

Abstract
Using the cell-attached recording configuration, we found that in adult bovine chromaffin cells there exists a direct membrane-delimited inhibition of single Bay K-modified L-channels mediated by opioids and ATP locally released in the recording pipette.
This autocrine modulation is mediated by pertussis toxin (PTX)-sensitive G-proteins and causes a 50 % decrease of the open channel probability (Po) and an equivalent percentage increase of null sweeps at +10 mV with no changes to the activation kinetics, single channel conductance and mean open time. The decrease in Po is mainly due to an increase in the occurrence and duration of slow closed times (> 40 ms).
Addition of purinergic and opioidergic antagonists (suramin and naloxone) or cell pre-treatment with PTX removes the inhibition while addition of ATP and opioids inside the pipette, but not outside, mimics the effect.
Strong pre-pulses (+150 mV, 280 ms) followed by short repolarizations are unable to remove the inhibition at test potential (+10 mV).
Increasing the level of cAMP by either direct application of 8-(4-chlorophenylthio)-cAMP (8-CPT-cAMP) or mixtures of forskolin and 1-methyl-3-isobutylxanthine (IBMX) potentiates the activity of L-channels by increasing the mean open time and decreasing the mean closed time and percentage of null sweeps.
The cAMP-induced potentiation occurs regardless of whether the G-protein-mediated inhibition is activated by ATP and opioids or inactivated by PTX. Protein kinase inhibitors (H7 and H89) prevent the effects of cAMP without altering the basal autocrine modulation associated with PTX-sensitive G-proteins.
Our results provide new evidence for the coexistence of two distinct modulations that may converge on the same neuroendocrine L-channel: a direct G-protein-dependent inhibition and a cAMP-mediated potentiation, which may work in combination to regulate Ca2+ entry during neurosecretion.
Gating modifications of L-channels induced by neurotransmitters are often associated with cAMP-dependent phosphorylation and may result in drastic alteration of excitable cell functioning (Reuter, 1983). In heart cells, the increased muscle strength following β-adrenergic stimulation derives from elevations of intracellular protein kinase A (PKA) activity which enhances the open probability (Po) and induces recruitment of functioning L-channels (Bean et al. 1984). Modulation of L-channels, however, appears not to be affected only by diffusible second messengers. In some case, prolonged and strong depolarizations produce a switching of the channel from a low- to a high-Po gating mode (Hoshi & Smith, 1987; Pietrobon & Hess, 1990). In hippocampal neurons, moderate depolarizations induce L-channel re-openings, which persist without the involvement of cAMP (Kavalali et al. 1997). There are also examples of voltage-dependent facilitation of cardiac and neuronal L-channels associated with cAMP elevations, but their interpretation and molecular features differ substantially from cell to cell (Artalejo et al. 1990; Sculptoreanu et al. 1993; Bourinet et al. 1994).
In contrast to the overwhelming data supporting the up-modulation of L-channels, little is known about the inhibition of these channels by neurotransmitters, despite its indisputable occurrence in neurons and neuroendocrine cells (Scholz & Miller, 1991; Kleppisch et al. 1992; Haws et al. 1993; Amico et al. 1995; Elhamdani et al. 1995). This inhibition has common features to that of non-L-channels. It is fast and pertussis toxin (PTX) sensitive but, unlike non-L-channels, is voltage independent, implying that strong depolarizations do not alter the percentage of channel inhibition (Pollo et al. 1993; Albillos et al. 1996a). In bovine (Carabelli et al. 1998) and rat chromaffin cells (Hernández-Guijo et al. 1999) this modulation has autocrine origins and causes marked Ca2+ current depressions, with a likely functional rationale in the long-term control of neurosecretion. The presence of a significant inhibition of L-channels mediated by Go/Gi-proteins and the reported existence of cAMP-dependent up-modulation on a similar channel (Artalejo et al. 1990) prompted us to assay whether the two processes converge on the same target. Since one of the possible major limitations for observing potentiation of L-channels is the loss of intracellular components during whole-cell recordings (Carbone & García, 1997), we analysed the modulation of L-channels in cell-attached patches of chromaffin cells.
Here we show that as for N- and P/Q-channels, inhibition of single L-channels in intact bovine chromaffin cells is fully defined in membrane microareas and induced by locally released ATP and opioids. Unlike N- and P/Q-channels, however, the inhibition decreases Po without slowing channel activation. The scaling down of Po is mediated by PTX-sensitive G-proteins and does not require diffusible second messengers. Surprisingly, the inhibition coexists with a cAMP-mediated potentiation of channel gating which differs from the cAMP-dependent facilitation observed in chromaffin cells of young cows (Artalejo et al. 1991). cAMP shifts channel gating from a low- to a high-Po gating mode regardless of the inhibition induced by either released or applied neurotransmitters. Thus, inhibition and potentiation proceed along distinct pathways but converge on the same effector thus furnishing a singular way for rapid channel switching from marked inhibition to maximal potentiation.
METHODS
Cell preparation and culture
Bovine chromaffin cells were isolated from the adrenal glands of 6- to 18-month-old cows as described by García et al. (1984) and Albillos et al. (1996b). Adrenal glands were obtained from the city slaughter-house under the supervision of the local veterinary service (ASL n.3, Turin). The overall procedures and solutions used for maintaining the cells in primary culture are detailed in Carabelli et al. (1998). Cells were plated at a density of 105 ml−1 in plastic dishes and incubated at 37 °C in a water-saturated 5 % CO2 atmosphere. Plastic dishes were pre-treated with poly-l-ornithine (1 mg ml−1) and laminin (5 μg ml−1 in L-15 carbonate). The culture medium contained Dulbecco's modified Eagle's medium (DMEM), fetal calf serum (10 %) (Gibco), penicillin 50 i.u. ml−1, streptomycin 50 μg ml−1 (Gibco), gentamycin 2.5 μg ml−1 (Sigma Chemical Co.), 10 μm cytosine arabinoside and fluorodexyuridine (Sigma).
Solutions
The pipette control solution contained (mm): 100 BaCl2, 10 TEA-Cl, 1 MgCl2, 10 Na-Hepes, plus ω-CTX-MVIIC (5-10 μm), tetrodotoxin (TTX) (300 nm) and Bay K 8644 5 μm (pH 7.3 with TEAOH). Membrane potential was zeroed with a solution containing (mm): 135 potassium aspartate, 1 MgCl2, 10 Hepes, 5 EGTA and TTX (300 nm) (pH 7.3 with KOH). For the experiments with receptor agonists in the pipette, ATP (100 μm), [d-Pen2-Pen5]-enkephalin (DPDPE) (1 μm) and [d-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin (DAMGO) (10 μm) were added to the control solution. For the experiments with receptor antagonists in the pipette, we added naloxone (100 μm) and suramin (10 μm). Receptor agonists (ATP, DPDPE and DAMGO) and antagonists (naloxone and suramin), 8-(4-chlorophenylthio)-cAMP (8-CPT-cAMP), H7 and forskolin were purchased from Sigma. H89 was obtained from Calbiochem and 1-methyl-3-isobutylxanthine (IBMX) from RBI. Forskolin was first diluted in ethanol and then added to the external medium to the final concentration (10 μm). Protein kinase inhibitors (H7 and H89) and IBMX were dissolved in distilled water and kept frozen in aliquots.
The Ca2+ channel agonist (±)-Bay K 8644 was a gift of Bayer A.G. (Wuppertal, Germany). Stock solutions of (±)-Bay K 8644 (1 mm) were prepared in 100 % ethanol and stored light protected at 4 °C. The dihydropyridine (DHP) was diluted daily to a final concentration of 5 μm. The racemic mixture (±)-Bay K 8644 (5 μm) was used because it produced very similar agonistic effects to the (-)-enantiomer: (1) large current amplitudes and slow tails comparable to those induced by the pure (-)-enantiomer, and (2) long-lasting openings of 8-15 ms mean open times at +10 mV, as we could verify by testing the compound on the cardiac α1C subunits expressed in heterologous systems (HEK 293 cells). Direct comparison of the effects of (±)-Bay K 8644 and the corresponding (-)-enantiomer (Sigma) on single L-channel recordings showed that the latter increased the mean open time only slightly (from ≈3.5 to ≈4.1 ms at +10 mV) (data not shown).
Current recordings and data analysis
Single channel activity was recorded with either an EPC-7 or an EPC-9 amplifier (Heka). Patch electrodes were fabricated from thick borosilicate glass (Hilgenberg, Mansfield, Germany) as previously described (Carabelli et al. 1996). The pipette resistance ranged between 4 and 8 MΩ. With the solutions used, the junction potential was between 3 and 4 mV and was not corrected since the ionic content of the pipette and bath solutions remained unchanged in most experiments. All the experiments were performed at room temperature (22-24 °C). Data are given as means ±s.e.m. for n number of patches. Statistical significance was calculated using Student's paired t test and P values smaller than 0.05 were considered significant.
Current traces were acquired at 5-10 kHz and filtered at 1 kHz with an 8-pole low-pass Bessel filter. Membrane stimulation and data acquisition were performed by either using pCLAMP (Axon Instrument Inc.) or PULSE programs (Heka). Data analysis was performed using TAC and TACFIT software (version 3.04; Bruxton Corporation, Seattle, WA, USA).
Fast capacitative transients were minimised on line by the patch-clamp analog compensation. Residual capacitative and leak currents were removed by subtracting from each active sweep an averaged current obtained from silent traces (nulls) or a polynomial function approximating the baseline current with no channel activity. Event detection was performed with the 50 % threshold detection method and limited to those patches containing only one channel. Such patches were identified for the presence of unitary openings at +30 mV at which the probability of channel opening is nearly maximal and multiple events could be clearly resolved. Patches containing more than one channel were either discarded or employed for testing the voltage dependence of autocrine inhibition, in which the comparison of averaged currents before and after the pre-pulse is not biased by the number of active channels.
