

Abstract
Western blotting revealed the presence of five somatostatin receptor types, sst1, sst2, sst3, sst4 and sst5, in the mouse pancreatic β-cell line MIN-6.
In MIN-6 cells, glucose-induced electrical activity was potently (pEC50= 12.7) and irreversibly reduced by somatostatin (SRIF-14); this was associated with hyperpolarization of the membrane potential (pEC50= 11.2) and a decrease in the input resistance (pEC50= 12.7).
The effects of SRIF-14 were mimicked by 100 nm L-362,855 (a partial agonist at sst5 receptors), but not BIM-23027 or NNC-26,9100 (selective agonists at sst2 and sst4 receptors, respectively). CH-275 at 100 nm (a selective agonist at sst1 receptors) partially inhibited electrical activity but without membrane potential hyperpolarization.
One hundred nanomolar SRIF-28 activated an inwardly rectifying K+ current (ISRIF). ISRIF was activated neither by 1 μm BIM-23056 nor CYN-154806 (antagonists at sst5 and sst2 receptors, respectively). The activation of ISRIF by 100 nm SRIF-28 was, however, inhibited 93 % by BIM-23056; CYN-154806 had no effect.
Both 100 nm glibenclamide and 200 μm tolbutamide, blockers of the β-cell ATP-sensitive K+ channel (K-ATP), reduced ISRIF by ≈44 %, whereas 1 mm Ba2+ abolished ISRIF.
In cell-attached patches, 100 nm SRIF-14 activated two types of single-channel currents whose properties were consistent with those of K-ATP and GIRK channels.
In conclusion, somatostatin can inhibit glucose-induced electrical activity in MIN-6 cells by the combined activation of K-ATP and GIRK channels. Studies with selective agonists and antagonists are consistent with this effect being mediated by the sst5 receptor.
A rise in the intracellular free calcium concentration, [Ca2+]i, is a key signal in the initiation of insulin secretion from the pancreatic β-cell. This increase principally results from Ca2+ influx through voltage-gated L-type Ca2+ channels located in the plasma membrane which open in response to membrane depolarization evoked by secretagogues, primarily glucose (Ashcroft & Rorsman, 1989). These events are manifest as glucose-induced electrical activity that consists of Ca2+-dependent action potentials.
The peptide somatostatin is a potent inhibitor of insulin secretion. Analogues of somatostatin are used to inhibit the secretion of insulin in a variety of hyperinsulinaemic clinical conditions. Such pathologies can arise from endocrine tumours, genetic disorders or the side effects of medication. For these reasons, plus the possibility that antagonism of tonic somatostatin inhibition could be used to promote insulin secretion in type-2 diabetes, understanding the mechanisms that underlie the actions of somatostatin is an important issue (Reisine & Bell, 1995).
One mechanism by which somatostatin inhibits insulin secretion is via a pertussis toxin (PTX)-sensitive hyperpolarization of the β-cell membrane potential (Pace & Tarvin, 1981) which acts to reduce Ca2+-dependent electrical activity and Ca2+ influx (Sussman et al. 1987; Nilsson et al. 1989). Somatostatin can also inhibit insulin secretion by PTX-sensitive mechanisms distal to Ca2+ influx, e.g. by reducing cytoplasmic levels of cyclic AMP, a permissive sensitizer of insulin release (Malm et al. 1991), and/or by direct G-protein interactions with the exocytotic machinery (Ullrich et al. 1990).
The ionic mechanisms that underlie the electrophysiological changes produced by somatostatin in the pancreatic β-cell are poorly understood. An increase in K+ conductance is implicated in the action of this peptide (Pace & Tarvin, 1981). This may result from an increase in the activity of K-ATP channels (De Weille et al. 1989). However, the viability of this pathway is questionable in the light of data from subsequent studies in which somatostatin failed to activate K-ATP channels at concentrations of glucose above the threshold for electrical activity (Hsu et al. 1991; Ribalet & Eddlestone, 1995). Furthermore, somatostatin has been shown to hyperpolarize the membrane potential and inhibit both glucose-induced electrical activity and insulin release in the presence of sulphonylureas (Seaquist et al. 1992; Abel et al. 1996), drugs which specifically block the β-cell K-ATP channel (Ashcroft & Rorsman, 1989). These data argue against a sole role for the K-ATP channel in mediating the electrophysiological actions of somatostatin. To clarify this situation we have further investigated the identity of the K+ channels that underlie the somatostatin-induced hyperpolarization of the membrane potential and inhibition of glucose-induced spiking electrical activity.
