

Abstract
Over the last decade, a debate has developed about the mechanism of the passive or ‘diffusive’ component of intestinal glucose absorption and, indeed, whether it even exists. Pappenheimer and colleagues have proposed that paracellular solvent drag contributes a passive component, which, at high concentrations of sugars similar to those in the jejunal lumen immediately after a meal, is severalfold greater than the active component mediated by the Na+-glucose cotransporter SGLT1. On the other hand, Ferraris & Diamond maintain that the kinetics of glucose absorption can be explained solely in terms of SGLT1 and that a passive or paracellular component plays little, if any, part. Recently, we have provided new evidence that the passive component of glucose absorption exists, but is in fact facilitated since it is mediated by the rapid, glucose-dependent activation and recruitment of the facilitative glucose transporter GLUT2 to the brush-border membrane; regulation involves a protein kinase C (PKC)-dependent pathway activated by glucose transport through SGLT1 and also involves mitogen-activated protein kinase (MAP kinase) signalling pathways. This topical review seeks to highlight the significant points of the debate, to show how our proposals on GLUT2 impact on different aspects of the debate and to look at the regulatory events that are likely to be involved in the short-term regulation of sugar absorption during the assimilation of a meal.
The debate over the mechanism of the passive component of glucose absorption
Following the initial proposal of Crane et al. (1961), the mechanism of Na+-glucose cotransport became firmly established over the next 25 years; for a review, see Stevens et al. (1984). One simple, but vital, point for the debate over the passive component is that there was and remains common agreement that Na+-glucose cotransport saturates at 30-50 mm glucose in vivo. Yet there were numerous reports that glucose absorption increases almost linearly from 50 mm to several hundred millimolar (see Fordtran & Inglefinger, 1968). It appeared that there must be other mechanisms for the absorption of glucose.
Indeed, there was already much evidence, accruing alongside and even predating this intense period of work on Na+-glucose cotransport, that there is also a significant passive component of glucose absorption. Many papers referred to Km values for glucose absorption of 100 mm or more (see Parsons & Prichard, 1966); typical data are to be found in Holdsworth & Dawson (1964), showing that glucose and fructose absorption increased almost linearly up to a concentration of 280 mm. As early as 1956, Fullerton & Parsons reported that glucose absorption in vivo comprised two components: one was constant and not associated with water absorption (now identified with SGLT1); the other was variable and approximately proportional to water absorption up to 246 mm. Manome & Kuriaki (1961) used phloridzin to inhibit Na+-glucose cotransport and showed that glucose absorption in vivo comprised phloridzin-sensitive and phloridzin-insensitive components. Using this approach, Debnam & Levin (1975) defined glucose absorption as the sum of an active component saturating around 30-50 mm glucose, and a phloridzin-insensitive component described as ‘passive’ or ‘diffusive’, since it was broadly linear and appeared non-saturable; the passive component equalled the active one at about 26 mm and continued to increase up to 128 mm glucose. Ilundain et al. (1979) and Lostao et al. (1991) reported that the passive component was some 3-5 times greater than the active component at high glucose concentrations. One significant point is that all studies showing clear evidence for a significant passive component are in vivo studies. That suggests (with hindsight - see below) that there is a significant difference between in vivo and in vitro preparations.
Nineteen eighty-seven was a pivotal year for studies of intestinal glucose absorption. Pappenheimer & Reiss (1987) proposed the theory of paracellular solvent drag as an explanation for the passive component of glucose absorption, and Hediger et al. (1987) cloned the Na+-glucose cotransporter SGLT1. The molecular era of intestinal glucose absorption had arrived and produced a profound change in the intellectual climate of the field, which was to ensure that the molecular biology of SGLT1 became almost totally dominant for the next 13 years (Hediger & Rhoads, 1994). It was against this background that the debate over the passive component took place.
In an attempt to explain the in vivo experiments, Pappenheimer & Reiss (1987) proposed the theory of paracellular solvent drag, based on the fact that the passive (also termed ‘paracellular’) component of glucose absorption is associated with high rates of water absorption (Fullerton & Parsons 1956). They envisaged that concentration of glucose in the intercellular spaces (up to 425 mm - Pappenheimer, 1993) by Na+-coupled transport from the lumen (300 mm - then thought to be the physiological value after a meal) provides an osmotic force for fluid flow. The latter results in bulk absorption of nutrients against the concentration gradient by literally dragging glucose from the lumen through opened tight junctions into the intercellular spaces by non-ideal solvent-solute interactions. In support of this view, Madara & Pappenheimer (1987) showed that transport of glucose through SGLT1 causes contraction of the peri-junctional actomyosin ring and so promotes dilatation of the intercellular tight junctions, suggesting the passive component occurs by paracellular solvent drag and is SGLT1 dependent.
The kinetic relationship between glucose absorption and fluid flow is defined by non-equilibrium thermodynamics as the sum of two terms, namely, the flux due to back-diffusion down the concentration gradient from intracellular spaces to lumen and the flux from lumen to intracellular spaces due to solvent drag. Pappenheimer & Reiss (1987) determined the relationship between glucose absorption and fluid flow in vivo at a single glucose concentration, approximately 23 mm. The contribution of solvent drag to overall glucose flux at 23 mm was small; however, they then applied the diffusional permeability and solvent drag coefficients so measured to calculate the relevant contributions to the overall glucose flux at 300 mm. They concluded that back-diffusion is negligible and that flux due to solvent drag exceeds that due to active transport severalfold.
The alternative view to paracellular flow, advanced by Ferraris & Diamond, is that adaptation of brush-border membrane transporters (that is, SGLT1) is matched to dietary intake (Ferraris & Diamond, 1989, 1997; Ferraris et al. 1990). In Table 1 of their paper, Ferraris et al. (1990) give a list of ‘modern’Km values for SGLT1-mediated transport, which are in the range 6-23 mm for in vivo and 2-6 mm for in vitro studies, the difference being attributed to unstirred layer effects. They maintain that the currently known kinetic properties of SGLT1 can account for the observed rates of glucose absorption and that paracellular flow, and by implication any passive component, is negligible. These conclusions depend crucially on what the luminal glucose concentrations are after a meal. Pappenheimer & Reiss (1987) used the high values of the early literature in calculating paracellular fluxes of glucose. However, improvement of analytical techniques has resulted in continual revision of these values downwards. Using modern analytical techniques, therefore, Ferraris et al. (1990) undertook a detailed study of the concentrations of free glucose present throughout the day in the lumen of rats on different diets. For rats on the most nearly physiological diets, concentrations ranged with time and small intestinal region from 0.2 to a maximum of 48 mm. These figures now span the effective concentration range of SGLT1 activity.