Open probability (Po) was evaluated excluding the first and last closure of the channel and null sweeps. This method of calculating Po ignores the first latency and the rate of channel inactivation and is preferred for looking at changes in Po when the channel is actively gating. It furnishes also an estimate of Po independent of the length of the pulse. Notice that in our case, the agonists, antagonists and PTX do not significantly affect the first latency and mean inactivation (Fig. 3). Thus, this method is expected to give substantially similar results to the alternative one in which Po is calculated by dividing the sum of the open times by the entire step duration. The mean Po (±s.e.m.) at each potential was calculated by averaging the Po measured from each single patch over a variable number of sweeps. To construct Po(V) curves, mean Po values were plotted versus potential and fitted to a Boltzmann equation.
Figure 3. Single L-channel parameters in control, with the agonists and antagonists and in PTX-treated cells at +10 mV.
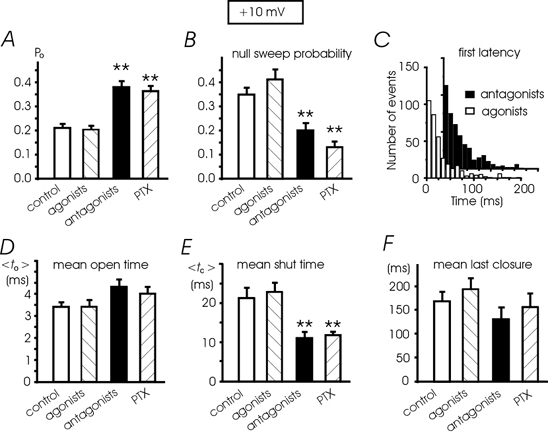
A, Po values calculated from n = 29 patches (control), n = 27 (agonists), n = 28 (antagonists) and n = 25 (PTX). The increment of mean Po with the antagonists (or PTX) with respect to control cells (or agonists) was statistically significant (**P < 0.01). B, mean percentage of nulls derived from 25-29 patches (see above). The values with PTX and antagonists were significantly different from control patches (**P < 0.01). C, the distributions of first latencies (+10 mV) were not significantly different in the presence of receptor agonists and antagonists; mean latency was 31.9 ± 2.0 ms (27 patches) and 33.8 ± 1.6 ms (28 patches), respectively (P < 0.5). D and E, mean to and tc (< to > and < tc >) ±s.e.m.) calculated as described in Fig. 1E. The mean tc with antagonists and PTX is significantly different from control and agonists (**P < 0.01). F, antagonists and PTX do not statistically change the mean inactivation (calculated as the mean duration of the last closure).
To estimate the mean open and mean closed times, the single channel events were log binned into open and closed time histograms, excluding the first and the last closures and limiting the analysis to events longer than twice the dead time (360 μs) (Colquhoun & Hawkes, 1995). Openings were long enough and well resolved (mean open time ≈4 ms). Missed openings were below 2 % of total events and data were not corrected for them. This implies that mean close times (11-21 ms) were only slightly overestimated (within the time resolution of our apparatus, see pp. 455-457 in Colquhoun & Hawkes, 1995), while missing gaps could have induced an approximately 12 % overestimate of the observed mean open time. Thus uncorrected missed events introduced minor errors which were not relevant to the present analysis.
We followed two ways to determine mean open and mean closed times, obtaining very similar results. In one case, the two parameters were estimated by averaging the arithmetic mean of the open (to) and closed times (tc) of each patch. This gave an estimate of the two parameters independently of the fitting procedure and number of exponentials used for the fit and allowed the derivation of values at various potentials when, for practical reasons, a limited number of sweeps at each potential could be collected (Fig. 1E, 3D and E, and 5C and D). In the second case, all single channel events at +10 mV were pooled together and plotted on square root-log coordinates to construct a single open and closed time distribution which was best fitted with either two or three exponentials using the maximum likelihood method (Sigworth & Sine, 1987). In this case the distributions were constructed from a large number of traces either coming from many patches with a small number of traces per patch (≈12) or from fewer patches with many more traces per patch (≈51). The second set of recordings was undertaken to ensure the detection of single channel activity and was performed using shorter pulses (200 versus 320 ms) to allow the collection of a large number of traces. The mean to and mean tc obtained from the total fit had a nice correspondence with those obtained with the first method at the same potential (+10 mV). The open times were well fitted with two exponentials while the fit of the closed times would have required a fourth component with very short tc (< 0.5 ms, Fig. 4). This fast component, however, contributed little to mean tc. The unfitted area limited to the first three bins contributed to about 10 % of the total and was visibly unaltered by the presence of agonists, antagonists or PTX treatment (Fig. 4). On the other hand, the goodness of the fit did not improve when using four rather than three exponentials. Thus, the value of the fast tc component of our fit (1.3-1.5 ms) contributing up to 66 % of the distribution must be considered only partially biased by the missed fourth component.
Figure 1. Elementary properties of single Bay K-modified L-channels.
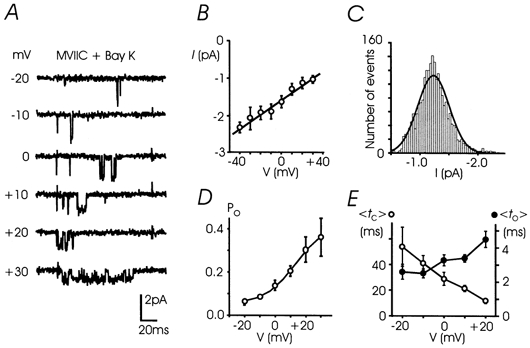
A, unitary L-currents were recorded from -20 to +30 mV with 5 μm Bay K 8644 and 10 μmω-CTX-MVIIC in the pipette control solution; holding potential (Vh) was -40 mV. B, mean unitary current amplitudes plotted versus potential. The linear regression through data points has a mean slope conductance of 20.0 ± 0.6 pS (n = 5-29 patches). C, distribution of single L-current amplitudes at +10 mV best fitted by a Gaussian function with mean -1.24 ± 0.14 pA. D, mean open probability plotted versus potential obtained from 5 to 29 patches. Data points were fitted to a Boltzmann function Po= Pmax/(1 + exp (V1/2 - V)/k), where Pmax= 0.43, V1/2=+13.7 mV and k = 10.1 mV. E, mean to (•, < to >, right scale) and mean tc (○, < tc >, left scale) plotted between -20 and +20 mV derived from 5-29 patches. The two parameters were estimated by averaging the arithmetic mean of the open and closed times of each patch.
Figure 5. Voltage dependence of single L-channels in different conditions: control, agonists, antagonists and PTX treatment.
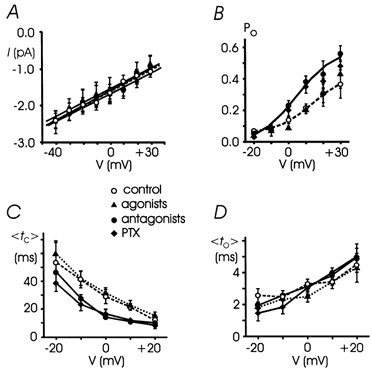
A, mean unitary current versus potential compared with control values taken from Fig. 1 (open symbols and dashed lines). Receptor agonists, antagonists and PTX treatment did not affect the single channel conductance. Linear regression on experimental data points gave 20.9 ± 0.1 pS (agonists, 4-27 patches), 20.1 ± 0.1 pS (antagonists, 4-28 patches) and 19.9 ± 0.7 pS (PTX, 4-25 patches). B, mean Poversus potential was fitted to a Boltzmann equation: V1/2=+4.7 mV; k = 11.0 mV; Pmax= 0.59 (antagonists, continuous line). Notice the enhanced Po in the presence of the antagonists or PTX. C and D, mean to and tc (< to > and < tc >) versus potential obtained from 4-28 patches. The receptor antagonists (•) and PTX (♦) decreased the mean tc between -20 and +20 mV, without significantly affecting mean to.
Figure 4. Open and closed time distributions at +10 mV.
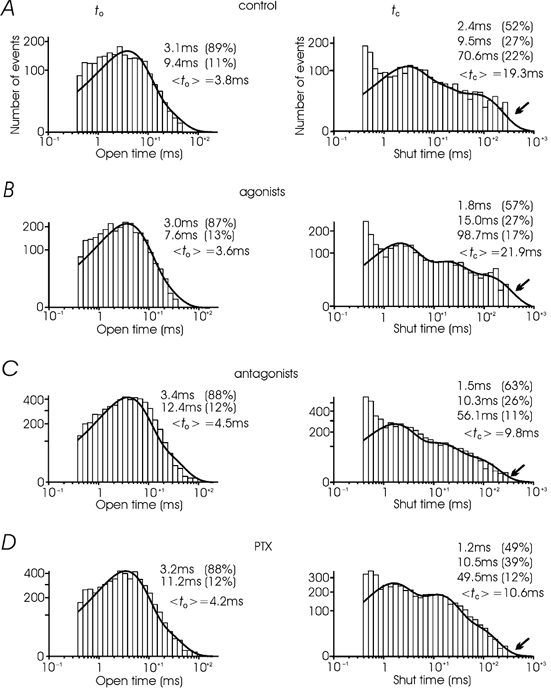
The two distributions were obtained in control from 634 sweeps and 29 patches (A), 728 sweeps and 27 patches with the agonists (B), 580 sweeps and 28 patches with the antagonists (C) and 576 sweeps and 25 patches in PTX-treated cells (D). Open and closed distributions were fitted by two and three exponential functions, respectively (continuous curves, see Methods). To the right of each distribution are indicated the value and the percentage of each component of the fit and the average to (left) and tc (right). The arrows indicate the tail of the closed time distribution, which appears fully resolved in the case of the antagonists and PTX but is incomplete in control and with the agonists (see text). Notice also the minor changes of mean to in the four cases while, compared with control and the agonists, mean tc nearly halves with the antagonists and PTX.
