

Abstract
One popular model for the activation of store-operated Ca2+ influx is the secretion-like coupling mechanism, in which peripheral endoplasmic reticulum moves to the plasma membrane upon store depletion thereby enabling inositol 1,4,5-trisphosphate (InsP3) receptors on the stores to bind to, and thus activate, store-operated Ca2+ channels. This movement is regulated by the underlying cytoskeleton. We have examined the validity of this mechanism for the activation of ICRAC, the most widely distributed and best characterised store-operated Ca2+ current, in a model system, the RBL-1 rat basophilic cell line.
Stabilisation of the peripheral cytoskeleton, disassembly of actin microfilaments and disaggregation of microtubules all consistently failed to alter the rate or extent of activation of ICRAC. Rhodamine-phalloidin labelling was used wherever possible, and revealed that the cytoskeleton had been significantly modified by drug treatment.
Interference with the cytoskeleton also failed to affect the intracellular calcium signal that occurred when external calcium was re-admitted to cells in which the calcium stores had been previously depleted by exposure to thapsigargin/ionomycin in calcium-free external solution.
Application of positive pressure through the patch pipette separated the plasma membrane from underlying structures (cell ballooning). However, ICRAC was unaffected irrespective of whether cell ballooning occurred before or after depletion of stores.
Pre-treatment with the membrane-permeable InsP3 receptor antagonist 2-APB blocked the activation of ICRAC. However, intracellular dialysis with 2-APB failed to prevent ICRAC from activating, even at higher concentrations than those used extracellularly to achieve full block. Local application of 2-APB, once ICRAC had been activated, resulted in a rapid loss of the current at a rate similar to that seen with the rapid channel blocker La3+.
Studies with the more conventional InsP3 receptor antagonist heparin revealed that occupation of the intracellular InsP3-sensitive receptors was not necessary for the activation or maintenance of ICRAC. Similarly, the InsP3 receptor inhibitor caffeine failed to alter the rate or extent of activation of ICRAC. Exposure to Li+, which reduces InsP3 levels by interfering with inositol monophosphatase, also failed to alter ICRAC. Caffeine and Li+ did not affect the size of the intracellular Ca2+ signal that arose when external Ca2+ was re-admitted to cells which had been pre-exposed to thapsigargin/ionomycin in Ca2+-free external solution.
Our findings demonstrate that the cytoskeleton does not seem to regulate calcium influx and that functional InsP3 receptors are not required for activation of ICRAC. If the secretion-like coupling model indeed accounts for the activation of ICRAC in RBL-1 cells, then it needs to be revised significantly. Possible modifications to the model are discussed.
In many non-excitable cells, a Ca2+ influx pathway is activated following the process of store emptying, and has been called store-operated or capacitative Ca2+ entry (Putney, 1986). Store-operated Ca2+ entry can give rise to an inward Ca2+ current, the best characterised and most widely distributed of which is ICRAC (Ca2+ release-activated Ca2+ current; Hoth & Penner, 1992; Parekh & Penner, 1997). Despite the importance of ICRAC in physiological responses (Parekh & Penner, 1997), the mechanism whereby the process of Ca2+ store emptying activates Ca2+ influx is not clear.
To date, three models have been put forward to account for store-operated Ca2+ entry: (i) the diffusible messenger hypothesis, in which a mobile cytosolic Ca2+ influx factor is released from the stores and opens Ca2+ channels in the plasma membrane (Randriamampita & Tsien, 1993; Csutora et al. 1999), (ii) the conformational-coupling hypothesis in which InsP3 receptors that span the stores physically attach to, and thus activate, the Ca2+ channels in the plasma membrane (Irvine, 1990; Berridge, 1995; Kiselyov et al. 1998, 1999; Boulay et al. 1999; Ma et al. 2000) and (iii) the fusion model in which CRAC channels are stored in vesicles and then somehow inserted into the plasma membrane following store depletion (Yao et al. 1999).
Recently, evidence has been presented in support of a modified conformational-coupling model (Putney, 1999; Berridge et al. 2000). It is now thought that, upon store depletion, a portion of the endoplasmic reticulum (ER) moves to the plasma membrane and this enables InsP3 receptors to couple directly to store-operated Ca2+ channels (Patterson et al. 1999). The ability of the ER to approach the plasma membrane is impeded by the cortical actin network, and pharmacological manoeuvres that stabilise this network prevent activation of store-operated Ca2+ entry (Patterson et al. 1999; Rosado et al. 2000). This modified conformational-coupling model has been called the secretion-like coupling model. It has also been concluded, using the novel InsP3 receptor inhibitor 2-APB, that the intracellular InsP3 receptors need to be functional at all stages of store-operated Ca2+ entry (Ma et al. 2000).
Although this modified version of the coupling model provides an attractive explanation for store-operated Ca2+ influx, to date the model has not been tested under conditions where the store-operated Ca2+ current has been directly and unambiguously measured. In the previous reports, Ca2+ entry was monitored only indirectly from the size of the global Ca2+ fluorescence signal and factors other than, or in addition to, activity of store-operated Ca2+ channels could have shaped the Ca2+ response (see Parekh & Penner, 1997).
To strengthen the secretion-like coupling model, we have examined the consequences of interfering with the cytoskeleton on the activation and inactivation of store-operated Ca2+ influx using the whole-cell patch-clamp technique to record the store-operated Ca2+ current ICRAC in RBL-1 cells. We have also investigated the effects of InsP3 receptor antagonists on the activation and maintenance of ICRAC. Unexpectedly, we found that a variety of manoeuvres that dramatically altered cell shape and the peripheral actin network failed to affect ICRAC at all. These agents also failed to affect calcium influx when the latter was measured indirectly by re-admission of external calcium to thapsigargin-treated cells loaded with fura 2. Furthermore, we found that occupation of InsP3 receptors did not seem to be necessary for either the activation or maintenance of the store-operated current. Our results suggest that the secretion-like coupling model needs to be modified significantly in order to explain the activation of ICRAC in RBL-1 cells. Alternatively, ICRAC might not be activated by a coupling-type mechanism.
METHODS
Rat basophilic leukaemia (RBL-1) cells, which were bought from Cell Bank at the Sir William Dunn School of Pathology, Oxford University, were cultured on coverslips as previously described (Parekh et al. 1997; Fierro & Parekh, 1999).
Patch-clamp experiments were conducted in the tight-seal whole-cell configuration at room temperature (20-25 °C) as previously described (Parekh et al. 1997; Fierro & Parekh, 1999). Sylgard-coated, fire-polished pipettes had DC resistances of 2.5-4 MΩ when filled with standard internal solution that contained (mm): caesium glutamate 145, NaCl 8, MgCl2 1, EGTA 10 and Hepes 10, pH 7.2 with CsOH. A correction of +10 mV was applied for the subsequent liquid junction potential that arose from this glutamate-based internal solution. In some experiments (described in the text), cells were dialysed with a pipette solution in which Ca2+ was strongly buffered at 140 nm (10 mm EGTA, 4.5 mm CaCl2) or 225 nm (10 mm EGTA, 6 mm CaCl2). In the experiments of Fig. 2B and C, 2 mm Mg-ATP was included in the pipette solution. Extracellular solution contained (mm): NaCl 145, KCl 2.8, CaCl2 10, MgCl2 2, CsCl 10, glucose 10 and Hepes 10, pH 7.4 with NaOH. ICRAC was measured by applying voltage ramps (-100 to +100 mV in 50 ms) at 0.5 Hz from a holding potential of 0 mV as previously described (Parekh et al. 1997). Currents were filtered using an 8-pole Bessel filter at 2.5 kHz and digitised at 100 μs. Currents were normalised by dividing the amplitude (measured from the voltage ramps at -80 mV) by the cell capacitance. Capacitative currents were compensated before each ramp by using the automatic compensation of the EPC 9-2 amplifier. All leak currents were subtracted by averaging the first few ramp currents (usually two), and then subtracting this from all subsequent currents. Data are presented as means ±s.e.m., and statistical evaluation was carried out using Student's unpaired t test.
Figure 2. The cytoskeletal restructuring agent calyculin A does not affect the activation or inactivation of ICRAC.
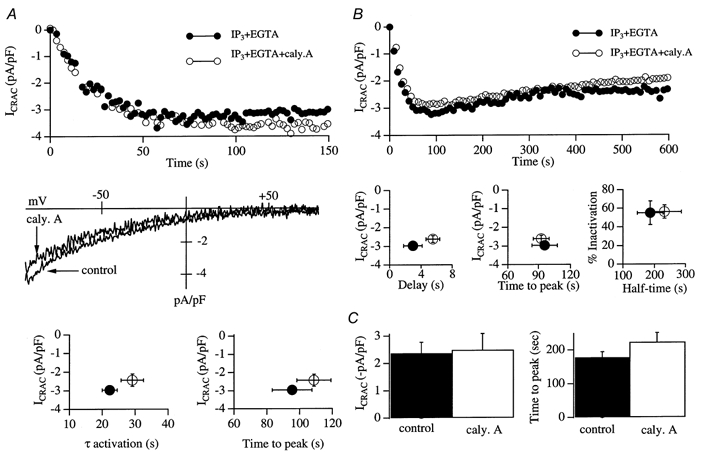
A, upper panel, development of ICRAC (measured at -80 mV from the voltage ramps) in control conditions (•) and following pre-incubation with 100 nm calcyculin A for 30 min (○). The I-V relationship for each cell is shown in the middle panel (taken at 76 and 82 s for control and calyculin A, respectively). The lower panel depicts graphs which summarise the relationship between current amplitude and the time constant of activation, and the time to peak of the current. B, intracellular dialysis with 1 μm calyculin A fails to alter ICRAC. The upper panel shows a typical control recording (InsP3+ 10 mm EGTA + 2 mm Mg-ATP) and one from a cell dialysed with this solution supplemented with calyculin A. The left and centre plots in the lower panel show that the delay, time to peak and amplitude of ICRAC were similar for the two conditions (control and calyculin A treated). The graph on the right shows that the extent of inactivation of ICRAC (measured at 600 s) as well as the time at which half-inactivation had been reached (half-time) were not different between control and calyculin A-treated cells. Hence calyculin A fails to affect the activation of ICRAC, nor does it reverse activation. C, cells were dialysed with an internal solution containing buffered Ca2+ (140 nm) and 2 mm Mg-ATP alone (control) or supplemented with 1 μm calyculin A. After 300 s of dialysis, thapisgargin was applied externally. The histograms compare the extent of activation and the time course of ICRAC for control versus calyculin A-treated cells. Neither the extent of activation nor the time to peak was significantly different between the two conditions.