To date, five somatostatin receptors (sst1, sst2, sst3, sst4 and sst5; with sst2 existing as two splice variants sst2(a) and sst2(b)) have been identified and cloned, all of which belong to the seven-transmembrane, G-protein-coupled receptor superfamily (Reisine & Bell 1995; Meyerhof, 1998). Recombinant gene expression in heterologous systems has allowed the pharmacological and physiological properties of the different receptor subtypes to be explored in isolation (Reisine & Bell 1995; Meyerhof, 1998). Importantly, such studies have permitted the development of high affinity selective agonists and antagonists for the different receptor subtypes (Raynor et al. 1993a, b; Liapakis et al. 1996; Ankersen et al. 1998).
Somatostatin exists in two natural forms, as a cyclic tetradecapeptide, SRIF-14, and as the N-terminally extended form, SRIF-28. Of the different receptor types, only sst5 receptor shows any marked preference in binding between the two forms of somatostatin, expressing a higher affinity towards SRIF-28 (Reisine & Bell 1995; Meyerhof, 1998). In rodents, the inhibition of insulin release by somatostatin in vitro appears to be mediated by the somatostatin receptor subtype 5 (sst5). Several lines of evidence support this claim. (1) SRIF-28 has a higher potency than SRIF-14 in its ability to displace bound somatostatin analogues from β-cell membranes (Thermos et al. 1990), to inhibit insulin release (Mandarino et al. 1981) and to reduce forskolin-stimulated adenylate cyclase activity (Schuit et al. 1989). (2) Agonists selective for sst5 receptors have the greatest potency to inhibit glucose-induced insulin release (Fagan et al. 1998; Strowski et al. 2000). Immunohistochemistry (Mitra et al. 1999) and RT-PCR (Fagan et al. 1998) have demonstrated the presence of the sst5 receptor in β-cells. In sst2 receptor knockout mice, neither the potency nor the efficacy of the inhibition of insulin secretion by somatostatin was affected (Strowski et al. 2000), a finding that rules out an involvement of the sst2 receptor in the secretory actions of somatostatin in the mouse β-cell. What is unknown is whether the electrophysiological changes produced by somatostatin are also caused by activation of the sst5 receptor. If not, this would imply that it is events distal to the reduction of Ca2+ influx, e.g. a reduction in cytosolic cyclic AMP, which are primarily responsible for the inhibition of insulin release by somatostatin working via the sst5 receptor. In a previous study, 100 nm somatostatin reduced the rate of action potential firing in β-cells (Pace & Tarvin, 1981), but it is unknown if this phenomenon occurs only at high concentrations of peptide and whether it results from the activation of non-sst5 receptors. In order to investigate the receptor subtype responsible for the electrophysiological changes that precede insulin secretion we have therefore examined the effects of some somatostatin receptor-selective ligands on the electrical activity of the β-cell.
For these studies we used the endogenous insulin-secreting cell line MIN-6 (Miyazaki et al. 1990), a well-established cell model, for studying the physiology of the pancreatic β-cell (Ishihara et al. 1993). MIN-6 cells possess a robust glucose metabolism and glucose-stimulated insulin release that closely resembles that found in β-cells of normal mouse islets (Ishihara et al. 1993). For these reasons, their unlimited availability and homogeneous responses, MIN-6 cells are ideal for studying the effects of hormones on β-cells.
METHODS
Cells
MIN-6 cells were kindly supplied by Dr Steve Ashcroft (John Radcliffe Hospital, Oxford, UK) and were used with the permission of Professor Jun-Ichi Miyazaki (Osaka University Medical School, Osaka, Japan) who established this cell line (Miyazaki et al. 1990). Cells were grown in RPMI 1640 tissue culture media with 11 mm glucose, 25 mm Hepes and 10 % fetal calf serum. They were kept at 37 °C in a humidified atmosphere containing 5 % CO2.
Western blotting
Western blotting, as described previously, was used to identify somatostatin receptor proteins (Sellers et al. 1999). Briefly, antibodies were raised to fusion proteins incorporating a region from the predicted C-terminus of the sst1, sst2, sst3, sst4 or sst5 receptors in conjunction with glutathione-S-transferase and were expressed using the pGEX-system (Pharmacia). The cDNA sequences chosen from the cloned human receptors included base pairs 1120-1275 from the sst1 receptor, 1070-1192 from sst2, 1288-1354 from sst3, 1035-1217 from sst4 and 1014-1145 from sst5. The membrane proteins of MIN-6 cells were solubilized, electrophoretically resolved on 10 % (w/v) polyacrylamide gels and transferred onto nitrocellulose membranes. Primary incubations with antibodies were made at a 1:1000 dilution for 1 h at 22 °C. Samples were then further incubated after washing for 1 h at 22 °C with a 1:3000 dilution of the appropriate horseradish peroxidase-conjugated secondary antibody. Excess antibody was removed by washing and immunocomplexes were visualised using enhanced chemiluminescence (ECL) detection according to the manufacturer's instructions (APBiotech, Amersham, UK).