RESULTS
L-channel activity in cell-attached patches
L-channels in bovine chromaffin cells contribute only 10-20 % of the total Ca2+ currents and, thus, the recording of their activity is expected to be affected by the presence of N- and P/Q-channels, which carry the remaining fraction of the current (Albillos et al. 1996a). To avoid the contribution of non-L-types (N, P/Q and R) and to increase the activity of single L-channels, all the experiments were carried out in cell-attached patches with the recording pipette containing 100 mm Ba2+, 5-10 μmω-CTX-MVIIC and 5 μm (±)-Bay K 8644 and keeping the holding potential to -40 mV. L-channel activity could be well resolved only in the presence of the DHP agonist and was evident in most of the patches (76 %). The channel conductance (γ) was 20.0 ± 0.6 pS (Fig. 1A and B) and the amplitudes of unitary events were distributed around a single Gaussian function with mean amplitude of -1.24 pA at +10 mV (Fig. 1C), in good agreement with single L-channel recordings in the same preparation (Bossu et al. 1991). Despite the presence of Bay K 8644, openings were usually shorter than 20 ms and followed by long closures. Longer openings were also observed but occurred less frequently and were accompanied by brief closures.
Probability of openings (Po) was strictly voltage dependent with half-maximum at +13.7 mV and a maximal value of 0.43 above +40 mV (Fig. 1D). The mean open times (to) and mean closed times (tc) at various voltages obtained from the arithmetic means of open and closed times of each patch were also voltage dependent (see Methods and Fig. 1E). Mean to increased weakly with voltage (an e-fold change for 63 mV) while mean tc decreased more sharply with increasing voltage (an e-fold change for 27 mV). At +10 mV, mean to was 3.4 ms, which is a factor of ≈1.5 smaller than that of cardiac L-channels and 3-6 times larger than that of N-, P/Q- and R-channels (Carabelli et al. 1996; Tottene et al. 1996; Lee & Elmslie, 1999). This suggests little or no contamination of non-L-channels in our recordings despite the fact that the blocking potency of ω-CTX-MVIIC is severely decreased in high Ba2+ solutions (McDonough et al. 1996). It is most likely that part of the reduced activity of N-, P/Q- and R-channels is also due to the low value of Vh (-40 mV), which inactivates steadily the non-L channels.
In control conditions the L-channel displayed a variable degree of inactivation above 0 mV and relatively fast deactivation on return to -40 mV, which was complete in 20-30 ms (Fig. 2A). Channel openings were most evident soon after the onset rather than at the end of the pulse and were absent during repolarization. In some cases, brief openings appeared immediately on repolarization to -40 mV while prolonged channel re-openings (> 100 ms), as reported in central neurons, were absent. This excluded the presence of ‘cardiac-like’ (Hess et al. 1984) and ‘re-opening’ L-channels (Kavalali et al. 1997) in our records. The former usually displays long-lasting openings in the presence of Bay K 8644 while the latter gives rise to bursts of openings on repolarisation. In terms of single channel conductance, mean to, Po and inactivation time course, the L-channel of bovine chromaffin cells appeared as a homogeneous population with some similarity to the inactivating L-channel with low-Po of GH3 cells (Mantegazza et al. 1995).
Figure 2. L-channel autocrine inhibition is mimicked by ATP and opioids and is removed by receptor antagonists or PTX treatment.
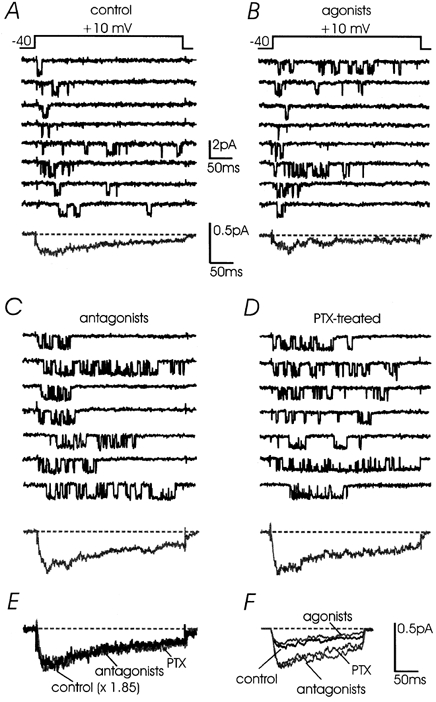
Representative traces of Bay K-modified L-channel activity recorded at +10 mV in different experimental conditions; Vh -40 mV. A, low-Po channel activity recorded in control conditions with pulses of 320 ms. The pipette solution was the same as in Fig. 1. The averaged current at the bottom was calculated over 150 sweeps of 320 ms including null sweeps pooled from 16 patches. B, ATP (100 μm), DAMGO (10 μm) and DPDPE (1 μm) added to the control solution mimicked the low-Po activity of control patches. The averaged current had slightly lower amplitude but similar kinetics to the control (n = 188 sweeps, 12 patches). C and D, sustained L-channel activity recorded at +10 mV with the antagonists in the pipette (10 μm naloxone and 100 μm suramin) (n = 166 sweeps, 12 patches) or after incubation with 100 ng ml−1 PTX for 12 h (n = 125 sweeps, 13 patches). Notice the increased amplitude of averaged currents but similar activation/inactivation kinetics. E, overlapped averaged currents taken from above. The control current was scaled by a factor of 1.85 and compared with that with the antagonists and PTX. F, overlapped averaged currents obtained from a second series of experiments using pulses of 200 ms to +10 mV; Vh -40 mV. The currents were derived from a large number of sweeps per patch: control, 415 sweeps, 8 patches; agonists, 495 sweeps, 10 patches; antagonists, 400 sweeps, 8 patches; and PTX, 415 sweeps, 8 patches.
Po increases with purinergic and opioidergic antagonists or cell treatment with PTX
In chromaffin cells, L-channels are endogenously inhibited by ATP and opioids released during cell activity (Hernández-Guijo et al. 1999). The same is expected to occur at the single channel level if enough material is released inside a patch pipette during local stimulation (Carabelli et al. 1998). Under these conditions, the activity of L-channels in control patches is expected to be little affected by the addition of exogenous ATP and opioids and to increase significantly in the presence of purinergic and opioidergic antagonists. This is what we observed in a double series of experiments. In one case we used recordings of 320 ms at +10 mV from 53 patches with a low number of traces per patch (≈12, Fig. 2A-D). In the second case we used recordings of 200 ms from 34 patches with many more traces per patch (≈51, Fig. 2F). In both cases, control patches had single channel activity comparable to that recorded in the presence of ATP and the μ/δ opioid agonists DAMGO and DPDPE (Fig. 2). Single channel conductance, Po, mean to and mean tc were not significantly affected by the agonists (Fig. 3 and 5A). Ensemble currents had the same activation-inactivation time course but 22 % smaller amplitude, very probably due to a proportional increase of nulls (from 34 to 41 %, Fig. 3B). Time to peak was between 20 and 25 ms in control and with agonists and the percentage of inactivation at the end of the pulse varied little (from 60 to 75 %). The inactivation time course was better estimated from traces of 320 ms by averaging the mean duration of the last channel closure, which changed from 169 to 194 ms, with no significant difference (Fig. 3F).
Channel activity, but not the amplitude of elementary events, increased significantly if the patch pipette contained mixtures of suramin and naloxone, which prevented the activation of P2xy and μ/δ opioid autoreceptors (Fig. 2C). The same occurred if the cells were pre-treated with PTX (Fig. 2D). In both cases, the mean percentage of nulls at +10 mV decreased from 41 % with the agonists to 20 and 13 % with the antagonists and PTX, respectively (Fig. 3B). Mean Po increased by nearly a factor 2 (from 0.2 to 0.38, Fig. 3A) while mean to showed no significant changes (from 3.4 to 4.3 ms, Fig. 3D). The same was true for the mean to derived from the fit of all the pooled data (Fig. 4). On the contrary, the mean tc at +10 mV decreased significantly from 22.8 ms (agonists) to 11.0 ms (antagonists) (Fig. 3E), which is comparable to the mean tc derived from the fit of the pooled data (Fig. 4B and C). Thus, the increment of Po in the presence of the antagonists and PTX was mainly due to a decrease of mean tc rather than an increase of mean to. Looking more closely at the distributions of open and shut times it is evident that the most substantial changes of mean tc are due to the slower tc component which decreases from 70.6 ms (22 %) to 56.1 ms (11 %). Notice also that the tc distribution with the antagonists and PTX is fully resolved with pulses of 320 ms (arrows in Fig. 4C and D), while in control and with the agonists the distribution is clearly truncated (arrows in Fig. 4A and B). This indicates that with the agonists tc might attain longer values than 320 ms and that its observed mean value is most probably underestimated.
The effects of antagonists and PTX on γ, Po, to and tc observed at +10 mV were also evident at different potentials (Fig. 5). Between -10 and +20 mV, Po(V) showed a proportional increase while tc(V) showed a significant decrease between -10 and +10 mV. to(V) and γ remained unaltered.
L-channel inhibition is not reverted by pre-depolarizations
As shown in Fig. 2, the endogenous modulation of L-channels mediated by purinergic and opioidergic autoreceptors and PTX-sensitive G-proteins (Go/Gi) appears different from the autocrine modulation reported for single non-L-channels (Carabelli et al. 1998). N- and P/Q-channel modulation by neurotransmitters or secreted material is characterised by slowly activating averaged currents of low amplitude which originate from drastically delayed single channel openings (Carabelli et al. 1996). In the case of L-channels the endogenous inhibition did not produce significant delays to the first latency of channel openings. First latency distributions were indistinguishable in the presence of agonists and antagonists (Fig. 3C) and averaged control currents could overlap those recorded with the antagonists (or PTX) by a simple scaling up (× 1.85, Fig. 2E). This was true also at different voltages (-10 and 0 mV), suggesting that the endogenous inhibition was largely voltage independent (Hernández-Guijo et al. 1999).