For calcium fluorescence measurements, cells were loaded with the acetoxymethyl ester form of the dye fura 2 (Molecular Probes) for 40 min at room temperature (in culture medium) and then the cells were washed 5 times in culture medium to remove external dye. Cells were then left for 20 min to allow further de-esterification. Coverslips were mounted onto the stage of an upright microscope (Zeiss Axiovert) and perfused with Ca2+-free external solution (composition identical to the extracellular solution stated above for patch-clamp recordings except that 10 mm Ca2+ was omitted and 0.2 mm EGTA added) just prior to recording. Excitation light was applied through the epi-fluorescence port of the microscope using a fibre optic and Zeiss objective (×60, water immersion, NA 1.3), and excited at 360 and 380 nm (25 ms exposures) using a monochromator-driven system (TILL Photonics), and images were collected every 2-4 s at > 500 nm. Regions of interest as well as background areas of similar size were obtained using custom-made software (TILL Photonics) and then exported to IGOR Pro for analysis. Fluorescence signals are presented as ΔF/F0, where F0 denotes the ratio (360 nm/380 nm) prior to application of thapsigargin/ionomycin in Ca2+-free external solution (averaged over 10 s) and ΔF represents the peak increase in the ratio (for either the calcium release or calcium influx components) subtracted from F0.
Rhodamine-phalloidin labelling was carried out as follows. Coverslips with growing RBL cells were incubated for 30 min with 100 nm calyculin A or for 45 min with 2 μm cytochalasin D in external solution and then were added to an equal volume of paraformaldehyde solution (6.4 %) for 10 min, then washed 3 times in phosphate-buffered saline (PBS, composition (mm): NaCl 137, KCl 2.7, Na2PO4 8 and K2PO4 1.5, pH 7.5 with NaOH)). Cells were subsequently permeabilised with Triton X-100 (0.02 %) for a further 10 min then washed 3 times in PBS. Coverslips were then incubated with rhodamine- phalloidin (2 U ml−1; Molecular Probes) for 60 min and then washed 5 times in PBS. Coverslips were mounted onto slides with ImmunoFluor mounting medium and allowed to set for 24-48 h. The dye was excited at 554 nm and emission was collected at 573 nm.
Drugs were applied locally by means of positive pressure applied to an application pipette placed within 20 μm of the cell. Thapsigargin was purchased from Alomone Laboratories. Jasplakinolide was bought from Molecular Probes. Calyculin A and cytochalasin D were from Calbiochem. All other chemicals were purchased from Sigma.
RESULTS
The phosphatase blocker calyculin A does not interfere with the activation or inactivation of ICRAC
In the presence of calyculin A, actin becomes tightly associated with the plasma membrane and a more stable cortical actin network is thought to form (Downey et al. 1993; Shinoki et al. 1995; Patterson et al. 1999). Formation of this network seems to regulate store-operated Ca2+ entry because pre-treatment with 100 nm calyculin A for just 10 min is sufficient to inhibit thapsigargin-evoked Ca2+ entry (Patterson et al. 1999). Pre-treatment with calyculin A had quite a marked effect on RBL-1 cell morphology compared with that of control cells. In transmitted light images, cells adopted a ruffled, roundish shape (Fig. 1A), consistent with the cytoskeletal changes induced by calyculin A that have been described by others (Downey et al. 1993; Shinoki et al. 1995; Patterson et al. 1999). Using rhodamine- phalloidin, we directly determined the sub-cellular distribution of actin (Fig. 1B). Heavy staining was consistently seen just below the plasma membrane in control cells, with a little punctate distribution within the cytosol. This pattern was dramatically altered by calyculin A. Now, very dense staining occurred close to the plasma membrane, but this tended to condense at a ‘hot-spot’ (Fig. 1B). The punctate distribution within the cytoplasm also increased. This change in pattern of actin distribution in response to calyculin A in RBL cells is remarkably similar to that described for HEK-293 cells by Ma et al. (2000; see their Fig. 1H). Despite these sub-plasmalemmal changes, ICRAC was unaffected (Fig. 2). The upper panel of Fig. 2A shows a control recording (•) and one taken from a cell pre-treated with 100 nm calyculin A (○). The rate and extent of development of ICRAC (evoked by dialysis with 30 μm InsP3+ 10 mm EGTA) were very similar for the two conditions. The middle panel of Fig. 2A shows the I-V relationship for both treatments, taken at 76 and 82 s for the control and calyculin A-treated cell, respectively, which reveals the hall-marks of ICRAC (inward rectification, voltage-independent gating and reversal potential (Vrev) > +50 mV). The graphs in the lower panel of Fig. 2A plot the averaged time constant and time to peak versus the amplitude of ICRAC for control cells (n = 7) and cells pre-exposed to calyculin A (n = 7). There were no significant differences between any of these parameters for control and calyculin A-treated cells (P > 0.25). Raising the calyculin A concentration to 1 μm did not affect ICRAC either (4/4 cells, not shown).
Figure 1. Cytoskeletal agents evoke quite marked changes in cell shape.
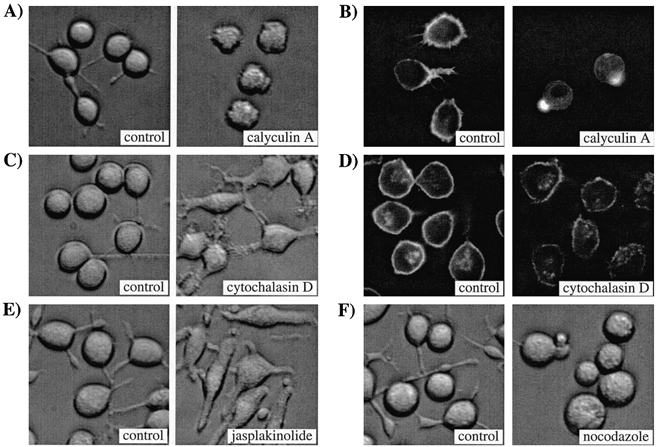
A, a control transillumination image of control (non-treated) RBL-1 cells is shown on the left and one taken after 12 min exposure to 100 nm calyculin A is shown on the right. The cells were from the same coverslip but from different fields of view. Note the rounded, shrivelled appearance following the brief exposure to calyculin A. All cells took on this appearance within 10 min of calyculin A treatment. B, rhodamine-phalloidin labelling of actin in control (left) and calyculin A-treated cells (right). C, a control transillumination image of control cells (left) and of cells exposed to the microfilament-disrupting agent cytochalasin D (1 μm for 45 min). Cells were from the same coverslip but different fields of view. D, distribution of actin in control (left) and cytochalasin D-treated cells using rhodamine-phalloidin labelling. E, transillumination images of control cells and cells exposed to the microfilament stabiliser jasplakinolide (1 μm for 30 min). F, a control transillumination image of cells before (left) and then 18 h after exposure to the microtubule-disaggregating agent nocodazole (5 μm; right). Cells were from different coverlips but from the same preparation. Note the rounded appearance and reduction in cell processes with nocodazole. All the experiments described above were carried out at room temperature and were repeated on at least two different cell preparations. Magnification, × 30.
Calyculin A has been reported to reverse the activation of thapsigargin-meditated Ca2+ entry (Patterson et al. 1999). To see whether ICRAC could be switched off by calyculin A, we dialysed cells with InsP3 and 10 mm EGTA with (n = 7) or without (n = 6) 1 μm calyculin A and followed the current over 10 min. Representative recordings are shown in Fig. 2B (upper panel). The recordings were almost identical over the entire time course of the experiment. There were no significant differences in the delay before ICRAC was activated, in the time to peak, in the size of ICRAC, in the extent of inactivation after 10 min or in the rate at which this inactivation was attained (see graphs in Fig. 2B, lower panel). With our mean series resistance of 7 MΩ and cell size of 15 pF in these experiments, we estimate that the time constant of wash-in of calyculin A was 280 s. Nevertheless, the time course of ICRAC was unaffected over 600 s. In two cells dialysed with calyculin A, we recorded ICRAC for 20 and 27 min, yet the extent of inactivation was similar to that seen after 10 min.
Nevertheless, we carried out a further type of experiment in which we dialysed the cell with 1 μm calyculin A for 300 s before activating ICRAC by external application of the SERCA (sarco/endoplasmic reticulum Ca2+-ATPase) inhibitor thapsigargin. In these experiments, pipette Ca2+ was strongly buffered at 140 nm and ATP was present in order to prevent passive depletion of stores. ICRAC was activated in both conditions, and reached the same overall extent (left-hand histogram of Fig. 2C; n = 5 for control and n = 7 for calyculin A-treated cells). There was a tendency for the current activation to occur slightly more slowly in the presence of calyculin A, but this was not significant (right-hand histogram of Fig. 2C, P > 0.1). Note that the size of ICRAC following exposure to thapsigargin after 300 s dialysis was very similar to that seen when the current was rapidly activated following break-in with InsP3 (-2.5 ± 0.4 versus -3.0 ± 0.2 pA pF−1, respectively).