Electrophysiology
Cells were plated onto 22 mm glass coverslips and maintained for 1-4 days in tissue culture prior to use. For experiments, cells were chosen that appeared round and single. In order to maintain cellular metabolism and second-messenger systems intact, whole-cell membrane currents and potentials were recorded using the perforated-patch technique as described previously (Smith et al. 1990).
Membrane potential and currents were measured using either an Axopatch 1D or 1B patch clamp amplifier (Axon Instruments, Foster City, CA, USA). The zero-current potential of the pipette was adjusted with the pipette in the bath just before establishment of the seal. No corrections have been made for either liquid junction potentials (< 4 mV) or series resistance errors (< 2 mV). Data were stored on DAT tape for subsequent playback and analysis.
Current clamp
The effects of somatostatin were assayed on the glucose-induced action potential firing rate (APFR), input resistance (Rin) and membrane potential (Vm) using current-clamp mode. The APFR was measured by computing the average number of action potentials recorded over 1 min intervals. Input resistance was measured at 10 s intervals as the deflection of the membrane potential evoked by current pulses -10 pA in amplitude, 250-500 ms in duration. Membrane potential measurements were taken from the flattest region of the interspike period.
Estimation of membrane parameters
If it is assumed that the membrane conductances that contribute to the interspike potential, and also those activated by somatostatin, are ohmic in behaviour then the conductance activated by somatostatin, GSRIF (1/Rin,SRIF), can be estimated from:
| (1) |
where Rin,c and Rin,SRIF are the input resistances in the absence and presence of somatostatin respectively. The reversal potential for the current activated by somatostatin (Vr,SRIF) can then be estimated from:
| (2) |
where Vm,c and Vm,SRIF are the membrane potentials in the absence and presence of somatostatin. Vr,SRIF and Rin,SRIF were also estimated by fitting the equation:
| (3) |
to the relationship between Vm,SRIF and the independent variables, Vm,c and Rin,c.
Voltage clamp
The effects of somatostatin on membrane currents were studied using voltage-clamp mode. Current-voltage relationships (I-V) were determined by initially stepping the membrane potential from a holding potential of -50 mV to 0 mV for 1 s and then ramping down to -100 mV at 0.1 mV ms−1. To improve the signal-to-noise ratio, averages were formed of the current responses to 10 consecutive ramps applied at a frequency of 0.3 Hz. For display, records were further low pass filtered at 20 Hz (-3 db, 4 pole Bessel).
The currents activated by somatostatin and test compounds were measured as the difference between the currents recorded in test and pre-test control solutions. If the effects of a drug were reversible, the average of the pre- and post-test currents was used as the control. Slope conductance was determined by linear regression analysis of the I-V relationship between -80 and -100 mV and between -60 and -80 mV for 5.6 mm and 56 mm extracellular K+ ([K+]o), respectively. To test the effects of channel inhibitors and receptor antagonists on the currents activated by somatostatin the following protocol was used. Somatostatin was applied three times, each for a duration of 1 min separated by a wash period of 4 min. Test compounds were presented from 2 min before, until the end of, the second peptide application, when they were simultaneously removed with somatostatin. To control for desensitization of the somatostatin response, the effect of the test compounds on the currents activated by somatostatin was compared to time matched controls.
Single-channel studies
To further identify the K+ channels activated by somatostatin, single-channel currents were studied using the cell-attached patch-clamp configuration. Currents were low pass filtered at 2 kHz (-3 db, 8 pole Bessel) and digitized at 10 kHz using pCLAMP 5 (Axon Instruments). Single-channel currents were analysed with half-amplitude threshold techniques as discussed by Colquhoun & Sigworth (1983). These were performed using in-house software using a minimum time resolution of 100 μs on 60 s periods of record (Smith et al. 1994). Single-channel chord conductances were calculated on the assumption that the reversal potential was ≈0 mV with the near-identical [K+] of the cell interior and pipette solution.
Concentration-response relationships
Concentration-response relationships were constructed from the effects of one concentration of somatostatin applied on one cell per coverslip. In only a few cases, where the responses to somatostatin entirely reversed, was a second concentration of somatostatin tested. The concentration-response relationships were quantified by fits of the following equation to the data:
| (4) |
where [SRIF-14] is the concentration of somatostatin, EC50 is the concentration that produces the half-maximal effect, Ymax is the maximal response and h is an index of the slope.