To strengthen this issue, we checked whether strong pre-pulses of different amplitude and duration could alter the activation kinetics and the time course of averaged currents. Figure 6 shows examples of traces recorded at +10 mV before and after pre-pulses of 280 ms to +150 mV in the presence of agonists. There was no facilitation (or removal of inhibition) either with the agonists (top traces) or with the antagonists in the patch (bottom traces). In both cases, the averaged currents had nearly identical peak values before and after the pre-pulse: 0.39 and 0.35 pA with the agonists and 0.64 and 0.60 pA with the antagonists. There were no shortenings of the first latency with the pre-pulse and ensemble currents had identical activation kinetics with and without pre-pulse. We tested various conditions of pre-pulses followed by brief repolarisation to the holding potential (-40 mV) or to more negative voltages. Pre-pulses of 50-300 ms with amplitudes of +100 to +150 mV were unable to recover the inhibition and to accelerate the kinetics of channel openings, providing that a 20 ms pulse to -60 or -80 mV was interposed between test and pre-pulse. This allowed the deactivation of Bay K-modified channels which were maximally open at very positive potentials and produced tail currents on return to Vh, which were complete within 20-30 ms.
Figure 6. L-type channel inhibition is insensitive to conditioning pre-pulses.
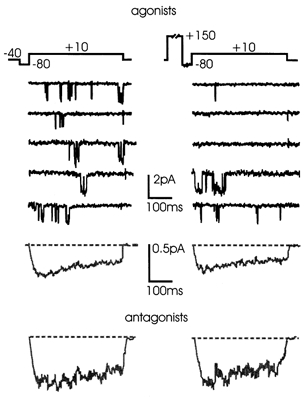
Low-Po channel activity recorded during pairs of test depolarizations at +10 mV before and after a sequentially applied pre-pulse (+150 mV, 280 ms) in the presence of opioidergic and purinergic agonists in the pipette solution (Vh -40 mV). The corresponding mean currents calculated from 249 sweeps pooled from 14 patches are shown below. The bottom traces are averaged currents at +10 mV calculated from 169 sweeps pooled from 10 patches in the presence of naloxone and suramin. Notice the larger amplitude of the currents under these conditions and the absence of any pre-pulse-induced facilitation.
Some degree of facilitation was observed only with interposed hyperpolarisations shorter than 10 ms and at test potentials below 0 mV (not shown), which correspond to test potentials more negative than -40 mV in 2 mm Ba2+ solutions. This occurred only in the presence of Bay K 8644 and we attributed part of this facilitation to a direct action of the DHP on Ca2+ channel gating for two reasons. First, there was no sign of facilitation in the presence or absence of Ca2+ channel agonists even using strong and long pre-pulses (Carabelli et al. 1998). Second, Bay K 8644 and other DHP agonists are able to induce on their own a variable delay of L-channel activation and exhibit significant voltage-dependent facilitation with double-pulse stimulation (Visentin et al. 1999). A voltage-dependent facilitation with inter-pulses shorter than 10 ms is in agreement with the voltage-dependent facilitation of cardiac L-channels (Pietrobon & Hess, 1990) which is shown to decay quickly during repolarisations to the holding potential (Kavalali & Plummer, 1996). In our case, however, the facilitation was observed only in the presence of Bay K 8644.
L-channel inhibition by ATP and opioids is direct and does not require diffusible second messengers
A significant body of evidence supports the idea that the endogenous inhibition of neuroendocrine L-channels is direct, probably due to the close proximity of L-channels to autoreceptors and G-proteins. L-channel inhibition by opioids and ATP in chromaffin cells is fast and fully reversible. The onset of the inhibition develops with a time constant of 0.9 s and recovers fully within 10 s, suggesting close coupling between autoreceptors, G-proteins and Ca2+ channels (Hernández-Guijo et al. 1999). On the other hand, since neuroendocrine L-channels are effectively modulated by changes in cAMP levels and PKA activity (Artalejo et al. 1991), the inhibition may alternatively develop through a down-regulation of cAMP levels mediated by PTX-sensitive G-proteins negatively coupled to adenylate cyclase. We therefore tested whether ATP and opioids could inhibit the activity of L-channels through diffusible second messengers (cAMP), by applying mixtures of the two agonists outside the patch while keeping the Po high with purinergic and opioidergic antagonists inside the pipette. Figure 7 illustrates an example, which is representative of another six patches, where L-channel activity remained high (mean Po 0.39) for more than 8 min during continuous superfusion of ATP and opioids outside the patch. This observation, and the fact that local inhibition of L-channels was effective in 89 % of patches with the agonists inside the pipette, can be taken as evidence that the down-modulation of L-channels by released neurotransmitters is mainly direct and does not require diffusible second messengers.
Figure 7. Opioid and purinergic agonists inhibit single L-channel activity through a membrane-delimited pathway.
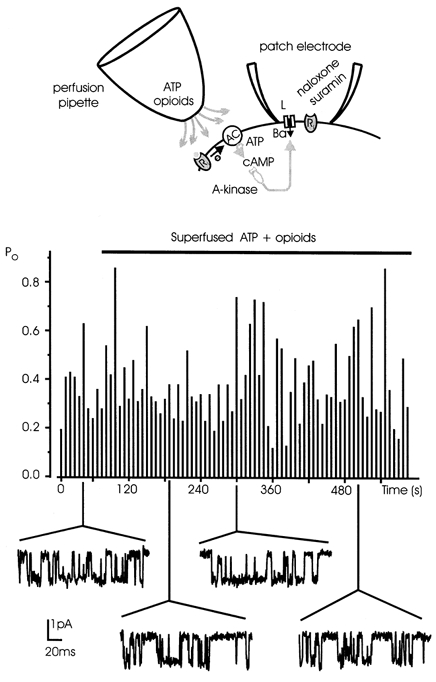
To assess whether the L-channel inhibition requires any diffusible second messenger, ATP and opioid agonists were superfused outside the patch, while the naloxone and suramin were added inside the pipette solution to avoid the direct inhibition by G-proteins (top panel). Po at +10 mV was monitored sweep by sweep and plotted versus time (central panel). The cell was initially perfused with a standard external solution (mean Po 0.36) and then switched to a solution containing ATP and opioid agonists for the time indicated by the horizontal bar. Even in these conditions Po remained high, varying between 0.13 and 0.82 (mean Po 0.39). Some representative traces are shown corresponding with their Po value. The inability of externally applied agonists to lower the Po excludes the involvement of a second messenger pathway in the L-channel inhibition.
cAMP potentiates L-channel activity independently of the endogenous inhibition
cAMP increases L-channel activity in cardiac and some neuronal tissue by mainly increasing the Po with no effects on single channel conductance (Reuter, 1983). We therefore tested whether the Bay K-modified L-channels of adult bovine chromaffin cells also were sensitive to elevations of cAMP levels and whether a cAMP-mediated phosphorylation of the channel could interfere with the endogenous inhibition by ATP and opioids. We found that in 12 out of 16 patches pre-incubated for 30 min with 1 mm 8-CPT-cAMP and bathed in a solution containing the cAMP analogue, the activity of the channel at +10 mV was drastically enhanced (Fig. 8A). 8-CPT-cAMP increased the mean Po from 0.2 to 0.42 (Fig. 8D) by increasing both the number of sweeps with high Po (Fig. 8B) and mean to (from 3.4 to 5.4 ms, Fig. 8F). 8-CPT-cAMP reduced also the number of nulls from 41 to 13 % (Fig. 8E) and mean tc from 22.8 to 10.1 ms (Fig. 8G). At +10 mV, the distribution of open and shut times with cAMP was well fitted by multi-exponential functions (Fig. 8C) and the average values from the fit compared well with the mean to and mean tc given in panels F and G. Mean currents pooled from 16 patches were larger than those at control without cAMP. They displayed less complete inactivation with respect to control (Fig. 8A). Deactivation on return to -40 mV was fast and complete within 20-30 ms. There was little sign of bursting activity on return to -40 mV following mild depolarizations (+10, +20 mV). This suggested that also in the presence of cAMP long lasting re-openings on return to Vh were absent (Kavalali et al. 1997).
Figure 8. cAMP removes the agonist-induced inhibition and potentiates the Bay K-modified L-channels.
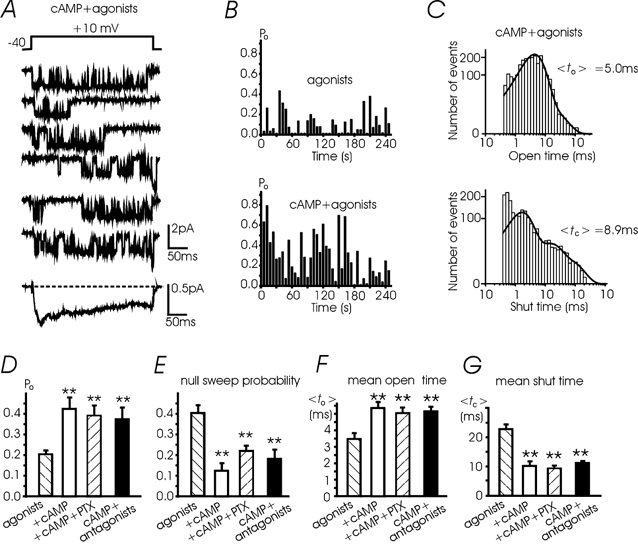
A, channel activity with high Po during consecutive depolarizations to +10 mV after incubation with 1 mm 8-CPT-cAMP (30 min) and in the presence of ATP and opioid agonists in the pipette; Vh -40 mV. The mean current to the bottom was calculated from 164 sweeps pooled from 16 patches. B, with ATP and opioids in the pipette the L-channel activity was low (top): mean Po was 0.13 and the nulls (indicated by spaces) were quite frequent (26 %). Following 8-CPT-cAMP (50 μm) incubation (with agonists in the pipette) Po increased to 0.30 and the percentage of nulls was reduced to 9 %. C, open and close time distributions at +10 mV derived from n = 164 sweeps pooled from 16 patches incubated with 1 mm 8-CPT-cAMP. Data were plotted on a square root-log binned histogram and fitted with two and three exponentials with the following values: 3.9 ms (86 %) and 11.7 ms (14 %) for to and 1.64 ms (71 %), 13.6 ms (21 %) and 61 ms (8 %) for tc. D-G, mean Po, null sweeps, mean to and tc at +10 mV in the presence of the agonists (27 patches), agonists + 1 mm cAMP (16 patches), agonists + cAMP + PTX (7 patches) and cAMP + antagonists (n = 10 patches) (**P < 0.01).