Collectively, these results demonstrate that activation and reversal of activation of ICRAC are not altered by calyculin A, an agent that restructures the cortical actin network.
The microfilament disaggregating agent cytochalasin D does not alter ICRAC
We then examined the effects of disrupting the cytoskeleton by pre-treatment with cytochalasin D, since this manoeuvre has been reported to inhibit capacitative Ca2+ entry in endothelial cells (albeit at very high concentrations; Holda & Blatter, 1997), and to physically separate peripheral InsP3-sensitive stores from the plasma membrane in hepatocytes (Rossier et al. 1991). Pre-incubation with 2 μm cytochalasin D for > 45 min induced marked alterations in cell shape (see Fig. 1C for transmitted light images). Using rhodamine-phalloidin labelling, we confirmed that actin was extensively redistributed following cytochalasin D exposure (Fig. 1D). Whereas all control cells had a prominent cortical actin ring, this was extensively disaggregated by cytochalasin D (right-hand image). Nevertheless, remodelling of the actin microfilaments failed to affect the rate or extent of activation of ICRAC in response to 30 μm InsP3+ 10 mm EGTA (data not shown). In pancreatic acinar cells, it has been reported that cytochalasin-induced disruption of peripheral actin filaments alone is sufficient to enable secretory vesicles to fuse with the plasma membrane and this is thought to reflect removal of spatial restrictions between the vesicles and the surface membrane (so-called ‘fusion-clamp’; Muallem et al. 1995). The prominent disaggregation of the cortical actin network by cytochalasin D (Fig. 1D) should facilitate the movement of peripheral ER to the plasma membrane and this, according to the conformational-coupling model, would accelerate the activation of ICRAC. To test this, we took advantage of the slow, biphasic development of ICRAC that arises when stores are depleted passively by dialysis with 10 mm EGTA alone (Fierro & Parekh, 1999). Following a delay of around 70 s after break-in, ICRAC initially develops slowly at a mean rate of -5.0 ± 0.5 fA (pF s)−1, which lasts for around 50 s and accounts for 20 % of steady-state ICRAC before a secondary rapid increase in current occurs (duration 75 s) at a mean rate of -20.0 ± 2 fA (pF s)−1, which accounts for 68 % of steady-state ICRAC (Fierro & Parekh, 1999). This multi-phasic development of ICRAC enabled us to examine in detail whether any of these parameters was altered following the extensive disruption of the peripheral cytoskeleton with cytochalasin D. The results are summarised in Fig. 3A. The left-hand panel shows the time course of development of ICRAC following dialysis with 10 mm EGTA for a non-treated (control) cell (•) and one that was exposed to cytochalasin D for 60 min (○). The biphasic time course and overall extent of current activation are clearly similar. More detailed analysis is presented in the adjacent plots. The delay, time to peak, percentage of steady-state ICRAC and rates of development as well as the overall extent of the current were not significantly different between control and cytochalasin D-treated cells (P > 0.15 for all cases; n = 5 for control and n = 6 for cytochalasin D-treated cells). Hence disruption of the sub-plasmalemmal microfilamentous network does not alter any of the components involved in the activation of ICRAC following passive store depletion.
Figure 3. Pharmacological tools that disaggregate or stabilise the cytoskeleton fail to affect the activation of ICRAC.
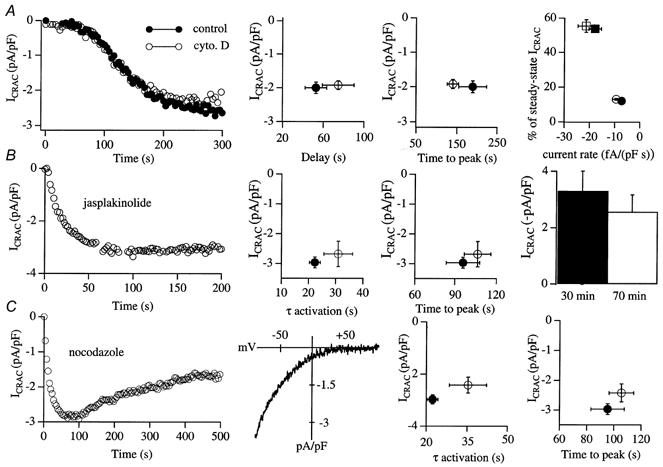
A, activation of ICRAC by passive depletion of stores is not altered by cytochalasin D pre-treatment. The left panel depicts a control recording and one taken after 60 min exposure to 2 μm cytochalasin D. ICRAC was activated by dialysing the cells with a pipette solution containing 10 mm EGTA. None of the parameters we measured were affected by cytochalasin D (right panels; see text for further details). The filled symbols in the right-hand graph represent control and the open symbols data from cytochalasin D-treated cells. Circles represent the initial slow component of the current and squares represent the secondary large phase (see Fierro & Parekh, 1999, for further details). B, ICRAC activates normally despite pre-treatment with the microfilament-stabilising peptide jasplakinolide. The left panel shows the time course of development of ICRAC in a cell pre-exposed to 1 μm jasplakinolide for 34 min. Neither the activation time constant nor time to peak was significantly different between control and jasplakinolide-treated cells (centre plots; •, controls (non-treated); ○, jasplakinolide treated). Increasing the exposure to jasplakinolide to 70 min still did not prevent ICRAC from activating (histogram in right-hand panel). C, disrupting microtubules by an overnight incubation with 5 μm nocodazole also did not affect ICRAC activation. The left panel shows a recording following an 18 h exposure to the drug and the I-V relationship (taken at 82 s) is shown. The activation time constant, time to peak and extent of the current were similar between control and nocodazole-treated cells (right-hand graphs; •, control; ○, nocodazole treated).
Stabilisation of the peripheral cytoskeleton with jasplakinolide does not impede activation of ICRAC
The membrane-permeable peptide jasplakinolide stabilises the peripheral cytoskeleton by inducing actin polymerisation (Bubb et al. 1994). Pre-treatment with jasplakinolide strongly suppressed thapsigargin-evoked Ca2+ influx in the study of Patterson et al. (1999). To see whether a similar action was exerted on ICRAC, we pre-incubated cells with 1 μm jasplakinolide for > 25 min and then examined whether activation of ICRAC was prevented. Despite inducing quite marked changes in cell morphology (Fig. 1E), ICRAC could be routinely activated following dialysis with InsP3+ 10 mm EGTA (Fig. 3B, 9/9 cells). The activation time constants as well as the times to peak were not significantly different between control and jasplakinolide-treated cells (Fig. 3B middle panels, P > 0.2). Even after 70 min exposure to the drug, robust ICRAC could still be activated (Fig. 3B panel, 4/4 cells). We tried to observe the redistribution of actin following treatment with jasplakinolide. Because jasplakinolide binds to the same site on actin as phalloidin, we raised the concentration of rhodamine- phalloidin 10-fold. The reorganisation of actin seemed similar to that induced by calyculin but the images were very dim and the resolution was poor.
Effects of disrupting microtubules with nocodazole
It is well established that the morphology of the ER is maintained through a tight interaction with microtubules (Dabora & Sheetz, 1988). Disruption of the microtubular network with nocodazole results in retraction of the peripheral ER that lies close to the plasma membrane towards the cell centre (Graier et al. 1998). Such changes, which occur over the time course of a few hours, have been seen in a variety of cell types using dyes to directly visualise the location of the ER (Terasaki et al. 1986; Lee & Chen, 1988). According to the conformational-coupling model, such a profound collapse of peripheral ER should prevent ICRAC from activating, since InsP3 receptors would now be spatially far away from CRAC channels in the plasma membrane. To see whether this prediction was met, we incubated RBL-1 cells overnight (16-26 h) in 5 μm nocodazole and then tried to activate ICRAC. Nocodazole-treated cells were very round in shape, and were only loosely attached to the coverslips (Fig. 1F). Nevertheless, we were able to routinely activate a robust ICRAC after dialysis with InsP3+ 10 mm EGTA (Fig. 3C; 8/8 cells). The activation time constant was slightly slower but this and the time to peak were not significantly different between nocodazole-treated and control cells (P > 0.07 and > 0.15, respectively). The extent of inactivation in the presence of nocodazole was 38.6 ± 2 % and the half-time was 130.5 ± 8 s. Neither parameter was significantly different from non-nocodazole-treated cells.
Interfering with the cytoskeleton does not affect store-operated calcium influx in intact RBL-1 cells
Although ICRAC is the major store-operated Ca2+ entry pathway in RBL-1 cells in both whole-cell and perforated-patch configurations (Parekh & Penner, 1997), we felt it necessary to examine the effects of interfering with the cytoskeleton on store-operated Ca2+ influx in intact cells. In these experiments, we loaded cells with the fluorescent calcium dye fura 2 (see Methods). Stores were depleted by local application of the SERCA pump blocker thapsigargin (2 μm) together with the calcium ionophore ionomycin (50-100 nm) in Ca2+-free external solution (containing 0.2 mm EGTA). We used a combination of these agents because thapsigargin alone sometimes fails to evoke calcium influx despite calcium release (Parekh et al. 1997). Note that, with these low concentrations of ionomycin, calcium influx is entirely due to store depletion and not to calcium transport into the cell directly by the ionophore (Morgan & Jacob, 1994). A typical control recording is shown in Fig. 4A. The combination of thapsigargin and ionomycin resulted in robust calcium release due to store emptying, and then calcium levels returned to basal values. The mean increase in cytosolic Ca2+ due to release from the stores is summarised in Fig. 4C. Re-admission of external calcium now resulted in a further elevation of intracellular calcium since calcium influx occurred through open store-operated calcium channels (Fig. 4A). Although the size of this response is not an accurate assessment of the extent of calcium influx (since the activity of other calcium removal mechanisms also contributes), it is nevertheless a rough estimate of the amount of store-operated calcium influx. We therefore measured the peak amplitude of this signal, as well as the time to peak. Pooled data for Ca2+ influx from 14 control cells are summarised in Fig. 4D and E. We then examined the effects of the various cytoskeletal modifying agents. Cells were pre-treated with each drug and then maintained in the presence of the drug throughout the entire experiment. Pre-treatment with jasplakinolide (1 μm for > 25 min) failed to alter the rate or extent of the calcium signal following re-admission of external calcium (Fig. 4B, pooled data in Fig. 4D and E). Similarly, cytochalasin D (2 μm for > 45 min) also failed to affect the calcium signal (Fig. 4D and E). With calyculin A, we found that the size of the calcium signal upon re-admission of calcium tended to vary more than in the control, and some cells clearly had smaller responses. However, the size of the averaged responses was not significantly different from that of the control (Fig. 4D and E). Because ICRAC can be inactivated by protein kinase C (Parekh & Penner, 1995a), we think that the reduction seen in some cells is probably due to phosphatase inhibition by calyculin A, resulting in greater phosphorylation and thus inactivation of CRAC channels. Note that none of the manoeuvres that interfered with the cytoskeleton significantly altered the extent of Ca2+ release from the stores (Fig. 4C). Collectively, these results indicate that interfering with the cytoskeleton fails to alter store-operated calcium influx in intact cells and are in good agreement with the direct recordings of the store-operated calcium current ICRAC.