Solutions
Whole-cell recordings
Pipettes were filled with (mm): 76 K2SO4, 10 KCl, 10 NaCl, 55 sucrose, 10 Hepes (pH 7.2 with KOH) and 1 MgCl2. Perforation was produced by the addition of 100 μg ml−1 amphotericin B to the pipette solution (Smith et al. 1990). Perforation was considered adequate when the series conductance exceeded 40 nS. For current-clamp recording the extracellular solution consisted of a modified Hanks' solution composed of (mm): 5.6 KCl, 138 NaCl, 4.2 NaHCO3, 1.2 NaH2PO4, 2.6 CaCl2, 1.2 MgCl2 and 10 Hepes (pH 7.4 with NaOH). In voltage-clamp recordings the extracellular solution was composed of (mm): 5.6 KCl, 128 NaCl, 4.2 NaHCO3, 1.2 NaH2PO4, 3.8 MgCl2, 10 Hepes (pH 7.4 with NaOH), 10 glucose and 10 TEACl. Extracellular Ca2+ was omitted to reduce contamination of records by inward Ca2+ currents and TEA was included to block voltage-gated K+ currents. In some experiments 50 mm NaCl was replaced with 50 mm KCl to give 56 mm KCl. The extracellular solutions contained 0.01 % w/v. BSA to limit non-specific binding of drugs. The bath had a volume of ≈0.2 ml and was continuously perfused at a rate of 1-2 ml min−1. Drug additions were added when the electrical responses, either action potential firing rate or membrane current, were at steady state. All whole-cell experiments were carried out at 31-33 °C.
Cell-attached recordings
To control the cell resting membrane potential, cells were bathed in a solution which contained (mm): 143 KCl, 4.2 NaHCO3, 1.2 NaH2PO4, 3.8 MgCl2, 10 Hepes (pH 7.4 with NaOH) and 10 glucose. This solution clamped Vm to 0 mV, as confirmed by current-clamp measurements. The pipette solution contained (mm): 140 KCl, 2.6 CaCl2, 1.2 MgCl2 and 10 Hepes (pH 7.4 with NaOH). Somatostatin (100 nm SRIF-14) was added either to the bath or pipette solution. To reduce noise generated by perfusion, single-channel current recordings were made during periods of stopped flow at 21-22 °C.
Drugs
SRIF-14 and SRIF-28 came from Peninsula Laboratories (St Helens, UK). CH-275 (c[Cys-Lys-Phe-Phe-d-Trp-Iamp-Thr-Phe-Thr-Ser-Cys]-OH), BIM-23027 (c[N-Me-Ala-Try-d-Trp-Lys-Abu-Phel]), BIM-23056 (d-Phe-Phe-Tyr-d-Trp-Lys-Val-Phe-d-Na1-NH2), L-362,855 (c[Ala-Phe-Trp-d-Trp-Lys-Val-Phen]), NNC-26,9100 (for structure see Ankersen et al. 1998) and CYN-154806 (Ac-4-NO2-Phe-c [d-Cys-Tyr-d-Trp-Lys-Thr-Cys]-d-Tyr-NH2) were purchased from commercial suppliers of synthetic peptides (Peptide and Protein Research Consultants, Exeter, UK; Neosystems Laboratories, Strasbourg, France). All other drugs and reagents were obtained from Sigma.
Statistical analyses
For statistical comparison of unpaired sample populations Student's t test (two-tailed) was used; for multiple, unpaired comparisons a one-way ANOVA was conducted with Dunnet's post test. Skewed data distributions were compared using either the Mann-Whitney (unpaired) or Kruskal-Wallis test with Dunn's post test (multiple, unpaired). Spearman's non-parametric rank correlation coefficient, R, was used to investigate whether variables were correlated. These procedures were performed using GraphPad Prism 3 (GraphPad Software, Inc., San Deigo, CA, USA). All other mathematical analyses were performed using Origin 5.0 (OriginLab Corp., Northampton, MA, USA). Data are given as means ±s.e.m., with n as the number of determinations. Statistical significance is defined as P < 0.05.
RESULTS
Somatostatin receptor complement of MIN-6 cells
Whole-cell extracts prepared from MIN-6 cells showed strong immunoreactive products following Western analysis using all five somatostatin receptor-specific antibodies (Fig. 1). The products detected ranged in apparent molecular mass from 45 kDa to approximately 90 kDa, with several distinct entities cross-reacting with the anti-sst1, -sst2 and -sst3 receptor antibodies. The expected size for the receptors based on their primary sequences is between 41 and 53 kDa, consistent with the apparent molecular mass of those products, with greatest mobility for the sst1, sst2 and sst3 receptor types. The multi-banding is attributed to differential covalent modification that is well established to occur within the somatostatin receptor family (Meyerhof, 1998).
Figure 1. Western blot of MIN-6 whole cell extract.