There are, however, two serious questions prompted by their subsequent conclusion that everything can be explained in terms of SGLT1. The first is whether the measured concentration was the relevant one. In making their measurements of luminal glucose concentration, Ferraris et al. (1990) simply emptied the contents of a gut segment into a vial for analysis. Such a method can only measure the free glucose that escapes into the lumen down the concentration gradient or, at best, an average over all luminal contents. As Pappenheimer (1993, 1998) has pointed out, this will inevitably be much less than the effective concentration at the membrane resulting from the hydrolysis of α-limit dextrins and disaccharides. It is, of course, not possible to measure the relevant glucose concentration directly, since glucose from hydrolysis is present only transiently in a local environment. Nevertheless, indirect measurements from the rate of membrane hydrolysis of maltose suggest that the local concentration is of the order of 300 mm (see Pappenheimer, 1998). In recognition of the importance of local concentration, Lane et al. (1999) measured paracellular flow directly in the unanaesthetised dog in the presence of 150 mm maltose. They convincingly demonstrated that paracellular flow, monitored by the transport of l-glucose, accounts for no more than 2-7 % of overall absorption, even at these very high local maltose concentrations, which were associated with a high rate of water absorption. Thus these direct measurements provide support for the closely argued view of Ferraris & Diamond (1997) that such paracellular flow of nutrients as occurs in the experiments of Pappenheimer & Reiss (1987) does so at very low rates in long-lasting experiments.
The second question is how can SGLT1 possibly account for the continued increase in absorption up to several hundred millimolar glucose, when there is general agreement that SGLT1 is saturated at 30-50 mm? Ferraris & Diamond make no comment on this in any of their articles (Ferraris & Diamond, 1989, 1997; Ferraris et al. 1990), even though in the latest in vivo experiments of Lane et al. (1999) the rate of glucose absorption increased about 3.6-fold between 50 and 150 mm glucose. In fact they appear to have systematically discounted the existence of any passive component. For example, in Table 1 of Ferraris et al. (1990), the Km of active transport reported by Debnam & Levin (1975) is quoted as 23 mm for the phloridzin-sensitive component, but there is no mention at any point of a passive component. This is true for all other quoted references to in vivo work, where the concentration is high enough to detect a passive component, which can be as high as 3-5 times the active. The ensuing disjunction of their work and Pappenheimer's meant that the two sides could not agree on the passive component of glucose absorption.
Two final points, apparently overlooked in the debate so far, are of note. Firstly, glucose absorption by solvent drag is proposed to depend on the activity of SGLT1. If, then, Na+-glucose cotransport is inhibited in vivo, most, if not all, of the passive component should be inhibited. Yet the data of Debnam & Levin (1975) showed this was not the case, for an independent passive component remained after inhibition of Na+-glucose cotransport by phloridzin that was as much as 1.5 times greater than the active component. However, see below.
The second point concerns fructose absorption, which was singled out by Pappenheimer & Reiss (1987) as being similar to that of glucose. We know now that fructose absorption is mediated by GLUT5 in the brush-border membrane, which has a Km of 6 mm (Gould & Holman, 1993); GLUT2 is also involved (see below). The significant point here is that, to the best of current knowledge, only facilitative transporters are involved. There can be no concentration of fructose in the intercellular spaces and therefore no sugar-induced paracellular solvent drag.
At this point in the debate, glucose absorption at high concentrations cannot be explained in terms of either paracellular flow or the currently known properties of SGLT1.
Evidence that the passive component of sugar absorption is mediated by the glucose- and hormone-dependent regulation of GLUT2 at the brush-border membrane
The current dogma of intestinal absorption is that glucose enters the absorptive cell by SGLT1 in the brush-border membrane and exits into the blood by GLUT2, a member of the facilitative glucose transporter family, in the basolateral membrane (see Fig. 2 of Hediger & Rhoads, 1994). Fructose enters by GLUT5, which is highly specific for fructose, and exits by GLUT2, so that glucose and fructose share a common exit pathway. We have now provided evidence for an alternative mechanism for the passive component of absorption, namely that it is mediated by the glucose- and hormone-dependent regulation of GLUT2 at the brush-border membrane: rapid trafficking of GLUT2 to the brush-border membrane is controlled by the SGLT1-dependent activation of a protein kinase C (PKC)-dependent pathway and also by MAP kinase intracellular signalling pathways (Helliwell et al. 2000a, b; Kellett & Helliwell, 2000).
Figure 2. Brush-border membrane glucose absorption in rat jejunum in vivo comprises an active and a passive component.

Total glucose absorption by rat jejunum in vivo (▪) comprises a phloretin-sensitive, GLUT2-mediated component (•) and a phloretin-insensitive, SGLT1-mediated component (▴). The value of [G1/2] for the passive component was determined to be 56 ± 14 mm by fitting the data by non-linear regression analysis to a Hill-type equation: v = (Vmax[G]h)/(K+[G]h), where Vmax is the maximal rate of transport, [G1/2] is the glucose concentration at half Vmax, K =[G1/2]h and h is the Hill coefficient. Data for the active component were fitted to a simple Michaelis-Menten equation to give a Km of 27 ± 7 mm. The corresponding theoretical lines for the passive and active components are shown together with the line for the total rate given by their sum. Reproduced with permission from Kellett & Helliwell (2000), Biochemical Journal 350, 155-162. © The Biochemical Society.
Adaptation of fructose absorption to diabetes
Although GLUT2 has been considered to be located solely at the basolateral membrane, we first reported the potential role of GLUT2 at the brush-border membrane in the adaptation of fructose absorption to diabetes (Corpe et al. 1996). After 10 days of streptozotocin diabetes, the brush-border level of GLUT5 was increased almost 3-fold and GLUT2 was readily detectable at the brush-border membrane in contrast to normal jejunum. GLUT5 in brush-border membrane is highly specific for fructose and is not inhibited by glucose at ratios as high as 100:1, nor by phloretin. In contrast, GLUT2-mediated fructose transport displays complete and mutual inhibition with glucose and is inhibited by phloretin. We were therefore able to use the difference in phloretin specificity to separate the transport contributions of GLUT5 and GLUT2 to fructose absorption determined by perfusion of isolated jejunal loops in vitro. At 5 mm fructose, all fructose is metabolised in this preparation; none appears in the serosal medium. Inhibition of absorption by luminal phloretin therefore reflected events only at the brush-border membrane. In complementary experiments with vesicles, we used 100 mm glucose to inhibit the transport of fructose by GLUT2. Perfusion and vesicle experiments gave the same results. They revealed that GLUT2 was fully functional at the brush-border membrane. The 1.6-fold enhancement of fructose absorption in diabetes comprised a phloretin- or glucose-insensitive component mediated by GLUT5 and a phloretin- or glucose-sensitive component mediated by GLUT2. Diabetes diminished the intrinsic activity (the catalytic activity per molecule) of GLUT5 to 20 % of its value in normal rat jejunum. Although in these experiments we did observe some GLUT2 at the brush-border membrane of normal rats, the amounts were variable and much lower than in diabetic jejunum; therefore, at that point, we did not ascribe to them the significance demanded by subsequent investigations.