Figure 4. Store-operated calcium influx is unaffected by interfering with the cytoskeleton.
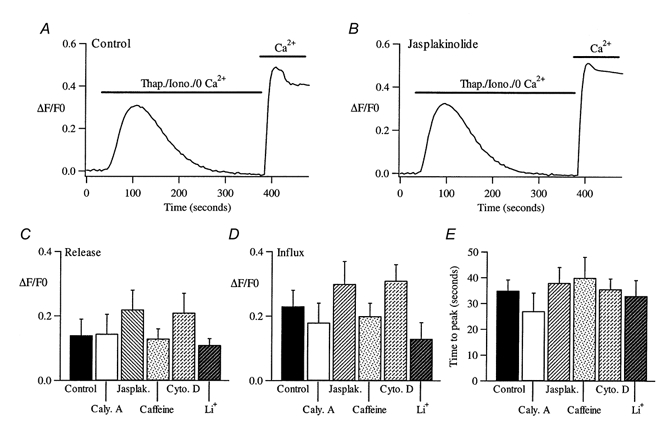
A, a control response. Stores were depleted by applying thapsigargin (Thap, 2 μm) together with ionomycin (Iono, 100 nm) in calcium-free external solution (0.2 mm EGTA) and then 10 mm calcium was applied to the cell. B, the cell was pre-treated with 1 μm jasplakinolide for 45 min and the above protocol repeated. Jasplakinolide was present in all solutions. C-E, pooled data from many cells for the different treatments carried out. C, summary of the amount of Ca2+ released from the stores (measured indirectly as the peak of the Ca2+ release event). D, the mean amplitude of the Ca2+ signal obtained when external Ca2+ was re-admitted into the cells. E, the time taken for this Ca2+ signal to peak (time to peak), shown for the different treatments. Ca2+ influx in cytochalasin D-treated cells tended to develop more slowly than in control, although this was not significant (P = 0.09). In the presence of Li+, Ca2+ influx tended to be smaller than in control but this again was not quite significant (P = 0.08). For all other conditions, P > 0.15.
Effect of balloon patching on activation and reversal of activation of ICRAC
Studies on mechano-sensitive cation channels have revealed that a very effective way to separate the plasma membrane from underlying structures in the whole-cell configuration is to apply positive pressure through the patch pipette (Hamill & McBride, 1997). This results in a dramatic increase in cell volume, as the cell inflates like a balloon. The inflation is thought to be the whole-cell equivalent of membrane blebs, which reflect local uncoupling of the plasma membrane from the underlying cytoplasm (Sokabe & Sachs, 1990). If the ER has to move up close to the plasma membrane in order for ICRAC to be activated, one might expect cell inflation to interfere with activation of the current since the plasma membrane would move further away. The upper panel of Fig. 5 shows a typical balloon-patch experiment. The left-hand transmitted image shows the cell shortly after the onset of whole-cell recording and the lower left panel plots the time course of ICRAC corresponding to this experiment. After around 50 s, positive pressure was applied through the patch pipette to inflate the cell and this pressure (3 kPa) was maintained. The cell ballooned and then stabilised within 20 s, as shown in the upper right panel of Fig. 5. Interestingly, the cell capacitance (index of cell surface area) did not change after the inflation, indicating that the increase in cell size did not reflect insertion of new membrane but rather opening out of the numerous infoldings of the plasma membrane, consistent with Solsona et al. (1998). The I-V relationship taken after inflation (Fig. 5, lower left-hand panel, marked i) shows that cell inflation did not alter membrane conductance. At 75 s, thapsigargin was applied to the cell in order to see whether ICRAC could now be activated. The current developed normally to reach a steady-state value of -1.8 pA pF−1 with a time to peak similar to that of control cells (Fig. 5, lower left-hand panel; compare with that of Fig. 2C); the I-V relationship is shown in the inset, taken at the time point marked ii. Similar results were seen in three additional cells. It is important to note that we applied an amount of pressure to inflate the cells such that the cells did not deflate when the pressure was removed. This indicates that the elastic modulus of the cytoskeletal strings that are responsible for retracting the cell membrane has been exceeded, and therefore that these elements are no longer operational.
Figure 5. Separation of plasma membrane from underlying structures using balloon patching does not affect the activation nor does it induce reversal of activation of ICRAC.
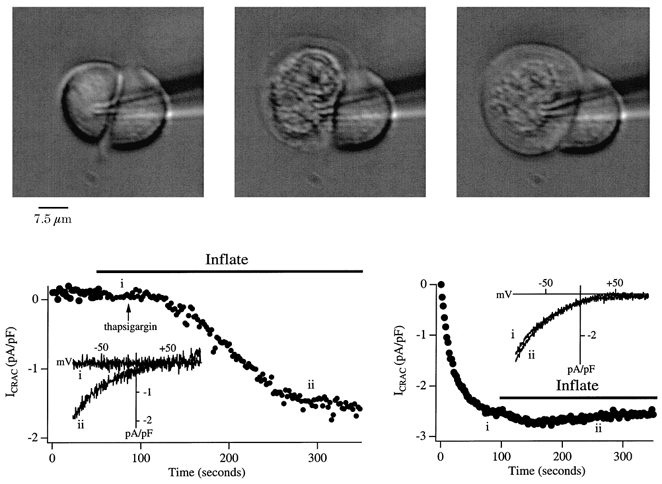
The upper panel shows the development of the ballooned cell and the activation of ICRAC is shown below. The upper left image shows the cell a few seconds following break-in with a pipette solution containing buffered Ca2+ and ATP. The cell was then inflated by applying 3 kPa pressure via a manometer connected to the pipette and the upper middle image shows the cell in the process of ballooning. After around 10 s, the cell reached its maximum inflation (upper right panel) and maintained this shape for the duration of the recording. Note that the intracellular components seem to aggregate at the cell centre and there is a transparent space below the plasma membrane. Following stabilisation, thapsigargin was applied and ICRAC could still be activated to the same extent and with a similar time course to control (see Fig. 2C). The inset shows I-V relationships 30 s after inflation (i) and then once ICRAC had peaked (ii). The background current upon break-in has been subtracted. Note that inflation per se did not change membrane conductance. The lower right panel shows the effect of inflation when applied after ICRAC had been activated (InsP3+ 10 mm EGTA + thapsigargin). There was a small, reversible further increase in the current, but this was not always seen. Clearly, cell ballooning did not affect ICRAC significantly in spite of the large structural changes in the vicinity of the plasma membrane.
In a second set of experiments, we activated ICRAC by dialysis with InsP3+ 10 mm EGTA and then inflated the cell as the current approached steady state. A typical recording is shown in the lower right-hand panel of Fig. 5. Despite cell ballooning, ICRAC was largely unaffected. Similar results were seen in all eight other cells in which this manoeuvre was carried out. We tried to obtain direct evidence that the plasma membrane had moved away from the ER after ballooning by loading the latter with the fluorescent dye mag-fura 2 and then imaging its distribution. However, every time we applied inflation, the cell became slightly out of focus and it was therefore not possible to follow this approach.
2-APB, an inhibitor of intracellular InsP3 receptors, seems to block CRAC channels directly
Fundamental to both versions of the conformational-coupling model is the requirement for InsP3 to be bound to the InsP3 receptor at all stages of Ca2+ influx. In support of this, Ma et al. (2000) have recently reported that the membrane-permeable InsP3 receptor antagonist 2-APB (20 μm) rapidly reverses activation of store-operated Ca2+ influx within a few seconds. They interpreted this as evidence that InsP3 receptors need to be functional in order to initiate and then sustain store-operated Ca2+ influx. In agreement with this, Fig. 6A shows that pre-incubation with 20 μm 2-APB (> 15 min) prevented ICRAC from being detected, despite dialysis with a pipette solution that would normally activate ICRAC rapidly and to its maximal extent (InsP3+ 10 mm EGTA + 2 μm thapsigargin; Fig. 6A). If 2-APB were working through the InsP3 receptor and occupancy of this receptor by InsP3 was required for sustained activation of ICRAC, then one would expect that intracellular dialysis with 2-APB should reverse the activation of the current. To test this, we dialysed cells with InsP3, 10 mm EGTA and 50 μm 2-APB. This higher concentration of 2-APB was chosen to induce a rapid block of the InsP3 receptors and to ensure that the cytosolic concentration of the drug was reasonably high (2-APB is membrane permeable and therefore can diffuse out of the cell). However, ICRAC could still be activated to more than 70 % of control and the current was sustained for up to 10 min (our longest recording time). Because 2-APB is membrane permeable, one could argue that its rate of diffusion out of the cell is faster than the rate of entry into the cytoplasm via dialysis through the patch pipette. This would explain the ineffectiveness of 2-APB when applied intracellularly. To examine this, we determined the lowest concentration of 2-APB, applied externally, that was necessary to fully inhibit ICRAC. Five micromolar 2-APB was sufficient to completely suppress the activation of the current (Fig. 6A, right-hand panel). Even at 2 μm, many cells failed to respond (data not shown). Hence dialysis with a concentration of 2-APB that is 10-25 times higher than the lowest external concentration needed to block ICRAC consistently fails to substantially reduce (let alone block) the current.