Single L-channel conductance remained unchanged with cAMP (Fig. 9A and B) while mean Po increased at all the potentials tested (Fig. 9C). In parallel there was a sharp increase of mean to, with an e-fold change for 25 mV from -10 to +20 mV (Fig. 9D), and a decrease of mean tc (Fig. 9E). Po reached maximal values of 0.73 at +30 mV, which were larger than the maximal Po attained after removal of the endogenous inhibition with the antagonists (0.56, Fig. 5B).
Figure 9. The voltage dependence of cAMP-potentiated L-channels.
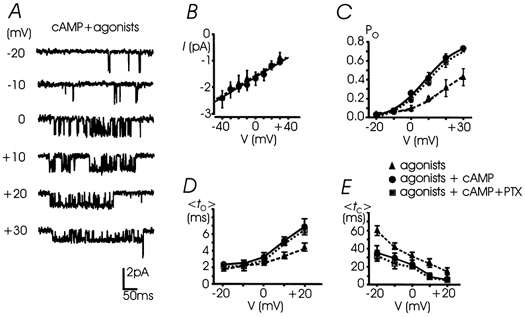
A, unitary L-currents recorded from -20 to +30 mV after incubation with 1 mm 8-CPT-cAMP (30 min) and in the presence of ATP and opioid agonists in the pipette; Vh -40 mV. B, cAMP incubation did not significantly modify the unitary L-channel conductance (20.1 ± 0.6 pS) (•, continuous line). Data with the agonists (▴, dashed line) were taken from Fig. 5. C, cAMP induced a remarkable Po increase between 0 and +30 mV (•). The Boltzmann function fitted through data points gave V1/2=+8.0 mV, k = 9.8 mV, Pmax= 0.82 (continous curve). Cell pre-treatment with PTX caused no significant change to Po(V), which appears only slightly scaled down (▪, dotted curve) with respect to that with cAMP alone (n = 4-7 patches). D and E, mean to and mean tc plotted versus potential. cAMP incubation (•, continuous line) produced a drastic increase of mean to(V) above 0 mV and a minor but significant reduction of mean tc between -20 and +20 mV.
The cAMP-induced potentiation of L-channels occurred in the presence of agonists in the pipette and persisted in patches either containing the antagonists or pre-treated with PTX (Po 0.38 and 0.39, respectively; Fig. 8D). In both cases, the percentage of null sweeps and the values of mean to and mean tc at +10 mV were not statistically different from those derived in the presence of cAMP alone (Fig. 8E-G). The same was true for the Po(V), mean to(V) and mean tc(V) if comparing the data with cAMP + PTX and cAMP alone, which were statistically indistinguishable (dotted vs. continuous lines, Fig. 9C-E). This suggests that cAMP potentiates L-channel gating regardless of the inhibitory action of G-proteins and that the two modulations (inhibition and potentiation) may converge on a unique channel.
cAMP action is mediated by protein kinase A
As for other L-channels, the potentiating effects of 50 μm 8-CPT-cAMP were prevented by cell pre-treatment with the unspecific protein kinase inhibitor H7 (Fig. 10A) or the more specific protein kinase A inhibitor H89. Sequential pre-incubations of either one of the two inhibitors and 8-CPT-cAMP reduced the probability of L-channel openings and increased the number of nulls. In the case of H89 the effects were more evident. Mean Po decreased from 0.33 to 0.18 (Fig. 10A, right) while the number of nulls increased from 13 ± 4 to 35 ± 5 % (P < 0.01; not shown). Averaged currents pooled from many patches were smaller but had comparable activation-inactivation kinetics. A similar L-channel potentiation was observed by applying forskolin and IBMX at concentrations capable of elevating the levels of intracellular cAMP, by activating adenylate cyclase and inhibiting the phosphodiesterase (Fig. 10B). Despite the presence of the agonists in the pipette, there was a significant increase of L-channel activity per sweep (mean Po 0.36 at +10 mV) and a drastic reduction in the number of null sweeps (13 ± 7 %). Ensemble currents were comparable to those with 8-CPT-cAMP (bottom trace in Fig. 10B, left). The combined action of forskolin and IBMX was prevented by pre-incubation with either H89 (Fig. 10B, middle) or H7, while the two inhibitors alone did not alter the activity of L-channels (Fig. 10B, right). Mean Po at +10 mV decreased from 0.36 with forskolin + IBMX to 0.20 and 0.23 after adding H7 and H89, respectively. Notice that mean Po with H7 and H89 alone was low in both cases (0.19, Fig. 10B, right) and comparable to the Po with the agonists in the absence of cAMP (0.2, Fig. 3A). This indicates that the basal activity of L-channels was not affected by PKA, most probably because of the low basal level of cAMP in control conditions.
Figure 10. cAMP-induced potentiation of L-channels is mediated by PKA: coexistence with the G-protein-dependent inhibition.
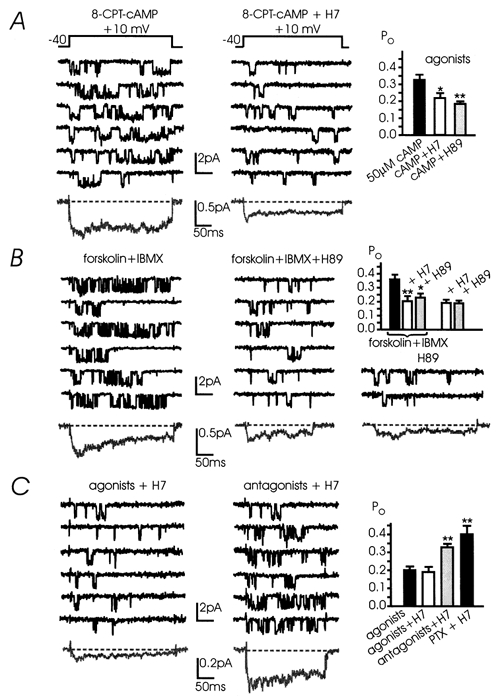
A, the protein kinase inhibitors H7 and H89 antagonise the cAMP-induced potentiation. The figure shows current traces recorded at +10 mV in two patches with ATP and opioid agonists in the pipette following incubation with 50 μm 8-CPT-cAMP or 8-CPT-cAMP + 200 μm H7; Vh -40 mV. In the left panel the cAMP-induced potentiation was evident at the low 8-CPT-cAMP concentration (50 μm; Po 0.33 ± 0.03; n = 11 patches). In another 9 cells incubated with 50 μm 8-CPT-cAMP + 200 μm H7 (centre panel) and 6 cells with 1 μm H89 (right panel) the channel activity was depressed and Po was reduced to 0.22 (*P < 0.05) and 0.18 (**P < 0.01), respectively. The two averaged currents to the bottom were calculated over 48 sweeps (cAMP, 4 patches) and 195 sweeps (cAMP + H7, 8 patches). B, H7 and H89 prevent L-channel potentiation induced by forskolin and IBMX. The simultaneous external application of forskolin (10 μm) and IBMX (20 μm) was sufficient to induce L-channel potentiation (left panel). Channel activity at +10 mV was sustained and mean Po increased to 0.36 (11 patches; top right panel). The potentiating effect of forskolin + IBMX was reversed and the agonist-induced inhibition restored in cells incubated with H7 (n = 9) or H89 (n = 6): mean Po was 0.20 (**P < 0.01) and 0.23 (*P < 0.05), respectively (centre panel). H89 and H7 had no effect on basal channel activity: mean Po was 0.19 (n = 8), in both cases. Mean currents were calculated over 67 (left), 69 (centre) and 167 sweeps (right). The top right panel summarises the mean Po obtained with forskolin + IBMX or with H7 and H89 alone. C, the agonist-induced L-channel inhibition persists independently of the cAMP pathway activation. Single L-channel activities at +10 mV (Vh -40 mV) were recorded from patches pre-treated with 200 μm H7 with either the agonists (ATP + opioids) (left) or the antagonists (suramin + naloxone) in the pipette (centre). The mean currents to the bottom were calculated over 180 and 153 sweeps pooled from 7 and 4 patches, respectively. Right, mean Po at +10 mV calculated from n = 27 patches (agonists), n = 11 (agonists + H7), n = 4 (antagonists + H7) (**P < 0.01) and n = 5 (PTX + H7) (**P < 0.01).