Rapid trafficking of GLUT2 at the brush-border membrane
We then observed that perfusion of rat jejunum in vitro with the PKC agonist phorbol 12-myristate 13-acetate (PMA) for 30 min increased overall fructose transport by 70 % (Helliwell et al. 2000a). Interestingly, PMA caused a 4-fold increase in the level of GLUT2 at the brush-border membrane, but no changes in the level of GLUT5 or SGLT1. The increase in GLUT2 level was matched by a 4-fold increase in the phloretin-sensitive, GLUT2-mediated component of absorption and trafficking of GLUT2 correlated with activation of PKC βII. The increases in overall fructose transport, in GLUT2 level and in PKC βII activation were all blocked by the PKC inhibitor chelerythrine.
In untreated jejunum, PKC βII was activated in vivo compared with in vitro; the level of brush-border GLUT2 in untreated jejunum in vivo was the same as that for PMA-treated jejunum in vitro. Thus we concluded that excision of jejunum for in vitro experiments of any kind must be associated with inactivation of PKC βII and the rapid loss of GLUT2 from the brush-border membrane, because of the loss of influence of stimulating hormones and nutrients. This conclusion was confirmed in the time course experiments shown in Fig. 1. In the absence of PMA, perfusion in vitro results in diminution of GLUT2 to about 25 % of in vivo levels within 25 min (the earliest measured time), implying a t1/2 for loss of no more than a few minutes. PMA prevented the loss by permanently activating PKC βII. That GLUT2 was indeed at the brush-border membrane was confirmed by cell-surface biotinylation. We therefore concluded that GLUT2 plays an important role in fructose absorption across the brush-border membrane in normal jejunum. Unpublished experiments show that PKC βII remains activated in diabetic jejunum even in vitro. In this respect, diabetes has the same effect as PMA in normal jejunum (Fig. 1) and explains why we readily detected large amounts of brush-border GLUT2 in diabetic but not normal rats in the earlier work.
Figure 1. Excision of jejunum results in rapid loss of GLUT2 from the brush-border membrane.

Three pairs of normal jejuna were each perfused with 5 mm fructose in vivo for 30 min and then for different times in vitro; pair 1 was not perfused further (0 min in vitro), pair 2 was perfused for 25 min in vitro and pair 3 was perfused for 60 min in vitro. Three pairs were perfused in the absence of PMA (no PMA, left-hand side) and three pairs in the presence of PMA (with PMA, right-hand side). After perfusion, trafficking was stopped by flushing with ice-cold perfusate, and brush-border membrane vesicles were prepared and blotted for GLUT2. As soon as jejunum from normal rats is excised, PKC is inactivated because of the loss of influence of endogenous hormones, and about 75 % of the GLUT2 is lost from the membrane within minutes. If, however, PKC remains active because the jejunum is perfused with PMA, then GLUT2 is not lost. All types of in vitro preparations from normal, untreated jejunum suffer loss of GLUT2 from the brush-border membrane and the passive component is not readily detectable; see text. Reproduced with permission from Helliwell et al. (2000a), Biochemical Journal 350, 149-154. © The Biochemical Society.
GLUT2 mediates the passive component of glucose absorption and is regulated by glucose
GLUT2 is the only GLUT family member known to transport both glucose and fructose. Since GLUT2 is a high-capacity, high-Km transporter, it follows that brush-border membrane GLUT2 should provide a passive component of glucose absorption detectable at high concentrations. Single-pass perfusions of jejunum with glucose in the presence and absence of phloretin confirmed this supposition (Fig. 2; Kellett & Helliwell et al. 2000). Glucose absorption comprises a phloretin-sensitive, GLUT2-mediated component and a phloretin-insensitive, SGLT1-mediated component. The phloretin-insensitive component shows simple saturation kinetics with a Km of 27 ± 7 mm, similar to that reported for the phloridzin-sensitive component by Debnam & Levin (1975).
Consistent with previous reports, the passive component was superficially linear, suggesting simple diffusion. Being carrier mediated, however, it was in fact cooperative withh = 1.6 and [G1/2]= 56 ± 14 mm, where h is the Hill coefficient and [G1/2] is the glucose concentration at half Vmax (see legend to Fig. 2). Transport of glucose through SGLT1 activates PKC βII with simple saturation kinetics and a [G1/2] of 20 mm. The activation of PKC βII correlated with a doubling of brush-border GLUT2 at high glucose concentrations (100 mm and more); the level of SGLT1 was unchanged. Nevertheless, when the rate of absorption of the passive component was normalised for the increase in GLUT2, the dependence of rate on glucose concentration remained sigmoidal (h = 1.7). It therefore appears that two factors are responsible for the cooperativity of the passive component, the glucose-induced recruitment of GLUT2 and the stimulation of GLUT2 intrinsic activity. Dependence of the passive component on SGLT1 was demonstrated in four ways: (i) the kinetics of activation of PKC βII activation showed a saturation response with a Ka for glucose similar to the Km of SGLT1; (ii) inhibition of SGLT1 with phloridzin or by abolition of the Na+ gradient inactivated PKC βII; (iii) inhibition of SGLT1 resulted in loss of GLUT2 from the brush-border membrane and therefore (iv) in inhibition of the passive component of absorption.