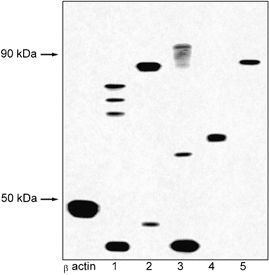
Representative example (n = 3) of a Western blot of MIN-6 whole cell extract. The samples have been screened with primary antibodies for β-actin, sst1 (1), sst2 (2), sst3 (3), sst4 (4) and sst5 (5) receptors.
Characterisation of the electrophysiological response of MIN-6 cells to glucose
A typical membrane potential recording from a single MIN-6 cell is illustrated in Fig. 2. In the absence of glucose, the cell was electrically silent with a membrane potential (Vm) of -66 ± 0.5 mV (n = 108) and an input resistance (Rin) of 0.9 ± 0.1 GΩ (n = 83). On addition of 10 mm glucose, after a delay of 43 ± 3 s (n = 106) the membrane potential depolarized and the input resistance increased. On average, glucose depolarized Vm by 20 ± 1 mV to -44 ± 1 mV, and increased Rin by 2.4 ± 0.2 GΩ to 3.5 ± 0.2 GΩ (n = 31). Similar responses were observed in 123 cells out of 129 tested. In 101of the responsive cells, electrical activity was evoked (Fig. 2A). This was characterised by a continuous train of action potentials which peaked at ≈+20 mV and varied widely in shape and form. Although the action potential firing rate (APFR) was variable between cells, it was normally distributed with a mean of 3.5 ± 0.3 spikes s−1 (n = 56). In the presence of glucose, no correlation could be detected between APFR, Rin and Vm. In 95 % of electrically active cells the membrane potential threshold for an action potential was >-54 mV. These results are consistent with those previously reported for single β-cells isolated from both mouse (Smith et al. 1990) and rat (Falke et al. 1989) pancreatic islets.
Figure 2. Effects of glucose and somatostatin on the membrane potential of MIN-6 cells.
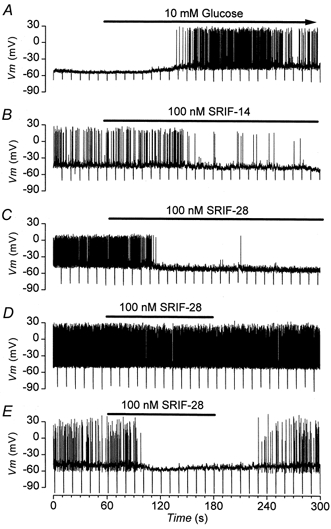
Current clamp recordings of the membrane potential from three different cells. Downward deflections are the voltage responses to -10 pA current pulses, 500 ms in duration. Additions are present for the duration of the bars. A, electrical activity stimulated by 10 mm glucose. B, subsequent response of the cell showed in A to 100 nm SRIF-14 in the continued presence of glucose. C, the irreversible effect of SRIF-14 is mimicked by 100 nm SRIF-28 added to a different cell that is electrically active in 10 mm glucose. D, cell electrically active in 10 mm glucose to which the addition of 100 nm SRIF-28 has little effect. E, effect of a second addition of 100 nm SRIF-28 to the same cell shown in D but in which the activity is reduced by injection of a -4 pA current.
Inhibition of electrical activity by somatostatin
In the majority of studies that have investigated the action of SRIF-14 on β-cell function, maximal effects are always observed at ≈100 nm. For this reason, as is the case in many other studies, 100 nm was used as the test concentration for the peptide. Within 60 s of the addition of 100 nm SRIF-14 to the bath, the glucose-induced electrical activity was reduced (Fig. 2B). This was associated with a decrease in Rin and hyperpolarization of Vm, which in the cell illustrated were sufficient in magnitude to abolish electrical activity. In 9 out of 30 cells tested, 100 nm SRIF-14 abolished electrical activity permanently. Overall, 100 nm SRIF-14 inhibited the APFR by 57 ± 7 % (n = 29), hyperpolarized Vm by -4.5 ± 0.7 mV (n = 23) and decreased Rin by 0.5 ± 0.1 GΩ (n = 16). In 5 out of 5 cells tested which had failed to recover after the removal of somatostatin, 200 μm tolbutamide was able to depolarize the membrane potential and elicit electrical activity, data which indicate that electrical activity in the cell was still viable. The irreversible abolition of electrical activity by SRIF-14 was not due to rupture of the perforated patch, cell dialysis and consequent cell death. Application of suction to deliberately achieve the standard whole-cell configuration resulted in a reduced access resistance and wash-up of whole-cell K-ATP currents (Ashcroft & Rorsman, 1989), events that were not associated with the sustained loss of electrical activity caused by SRIF-14 (data not shown). Similar effects to those produced by 100 nm SRIF-14 were also observed with 100 nm SRIF-28 (Fig. 2C, n = 17).