Figure 6. The InsP3 receptor antagonist 2-APB appears to block CRAC channels from an external site.
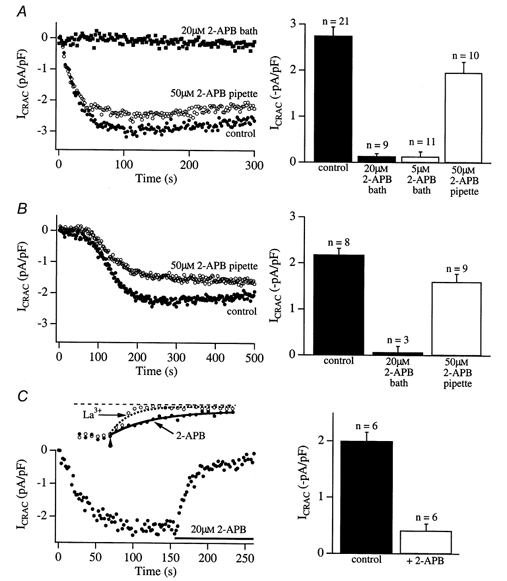
A, 2-APB blocks ICRAC activity when added to the bath but not when included in the recording pipette. Cells were dialysed with 30 μm InsP3+ 10 mm EGTA + 2 μm thapsigargin. Filled circles show control recordings, taken in the absence of 2-APB. Following incubation of the cells with 20 μm 2-APB for > 15 min, ICRAC could not be recorded. However, when cells were dialysed with InsP3+ 10 mm EGTA + thapsigargin and 50 μm 2-APB (i.e. 2-APB was not added to the bath), ICRAC could be activated. The histogram on the right summarises data from several cells. ICRAC was reduced by 27 % when 2-APB was included in the pipette and this was significant (P = 0.018). In B, stores were depleted passively by dialysis with 10 mm EGTA alone. Compared with control cells (•), dialysis with 50 μm 2-APB (○) reduced the extent of activation of ICRAC only slightly (histogram on the right, P > 0.07). In these experiments, around 70 s elapsed following break-in before the current started to develop slowly. The cytosol would therefore have been exposed to appreciable levels of 2-APB prior to the activation of ICRAC (see text). As with the InsP3 experiments in A, addition of 2-APB to the bath prevented ICRAC from being recorded using passive depletion (histogram in B). C, external application of 2-APB rapidly reduces ICRAC when applied after the current has reached steady state. The left panel shows a typical recording using this protocol and pooled data are summarised in the histogram on the right. The inset of C compares the kinetics of loss of ICRAC in response to 10 μm La3+ (a rapid Ca2+ channel blocker) and 20 μm 2-APB using our application system. La3+ (10 μm) fully inhibited ICRAC (3/3 cells) and did this at a rate only slightly faster (around 2-fold) than that seen with 20 μm 2-APB, a concentration that did not fully inhibit the current. In addition to its reported action on the InsP3 receptor, these results suggest that 2-APB may block CRAC channels directly.
A further explanation for the weaker effect of 2-APB when applied intracellularly could be that the high InsP3 concentrations prevent the inhibition by 2-APB. We felt this simple explanation was somewhat unlikely because we would then have expected to see ICRAC develop following dialysis with InsP3 in the experiments of Fig. 5A, where cells had been pre-incubated with 2-APB. Alternatively, 2-APB might irreversibly block the InsP3 receptors provided it accessed them before InsP3. This could explain why 2-APB was very effective when applied before InsP3 (pre-incubation) but much less so when applied together with InsP3 (dialysis). We tested this idea by dialysing cells with 50 μm 2-APB together with 10 mm EGTA to passively deplete the stores. Using this approach, the cytosol is extensively dialysed for around 70 s before activation of ICRAC starts in the presence of low (basal) levels of InsP3 (Fierro & Parekh, 1999). With our series resistance of around 6 MΩ in these experiments and a cell size of 15 pF, we calculate a time constant of wash-in of 2-APB of 146 s. Hence after 70 s dialysis there would be almost 15 μm 2-APB (slightly less since some drug would cross the plasma membrane) and at 200 s approximately 43 μm. 2-APB should therefore easily access the InsP3 receptors, irreversibly inactivate them and thereby prevent ICRAC from activating. However, as shown in Fig. 6B, intracellular dialysis of 2-APB only weakly reduced the extent of ICRAC, evoked by passive store depletion. On the other hand, pre-incubation with a lower concentration of 2-APB (20 μm) completely prevented passive depletion from evoking ICRAC, as was the case when the current was evoked by InsP3 dialysis (Fig. 5A).
Taken together, these results might suggest that 2-APB blocks ICRAC mainly from the external side rather than via an intracellular site. This would be compatible with the notion that 2-APB is actually a CRAC channel blocker. The experiments of Fig. 6C were designed to test this. Following maximal activation of ICRAC (dialysis with InsP3+ 10 mm EGTA + thapsigargin), we locally applied 20 μm 2-APB to the cell once the current had reached steady state. The current rapidly declined to a low level (21.7 ± 5.8 % of peak, n = 5). Pooled results are summarised in the right-hand panel of Fig. 6C.
Because intracellular 2-APB could not inactivate ICRAC when the current had been evoked by InsP3 or passive depletion, but rapidly blocked the current when applied extracellularly (at lower concentrations) before or after dialysis with high InsP3, we believe that one possible explanation of our results is that 2-APB is simply blocking the CRAC channels directly, probably at an extracellular site. The inset of Fig. 6C compares the time course of block of ICRAC when either 20 μm 2-APB or 10 μm La3+ was locally applied to two different cells, in which ICRAC had been evoked by dialysis with InsP3 and 10 mm EGTA. La3+ is considered to be a rapid and potent blocker of ICRAC (Hoth & Penner, 1993). With our somewhat slow local application system, La3+ fully blocked ICRAC with a time constant of 8 s (n = 3). This is only slightly faster than that seen with 20 μm 2-APB (14.4 s).
We think that the small reduction in ICRAC amplitude seen when cells are dialysed with 2-APB reflects channel block as the drug diffuses onto the cell when the patch pipette first approaches and then seals onto the membrane. Consistent with this is our observation that ICRAC did not recover quickly when 2-APB application was stopped in the experiments of Fig. 6C (not shown).
We do not know the exact site of action of 2-APB and it is conceivable that there is more than one target. For example, 2-APB could block both the intracellular InsP3 receptors and the CRAC channels over a similar concentration range. Regardless, our results suggest caution should be used in interpreting results on store-operated Ca2+ influx based on 2-APB.
Heparin inhibits InsP3 receptors but fails to prevent activation of ICRAC
We sought a more selective inhibitor of the InsP3 receptor. Heparin is a widely used antagonist of the InsP3 binding site on the receptor (Ghosh et al. 1988) but, because it is membrane impermeable, has been of only limited use. This problem is circumvented by whole-cell patch-clamp recording, because heparin can be loaded into the cytosol through the patch pipette. We first checked whether heparin blocked ICRAC from the outside, as seemed to be the case with 2-APB. However, pre-incubation with 1 mg ml−1 heparin (a concentration that fully prevents InsP3-mediated activation of ICRAC when included in the pipette solution; Parekh & Penner, 1995b) failed to alter the rate or extent of activation of the current (n = 5).
In the following experiments, we wanted to initially activate ICRAC via InsP3 and then displace the InsP3 that was bound to the InsP3 receptor by competition with heparin such that the stores would refill and hence ICRAC would be turned off. This way, we could be confident that InsP3 had indeed been dislodged from its binding site on the InsP3 receptors. Following store refilling in the presence of heparin, we wanted to subsequently see whether we could activate ICRAC a second time. If we could, then this would mean that the current can be evoked despite InsP3 receptors not being occupied with InsP3. To permit store refilling, we dialysed cells with a pipette solution containing Ca2+ buffered at 225 nm and ATP. The pipette solution also contained InsP3 and 1 mg ml−1 heparin, a concentration that rapidly reverses activation of ICRAC (Glitsch & Parekh, 2000). As shown in Fig. 7A, ICRAC was rapidly activated. But then the heparin started to accumulate in the cytoplasm (although more slowly than InsP3 due to its much larger molecular weight) and displaced the bound InsP3 from the InsP3 receptors. Since InsP3-mediated Ca2+ efflux from the stores was therefore attenuated, SERCA pumps were able to refill the stores due to the presence of the buffered Ca2+ and ATP. ICRAC deactivated fully (Fig. 7A; mean of 7 cells). Consistent with this interpretation were the findings that buffered Ca2+ and ATP were required for the decline of ICRAC (Fig. 7B; 5/5 cells; curve without error bars in this and subsequent graphs of the figure is that shown in Fig. 7A and is included for ease of comparison) and that ICRAC declined only partially when cells were dialysed with InsP3, buffered Ca2+ and ATP but without heparin (Fig. 7C; 7/7 cells). Some decay in ICRAC does occur (around 40 %), but this reflects both slow Ca2+-dependent (Zweifach & Lewis, 1995; Parekh, 1998) and kinase-mediated inactivation (Parekh & Penner, 1995a). Finally, inhibition of SERCA pumps (and hence refilling) with thapsigargin (included in the pipette together with InsP3, buffered Ca2+, ATP and heparin) reduced the decay of ICRAC (Fig. 7D; 6/6 cells) by around 40 %. This level of inactivation is very similar to that seen in Fig. 7C (without heparin and thapsigargin) and shows that very little store refilling occurs if InsP3 can access its receptors. Note that dialysis with heparin failed to inactivate ICRAC in the absence of Ca2+ and ATP, ruling out an action of heparin on either the CRAC channels themselves or the activation mechanism.