Regulation of the passive component by MAP kinase intracellular signalling pathways
The ERK, p38 and PI 3-kinase pathways play an important role in the regulation of many cellular processes by mitogens, such as insulin, which regulates intestinal sugar absorption (Kellett et al. 1984; Wollen & Kellett, 1988; Pennington et al. 1994), and by various stress factors such as cytokines and hyperosmolarity. The involvement of these intracellular signalling pathways in the regulation of the intrinsic activity of other members of the facilitative glucose transporter family has been reported (Gould et al. 1995; Barros et al. 1997). We have therefore investigated their involvement in the regulation of fructose transport by manipulating the activities of the different pathways using different combinations of anisomycin, PD98059 and wortmannin (Helliwell et al. 2000b). Activation of the p38 pathway by 2 μm anisomycin stimulated transport maximally. Inhibition of the ERK pathway by PD98059, which inhibits MEK, had little effect on fructose transport. However, it caused a dramatic 50-fold diminution in the Ka for anisomycin from 1 μm to 20 nm, demonstrating that the ERK pathway inhibits the p38 pathway. The Ka of anisomycin in diabetic jejunum was 30 nm and PD98059 had no effect. Such behaviour is consistent with the expected inhibition of the insulin-sensitive ERK pathway in diabetic jejunum. By using different combinations of drugs, it was possible to vary within minutes: fructose transport over a 3.4-fold range; GLUT2 levels over a 4-fold range with only minimal changes in GLUT5 or SGLT1 levels, demonstrating that GLUT2 trafficks by a rapid trafficking pathway distinct from that of GLUT5 and SGLT1; GLUT2 intrinsic activity over a 9-fold range; and GLUT5 intrinsic activity to the 20 % level seen in diabetic jejunum. Our experiments therefore demonstrated that there is extensive cross-talk between the ERK, p38 and PI 3-kinase pathways in their control of brush-border fructose transport by modulation of the levels and/or intrinsic activities of GLUT5 and GLUT2. The balance between the activities of the different pathways is all-important.
The assimilation of sugars after a meal
Our current working model for some of the events that occur during the assimilation of sugars after a meal is as follows. Before a meal, the lumen is empty and the concentration of glucose is lower than in the blood (Fig. 3A). The presence of a facilitative transporter at the brush-border membrane might be expected to compromise intestinal function by permitting the loss of glucose from the blood through the absorptive cell into the lumen. The potential loss is limited by three factors. Firstly the level and intrinsic activity of GLUT2 at the brush-border membrane are low compared with the situation when high concentrations are present in the lumen. Secondly, a large part of the glucose entering the absorptive cell from the blood will be metabolised at these low concentrations. Thirdly, any glucose that does escape will be recycled by SGLT1, which is capable of moving glucose against a concentration gradient. Note that this problem does not occur with fructose, since its circulating concentration is very low.
Figure 3. A working model for the regulation of the facilitated component of glucose absorption by glucose during the assimilation of sugars after a meal.
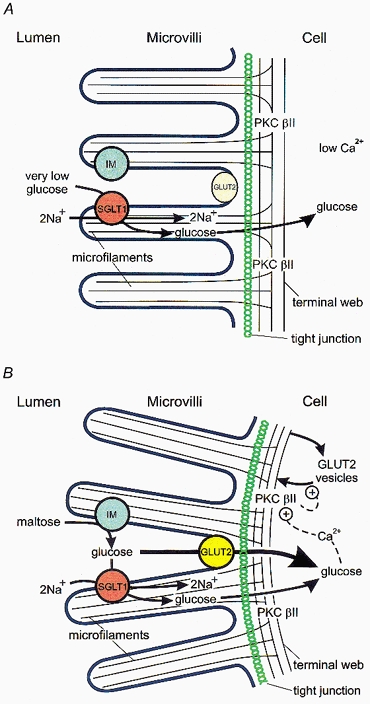
This cartoon of our current working model shows the absorptive epithelial cell before (A) and after (B) a meal. Before a meal, the glucose concentration in the lumen is very low, being less than that in the blood. The level and intrinsic activity of GLUT2 are low, indicated by the pale yellow ellipse for GLUT2; absorption of glucose against its concentration gradient occurs through SGLT1. After a meal, high local concentrations of glucose are present at the microvilli from the hydrolysis of dissacharides, for example, by the action of isomaltase (IM) on maltose. Transport of glucose through SGLT1 results in activation of PKC βII and activation and recruitment of GLUT2 to the brush-border membrane, indicated by the bright yellow circle for GLUT2. The rate of absorption through GLUT2 is then severalfold greater than that through SGLT1. Transport of glucose through SGLT1 also results in contraction of the peri-junctional actomyosin ring (just below the tight junction), causing subtle rounding of the surface of the absorptive cell. Both processes may be mediated by a glucose-induced increase in intracellular Ca2+ concentrations. For further explanation see text.
After a meal, the action of membrane-bound hydrolytic enzymes, such as isomaltase, in the microvilli on dissacharides and α-limit dextrins results in a high local concentration of glucose (Fig. 3B). Since both the brush-border level of GLUT2 and its intrinsic activity are low, transport will occur initially through SGLT1 to activate PKC βII; the latter is reported to be located in the terminal web of the mature enterocytes in the upper part of the villus (Saxon et al. 1994), that is those cells responsible for the majority of glucose absorption. Interestingly, glucose also causes condensation of the peri-junctional actomyosin ring and focal condensation of microfilaments at other sites within the terminal web (Madara & Pappenheimer, 1987). Rapid trafficking (t1/2 no more than about 5 min) of GLUT2-containing vesicles to the brush-border membrane, and insertion as well as activation of GLUT2 already at the membrane, would then occur as a probable consequence of PKC βII activation. These events result in the activated state of the absorptive cell shown in Fig. 3B, in which the flux through the GLUT2-mediated pathway of glucose absorption is several times greater (large arrow) than that through the SGLT1-mediated pathway (small arrow).
One possible mechanism for activation of PKC βII and the contraction of the peri-junctional actomyosin ring is that concentrative Na+-coupled glucose entry may trigger the entry of extracellular Ca2+ (Pappenheimer, 1993). Contraction of the peri-junctional actomyosin ring causes rounding of the apical surface of absorptive cells and alterations in tight junction structure (Madara & Pappenheimer, 1987). PKC promotes transcytosis and it may be that changes in tight junction structure and the regulation of associated scaffold/binding proteins alter the polarised targeting of transport vesicles so as to allow selective passage of GLUT2 from the basolateral to the apical side. Rounding of the apical surface might also increase access of luminal glucose and of complex sugars to the hydrolases and transporters of the brush-border membrane.
As sugars are assimilated after a meal and the luminal glucose concentration decreases, the sequence of signalling events and associated structural changes is rapidly reversed.