Inhibition by somatostatin is state dependent
The inhibitory effect of somatostatin was dependent on the magnitude of the APFR. This phenomena is demonstrated by the observation that reduction of the control APFR (Fig. 2D) from 7.1 to 1.7 spikes s−1 via injection of -4 pA of current potentiated the inhibition of the APFR produced by SRIF-28 from 24 % to 100 % (Fig. 2E). A plot of inhibition against control APFR illustrates that SRIF-14 produced greater inhibition and reduced the APFR by a larger extent in cells which were less electrically active with a smaller control APFR (Fig. 3A; R = -0.46; P < 0.02).
Figure 3. Aspects of inhibition of electrical activity produced by somatostatin.
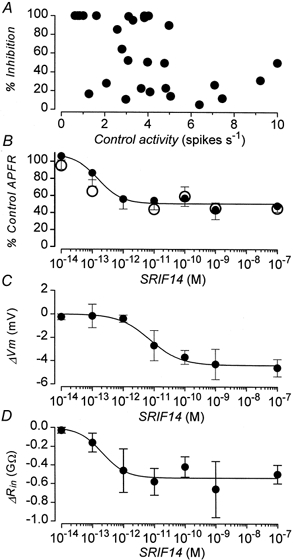
A, relationship between the percentage inhibition of electrical activity produced by 100 nm SRIF-14 and the preceding control activity. Data are from 29 cells. B, concentration-response relationship for the inhibition of electrical activity produced by somatostatin. •, in the presence of SRIF-14; ○, 5 min after the removal of the peptide. Data are means ±s.e.m. from 4-23 cells. C, concentration-response relationship for the hyperpolarization of Vm (ΔVm) produced by SRIF-14. Data are means ±s.e.m. from 5-8 cells. D, concentration-response relationship for the decrease in Rin (ΔRin) produced by SRIF-14. Data are means ±s.e.m. from 5-8 cells. Continuous lines in B, C and D are drawn with the parameters (given in the text) obtained from best fits of eqn (4) to the data shown.
Concentration-response relationship of inhibition caused by somatostatin
The concentration-response relationship for the inhibition of the glucose-induced APFR by SRIF-14 is shown in Fig. 3B. SRIF-14 was extremely potent, with concentrations as low as 0.1 pm able to inhibit electrical activity. The effect of the peptide was, however, highly variable (cf. Fig. 3A), some cells showing very little response to the peptide whilst in others electrical activity was abolished. The mean maximum inhibition of the APFR for the sample population was 51 %. The best fit of eqn (4) to the inhibition data (continuous line, Fig. 3B) was obtained with Ymax= 52 ± 4 %, pEC50= 12.7 ± 0.21 (13.3-12.1, 95 % confidence limits) and h = 1.6± 0.8.
Somatostatin decreased Rin as a function of concentration (Fig. 3D). The change in Rin was best fitted with eqn (4) (continuous line, Fig. 3D) with Ymax= -0.53 ± 0.1 GΩ, pEC50= 12.7 ± 0.3 (13.4-12, 95 % confidence limits) and h = 1.2± 14. Somatostatin also hyperpolarized Vm in a concentration-dependent fashion (Fig. 3C) although this effect was only observed at peptide concentrations > 1 pm. The hyperpolarization data were best fitted with eqn (4) (continuous line, Fig. 3C) with Ymax= -4.2 ± 0.2 mV, pEC50= 11.2 ± 0.12 (11.5-10.8, 95 % confidence limits) and h = 1.1± 0.2.
Reversibility of the inhibition caused by somatostatin
The effects of somatostatin were poorly reversible. In the majority of cells, recovery from the effects of a 3 min application of somatostatin were incomplete with up to 20 min of washing, the longest period tested. The APFR measured 5 min after the removal of somatostatin was almost identical to that produced in the presence of the peptide (Fig. 3B, open circles). The electrophysiological effects of somatostatin, including the poor reversibility, are similar to those previously reported for the action of the peptide on β-cells in intact mouse islets (Pace & Tarvin, 1981).
Pharmacology of the effect of somatostatin on electrical activity
To explore the identity of the somatostatin receptor involved in the inhibition of electrical activity the effects of a variety of agonists selective for different receptor subtypes were tested in order to construct a pharmacological profile (Meyerhof, 1998). To control for possible differences in efficacy between receptor subtypes and also to standardize responses, compounds were used at 100 nm, a maximal functional concentration for SRIF-14. For each compound used an EC50 obtained from functional assays in recombinant systems is given. However, since these values are dependent on receptor expression levels, receptor reserve and coupling efficiency, they will vary between systems and can only be used as a guide to their potency.