Figure 7. InsP3 bound to the InsP3 receptor is not necessary for the activation and maintenance of ICRAC: studies with the InsP3 receptor antagonist heparin.
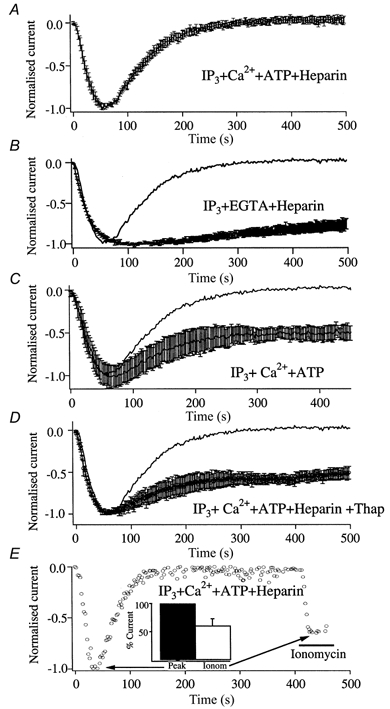
A, cells were dialysed with InsP3, 225 nm Ca2+ (buffered with 10 mm EGTA), 2 mm Mg-ATP and 1 mg ml−1 heparin. Following activation of ICRAC, the current deactivated fully within a further 250 s. This occurred because heparin diffused into the cell relatively slowly but then displaced InsP3 from the InsP3 receptors. In the absence of continuous Ca2+ release from the stores, SERCA pumps were able to refill the stores in the presence of buffered Ca2+ and ATP, thereby deactivating the current. Consistent with this were the findings that the current did not decline much in the presence of heparin when Ca2+ and ATP were omitted from the pipette (B), that the decline was only partial when heparin was omitted from the pipette (C) and that the decline was only partial when the SERCA pump blocker thapsigargin (2 μm) was added to the pipette solution (D). The partial loss of ICRAC reflects Ca2+- and phosphorylation-mediated inactivation (see text for references). In E, ICRAC could be subsequently reactivated by applying the Ca2+ ionophore ionomycin to deplete the stores, following full deactivation of the current in the presence of heparin. Because InsP3 has been displaced from its receptors, this indicates that the current can be activated even though InsP3 is not bound to the InsP3 receptor.
The key experiment is shown in Fig. 7E. Following full deactivation of ICRAC in the presence of InsP3, heparin, buffered Ca2+ and ATP, subsequent store depletion by local application of 10 μm ionomycin (to bypass the non-functional InsP3 receptors) re-activated ICRAC. Hence ICRAC can still be activated under conditions where InsP3 is no longer bound to the InsP3 receptors. The current obtained following exposure to ionomycin was smaller than that obtained with InsP3 (61 ± 10 %, n = 5, P < 0.05, Fig. 7E, inset), the reduction reflecting the Ca2+- and kinase-dependent inactivation described above.
Pre-exposure to Li+ does not alter the rate or extent of ICRAC
By inhibiting inositol monophosphatase, Li+ gradually depletes cells of InsP3 by reducing the synthesis of phosphatidyl inositol. Rosado & Sage (2000) have reported that exposure to 10 mm Li+ reduces the extent of thapsigargin-evoked calcium influx in platelets, which would be consistent with a role for InsP3 receptors in the activation of store-operated calcium influx (Rosado & Sage, 2000). To examine whether Li+ interferes with the activation of ICRAC, we pre-incubated RBL-1 cells with 15 mm Li+ (for 1-2 h). We then broke in with a pipette solution containing 10 mm EGTA in order to activate ICRAC passively. Passive depletion evokes ICRAC slowly and with a biphasic pattern (described above), which enabled us to examine whether Li+ interfered with any stage of current activation. In these experiments, intracellular solution was supplemented with 15 mm LiCl to ensure that the cytosol was continuously exposed to Li+. A control recording and one taken in the presence of Li+ are shown in Fig. 8A and B, respectively. The dashed lines in the control recording (upper panel of Fig. 8A) show linear fits to the slow and fast phases of ICRAC development (Fierro & Parekh, 1999). Corresponding I-V curves, taken when the macroscopic current had peaked, are depicted in the lower panel of Fig. 8A. In the presence of Li+, ICRAC was activated normally. The presence of Li+ failed to significantly alter the amplitude (Fig. 8D), delay (Fig. 8E), time to peak (Fig. 8E), rates of development of the slow and fast phases of the current (Fig. 8F) or their relative fractions of steady-state ICRAC (Fig. 8F) when compared with controls.
Figure 8. Neither Li+ nor caffeine interferes with the rate or extent of activation of ICRAC.
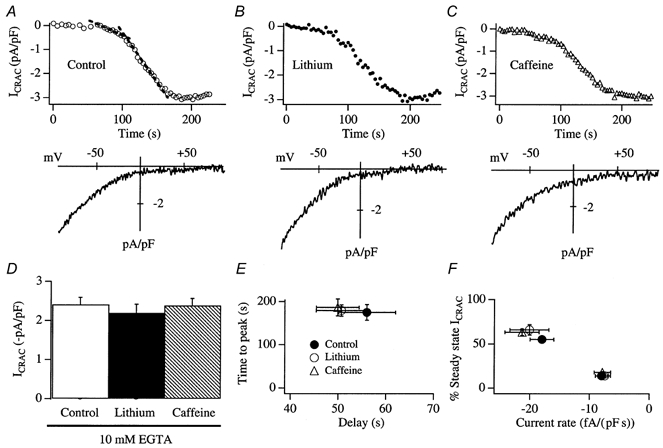
A, a control recording from a cell dialysed with a pipette solution containing 10 mm EGTA, to activate ICRAC passively. The upper panel shows the time course of the current and the lower panel the I-V relationship, taken as the current peaked. The dashed lines in the upper panel represent linear fits to the slow and fast components of development of the current (Fierro & Parekh, 1999). B, a typical recording from a cell that was pre-incubated in 15 mm Li+-containing external solution for 105 min and then dialysed with a pipette solution containing 15 mm Li+. ICRAC was evoked passively. C, a representative recording from a cell that was exposed to 10 mm caffeine for 22 min and then dialysed with 10 mm caffeine. ICRAC was evoked passively. D, summary of the size of the currents for the different conditions (6 control cells, 7 for Li+ and 7 for caffeine). E, the delay versus time to peak is plotted for the various conditions. F, the rate of development of ICRAC for the slow and fast components is plotted against the relative fractional amplitude of ICRAC. There were no significant differences between any of the parameters shown in D-F for control, Li+- or caffeine-treated cells.
We also examined whether pre-exposure to Li+ affected store-operated calcium influx in fura 2-loaded cells. Cells were pre-incubated with 15 mm Li+ for > 90 min and then stores were depleted by applying thapsigargin (2 μm) and ionomycin (100 nm) in Ca2+-free external solution (0.2 mm EGTA). Re-admission of external Ca2+ resulted in Ca2+ influx. Neither Ca2+ release nor the rate or extent of this Ca2+ entry was affected by Li+ (Fig. 4C-E; 6 cells).
The InsP3 receptor antagonist caffeine does not affect the rate or extent of activation of ICRAC
Caffeine is a widely used competitive inhibitor of InsP3 receptors in non-excitable cells (Parker & Ivorra, 1991; Ehrlich et al. 1994; Cancela et al. 2000). We therefore investigated whether caffeine interfered with the activation of ICRAC. We exposed cells to 10 mm caffeine for > 10 min and then broke in with a pipette solution containing 10 mm caffeine (to ensure that the cytosol was continuously exposed to the inhibitor). We depleted stores passively, using 10 mm EGTA. With this protocol, basal InsP3 levels are very low and therefore would not compete with the high concentrations of intracellular caffeine for binding to the InsP3 receptors. Despite the extracellular and intracellular presence of caffeine, ICRAC was activated in a manner indistinguishable from that of control cells (Fig. 8A and C). Again, the amplitude (Fig. 8D, 7 cells for caffeine), delay (Fig. 8E), time to peak (Fig. 8E), rates of development for the slow and fast phases of the current (Fig. 8F) and their relative fractions of steady-state ICRAC (Fig. 8F) were not significantly altered when compared with controls. In fura 2 experiments, store-operated Ca2+ influx was also unaffected by pre-exposure to caffeine (Fig. 4C-E, 10 cells).
DISCUSSION
In view of the fact that store-operated Ca2+ influx is found in a variety of species and within most cells of a given species, the underlying mechanisms are of great significance. Despite intense research, the mechanism that couples Ca2+ store emptying to the activation of Ca2+ influx remains enigmatic. The secretion-like coupling model, in which peripheral ER containing InsP3 receptors reversibly moves to the plasma membrane to bind to Ca2+ channels through a mechanism involving the cytoskeleton, is an attractive scheme to account for the activation of store-operated Ca2+ influx.
Our results reveal that manoeuvres that stabilise or disaggregate the cytoskeleton or that separate the plasma membrane from underlying structures all fail to interfere with the activation of ICRAC, nor can such treatments reverse the activation. It would appear that the store-operated Ca2+ current ICRAC is not modulated in any discernible way by the cytoskeleton nor does it seem to depend on a weak spatial interaction between the surface membrane and components of the underlying cytoplasm.