Potentiation by cAMP and inhibition by released neurotransmitters proceed through distinct but convergent pathways
Given the presence of two contrasting modulatory mechanisms, one potentiating (cAMP dependent) and the other inhibiting channel activity (G-protein dependent), we next assayed the degree of coupling between the two systems. Since activation of the cAMP pathway by elevation of intracellular cAMP increases the Po regardless of whether the inhibitory pathway was activated by ATP and opioids or prevented by the antagonists or PTX (Fig. 8D), this suggests that phosphorylation of L-channels is sufficient to prevent the endogenous inhibition. Alternatively, L-channel inhibition may proceed through the down-modulation of adenylate cyclase activity, challenging the hypothesis of a direct effect on the channels. We therefore checked whether the autocrine inhibition was still effective after the cAMP pathway was blocked by exposures to H7. Figure 10C (left) shows that, in cells treated with H7, exogenously applied agonists were still effective and produced a marked L-channel inhibition compared with patches containing the antagonists (Fig. 10C, middle). Mean Po with the antagonists + H7 and PTX + H7 was 0.33 and 0.4, respectively, and decreased to 0.19 with the agonists + H7 (Fig. 10C, right). Correspondingly, averaged currents pooled from many patches were larger with the antagonists and PTX than with the agonists. All this indicates that the endogenous L-channel inhibition is not mediated by cAMP pathways and that the two modulations (inhibition and potentiation) converge on the same target. cAMP elevation makes the agonist-induced inhibition of L-channels ineffective while the inhibition persists when the cAMP action is depressed by H7 or H89. Figure 10C shows also that H7 alone did not produce any significant change to channel activity if compared with H7-untreated patches (open bar, right). Thus, basal levels of cAMP are most likely low in control cells and inhibition of L-channels is mostly due to endogenously released neurotransmitters.
cAMP elevation does not produce voltage-dependent L-channel facilitation
cAMP and PKA play a critical role in the modulation of L-channels in young bovine chromaffin cells and in the maintenance of cardiac and neuronal L-channel facilitation (Sculptoreanu et al. 1993; Bourinet et al. 1994). Thus, we next tested whether cAMP elevations could introduce any voltage-dependent facilitation in our conditions. In control cells exposed to 8-CPT-cAMP we found that Po was not significantly altered by application of strong positive pre-pulses of various duration. Changing the amplitude of the pre-pulse from +100 to +150 mV and the duration from 100 to 300 ms we found systematically no sign of voltage-dependent facilitation. Channels opened quickly soon after the onset of the pulse and maintained their activity either for a brief period and then inactivated or remained active throughout the entire pulse. This also occurred after strong and sustained pre-pulses to +150 mV. Averaged currents at +10 mV before and after pre-pulses had a comparable amplitude and activation time course (Fig. 11, top traces). Similar findings were obtained in another set of 16 patches where ATP and opioids were added to the pipette solution (Fig. 11, bottom traces) and in 10-15 patches at 0 and +20 mV (not shown), suggesting complete absence of voltage-dependent facilitation in the presence of cAMP. Cells pre-treated with PTX and exposed to 8-CPT-cAMP had similar behaviour (not shown).
Figure 11. Conditioning pre-pulses do not facilitate cAMP-potentiated L-channels.
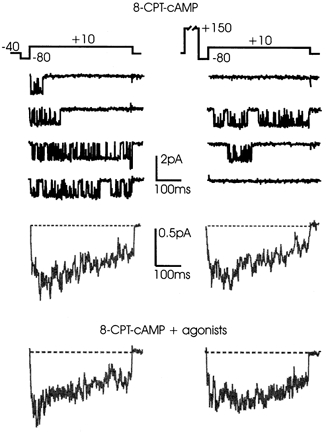
L-channel activity recorded before and after a conditioning pre-pulse (+150 mV, 280 ms) in control patches pre-incubated with 1 mm 8-CPT-cAMP for 30 min. Repolarisations to -80 mV lasted 40 ms. Most traces displayed high-Po activity before the conditioning pulse. Pre-pulses were ineffective in either accelerating the activation kinetics or increasing the Po of L-channels. The mean currents below were calculated over 55 sweeps pooled from 10 patches. The bottom traces were obtained from 37 sweeps pooled from 4 patches pre-incubated with 8-CPT-cAMP and with the agonists in the pipette.
We concluded therefore that elevation of cAMP levels produced potentiation of the L-channel activity independently of whether the endogenous inhibition induced by released neurotransmitters was active or prevented by PTX. cAMP action, however, resulted neither in the recruitment of long-lasting ‘facilitation channels’ as in young bovine chromaffin cells (Artalejo et al. 1991) nor in the development of a fast voltage-dependent and cAMP-mediated facilitation as for neuronal and cardiac α1C channels (Sculptoreanu et al. 1993; Bourinet et al. 1994).
Changes of L-channel gating reveal locally released neurotransmitters
Changes of single N- and P/Q-channel gating induced by neurotransmitters can be used to reveal the secretory activity of chromaffin cells (Carabelli et al. 1998). The idea is based on the fact that locally released granule content (ATP and opioids) can quickly activate some PTX-sensitive G-protein and inhibit the non-L-channels in a voltage-dependent manner by delaying their openings. Something similar is likely to occur with L-channels except that the inhibition by released neurotransmitters is not accompanied by delayed activation (Fig. 2) and strong pre-pulses are unable to remove it (Fig. 6 and 11). In the absence of these effects the only way to quantify the percentage of modulated and unmodulated patches was to separate them into two populations based on their Po at +10 mV: one possibly distributed around a mean low Po (0.2) and the other around a mean high Po (0.4). However, in the four conditions studied (control, agonists, antagonists and PTX) we failed to resolve clear bimodal distributions around two well-separated Po values (not shown). Consequently, any criteria to set a limiting value for Po, above which L-channels were unmodulated and below modulated, became arbitrary. Thus, the only reasonable conclusion on this issue can be based only on the size of the averaged currents of Fig. 2, which represents a useful way to compare Po values and percentage of null sweeps simultaneously. The control current is comparable to that with agonists and nearly half of that with the antagonists and PTX, suggesting that a large fraction of control patches are endogenously inhibited by the released neurotransmitters (> 80 %). This is somehow different from the 35-40 % of locally modulated patches associated with N- and P/Q-channels (Carabelli et al. 1998) but it might just represent a preliminary estimate which should wait for more accurate measurements under various modulatory conditions.
DISCUSSION
We have provided evidence for the existence of a direct endogenous inhibition of L-channels in bovine chromaffin cells induced by ATP and opioids locally released inside the patch pipette. The inhibition is prevented by receptor antagonists, mediated by PTX-sensitive G-proteins and delimited to membrane patches of ≈1 μm2. External applications of agonists are ineffective, thus excluding the need of diffusible second messengers, which usually cause increased Po through cAMP elevations (Reuter, 1983; Bean et al. 1984).
Our data nicely fit recent observations on chromaffin cells showing that L-currents are scaled down rather than delayed either during application of external opioids (Albillos et al. 1996a) or after exposure to endogenously released neurotransmitters in stop-flow conditions (Carabelli et al. 1998). This inhibition is fast, voltage independent, and mediated by PTX-sensitive G-proteins (Hernández-Guijo et al. 1999). The same inhibition by neurotransmitters is also likely to occur in central neurons and other neuroendocrine cells in which a scaling-down of L-currents is evident (Scholz & Miller, 1991; Pollo et al. 1993; Haws et al. 1993; Chavis et al. 1994; Amico et al. 1995; Formenti et al. 1998). Thus, although not distinctive of all L-channels (see Dolphin, 1999), the voltage-independent and PTX-sensitive inhibition described here appears a recurrent mechanism of neuronal/neuroendocrine L-channel modulation. Notice that a voltage-independent muscarinic scaling down of L-currents is also evident in rat sympathetic neurons, but this modulation develops slowly and is PTX insensitive (Beech et al. 1992).
Voltage independence of direct L-channel inhibition
The feedback endogenous inhibition of L-channels analysed here has similar origins to, but molecular distinctiveness from, the voltage-dependent autocrine modulation of N- and P/Q-channels (Carabelli et al. 1998). The modulation of neuronal non-L-channels is membrane delimited (Hille, 1994) and regulated by the state-dependent interaction of Goβγ-subunits with the channel α1-subunit (Ikeda, 1996). Binding of Goβγ is favoured in the channel shut configuration. The slow unbinding of Goβγ from open channels during depolarization produces delayed channel openings (Carabelli et al. 1996) which reveal transitions from low- (reluctant) to high-Po (willing) gating modes (Elmslie et al. 1990; Boland & Bean, 1993). For the L-channel inhibition there is no delay of channel openings but rather a voltage-independent reduction of Po. A way of accounting for this is to assume that binding of the G-protein subunit is insensitive to the state of the channel. If the G-protein subunit binds to open or closed channels with the same affinity, the percentage of inhibited channels at rest will persist even during depolarization when channels are open. Consequently, there will be no delayed shifts from low- to high-Po gating modes but a simple scaling-down of mean currents.
We do not have any indication of the molecular mechanism and the G-protein subunit involved, but it is interesting to notice that the voltage-independent component of N-current inhibition in sympathetic neurons is associated with Giβγ-subunits while the voltage-dependent component is mediated by Goβγ (Delmas et al. 1999). The same could be true for the L-channel inhibition, but whether the action is mediated by Giβγ, Goβγ or even Goα and Giα remains to be proved.
cAMP-dependent potentiation and neurotransmitter-induced inhibition: two pathways converging on a common channel?
As shown in Fig. 8 and 9, cAMP does not simply remove the endogenous inhibition of L-channels but produces marked changes to the voltage dependence of mean to and elevates Po to maximal levels never reached with Bay K 8644 alone. Thus, PKA phosphorylation induces drastic changes to channel gating, which go beyond the simple removal of G-protein inhibition. A simple removal of inhibition, preventing the dissociation of Gα- and Gβγ-subunits (Imaizumi et al. 1991), would increase Po by decreasing the closed times and the number of nulls with little change to open times. Thus, the dominant action of PKA phosphorylation is most likely to be on channel subunits (α1 and β), which shift to a high-Po gating mode with reduced affinity for the inhibitory G-protein. Phosphorylation sites have been shown to exist either at the α1- and β-subunits of L-channels (Perez-Reyes et al. 1994), but PKA-mediated potentiation is already fully defined in heterologous systems expressing only α1S and α1C (Johnson et al. 1997; Costantin et al. 1999). It is thus possible that the α1-subunit is the common target for cAMP-induced potentiation and G-protein-mediated inhibition.