The implications of our findings for the debate over the passive component
The passive component exists
The first implication is that the passive component exists and is facilitated by GLUT2. The GLUT2-based mechanism provides an explanation for glucose absorption at concentrations much greater than 30-50 mm, by which SGLT1 is saturated, and therefore for the long-established in vivo data.
Paracellular flow and water transport
The conclusion that solvent drag was important rested in part on the calculation that back-diffusion across the tight junctions was negligible compared with solvent drag, even when glucose was proposed to be concentrated in the intracellular spaces to such an extent that it could generate significant fluid flow. Such concentration would be very unlikely given the efficiency of vascular clearance in vivo. Nevertheless, we have now demonstrated that GLUT2 is present at the brush-border membrane with a high level and increased intrinsic activity at high glucose concentrations (in contrast to the situation at low concentrations). There is thus a potential facilitated transport pathway from the basolateral to the brush-border membrane, which would prevent any significant concentration of glucose in the intracellular spaces. The very presence of a facilitative transporter at the brush-border membrane therefore invalidates the theory of paracellular solvent drag. In keeping with this conclusion, we observed that the passive GLUT2-mediated and active SGLT1-mediated components accounted within experimental error for total glucose absorption. As noted above, fructose cannot concentrate in the intercellular spaces.
If there is no paracellular flow of glucose, why is water transport proportional to glucose absorption at higher glucose concentrations? We noted that when the passive component was inhibited with phloretin, water transport also was inhibited. That permitted us to calculate that 342 ± 22 molecules of water are associated in some way with the transport of one molecule of glucose by GLUT2. Experiments in oocytes have led to the view that glucose transporters can act as low conductance water channels (Loo et al. 1999). Thus it has been reported that GLUT2 transports water (Fischbarg et al. 1990) and that SGLT1 transports 390 moles of water per mole of glucose (Zeuthen et al. 1997). Presumably all glucose transporters will have similar stoichiometries, since their stereochemistry will be similar. If, then, water were transported by GLUT2 in jejunum, it would explain the feature of the passive component emphasised by Pappenheimer & Reiss (1987) and observed by Fullerton & Parsons (1956), namely that water transport is proportional to absorption at high glucose concentrations. Fullerton & Parsons only observed the association of water with the passive component. The other component of glucose absorption they observed was not associated with water absorption. We now interpret the latter component to be SGLT1. Since SGLT1 was already partially saturated at the lowest concentration in their experiments, it would provide a constant background of water absorption rather than a variable component.
Demonstrating the dependence of the passive component on SGLT1 and the rapid trafficking of GLUT2 to the brush-border membrane
Our finding that the passive component of glucose absorption is dependent on SGLT1 is at variance with the independence implied by the observations of Debnam & Levin (1975), in which inhibition of SGLT1 with phloridzin leaves a large diffusive component; see also Ilundain (1979) and Lostao et al. (1991). In preliminary work, we have now identified a possible reason for this discrepancy. The earlier in vivo perfusions were recirculated perfusions performed at high flow rates (approx. 6 ml min−1) and pressure heads, which cause the perfused loop to blow up and distend. In contrast, our in vivo perfusions were single-pass perfusions in which no distension occurred, because the pressure head was zero and the flow rate only 0.75 ml min−1. By switching from a single-pass, low flow rate perfusion with no glucose (50 mm mannitol) to a recirculated, high flow rate perfusion with 50 mm glucose, and vice versa, we have found that trafficking appears to be blocked by mechanical stress (P. A. Helliwell & G. L. Kellett, unpublished data). We have further confirmed that the passive component is therefore independent of SGLT1 in high flow rate perfusions. That mechanical stress blocks GLUT2 trafficking at high glucose concentrations may point to an alteration in sensitivity to glucose. Alternatively, it might indicate an important role for the peri-junctional actomyosin ring in the regulation of the passive component, a view closely identified with Madara and colleagues (for a review, see Turner, 2000).
Why has the role of GLUT2 at the brush-border membrane been overlooked?
First, the vast majority of studies on sugar transport have been performed with in vitro preparations. As soon as jejunum is excised for in vitro investigations, however, the majority of GLUT2 is lost from the brush-border within minutes, because PKC βII is inactivated as a result of the loss of influence of endogenous hormones and/or nutrients. In many in vitro preparations GLUT2 or GLUT2-mediated transport would therefore be difficult to detect, especially since transport is dominated by SGLT1 at the low concentrations often used for uptake studies.
These considerations apply particularly to all preparations of intestinal tissue at physiological temperature in vitro, for example perfused isolated loops in vitro or in situ, everted sacs, tissue sheets in Ussing chambers or enterocytes. One preparation that escapes this problem straightforwardly is that of membrane vesicles, provided trafficking is first inhibited in vivo by flushing the lumen with ice-cold buffer and then performing isolation rigorously under ice-cold conditions (Helliwell et al. 2000a). However, it may prove possible to escape the difficulty in other preparations by providing the right hormones and/or nutrients. The luminally and vascularly perfused preparation of rat jejunum seems particularly suited in this regard. Indeed, Professor C. I. Cheeseman (personal communication) has recently observed that large amounts of GLUT2 are found at the brush-border membrane of this preparation under the perfusion conditions described by Hirsh & Cheeseman (1998).
Many differences between in vitro and in vivo preparations have been attributed to the presence of unstirred layers in vivo. However, we now see that a major difference between in vitro and in vivo preparations is the loss of GLUT2 in vitro. No doubt this loss accounts for some of the effects attributed to unstirred layers. It also explains why the passive component was seen almost exclusively with in vivo preparations.
The second reason that the role of GLUT2 has been overlooked is that the passive component of absorption is largely SGLT1 dependent in vivo, although, as we have seen this depends on the type of preparation. The universal assumption that blocking a process with phloridzin defines that process as being solely mediated by SGLT1 does not pertain in vivo, since it masks a contribution from the passive component.
For these reasons, then, we see that a large majority of the data in the literature can indeed be explained primarily in terms of SGLT1, as proposed by Ferraris & Diamond, but in vivo data above 50 mm glucose cannot.
The roles of SGLT1 - scavenger, transporter, regulator
It is important to remember that SGLT1 does not mediate active absorption; rather it mediates secondary active absorption. If the intracellular concentration of Na+ is maintained at low levels by the Na+,K+-ATPase, then, in vivo, when vascular clearance is very efficient and the concentration of glucose in the lumen exceeds that in the blood, SGLT1 must transport glucose down hill: that SGLT1 operates in a passive mode is demonstrated by the data of Leese (1974), who reported that when luminal and plasma glucose concentrations are 28 and 11 mm, respectively, the mucosal concentration is 18 mm. We can therefore recognise that a key role of SGLT1 is to act as a scavenger, recovering any glucose that might have escaped from the absorptive cell into the lumen at luminal concentrations less than blood glucose. This is because it is the one situation in which SGLT1 can truly use its ability as the only transporter capable of transporting glucose against its concentration gradient. It is interesting to note that the low Km analogue of SGLT1 in fructose absorption is the facilitative transporter GLUT5; a secondary active mechanism is not necessary, since the circulating concentration of fructose is very low.