Although ICRAC appears to be the dominant electrogenic store-operated calcium influx pathway in RBL-1 cells and can be recorded in the absence of exogenous calcium chelator (Fierro & Parekh, 2000) and in the perforated-patch configuration (Zhang & McCloskey, 1995), it is nevertheless possible that very weakly electrogenic store-operated calcium entry pathways (beyond the level of our detection) or even electroneutral influx mechanisms also contribute to the total store-dependent Ca2+ entry. However, our experiments using fura 2-loaded intact cells revealed that thapsigargin-evoked calcium influx was unaffected by the manoeuvres that interfered with the cytoskeleton. Collectively, these results indicate that ICRAC as well as any other store-operated calcium influx pathways in RBL-1 cells are not affected by the state of the cytoskeleton.
We also do not think that functional InsP3 receptors are required to support all stages of ICRAC, as initially suggested (Ma et al. 2000). This conclusion was based on the finding that the InsP3 receptor antagonist 2-APB rapidly abolished Ca2+ influx when applied after Ca2+ entry had been activated (Ma et al. 2000). However, our results suggest that 2-APB may not be a useful tool to address this issue, since it seems to directly inhibit CRAC channels.
Using the more conventional InsP3 receptor antagonist heparin, which does not directly block CRAC channels at either an extracellular or an intracellular site, we have now found that ICRAC can be activated even though InsP3 has been functionally displaced from its receptors. Previously, we and others have reported that dialysis with heparin fails to affect the activation of ICRAC in response to passive depletion (Hoth & Penner, 1992; Fierro & Parekh, 1999). However, in those studies, it was not clear whether heparin had indeed blocked the InsP3 receptors because the current activated before the cytosol had been adequately dialysed with the inhibitor.
Furthermore, using both Li+ (to reduce resting levels of InsP3) and caffeine, another inhibitor of InsP3 receptors, we failed to affect the rate or extent of activation of ICRAC at all. Taken together, our findings suggest that functional InsP3 receptors are not required for the activation of ICRAC, at least in RBL-1 cells.
Our conclusions differ from those drawn by Gill and co-workers, using vascular smooth muscle cell lines (Patterson et al. 1999; Ma et al. 2000), and Sage and colleagues, using platelets (Rosado & Sage, 2000a,b; Rosado et al. 2000), who have interpreted their findings as evidence for the coupling model. One possible explanation for the discrepancy in the conclusions reached relates to the methodologies used. We have used whole-cell patch-clamp recordings in order to measure the store-operated calcium current ICRAC directly, whereas the aforementioned studies used fluorescent dyes to monitor calcium influx indirectly, through changes in the whole-cell calcium signal. Both methods have advantages and disadvantages (reviewed in Parekh & Penner, 1997). However, we do not think that differences in methodology can account for our different results because we failed to see any effects of interfering with the cytoskeleton on thapsigargin-evoked calcium influx when we used fluorescent dyes to measure cytosolic calcium. Instead, different cell types may use different mechanisms to activate store-operated calcium influx. It is important to bear in mind that, although ICRAC is the best characterised and most widely distributed electrogenic store-operated calcium entry pathway, there are reports of store-operated currents that differ from ICRAC in ionic selectivity and single channel conductance (Parekh & Penner, 1997). In the A7r5 vascular smooth muscle cell line used by Gill and colleagues, dialysis with high calcium buffer and then exposure to thapsigargin failed to reveal any store-operated current (Iwasawa et al. 1997). Using InsP3 and 10 mm EGTA, our preliminary results also failed to reveal a detectable store-operated current. It is not clear therefore how much of the thapsigargin-evoked Ca2+ influx in these smooth muscle cells is due to a store-operated pathway or whether second messenger-operated channels also contribute. In platelets, due to their small size, the store-operated calcium current has not been characterised. Recently, Rosado & Sage (2000a) found that type II InsP3 receptors from platelets co-immunoprecipitated with the plasmalemmal cation channel htrp1, but only after stores had been depleted. htrp1 is a non-selective cation channel, and is quite distinct from ICRAC. In platelets, a secretion-like conformational-coupling interaction seems to be taking place, but the calcium entry channel is not ICRAC.
Can our findings with ICRAC be reconciled with the secretion-like coupling model? They can, provided some stringent constraints are imposed. First, the balloon patch experiments indicate that the peripheral ER must be tightly attached to the plasma membrane under resting conditions so that a portion of the ER, with InsP3 receptors, presumably remains coupled to the plasma membrane during cell inflation. Such a tight pre-formed complex would also explain why interfering with the cytoskeleton consistently failed to affect the activation of ICRAC. If the complex already exists, then this would obviate a regulatory role for the cytoskeleton. This could explain why disruption of the cytoskeleton failed to affect Ca2+ entry in NIH 3T3 cells (Pedrosa-Ribeiro et al. 1997). Second, the InsP3 receptor, which supposedly couples to the CRAC channels, would have to be insensitive to heparin (and caffeine) since ICRAC could be re-activated by ionomycin even after InsP3 had been displaced from the InsP3 receptors by heparin. Although several isoforms of InsP3 receptor have been described, to our knowledge all are inhibited by heparin. Hence a new form of InsP3 receptor would be required. An alternative explanation is that heparin (and caffeine) are unable to access a sub-plasmalemmal region where a conventional InsP3 receptor is located which couples to CRAC channels. Previously, we have provided evidence that the stores which activate ICRAC are spatially close to the plasma membrane (Parekh & Penner, 1995). Sensitization of InsP3 receptors on these stores activates ICRAC under conditions of low cytosolic InsP3. However, heparin can prevent this activation of ICRAC, indicating that is able to access those stores close to the plasma membrane which are involved in the activation of store-operated Ca2+ entry.
One major criticism of the classical conformational-coupling model has been that it predicts the rapid activation of ICRAC. However, the current develops relatively slowly with a time constant of 20-30 s. Patterson et al. (1999) have argued that their secretion-like coupling model resolves this issue, because the coupling process is induced only after the slow movement of peripheral ER to the plasma membrane. Our findings, which require the existence of a tightly formed complex, represent something of a conundrum for the coupling model. Why does ICRAC activate so slowly if the coupling is tight and pre-formed? One possibility is that there is a significant delay between the detection of the fall in intraluminal Ca2+ and the subsequent conformational change in the coupling protein that spans the stores. Alternatively, a mechanism other than conformational coupling might be responsible for the activation of ICRAC in RBL-1 cells.
Acknowledgments
D.B. is a British Heart Foundation Prize Student. M.D.G. holds a long-term fellowship from the Human Frontiers in Science Programme. A.B.P. is a Wellcome Trust Career Development Fellow and holds the Amersham Fellowship in Medical Cell Biology at Keble College, Oxford. We are grateful to Professor Alison Brading for critical comments on the work, Vicky Pank for preparing the cells, Drs R. Bobe, J. Wilde and S. P. Watson for help with rhodamine labelling, and B. Bagel for technical support.
References
- Berridge MJ. Capacitative Ca2+ entry. Biochemical Journal. 1995;312:1–11. doi: 10.1042/bj3120001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ, Lipp P, Bootman MD. The calcium entry Pas de Deux. Science. 2000;287:1604–1605. doi: 10.1126/science.287.5458.1604. [DOI] [PubMed] [Google Scholar]
- Boulay G, Brown DM, Qin N, Jiang M, Dietrich A, Xi M, Chen Z, Birnbaumer M, Mikoshiba K, Birnbaumer L. Modulation of Ca2+ entry by polypeptides of the inositol 1,4,5-trisphosphate receptor (IP3R) that bind transient receptor potential (TRP): Evidence for roles of TRP and IP3R in store depletion-activated Ca2+ entry. Proceedings of the National Academy of Sciences of the USA. 1999;96:14955–14960. doi: 10.1073/pnas.96.26.14955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubb MR, Senderowicz AM, Sausville EA, Duncan KL, Korn ED. Jasplakinolide, a cytotoxic natural product, induces actin polymerisation and competitively inhibits the binding of phalloidin to F-actin. Journal of Biological Chemistry. 1994;269:14869–14871. [PubMed] [Google Scholar]
- Cancela JM, Gerasimenko OV, Gerasimenko JV, Tepikin AV, Petersen OH. Two different but converging messenger pathways to intracellular Ca2+ release: the roles of nicotinic acid adenine dinucleotide phosphate, cyclic ADP-ribose and inositol trisphosphate. EMBO Journal. 2000;19:2549–2557. doi: 10.1093/emboj/19.11.2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csutora P, Su Z, Kim HY, Bugrim A, Cunningham KW, Nuccitelli R, Keizer JE, Hanley MR, Blalock JE, Marchase RB. Calcium influx factor is synthesized by yeast and mammalian cells depleted of organellar Ca2+ stores. Proceedings of the National Academy of Sciences of the USA. 1999;96:121–126. doi: 10.1073/pnas.96.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabora SL, Sheetz MP. The microtubule-dependent formation of a tubulovesicular network with characteristics of the ER from cultured cell extracts. Cell. 1988;54:27–35. doi: 10.1016/0092-8674(88)90176-6. [DOI] [PubMed] [Google Scholar]
