NOS inhibitor or the NO. scavenger oxyhaemoglobin and a factor interfering with the activity of the sodium pump such as high baseline K+, low baseline glucose or ouabain (Dreier et al. 1997, 1998, 2000). More combinations are currently under investigation. Candidate clinical conditions for CSI might be stroke syndromes related to a state of low cortical NO. and low sodium pump activity. In principle, NO. can decrease through inhibition of NO. synthase, through destruction or disturbance of NO.-producing sources such as endothelium or perivascular nerves, or through NO. scavenging by, for example, haemoglobin. Sodium pump function can be reduced by down-regulation, direct inhibition, disturbed energy metabolism or decreased oxygen and/or glucose supply. CSI was shown to lead to bell shaped or laminar infarcts of the brain cortex (Dreier et al. 2000). As discussed in previous communications (Dreier et al. 1998, 2000), delayed ischaemic neurological deficits (DINDs) after subarachnoid haemorrhage might represent the most interesting candidate. This applies to the lesion morphology, localisation and distribution and also to the pathophysiological situation in the cortex and subarachnoid space. DINDs occur in relation to the NO. scavenger oxyhaemoglobin and factors such as high [K+]o, low glucose, endogenous digitalis-like compounds and vasospasm (Ohta et al. 1983; MacDonald & Weir, 1991; Luùsic et al. 1999; Sarrafzadeh et al. 1999).
Basic data on migraine-induced stroke are sparse except for the probable link to spreading depression. Whether a local state of low cortical NO. and sodium pump activity is related to migraine-induced strokes is unknown. Interestingly, one of the genetically determined migraine-related stroke syndromes has been associated with a disturbance of neuronal-astroglial energy metabolism plus angiopathy with endothelial alteration (Ohama et al. 1987; Clark et al. 1996). This so called MELAS syndrome (mitochondrial encephalopathy with lactic acidosis and stroke-like episodes) typically leads to laminar necrosis of the brain cortex. However, direct data on sodium pump function and NO. metabolism in the cerebral cortex are not available for this syndrome.
Vasodilators such as nimodipine have proven valuable in the prophylactic treatment of DINDs (Feigin et al. 1998). If CSI is involved in the pathogenesis of migraine-induced stroke – as predicted from our model – vasodilators might be of similar prophylactic value in the prevention of migraine-induced stroke.
Acknowledgments
This study was supported by grants DFG-SFB 507 A1 (J.P.D and U.D.). The support of the Hermann and Lilly Schilling foundation (U.D.) is gratefully acknowledged.
References
- Back T, Kohno K, Hossmann K-A. Cortical negative DC deflections following middle cerebral artery occlusion and KCl-induced spreading depression: effect on blood flow, tissue oxygenation, and electroencephalogram. Journal of Cerebral Blood Flow and Metabolism. 1994;14:12–19. doi: 10.1038/jcbfm.1994.3. [DOI] [PubMed] [Google Scholar]
- Basarsky TA, Duffy SN, Andrew RD, MacVicar BA. Imaging spreading depression and associated intracellular calcium waves in brain slices. Journal of Neuroscience. 1998;18:7189–7199. doi: 10.1523/JNEUROSCI.18-18-07189.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingmann D, Kolde G. PO2-profiles in hippocampal slices of the guinea pig. Experimental Brain Research. 1982;48:89–96. doi: 10.1007/BF00239575. [DOI] [PubMed] [Google Scholar]
- Bogousslavsky J, Regli F, van Melle G, Payot M, Uske A. Migraine stroke. Neurology. 1988;38:223–227. doi: 10.1212/wnl.38.2.223. [DOI] [PubMed] [Google Scholar]
- Buring JE, Herbert P, Romero J, Kittross A, Cook N, Manson J, Peto R, Hennekens C. Migraine and subsequent risk of stroke in the physician’s health study. Archives of Neurology. 1995;52:129–134. doi: 10.1001/archneur.1995.00540260031012. [DOI] [PubMed] [Google Scholar]