At intermediate concentrations, where the ‘active’ component exceeds the GLUT2-mediated passive component, SGLT1 has a second established role as the major transporter, but is operating in passive mode. At very high local concentrations present at the brush-border membrane immediately after a meal, however, GLUT2-mediated absorption is the major route, some 3-5 times that mediated by SGLT1. Nevertheless, SGLT1 is now seen to have also a third, regulatory role - it can control the onset of the activation of PKC βII and is therefore instrumental in triggering the passive component so that absorption is matched to dietary intake on a time scale of minutes.
Although the short-term regulation in our experiments has concerned brush-border GLUT2, SGLT1 also has established regulatory functions. Long-term developmental and dietary regulation of SGLT1 have been documented by Ferraris & Diamond (1989, 1997) and Shirazi-Beechey et al. (1991). Cheeseman (1997) and Hirsh & Cheeseman (1998) have also reported rapid changes in SGLT1 and SGLT1-mediated transport in response to GLP-2 and CCK-8, respectively. Both types of SGLT1 regulation are important in their own right, but both also have the potential to control the passive component.
A role for regulatory proteins?
In the original work of Thorens et al. (1990), GLUT2 was detected by immunocytochemistry only at the basolateral membrane. Initially, we thought that this was because intestine was excised before fixation. However, we have not been successful in detecting brush-border membrane GLUT2 by immunocytochemistry in samples fixed before excision (J. A. Affleck & G. L. Kellett, unpublished data). Since we have detected GLUT2 by cell-surface biotinylation, vesicle blotting and a battery of functional tests, one possibility - not the only one - is that the C-terminal determinant detected by the antibody is in some way masked, perhaps by a docking protein at the brush-border membrane. It may be necessary to use an antibody that recognises an extracellular epitope of GLUT2 in order to resolve this question; and even that may not be successful if other explanations pertain.
A second important question is sugar specificity. For example, there is no evidence in the literature for transport of 2-deoxyglucose across the brush-border membrane, although it is a substrate for GLUT2 in the basolateral membrane (Kimmich & Randles, 1976). 2-Deoxyglucose behaviour might well be explained by loss of GLUT2 from the brush-border of in vitro preparations and/or by the fact that, since 2-deoxyglucose is not a substrate for SGLT1, the role of the latter in regulating GLUT2 trafficking is lost. But a more radical thought occurs in relation to the low affinity transport system in guinea-pig brush-border membrane vesicles, ‘Alvarado's system II’ so-called. The Km of system II for glucose is 24 mm compared with 0.4 mm for SGLT1 in the same vesicle preparation; moreover, system II is inhibited by cytochalasin B (Brot-Laroche et al. 1986). However, system II is not immediately identified with GLUT2, since it is not inhibited by 2-deoxyglucose. What if GLUT2 at the brush-border membrane were to have a different specificity for glucose analogues from that at the basolateral membrane, because it associates with different scaffold/regulatory proteins? Conventional assumptions about sugar specificity may not hold and an indication of how seriously this point should be considered is afforded by the studies of Miyamoto et al. (1994) on GLUT5, which is highly specific for fructose in intact intestine. When GLUT5 is expressed in oocytes by injection of cRNA, it transports glucose as well as fructose, but when expressed from total mRNA, it transports only fructose.
With regard to sugar specificity, it should also be noted that new transporters, or recently discovered transporters of unknown specificity such as GLUT8 (Doege et al. 2000), may well be involved.
The future
One feature of the literature and the debate over the mechanism of the passive component of glucose absorption has been the almost interchangeable usage of terms such as ‘passive’, ‘diffusive’ and ‘non-carrier mediated’. This has led at times to confusion, depending on context and author. Now that a clearly defined, GLUT2-based mechanism has been identified, the term ‘facilitated’ seems preferable for it is both accurate and unambiguous.
Glucose absorption is regulated by a PKC-dependent pathway in response to luminal glucose: the facilitated component is mediated by GLUT2 under the control of SGLT1. Fructose absorption is regulated by an intracellular signalling network comprising a PKC-dependent pathway, a PI 3-kinase-dependent pathway and the ERK and p38 MAP kinase pathways, which show extensive cross-talk. This network will also be involved in control of glucose absorption. We therefore expect that sugar absorption will be regulated by a range of endocrine and paracrine hormones that regulate the network pathways, such as nutrients, insulin, growth factors and cytokines. A large part of future work will therefore be to establish what hormones and sugars regulate which pathways and how they interact. However, these pathways are unlikely to be the only ones and our present view will undoubtedly be modified and/or extended.
Recently, Kawano et al. (1999) have reported that hyperglycaemia (25 mm 3-O-methylglucose) activates transport in rat soleus muscle. They show that activation of 3-O-methylglucose transport correlates with Ca2+-dependent activation of PKC βII and its translocation to the plasma membrane. Since there is no change in the cell surface content of GLUT1 or GLUT4, they propose that stimulation of 3-O-methylglucose transport may involve an increase of cell surface glucose transporter intrinsic activity. At 20-30 mm glucose, we do not see an increase in brush-border GLUT2 level, but do see an increase in intrinsic activity. Thus the two mechanisms are sufficiently similar that it appears likely the mechanism is common and will appear as specific variations in other tissues. Indeed, Marks et al. (2000) have very recently reported preliminary data showing that GLUT2 is present at the brush-border membrane of rat kidney; after incubation of cortical tubule suspensions with purinoceptor agonists, the GLUT2-facilitated component of transport, measured by the uptake of 20 mm glucose into vesicles, is doubled without any change in GLUT2 level.
The general pattern of regulation displayed by GLUT2 is that a protein constitutively located in the basolateral membrane can undergo rapid trafficking to the brush-border membrane to mediate the passive component of absorption. Future investigations of the absorption of other nutrients, such as peptides and amino acids, therefore seem likely to reveal a similar pattern of regulation involving similar intracellular signalling pathways.