- Downey GP, Takai A, Zamel R, Grinstein S, Chan CK. Okadaic acid-induced actin assembly in neutrophils: role of protein phosphatases. Journal of Cell Physiology. 1993;155:505–519. doi: 10.1002/jcp.1041550309. [DOI] [PubMed] [Google Scholar]
- Ehrlich BE, Kaftan E, Bezprozvannaya S, Bezprozvanny I. The pharmacology of intracellular Ca2+ release channels. Trends in Pharmacological Sciences. 1994;15:145–148. doi: 10.1016/0165-6147(94)90074-4. [DOI] [PubMed] [Google Scholar]
- Fierro L, Parekh AB. On the characterisation of the mechanism underlying passive activation of the Ca2+ release-activated Ca2+ current ICRAC. Journal of Physiology. 1999;520:407–416. doi: 10.1111/j.1469-7793.1999.00407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh TK, Eis PS, Mullaney JM, Ebert CL, Gill DL. Competitive, reversible, and potent antagonism of inositol 1,4,5-trisphosphate-activated calcium release by heparin. Journal of Physiology. 1988;263:11075–11079. [PubMed] [Google Scholar]
- Glitsch MD, Parekh AB. Ca2+ store dynamics determines the pattern of activation of the store-operated Ca2+ current ICRAC in response to InsP3 in rat basophilic leukaemia cells. Journal of Physiology. 2000;523:283–290. doi: 10.1111/j.1469-7793.2000.t01-2-00283.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graier WF, Paltauf-Doburzynska J, Hill BJ, Fleischhacker E, Hoebel BG, Kostner GM, Sturek M. Submaximal stimulation of porcine endothelial cells causes focal Ca2+ elevation beneath the cell membrane. Journal of Physiology. 1998;506:109–125. doi: 10.1111/j.1469-7793.1998.109bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, McBride DW., Jr Induced membrane hypo/hyper-mechanosensitivity: a limitation of patch-clamp recording. Annual Review of Physiology. 1997;59:621–631. doi: 10.1146/annurev.physiol.59.1.621. [DOI] [PubMed] [Google Scholar]
- Holda JR, Blatter LA. Capacitative calcium entry is inhibited in vascular endothelial cells by disruption of cytoskeletal elements. FEBS Letters. 1997;403:191–196. doi: 10.1016/s0014-5793(97)00051-3. [DOI] [PubMed] [Google Scholar]
- Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355:353–356. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- Hoth M, Penner R. Calcium release-activated calcium current in rat mast cells. Journal of Physiology. 1993;465:359–386. doi: 10.1113/jphysiol.1993.sp019681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine RF. Quantal Ca2+ release and the control of Ca2+ entry by inositol phosphates - a possible mechanism. FEBS Letters. 1990;263:5–9. doi: 10.1016/0014-5793(90)80692-c. [DOI] [PubMed] [Google Scholar]
- Iwasawa K, Nakajima T, Hazama H, Goto A, Shin WS, Toyo-Oka T, Omata M. Effects of extracellular pH on receptor-mediated Ca2+ influx in A7r5 rat smooth muscle cells: involvement of two different types of channel. Journal of Physiology. 1997;503:237–251. doi: 10.1111/j.1469-7793.1997.237bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiselyov K, Mignery GA, Zhu MX, Muallem S. The N-terminal domain of the IP3 receptor gates store-operated hTrp3 channels. Molecular Cell. 1999;4:423–429. doi: 10.1016/s1097-2765(00)80344-5. [DOI] [PubMed] [Google Scholar]
- Kiselyov K, Xu X, Mozhayeva G, Kuo T, Pessah I, Mignery G, Zhu X, Birnbaumer L, Muallem S. Functional interaction between InsP3 receptors and store-operated Htrp3 channels. Nature. 1998;396:478–482. doi: 10.1038/24890. [DOI] [PubMed] [Google Scholar]
- Lee C, Chen LB. Dynamic behavior of endoplasmic reticulum in living cells. Cell. 1988;54:37–46. doi: 10.1016/0092-8674(88)90177-8. [DOI] [PubMed] [Google Scholar]
- Ma H-T, Patterson RL, Van Rossum DB, Birnbaumer L, Mikoshiba K, Gill DL. Requirement of the inositol trisphosphate receptor for activation of store-operated Ca2+ channels. Science. 2000;287:1647–1651. doi: 10.1126/science.287.5458.1647. [DOI] [PubMed] [Google Scholar]
- Morgan AJ, Jacob R. Ionomycin enhances Ca2+ influx by store-regulated calcium entry and not by a direct action at the plasma membrane. Biochemical Journal. 1994;300:665–672. doi: 10.1042/bj3000665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muallem S, Kwiatkowska K, Xu X, Yin HL. Actin filament disassembly is a sufficient final trigger for exocytosis in nonexcitable cells. Journal of Cell Biology. 1995;128:589–598. doi: 10.1083/jcb.128.4.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB. Slow feedback inhibition of calcium release-activated calcium current by calcium entry. Journal of Biological Chemistry. 1998;273:14925–14932. doi: 10.1074/jbc.273.24.14925. [DOI] [PubMed] [Google Scholar]
- Parekh AB, Fleig A, Penner R. The store-operated calcium current ICRAC: nonlinear activation by InsP3 and dissociation from calcium release. Cell. 1997;89:973–980. doi: 10.1016/s0092-8674(00)80282-2. [DOI] [PubMed] [Google Scholar]
- Parekh AB, Penner R. Depletion-activated calcium current is inhibited by protein kinase in RBL-2H3 cells. Proceedings of the National Academy of Sciences of the USA. 1995a;92:7907–7911. doi: 10.1073/pnas.92.17.7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB, Penner R. Activation of store-operated calcium influx at resting InsP3 levels by sensitization of the InsP3 receptor in rat basophilic leukaemia cells. Journal of Physiology. 1995b;489:377–382. doi: 10.1113/jphysiol.1995.sp021058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB, Penner R. Store-operated calcium influx. Physiological Reviews. 1997;77:901–930. doi: 10.1152/physrev.1997.77.4.901. [DOI] [PubMed] [Google Scholar]
- Parker I, Ivorra I. Caffeine inhibits inositol trisphosphate-mediated liberation of intracellular calcium in Xenopus oocytes. Journal of Physiology. 1991;433:229–240. doi: 10.1113/jphysiol.1991.sp018423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson RL, Van Rossum DB, Gill DL. Store-operated Ca2+ entry: evidence for a secretion-like coupling model. Cell. 1999;98:487–499. doi: 10.1016/s0092-8674(00)81977-7. [DOI] [PubMed] [Google Scholar]
- Pedrosa Ribeiro C M, Reece J, Putney JW., Jr Role of the cytoskeleton in calcium signaling in NIH 3T3 cells. An intact cytoskeleton is required for agonist-induced [Ca2+]i signalling. but not for capacitative calcium entry. Journal of Biological Chemistry. 1997;272 doi: 10.1074/jbc.272.42.26555. [DOI] [PubMed] [Google Scholar]
- Putney JW., Jr A model for receptor-regulated calcium entry. Cell Calcium. 1986;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- Putney JW., Jr Kissin' Cousins’: intimate plasma membrane-ER interactions underlie capacitative calcium entry. Cell. 1999;99:5–8. doi: 10.1016/s0092-8674(00)80056-2. [DOI] [PubMed] [Google Scholar]
- Randriamampita C, Tsien RY. Emptying of intracellular Ca2+ stores releases a novel small messenger that stimulates Ca2+ influx. Nature. 1993;364:809–813. doi: 10.1038/364809a0. [DOI] [PubMed] [Google Scholar]
- Rosado JA, Jenner S, Sage SO. A role for the actin cytoskeleton in the initiation and maintenance of store-mediated calcium entry in human platelets. Evidence for conformational coupling. Journal of Biological Chemistry. 2000;275:7527–7533. doi: 10.1074/jbc.275.11.7527. [DOI] [PubMed] [Google Scholar]
- Rosado JA, Sage SO. Coupling between inositol 1,4,5-trisphosphate receptors and human transient receptor potential channel 1 when intracellular Ca2+ stores are depleted. Biochemical Journal. 2000a;350:631–635. [PMC free article] [PubMed] [Google Scholar]
- Rosado JA, Sage SO. The actin cytoskeleton in store-mediated calcium entry. Journal of Physiology. 2000b;526:221–229. doi: 10.1111/j.1469-7793.2000.t01-2-00221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossier MF, Bird GS, Putney JW., Jr Subcellular distribution of the calcium-storing inositol 1,4,5-trisphosphate-sensitive organelle in rat liver. Possible linkage to the plasma membrane through the actin microfilaments. Biochemical Journal. 1991;274:643–650. doi: 10.1042/bj2740643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinoki N, Sakon M, Kambayashi J, Ikeda M, Oiki E, Okuyama M, Fujutana K, Yano Y, Kawasaki T, Monden M. Involvement of protein phosphatase-1 in cytoskeletal organization of cultured endothelial cells. Journal of Cellular Biochemistry. 1995;59:368–375. doi: 10.1002/jcb.240590308. [DOI] [PubMed] [Google Scholar]
- Sokabe M, Sachs F. The structure and dynamics of patch-clamped membranes: a study using differential interference contrast light microscopy. Journal of Cell Biology. 1990;111:599–606. doi: 10.1083/jcb.111.2.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solsona C, Innocenti B, Fernandez JM. Regulation of exocytotic fusion by cell inflation. Biophysical Journal. 1998;74:1061–1073. doi: 10.1016/S0006-3495(98)74030-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terasaki M, Chen LB, Fujiwara K. Microtubules and the endoplasmic reticulum are highly interdependent structures. Journal of Cell Biology. 1986;103:1557–1568. doi: 10.1083/jcb.103.4.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Ferrer-Montiel AV, Montal M, Tsien RY. Activation of store-operated Ca2+ current in Xenopus oocytes requires SNAP-25 but not a diffusible messenger. Cell. 1999;98:475–485. doi: 10.1016/s0092-8674(00)81976-5. [DOI] [PubMed] [Google Scholar]
- Zhang L, McCloskey MA. Immunoglobulin E receptor-activated calcium conductance in rat mast cells. Journal of Physiology. 1995;483:69–66. doi: 10.1113/jphysiol.1995.sp020567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweifach A, Lewis RS. Slow calcium-dependent inactivation of depletion-activated calcium current. Store-dependent and independent mechanisms. Journal of Biological Chemistry. 1995;270:14445–14451. doi: 10.1074/jbc.270.24.14445. [DOI] [PubMed] [Google Scholar]