- Caggiano AO, Kraig RP. Eicosanoids and nitric oxide influence induction of reactive gliosis from spreading depression in microglia but not astrocytes. Journal of Comparative Neurology. 1996;369:93–108. doi: 10.1002/(SICI)1096-9861(19960520)369:1<93::AID-CNE7>3.0.CO;2-F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carolei A, Marini C, de Matteis G. History of migraine and risk of cerebral ischemia in young adults. The Italian National Research Council Study Group on Stroke in the Young. Lancet. 1996;347:1503–1506. doi: 10.1016/s0140-6736(96)90669-8. [DOI] [PubMed] [Google Scholar]
- Clark JM, Marks MP, Adalsteinsson E, Spielman DM, Shuster D, Horoupian D, Albers GW. MELAS: Clinical and pathologic correlations with MRI, xenon/CT, and MR spectroscopy. Neurology. 1996;46:223–227. doi: 10.1212/wnl.46.1.223. [DOI] [PubMed] [Google Scholar]
- Colonna DM, Meng W, Deal DD, Gowda M, Busija DW. Neuronal NO promotes cerebral cortical hyperemia during cortical spreading depression in rabbits. American Journal of Physiology. 1997;272:H1315–1322. doi: 10.1152/ajpheart.1997.272.3.H1315. [DOI] [PubMed] [Google Scholar]
- Dickie BGM, Lewis MJ, Davies JA. Potassium-stimulated release of nitric oxide from cerebellar slices. British Journal of Pharmacology. 1990;101:8–9. doi: 10.1111/j.1476-5381.1990.tb12078.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier JP, Ebert N, Priller J, Megow D, Lindauer U, Klee R, Reuter U, Imai Y, Einhäupl KM, Victorov I, Dirnagl U. Products of hemolysis in the subarachnoid space induce cortical spreading ischemia and focal necrosis in rats: a model for delayed ischemic neurological deficits after subarachnoid haemorrhage? Journal of Neurosurgery. 2000;93:668–676. doi: 10.3171/jns.2000.93.4.0658. [DOI] [PubMed] [Google Scholar]
- Dreier JP, Ebert N, Reuter U, Wolf T, Leistner S, Villringer A, Dirnagl U. Pharmacological transformation of spreading depression (SD) related hyperperfusion to an acute and longlasting hypoperfusion by NOS-inhibiton and ouabain. Society for Neuroscience Abstracts. 1997;23:1309. [Google Scholar]
- Dreier JP, Heinemann U. Regional and time dependent variations of low Mg2+ induced epileptiform activity in rat temporal cortex slices. Experimental Brain Research. 1991;87:581–596. doi: 10.1007/BF00227083. [DOI] [PubMed] [Google Scholar]
- Dreier JP, Körner K, Ebert N, Görner A, Rubin I, Back T, Lindauer U, Wolf T, Villringer A, Einhäupl KM, Lauritzen M, Dirnagl U. Nitric oxide scavenging by haemoglobin or nitric oxide synthase inhibition by N-nitro-L-arginine induce cortical spreading ischemia when K+ is increased in the subarachnoid space. Journal of Cerebral Blood Flow and Metabolism. 1998;18:978–990. doi: 10.1097/00004647-199809000-00007. [DOI] [PubMed] [Google Scholar]
- Duckrow RB. A brief hypoperfusion precedes spreading depression if nitric oxide synthesis is inhibited. Brain Research. 1993;618:190–195. doi: 10.1016/0006-8993(93)91265-t. [DOI] [PubMed] [Google Scholar]
- Fabricius M, Akgören N, Lauritzen M. Arginine-nitric oxide pathway and cerebrovascular regulation in cortical spreading depression. American Journal of Physiology. 1995;269:H23–29. doi: 10.1152/ajpheart.1995.269.1.H23. [DOI] [PubMed] [Google Scholar]
- Feigin VL, Rinkel GJ, Algra A, Vermeulen M, van Gijn J. Calcium antagonists in patients with aneurysmal subarachnoid hemorrhage: a systematic review. Neurology. 1998;50:876–883. doi: 10.1212/wnl.50.4.876. [DOI] [PubMed] [Google Scholar]
- Goadsby PJ, Kaube H, Hoskin KL. Nitric oxide synthesis couples cerebral blood flow and metabolism. Brain Research. 1992;595:167–170. doi: 10.1016/0006-8993(92)91470-y. [DOI] [PubMed] [Google Scholar]