With a few notable exceptions, most studies on regulation of nutrient absorption in the literature concern long-term dietary or developmental studies. The first detailed description of how the different intracellular signalling pathways, glucose and PKC regulate sugar absorption on a time scale of minutes opens up the whole field of short-term regulation of sugar absorption by hormones and nutrients; the molecular description of the fundamental physiological mechanisms of assimilation of a meal and how they are perturbed in diseased states is now experimentally accessible.
Acknowledgments
I am grateful to Dr C. A. R. Boyd, Professor J. R. Bronk and Dr P. A. Helliwell for critical reading of the manuscript. I thank The Wellcome Trust for support.
References
- Barros LF, Young M, Saklatvala J, Baldwin SA. Evidence of two mechanisms for the activation of the glucose transporter GLUT1 by anisomycin. Journal of Physiology. 1997;504:517–525. doi: 10.1111/j.1469-7793.1997.517bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brot-Laroche E, Serrano MA, Delhomme B, Alvarado F. Temperature sensitivity and substrate specificity of two distinct Na+-activated d-glucose transport systems in guinea-pig jejunal brush-border membrane vesicles. Journal of Biological Chemistry. 1986;261:6168–6176. [PubMed] [Google Scholar]
- Cheeseman CI. Upregulation of SGLT-1 transport activity in rat jejunum induced by GLP-2 infusion in vivo. American Journal of Physiology. 1997;273:R1965–1971. doi: 10.1152/ajpregu.1997.273.6.R1965. [DOI] [PubMed] [Google Scholar]
- Corpe CP, Basaleh MM, Affleck J, Gould GW, Jess TJ, Kellett GL. The regulation of GLUT5 and GLUT2 activity in the adaptation of intestinal brush-border fructose transport in diabetes. Pflügers Archiv. 1996;432:192–201. doi: 10.1007/s004240050124. [DOI] [PubMed] [Google Scholar]
- Crane RK, Miller D, Bihler I. In: Membrane Transport and Metabolism. Kleinzeller A, Kotyk A, editors. New York: Academic Press; 1961. pp. 439–449. [Google Scholar]
- Debnam ES, Levin RJ. An experimental method of identifying and quantifying the active transfer electrogenic component from the passive component during sugar absorption measured in vivo. Journal of Physiology. 1975;246:181–196. doi: 10.1113/jphysiol.1975.sp010885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doege H, Schurmann A, Bahrenberg G, Brauers A, Joost HG. GLUT8, a novel member of the sugar transport facilitator family with glucose transport activity. Journal of Biochemistry. 2000;275:16275–16280. doi: 10.1074/jbc.275.21.16275. [DOI] [PubMed] [Google Scholar]
- Ferraris RP, Diamond J. Specific regulation of intestinal nutrient transporters by their dietary substrates. Annual Reviews of Physiology. 1989;51:125–141. doi: 10.1146/annurev.ph.51.030189.001013. [DOI] [PubMed] [Google Scholar]
- Ferraris RP, Diamond J. Regulation of intestinal sugar transport. Physiological Reviews. 1997;77:257–302. doi: 10.1152/physrev.1997.77.1.257. [DOI] [PubMed] [Google Scholar]
- Ferraris RS, Yarshapour S, Lloyd KCK, Mirzayan R, Diamond J. Luminal glucose concentrations in the gut under normal conditions. American Journal of Physiology. 1990;259:G822–837. doi: 10.1152/ajpgi.1990.259.5.G822. [DOI] [PubMed] [Google Scholar]
- Fischbarg JK, Kuang K, Vera JC, Arant S, Siverstein SC, Loike JD, Rosen OM. Glucose transporters serve as water channels. Proceedings of the National Academy of Sciences of the USA. 1990;87:3244–3247. doi: 10.1073/pnas.87.8.3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fordtran JS, Inglefinger FJ. Absorption of water, electrolytes and sugars from human gut. In: Code CF, editor. Handbook of Physiology Alimentary Canal Intestinal Absorption. Washington, CD: Williams & Wilkins; 1968. pp. 1457–1490. section 6, vol. III. [Google Scholar]
- Fullerton PM, Parsons DS. The absorption of sugars and water from rat intestine in vivo. Quarterly Journal Experimental Physiology. 1956;41:387–397. [Google Scholar]
- Gould GW, Cuenda A, Thomson FJ, Cohen C. The activation of distinct mitogen-activated protein kinase cascades required for the stimulation of 2-deoxyglucose uptake by interleukin-1 and insulin-like growth factor in KB cells. Biochemical Journal. 1995;311:735–738. doi: 10.1042/bj3110735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould GW, Holman GD. The glucose transporter family: structure, function and tissue-specific expression. Biochemical Journal. 1993;295:329–341. doi: 10.1042/bj2950329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hediger MA, Coady MJ, Ikeda TD, Wright EM. Expression cloning and cDNA sequencing of the Na+/glucose co-transporter. Nature. 1987;330:379–381. doi: 10.1038/330379a0. [DOI] [PubMed] [Google Scholar]
- Hediger MA, Rhoads DB. Molecular physiology of sodium-glucose cotransporters. Physiological Reviews. 1994;74:993–1026. doi: 10.1152/physrev.1994.74.4.993. [DOI] [PubMed] [Google Scholar]
- Helliwell PA, Richardson M, Affleck JA, Kellett GL. Stimulation of fructose transport across the intestinal brush-border membrane by PMA is mediated by GLUT2 and dynamically regulated by protein kinase C. Biochemical Journal. 2000a;350:149–154. [PMC free article] [PubMed] [Google Scholar]
- Helliwell PA, Richardson M, Affleck JA, Kellett GL. Regulation of GLUT5, GLUT2 and intestinal brush-border fructose absorption by the ERK, p38 and PI 3-kinase intracellular signalling pathways: implications for adaptation to diabetes. Biochemical Journal. 2000b;350:163–169. [PMC free article] [PubMed] [Google Scholar]
- Hirsh AJ, Cheeseman CI. Cholecystokinin decreases intestinal hexose absorption by a parallel reduction in SGLT1 abundance in the brush-border membrane. Journal of Biological Chemistry. 1998;273:14545–14549. doi: 10.1074/jbc.273.23.14545. [DOI] [PubMed] [Google Scholar]
- Holdsworth CD, Dawson AM. The absorption of monosaccharides in man. Clinical Science. 1964;27:371–379. [PubMed] [Google Scholar]