Figure 4B shows that L-362,855 a partial agonist at the sst5 receptor (Raynor et al. 1993b; EC50≈0.1 nm, Thurlow et al. 1996; EC50≈10 nm, Williams et al. 1997; EC50≈0.1 nm, Carruthers et al. 1999) mimicked the action of SRIF-14. L-362,855 irreversibly inhibited electrical activity, which was accompanied by hyperpolarization of Vm and a reduction in Rin (Fig. 5). Of all the other agonists tested, only CH-275, which is specific at the sst1 receptor (Liapakis et al. 1996; EC50 not determined), consistently inhibited glucose-induced electrical activity. This effect was less pronounced than that produced by SRIF-14 (Fig. 4C and 5) and was not associated with a change in Vm. CH-275 did, however, increase Rin (Fig. 5), the opposite to what was observed with SRIF-14 (Fig. 4A and 5). Figure 5 summarises the effects of the different somatostatin receptor agonists on APFR, Rin and Vm. BIM-23056, another partial agonist at the sst5 receptor (Raynor et al. 1993b; Williams et al. 1997; EC50≈0.2 nm, Carruthers et al. 1999), could also mimic the effects of SRIF-14. However, BIM-23027, a specific agonist at the sst2 receptor (Raynor et al. 1993b; EC50≈1 nm) and NNC-26,9100, a specific agonist at the sst4 receptor (EC50≈2 nm, Ankersen et al. 1998) were both without effect (Fig. 5).
Figure 4. Inhibition of electrical activity produced by somatostatin analogues.
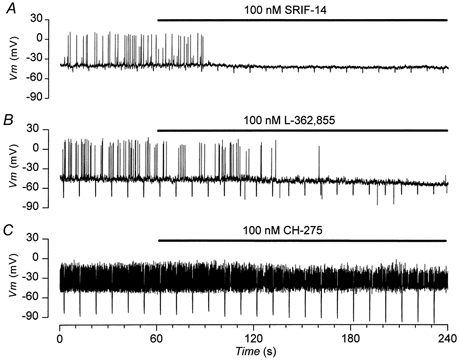
Current clamp recordings from three different cells in the continued presence of 10 mm glucose. Negative deflections in the membrane potential are the responses to -10 pA current steps, 500 ms in duration. Drugs are applied for the duration indicated by the bars. A, inhibition of electrical activity produced by 100 nm SRIF-14, which is associated with hyperpolarization of Vm and decrease in Rin. B, inhibition of electrical activity produced by 100 nm L-362,855, a partial agonist at the sst5, which produces similar electrophysiological effects to those caused by SRIF-14. C, inhibition of electrical activity by 100 nm CH-275, a selective agonist of sst1, which, unlike that observed in A and B, is associated with an increase in Rin and no change in Vm.
Figure 5. Summary of electrical responses produced by somatostatin analogues.
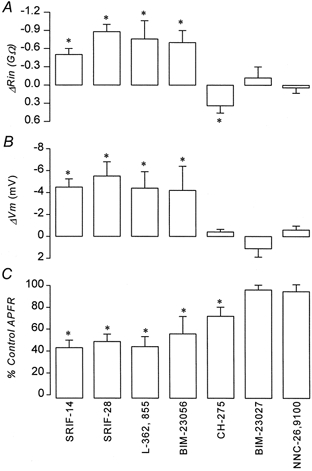
Comparison of the effects of somatostatin receptor agonists applied at 100 nm. Data are from 5-29 cells. A, change in input resistance Rin (ΔRin). B, hyperpolarization of membrane potential Vm (ΔVm). C, electrical activity (APFR) measured 2 min after the addition of drug as a percentage of control APFR. *P < 0.05.
Somatostatin activates an outward current
The electrophysiological effects of SRIF-14, SRIF-28, L-362,855 and BIM-23056 are consistent with these compounds activating an outward current (ISRIF). Using eqns (1) and (2), it is calculated from the values of, and changes in, Vm and Rin obtained with SRIF-14 that the peptide activates a conductance, GSRIF, of 164 ± 88 pS (n = 14) with a reversal potential, Vr,SRIF, of -71 ± 5 mV (n = 13). Similar values were obtained from the best fit of eqn (3) to the appropriate data (see Methods): 123 ± 188 pS and -66 ± 26 mV for GSRIF and Vr,SRIF, respectively. From these values it is estimated that somatostatin activates an outward current of ≈-3 pA at -50 mV (Vm,SRIF).