Our data exclude the possibility that the opposite actions of G-proteins and cAMP may occur on two different channels, one sensitive to cAMP and the other sensitive to G-proteins. If this were the case, one should have observed significant Po increases in the presence of cAMP + antagonists and cAMP + PTX with respect to patches with cAMP + agonists. On the contrary, we found comparably high Po values at +10 mV in all three cases (Fig. 8D) and remarkably similar Po(V) curves with PTX and the agonists (Fig. 9C). The present analysis based on unitary conductance, mean open time and activation- inactivation kinetics favours the idea of a rather homogeneous population of L-channels in bovine chromaffin cells. This channel class differs from the cardiac and neuronal α1C isoforms (Hess et al. 1984; Bourinet et al. 1994; Costantin et al. 1999) and from other neuronal L-channels displaying either anomalous gating (Forti & Pietrobon, 1993) or prolonged re-openings on return to the holding potential after mild depolarisations (Kavalali et al. 1997). The L-channel of bovine chromaffin cells has an unusually short mean open time with Bay K 8644 (≈3.5 ms at +10 mV) and significant inactivation (τinact≈100 ms). Long-lasting openings occurred only when Bay K 8644 and cAMP were simultaneously present and cAMP relieved part of the time-dependent inactivation. A similar channel with brief openings in the presence of DHP agonists and rapid inactivation has been reported in GH3 cells (Mantegazza et al. 1995).
Comparison with other L-channel modulations
The voltage-independent inhibition reported here is profoundly different from the voltage-dependent facilitation of cardiac, neuronal and skeletal muscle α1-subunits, which is regulated by cAMP and is insensitive to G-proteins (Sculptoreanu et al. 1993; Johnson et al. 1997). The role of cAMP in this modulation, however, is not clear. It is not always required (Pietrobon & Hess, 1990) and in neuronal α1C channels appears crucial only for potentiating channel gating rather than maintaining the voltage-dependent facilitation (Bourinet et al. 1994).
Concerning the ‘facilitation channel’ reported in chromaffin cells of young cows, we looked carefully for its presence in our preparation using the same protocols and conditions used in previous works (Artalejo et al. 1990, 1991). Detection of these channels must have been favoured in our recording conditions but we found no evidence for them, irrespective of the presence or absence of Bay K 8644 (Carabelli et al. 1998). Facilitation channels activated by pre-pulses last for hundreds of milliseconds at -40 mV and do not require Bay K 8644 and cAMP. Indeed, cAMP and Bay K 8644 potentiate these channels removing their voltage-dependent facilitation (Artalejo et al. 1990). We did not find voltage-dependent facilitation either in normal patches or in the presence of cAMP and cAMP plus Bay K 8644. Thus, a possible conclusion is that L-channels of bovine chromaffin cells of young and adult cows are functionally distinct as is their modulation. Whether this reflects intrinsically different α1-subunits or interactions with different β-subunits remains to be proved. On one point, however, there seems to be agreement, i.e. in the potentiating (not facilitating) action that cAMP exerts on L-channel gating. Future studies will indicate what kinds of receptors are more probably associated with the activation of adenylate cyclase through Gs-proteins in bovine chromaffin cells of adult animals.
Physiological relevance of the autocrine L-channel modulation
The existence of a localised autocrine inhibition of neuroendocrine L-channels by PTX-sensitive G-proteins opens new and interesting considerations about the role of these channels in the control of neurosecretion (García et al. 1984). The first consequence is that, besides being potentiated by a cAMP/PKA diffusible signalling pathway, neuroendocrine L-channels can also be down-modulated by a fast and direct G-protein pathway, which can halve the percentage of L-currents while secretion is taking place. The accumulation of secreted material is highly favoured in the packed columnar arrangement of chromaffin cells around small capillary vessels and thus the efficacy of an autocrine feedback inhibition of L-channels can be relevant to the local control of hormone release from the adrenal gland.
Given its inherent voltage independence, L-channel inhibition is likely to remain effective both at rest and during low or high frequency stimulation, possibly as a feedback protection to prevent hormonal oversecretion. How this effect may co-exist with the cAMP-mediated potentiation and the autocrine voltage-dependent facilitation of N- and P/Q-channels (Carabelli et al. 1998) is difficult to predict. A likely explanation is that while the cAMP-mediated action may originate from autocrine/paracrine signals mediated by membrane receptors coupled to adenylate cyclase, the autocrine modulation of non-L-channels may be critical during repeated cell activity. In both cases, the up-regulation of L-channels and the removal of inhibition of non-L-channels during repeated stimuli may furnish the high Ca2+ fluxes required for massive release of catecholamines under intense splanchnic stimulation. It cannot be excluded, however, that the right interplay between the various forms of L-channel modulation may critically depend on the cell recording conditions. In this case, a final conclusion should wait for measurements under more physiological constraints, i.e. trains of action potential stimulation, low external Ca2+ solutions, absence of Ca2+ agonists and in situ recordings from intact cells of adrenal gland slices.
Our present findings are likely to be applicable to other neuroendocrine cells in which L-channels play a critical role in hormone release. A basal voltage-independent feedback inhibition of L-channel gating may prove crucial for the long-term regulation of cell activity and neurosecretion. In pituitary melanotropes, the degree of gene expression of α1D channel subunits is strictly related to L-channel activity (Fass et al. 1999) and in central neurons, L-currents can trigger nuclear events associated with the activation of gene expression and cell function (Deisseroth et al. 1998).
Acknowledgments
We thank Dr T. Cesetti and R. Levi for helpful discussions. We also thank Dr H. Zucker for supplying new computer software for data analysis. J.M.H.-G. is a fellow of the Comunidad Autònoma de Madrid. This work was supported by the Italian MURST and NATO (grant no. CRG.972224).
References
- Albillos A, Carbone E, Gandía L, García AG, Pollo A. Opioid inhibition of Ca2+ channel subtypes in bovine chromaffin cells: selectivity of action and voltage-dependence. European Journal of Neuroscience. 1996a;8:1561–1570. doi: 10.1111/j.1460-9568.1996.tb01301.x. [DOI] [PubMed] [Google Scholar]
- Albillos A, Gandía L, Michelena P, Gilabert J-A, del Valle M, Carbone E, García AG. The mechanism of calcium channel facilitation in bovine chromaffin cells. Journal of Physiology. 1996b;494:687–695. doi: 10.1113/jphysiol.1996.sp021524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amico C, Marchetti C, Nobile M, Usai C. Pharmacological types of calcium channels and their modulation by baclofen in cerebellar granules. Journal of Neuroscience. 1995;15:2839–2848. doi: 10.1523/JNEUROSCI.15-04-02839.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artalejo CR, Ariano MA, Perlman RA, Fox AP. Activation of facilitation calcium channels in chromaffin cells by D1 dopamine receptors through a cAMP/protein kinase A-dependent mechanism. Nature. 1990;348:239–242. doi: 10.1038/348239a0. [DOI] [PubMed] [Google Scholar]
- Artalejo CR, Mogul DJ, Perlman RA, Fox AP. Three types of bovine chromaffin cell Ca2+ channels: facilitation increases the opening probability of a 27 pS channel. Journal of Physiology. 1991;444:213–240. doi: 10.1113/jphysiol.1991.sp018874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean BP, Nowycky MC, Tsien RW. β-Adrenergic modulation of calcium channels in frog ventricular heart cells. Nature. 1984;307:371–375. doi: 10.1038/307371a0. [DOI] [PubMed] [Google Scholar]
- Beech DJ, Bernheim L, Hille B. Pertussis toxin and voltage-dependence distinguish multiple pathways modulating calcium channels of rat sympathetic neurons. Neuron. 1992;8:97–106. doi: 10.1016/0896-6273(92)90111-p. [DOI] [PubMed] [Google Scholar]
- Boland L, Bean BP. Modulation of N-type calcium channels in bullfrog sympathetic neurons by luteinizing hormone-releasing hormone: kinetics and voltage-dependence. Journal of Neuroscience. 1993;13:516–533. doi: 10.1523/JNEUROSCI.13-02-00516.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossu J-L, De Waard M, Feltz A. Two types of calcium channels are expressed in adult bovine chromaffin cells. Journal of Physiology. 1991;437:621–634. doi: 10.1113/jphysiol.1991.sp018615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourinet E, Charnet P, Tomlinson WJ, Stea A, Snutch TP, Nargeot J. Voltage-dependent facilitation of a neuronal α1C L-type calcium channel. EMBO Journal. 1994;13:5032–5039. doi: 10.1002/j.1460-2075.1994.tb06832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carabelli V, Carra I, Carbone E. Localized secretion of ATP and opioids revealed through single Ca2+ channel modulation in bovine chromaffin cells. Neuron. 1998;20:1255–1268. doi: 10.1016/s0896-6273(00)80505-x. [DOI] [PubMed] [Google Scholar]