- Ilundain A, Lluch M, Ponz F. Kinetics of intestinal sugar transport, in vivo. Revista Espanola de Fisiologia. 1979;35:359–366. [PubMed] [Google Scholar]
- Kawano Y, Rincon J, Soler A, Ryder JW, Nolte LA, Zierath JR, Wallberg-Henriksson H. Diabetologia. 1999;42:1071–1079. doi: 10.1007/s001250051273. [DOI] [PubMed] [Google Scholar]
- Kellett GL, Helliwell PA. The passive component of intestinal glucose absorption is mediated by the glucose-induced recruitment of GLUT2 to the brush-border membrane. Biochemical Journal. 2000;350:155–162. [PMC free article] [PubMed] [Google Scholar]
- Kellett GL, Jamal A, Robertson JP, Wollen N. The acute regulation of glucose absorption, transport and metabolism in rat small intestine in vivo. Biochemical Journal. 1984;219:1027–1035. doi: 10.1042/bj2191027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmich GA, Randles J. 2-Dexoyglucose transport by intestinal epithelial cells isolated from the chick. Journal of Membrane Biology. 1976;27:363–379. doi: 10.1007/BF01869146. [DOI] [PubMed] [Google Scholar]
- Lane JS, Whang EE, Rigberg DA, Hines OJ, Kwan D, Zinner MJ, McFadden DW, Diamond J, Ashley SW. Paracellular glucose transport plays a minor role in the unanesthetized dog. American Journal of Physiology. 1999;276:G789–794. doi: 10.1152/ajpgi.1999.276.3.G789. [DOI] [PubMed] [Google Scholar]
- Leese HJ. Glucose accumulation by rat small intestine during absorption in vivo. Nature. 1974;251:512–513. doi: 10.1038/251512a0. [DOI] [PubMed] [Google Scholar]
- Loo DD, Hirayama BA, Meinild AK, Chandy G, Zeuthen T, Wright EM. Passive water and ion transport by cotransporters. Journal of Physiology. 1999;518:195–202. doi: 10.1111/j.1469-7793.1999.0195r.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lostao MP, Berjon A, Barber A, Ponz F. On the multiplicity of glucose analogues transport systems in rat intestine. Revista Espanola de Fisiologia. 1991;47:209–216. [PubMed] [Google Scholar]
- Madara JL, Pappenheimer JR. Structural basis for physiological regulation of paracellular pathways in intestinal epithelia. Journal of Membrane Biology. 1987;100:149–164. doi: 10.1007/BF02209147. [DOI] [PubMed] [Google Scholar]
- Manome SH, Kuriaki K. Effects of insulin, phlorizin and some metabolic inhibitors on the glucose absorption from the small intestine. Archives Internationale Pharmacodynamie et de Therapie. 1961;130:187–194. [PubMed] [Google Scholar]
- Marks J, Debnam ES, Unwin RJ, Srai SK. Potential role for purinoceptors in the regulation of glucose transport across isolated rat renal brush border membrane. Journal of Physiology. 2000:527.P–15.P. [Google Scholar]
- Miyamoto K, Tatsumi S, Morimoto A, Minami H, Yamomoto H, Sone K, Taketani Y, Nakabou Y, Oka T, Takeda E. Characterisation of the rabbit intestinal fructose transporter (GLUT5) Biochemical Journal. 1994;303:877–883. doi: 10.1042/bj3030877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappenheimer JR. On the coupling of membrane digestion with intestinal absorption of sugars and amino acids. American Journal of Physiology. 1993;265:G409–417. doi: 10.1152/ajpgi.1993.265.3.G409. [DOI] [PubMed] [Google Scholar]
- Pappenheimer JR. Scaling of dimensions of small intestines in non-ruminant eutherian mammals and its significance for absorptive mechanisms. Comparative Biochemistry and Physiology. 1998;A121:45–58. doi: 10.1016/s1095-6433(98)10100-9. [DOI] [PubMed] [Google Scholar]
- Pappenheimer JR, Reiss KZ. Contribution of solvent drag through intercellular junctions to absorption of nutrients by the small intestine of the rat. Journal of Membrane Biology. 1987;100:123–136. doi: 10.1007/BF02209145. [DOI] [PubMed] [Google Scholar]
- Parsons DS, Prichard MA. Properties of some model systems for transcellular active transport. Biochimica et Biophysica Acta. 1966;126:471–491. doi: 10.1016/0926-6585(66)90006-9. [DOI] [PubMed] [Google Scholar]
- Pennington AM, Corpe CP, Kellett GL. Rapid regulation of rat jejunal glucose transport by insulin in a luminally and vascularly perfused preparation. Journal of Physiology. 1994;478:187–193. doi: 10.1113/jphysiol.1994.sp020241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxon ML, Zhao X, Black JD. Activation of protein kinase C isozymes is associated with post-mitotic events in intestinal epithelial cells in situ. Journal of Cell Biology. 1994;126:747–763. doi: 10.1083/jcb.126.3.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirazi-Beechey SP, Hirayama BA, Wang Y, Scott D, Smith MW, Wright EM. Ontogenic development of lamb intestinal sodium-glucose co-transporter is regulated by diet. Journal of Physiology. 1991;437:699–708. doi: 10.1113/jphysiol.1991.sp018620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens BR, Kaunitz JD, Wright EM. Intestinal transport of amino acids and sugars: advances using membrane vesicles. Annual Review of Physiology. 1984;46:417–433. doi: 10.1146/annurev.ph.46.030184.002221. [DOI] [PubMed] [Google Scholar]
- Thorens B, Cheng ZQ, Brown D, Lodish HF. Liver glucose transporter: a basolateral protein in hepatocytes and intestine and kidney cells. American Journal of Physiology. 1990;259:C279–285. doi: 10.1152/ajpcell.1990.259.2.C279. [DOI] [PubMed] [Google Scholar]
- Turner JR. Show me the pathway! Regulation of paracellular permeability by Na+-glucose cotransport. Advanced Drug Delivery Reviews. 2000;41:265–281. doi: 10.1016/s0169-409x(00)00046-6. [DOI] [PubMed] [Google Scholar]
- Wollen N, Kellett GL. Regulation of glucose homeostasis in rat jejunum by despentapeptide-insulin in vitro. Gut. 1988;29:1064–1069. doi: 10.1136/gut.29.8.1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeuthen T, Meinild AK, Klaerke DA, Loo DD, Wright EM, Belhage B, Litman T. Water transport by the Na+/glucose cotransporter under isotonic conditions. Biologie Cellulaire. 1997;89:307–312. doi: 10.1016/s0248-4900(97)83383-7. [DOI] [PubMed] [Google Scholar]