- Hajek I, Subbarao KV, HertZ L. Acute and chronic effects of potassium and noradrenaline on Na+,K+-ATPase activity in cultured mouse neurones and astrocytes. Neurochemistry International. 1996;28:335–342. doi: 10.1016/0197-0186(95)00081-x. [DOI] [PubMed] [Google Scholar]
- Hansen AJ, Quistorff B, Gjedde A. Relationship between local changes in cortical blood flow and extracellular K+ during spreading depression. Acta Physiologica Scandinavica. 1980;109:1–6. doi: 10.1111/j.1748-1716.1980.tb06557.x. [DOI] [PubMed] [Google Scholar]
- Headache Classification Committee of the International Headache Society. Classification and diagnostic criteria for headache disorders cranial neuralgias and facial pain. Cephalgia. 1988;8(suppl. 7):1–96. [PubMed] [Google Scholar]
- Kunkler PE, Kraig RP. Calcium waves precede electrophysiological changes of spreading depression in hippocampal organ cultures. Journal of Neuroscience. 1998;18:3416–3425. doi: 10.1523/JNEUROSCI.18-09-03416.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauritzen M. Pathophysiology of the migraine aura. The spreading depression theory. Brain. 1994;117:199–210. doi: 10.1093/brain/117.1.199. [DOI] [PubMed] [Google Scholar]
- Leao AAP, Morison RS. Propagation of spreading cortical depression. Journal of Neurophysiology. 1945;8:33–45. [Google Scholar]
- Lipton SA, Choi YB, Pan ZH, Lei SZ, Chen HS, Sucher NJ, Loscalzo J, Singel DJ, Stamler JS. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature. 1993;364:626–632. doi: 10.1038/364626a0. [DOI] [PubMed] [Google Scholar]
- Lusic I, Ljutic D, Maŝkovic J, Jankovic S. Plasma and cerebrospinal fluid endogenous digoxin-like immunoreactivity in patients with aneurysmal subarachnoid haemorrhage. Acta Neurochirurgica. 1999;141:691–697. doi: 10.1007/s007010050363. [DOI] [PubMed] [Google Scholar]
- Lux HD, Neher E. The equilibration time course of [K+]o in cat cortex. Experimental Brain Research. 1973;17:190–205. doi: 10.1007/BF00235028. [DOI] [PubMed] [Google Scholar]
- McCulloch J, Edvinsson L, Watt P. Comparison of the effects of potassium and pH on the calibre of cerebral veins and arteries. Pflügers Archiv. 1982;393:95–98. doi: 10.1007/BF00582399. [DOI] [PubMed] [Google Scholar]
- MacDonald RL, Weir BKA. A review of hemoglobin and the pathogenesis of cerebral vasospasm. Stroke. 1991;22:971–982. doi: 10.1161/01.str.22.8.971. [DOI] [PubMed] [Google Scholar]
- Mayevsky A, Zeuthen T, Chance B. Measurements of extracellular potassium, ECoG and pyridine nucleotide levels during cortical spreading depression in rats. Brain Research. 1974;76:347–349. doi: 10.1016/0006-8993(74)90467-3. [DOI] [PubMed] [Google Scholar]
- Merikangas KR, Fenton BT, Cheng SH, Stolar MJ, Risch N. Association between migraine and stroke in a large-scale epidemiological study of the United States. Archives of Neurology. 1997;54:362–368. doi: 10.1001/archneur.1997.00550160012009. [DOI] [PubMed] [Google Scholar]
- Minato H, Hashizume M, Masuda Y, Hosoki K. Modulation of extraluminally induced vasoconstrictions by endothelium-derived nitric oxide in the canine basilar artery. Arzneimittelforschung. 1995;45:675–678. [PubMed] [Google Scholar]
- Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacological Reviews. 1991;43:109–142. [PubMed] [Google Scholar]
- Moskowitz MA, MacFarlane R. Neurovascular and molecular mechanisms in migraine headaches. Cerebrovascular and Brain Metabolism Reviews. 1993;5:159–177. [PubMed] [Google Scholar]
- Nedergaard M, Cooper AJ, Goldman SA. Gap junctions are required for the propagation of spreading depression. Journal of Neurobiology. 1995;28:433–444. doi: 10.1002/neu.480280404. [DOI] [PubMed] [Google Scholar]
- Nedergaard M, Hansen AJ. Spreading depression is not associated with neuronal injury in the normal brain. Brain Research. 1988;449:395–398. doi: 10.1016/0006-8993(88)91062-1. [DOI] [PubMed] [Google Scholar]