- Carabelli V, Lovallo M, Magnelli V, Zucker H, Carbone E. Voltage-dependent modulation of single N-type Ca2+ channel kinetics by receptor agonists in IMR32 cells. Biophysical Journal. 1996;70:2144–2154. doi: 10.1016/S0006-3495(96)79780-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone E, García AG. More on calcium channels. Trends in Neurosciences. 1997;20:448–450. [Google Scholar]
- Chavis P, Shinozaki H, Bockaert J, Fagni L. The metabotropic glutamate receptor types 2/3 inhibit L-type calcium channels via a pertussis toxin-sensitive G-protein in cultured cerebellar granule cells. Journal of Neuroscience. 1994;14:7067–7076. doi: 10.1523/JNEUROSCI.14-11-07067.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun D, Hawkes AG. The principles of the stocastic interpretation of ion-channel mechanisms. In: Sackmann B, Neher E, editors. Single Channel Recording. New York: Plenum Press; 1995. pp. 397–482. [Google Scholar]
- Costantin JL, Qin N, Waxham MN, Birnbaumer L, Stefani E. Complete reversal of run-down in rabbit cardiac Ca2+ channels by patch-cramming in Xenopus oocytes; partial reversal by protein kinase A. Pflügers Archiv. 1999;437:888–894. doi: 10.1007/s004240050859. [DOI] [PubMed] [Google Scholar]
- Deisseroth K, Heist EK, Tsien RW. Translocation of calmodulin to the nucleus supports CREB phosphorylation in hippocampal neurons. Nature. 1998;392:198–202. doi: 10.1038/32448. [DOI] [PubMed] [Google Scholar]
- Delmas P, Abogadie FC, Milligan G, Buckley NJ, Brown DA. βγ dimers derived from Go and Gi proteins contribute different components of adrenergic inhibition of Ca2+ channels in rat sympathetic neurones. Journal of Physiology. 1999;518:23–26. doi: 10.1111/j.1469-7793.1999.0023r.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin A. L-type calcium channel modulation. Advances in Second Messenger and Phosphoprotein Research. 1999;33:153–170. doi: 10.1016/s1040-7952(99)80009-3. [DOI] [PubMed] [Google Scholar]
- Elhamdani A, Bossu J-L, Feltz A. ATP and G-proteins affect the run up of the Ca2+ current in bovine chromaffin cells. Pflügers Archiv. 1995;430:410–419. doi: 10.1007/BF00373917. [DOI] [PubMed] [Google Scholar]
- Elmslie KS, Zhou W, Jones SW. LHRH and GTP-γ-S modify calcium current activation in bullfrog sympathetic neurons. Neuron. 1990;5:75–80. doi: 10.1016/0896-6273(90)90035-e. [DOI] [PubMed] [Google Scholar]
- Fass DM, Talimoto K, Mains RE, Levitan ES. Tonic dopamine inhibition of L-type Ca2+ channel activity reduces α1D Ca2+ channel gene expression. Journal of Neuroscience. 1999;19:3345–4452. doi: 10.1523/JNEUROSCI.19-09-03345.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formenti A, Martina M, Plebani A, Mancia M. Multiple modulatory effects of dopamine on calcium channel kinetics in adult rat sensory neurons. Journal of Physiology. 1998;509:395–409. doi: 10.1111/j.1469-7793.1998.395bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forti L, Pietrobon D. Functional diversity of L-type calcium channels in rat cerebellar neurons. Neuron. 1993;10:437–450. doi: 10.1016/0896-6273(93)90332-l. [DOI] [PubMed] [Google Scholar]
- García G, Sala F, Reig JA, Viniegra S, Frías J, Fonteriz RI, Gandía L. Dihydropyridine Bay-K-8644 activates chromaffin cell calcium channels. Nature. 1984;309:69–71. doi: 10.1038/309069a0. [DOI] [PubMed] [Google Scholar]
- Haws CM, Slesinger PA, Lansman JB. Dihydropyridine- and ω-conotoxin-sensitive Ca2+ currents in cerebellar neurons: persistent block of L-type channels by a pertussis toxin-sensitive G-protein. Journal of Neuroscience. 1993;13:1148–1156. doi: 10.1523/JNEUROSCI.13-03-01148.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Guijo JM, Carabelli V, Gandía L, García AG, Carbone E. Voltage-independent autocrine modulation of L-type channels mediated by ATP, opioids and catecholamines in rat chromaffin cells. European Journal of Neuroscience. 1999;11:3574–3584. doi: 10.1046/j.1460-9568.1999.00775.x. [DOI] [PubMed] [Google Scholar]
- Hess P, Lansman JB, Tsien RW. Different modes of Ca channel gating behaviour favoured by dihydropyridine Ca agonists and antagonists. Nature. 1984;311:538–544. doi: 10.1038/311538a0. [DOI] [PubMed] [Google Scholar]
- Hille B. Modulation of ion channel function by G-protein coupled receptors. Trends in Neuroscience. 1994;17:531–536. doi: 10.1016/0166-2236(94)90157-0. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Smith SJ. Large depolarization induces long openings of voltage-dependent calcium channels in adrenal chromaffin cells. Journal of Neuroscience. 1987;7:571–580. doi: 10.1523/JNEUROSCI.07-02-00571.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda SR. Voltage-dependent modulation of N-type calcium channels by G-protein βγ subunits. Nature. 1996;380:255–258. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- Imaizumi T, Watanabe Y, Yoshida H. Phosphorylation of Gi protein by cyclic AMP-dependent protein kinase inhibits its dissociation into α-subunits and βγ-subunits by Mg2+ and GTPγS. European Journal of Pharmacology. 1991;201:189–194. doi: 10.1016/0922-4106(91)90030-l. [DOI] [PubMed] [Google Scholar]
- Johnson BD, Brousal JP, Peterson BZ, Gallombardo PA, Hockerman GH, Lai Y, Scheuer T, Catterall WA. Modulation of the cloned skeletal muscle L-type Ca2+-channel by anchored cAMP-dependent protein kinase. Journal of Neuroscience. 1997;17:1243–1255. doi: 10.1523/JNEUROSCI.17-04-01243.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalali ET, Hwang KS, Plummer MR. cAMP-dependent enhancement of dihydropyridine-sensitive calcium channel availability in hippocampal neurons. Journal of Neuroscience. 1997;17:5334–5348. doi: 10.1523/JNEUROSCI.17-14-05334.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalali ET, Plummer MR. Multiple voltage-dependent mechanisms potentiate calcium channel activity in hippocampal neurons. Journal of Neuroscience. 1996;16:1072–1082. doi: 10.1523/JNEUROSCI.16-03-01072.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleppisch T, Ahnert-Hilger G, Gollasch M, Spicher K, Hescheler J, Schultz G, Rosenthalm W. Inhibition of voltage-dependent Ca2+ channels via α2-adrenergic and opioid receptors in cultured bovine adrenal chromaffin cells. Pflügers Archiv. 1992;421:131–137. doi: 10.1007/BF00374819. [DOI] [PubMed] [Google Scholar]
- Lee HK, Elmslie KS. Gating of single N-type calcium channels recorded from bullfrog sympathetic neurons. Journal of General Physiology. 1999;113:111–124. doi: 10.1085/jgp.113.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonough SI, Swartz KJ, Mintz IM, Boland LM, Bean BP. Inhibition of calcium channels in rat central and peripheral neurons by ω-conotoxin-MVIIC. Journal of Neuroscience. 1996;16:2612–2623. doi: 10.1523/JNEUROSCI.16-08-02612.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantegazza M, Fasolato C, Hescheler J, Pietrobon D. Stimulation of single L-type calcium channels in rat pituitary GH3 cells by thyrotropin-releasing hormone. EMBO Journal. 1995;14:1070–1083. doi: 10.1002/j.1460-2075.1995.tb07090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Reyes E, Yuan W, Wei X, Bers DM. Regulation of the cloned L-type cardiac calcium channel by cyclic-AMP-dependent protein kinase. FEBS Letters. 1994;342:119–123. doi: 10.1016/0014-5793(94)80484-2. [DOI] [PubMed] [Google Scholar]
- Pietrobon D, Hess P. Novel mechanism of voltage-dependent gating in L-type calcium channels. Nature. 1990;346:651–655. doi: 10.1038/346651a0. [DOI] [PubMed] [Google Scholar]
- Pollo A, Lovallo M, Biancardi E, Sher E, Socci C, Carbone E. Sensitivity to dihydropyridines, ω-conotoxins and noradrenaline reveals multiple high-voltage activated Ca2+ channels in rat insulinoma and human pancreatic β-cells. Pflügers Archiv. 1993;423:462–471. doi: 10.1007/BF00374942. [DOI] [PubMed] [Google Scholar]
- Reuter H. Calcium channel modulation by neurotransmitters, enzymes and drugs. Nature. 1983;301:569–574. doi: 10.1038/301569a0. [DOI] [PubMed] [Google Scholar]
- Scholz KP, Miller RJ. GABAB receptor-mediated inhibition of Ca2+ currents and synaptic transmission in cultured rat hippocampal neurones. Journal of Physiology. 1991;444:669–686. doi: 10.1113/jphysiol.1991.sp018900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sculptoreanu A, Scheuer T, Catterall W. Voltage-dependent potentiation of L-type Ca channels due to phosphorylation by cAMP-dependent protein kinase. Nature. 1993;364:240–243. doi: 10.1038/364240a0. [DOI] [PubMed] [Google Scholar]
- Sigworth FJ, Sine SM. Data transformations for improved display and fitting of single-channel dwell time histograms. Biophysical Journal. 1987;52:1047–1054. doi: 10.1016/S0006-3495(87)83298-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tottene A, Moretti A, Pietrobon D. Functional diversity of P-type and R-type calcium channels in rat cerebellar neurons. Journal of Neuroscience. 1996;16:6353–6363. doi: 10.1523/JNEUROSCI.16-20-06353.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visentin S, Amiel P, Fruttero R, Boschi D, Roussel C, Giusta L, Carbone E, Gasco A. Synthesis and voltage-clamp studies of methyl 1,4-dihydro-2,6-dimethyl-5-nitro-4-(benzofurazanyl)pyridine-3-carboxylate racemates and enantiomers and of their benzofuroxanyl analogues. Journal of Medicinal Chemistry. 1999;42:1422–1427. doi: 10.1021/jm980623b. [DOI] [PubMed] [Google Scholar]