- Nishiye E, Nakao K, Itoh T, Kuriyama H. Factors inducing endothelium-dependent relaxation in the guinea-pig basilar artery as estimated from the actions of haemoglobin. British Journal of Pharmacology. 1989;96:645–655. doi: 10.1111/j.1476-5381.1989.tb11864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohama E, Ohara S, Ikuta F, Tanaka K, Nishizawa M, Miyatake T. Mitochondrial angiopathy in cerebral blood vessels of mitochondrial encephalomyopathy. Acta Neuropathologica. 1987;74:226–233. doi: 10.1007/BF00688185. [DOI] [PubMed] [Google Scholar]
- Ohta O, Osaka K, Siguma M, Yamamoto M, Shimizu K, Toda N. Cerebral vasospasm following ruptured intracranial aneurysms, especially some contributions of potassium ion released from subarachnoid hematoma to delayed cerebral vasospasm. In: Bevan JA, editor. Vascular Neuroeffector Mechanisms. New York: Raven Press; 1983. pp. 353–358. [Google Scholar]
- Paulson OB, Newman EA. Does the release of potassium from astrocyte endfeet regulate cerebral blood flow? Science. 1987;237:896–898. doi: 10.1126/science.3616619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. London: Academic Press; 1986. [Google Scholar]
- Pellerin L, Magistretti P. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proceedings of the National Academy of Sciences of the USA. 1994;91:10625–10629. doi: 10.1073/pnas.91.22.10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read SJ, Parsons AA. Sumatriptan modifies cortical free radical release during cortical spreading depression. A novel antimigraine action for sumatriptan? Brain Research. 2000;870:44–53. doi: 10.1016/s0006-8993(00)02400-8. [DOI] [PubMed] [Google Scholar]
- Rosenthal M, Somjen G. Spreading depression, sustained potential shifts, and metabolic activity of cerebral cortex of cats. Journal of Neurophysiology. 1973;36:739–749. doi: 10.1152/jn.1973.36.4.739. [DOI] [PubMed] [Google Scholar]
- Sarrafzadeh AS, Unterberg AW, Lanksch WR. Bedside-microdialysis for early detection of vasospasm after subarachnoid hemorrhage. Case report and review of the literature. Zentralblatt für Neurochirurgie. 1999;59:269–273. [PubMed] [Google Scholar]
- Tzourio C, Tehindrazanarivelo A, Iglesias S, Alperovitch A, Chedru F, d’ Anglejan-Chatillon J, Bousser MG. Case-control study of migraine and risk of ischaemic stroke in young women. British Medical Journal. 1995;310:830–833. doi: 10.1136/bmj.310.6983.830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villringer A, Dirnagl U. Coupling of brain activity and cerebral blood flow: basis of functional neuroimaging. Cerebrovascular and Brain Metabolism Reviews. 1995;7:240–276. [PubMed] [Google Scholar]
- Wahl M, Schilling L, Parsons AA, Kaumann A. Involvement of calcitonin gene-related peptide (CGRP) and nitric oxide (NO) in the pial artery dilatation elicited by cortical spreading depression. Brain Research. 1994;637:204–210. doi: 10.1016/0006-8993(94)91234-3. [DOI] [PubMed] [Google Scholar]
- Wolf T, Lindauer U, Obrig H, Dreier JP, Back T, Villringer A, Dirnagl U. Systemic nitric oxide synthase inhibition does not affect brain oxygenation during cortical spreading depression in rats: A noninvasive near-infrared spectroscopy and laser-Doppler flowmetry study. Journal of Cerebral Blood Flow and Metabolism. 1996;16:1100–1107. doi: 10.1097/00004647-199611000-00003. [DOI] [PubMed] [Google Scholar]
- Woods RP, Iacobini M, Maiottazz JC. Bilateral spreading cerebral hypoperfusion during spontaneous migraine headache. New England Journal of Medicine. 1994;331:1689–1692. doi: 10.1056/NEJM199412223312505. [DOI] [PubMed] [Google Scholar]
- Zhang ZG, Chopp M, Maynard KI, MoskowitZ MA. Cerebral blood flow changes during cortical spreading depression are not altered by inhibition of nitric oxide synthesis. Journal of Cerebral Blood Flow and Metabolism. 1994;14:939–943. doi: 10.1038/jcbfm.1994.125. [DOI] [PubMed] [Google Scholar]