The possibility that such a small ionic current could bring about the electrophysiological changes produced by somatostatin is supported by the experiment illustrated in Fig. 6. Injection of ≈-5 pA of current produced electrophysiological changes (Fig. 6B) similar to those produced by 100 nm SRIF-28 (Fig. 6A). On switching from current clamp to voltage clamp with a holding potential of -50 mV, re-application of 100 nm SRIF-28 activated an outward current (Fig. 6C). This had a time course similar to that observed for the hyperpolarization induced by the peptide (Fig. 6A). The outward current peaked at ≈ 2 pA and was associated with an increase in the membrane conductance of 350 pS, a value comparable in magnitude to that estimated from the changes in Vm and Rin, i.e. 164 pS.
Figure 6. Somatostatin activates an outward current.
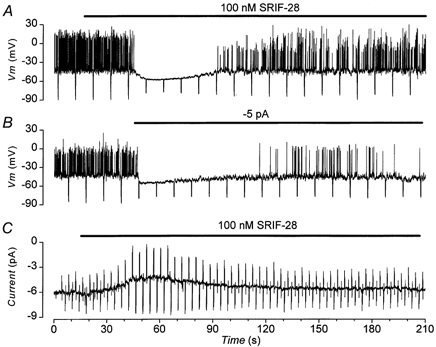
Electrophysiological records from the same cell in the presence of 10 mm glucose. Treatments were applied for the duration of the bars. A and B, current clamp; negative deflections in the membrane potential are the responses to -10 pA current pulses, 500 ms in duration. A, effect of 100 nm SRIF-28. B, effect of injection of -5 pA current. C, voltage clamp; current activated by 100 nm SRIF-28 at a holding potential of -50 mV; deflections in current are the responses to alternately applied ± 10 mV voltage pulses.
Somatostatin activates inwardly rectifying K+ currents
In 5.6 mm[K+]o, 100 nm SRIF-28 activated an inwardly rectifying current, ISRIF (Fig. 7A), which had a peak amplitude of -1.2 ± 0.3 pA (n = 5) at -50 mV. The associated I-V relationship had a slope conductance (GSRIF) of 90 ± 18 pS (n = 5) and a reversal potential (Vr,SRIF) of -76 ± 3 mV. When [K+]o was raised 10-fold to 56 mm, GSRIF increased ≈10-fold to (1.2 ± 0.1 nS, n = 67) and Vr,SRIF depolarised by 55 mV to -21 ± 1.5 mV (n = 42). These changes are consistent with ISRIF being carried by inwardly rectifying K+-selective ion channels as has been reported in other systems (Reisine & Bell 1995; Yamada et al. 1998; Yoshimoto et al. 1999). To enhance the measurement of ISRIF and GSRIF all subsequent voltage-clamp experiments were performed in 56 mm[K+]o.
Figure 7. Aspects of outward currents activated by somatostatin.
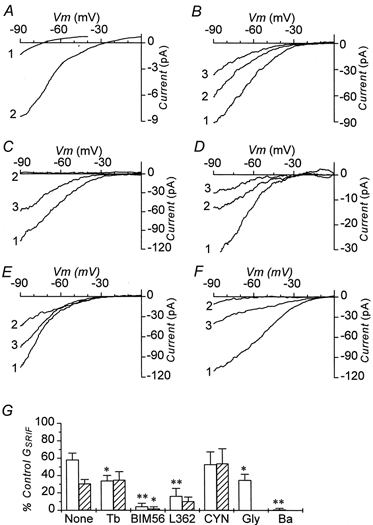
Difference currents between I-V relationships measured in the absence and presence of SRIF-28. Except in A, the currents were recorded in 56 mm extracellular K+. A, currents activated by 100 nm SRIF-28 in 5.6 mm (1) and 56 mm (2) extracellular K+. B-F, currents activated by three consecutive applications of 100 nm SRIF-28. Each was added for a duration of 1 min separated by a 4 min wash interval. 1, the response to the first application; 2, the response to the second application; 3, the response to the third application. B, currents recorded under control conditions alone. C, currents recorded in control conditions (1), in the presence of 1 mm extracellular Ba2+ (2) and after the washout of Ba2+ (3). D, currents recorded in the continued presence of 100 nm glibenclamide. 1, 2 and 3, responses to first, second and third applications of SRIF-28 respectively. Note the diminished amplitudes relative to the currents shown in B; E, currents recorded; 1, in control conditions; 2, in the presence of 200 μm tolbutamide and 3, after the washout of the sulphonylurea. F, currents recorded in control conditions (1), in the presence of 1 μm L-362,855 (2) and after washout of the sst5 receptor antagonist (3). G, conductance activated by SRIF-28 plotted as a percentage of that activated by the first application of peptide. □, responses to 2nd application of SRIF-28 in presence of drug indicated;  , responses to 3rd application of SRIF-28 after washout of drug. Data are means ±s.e.m. from 4-9 cells. Statistical significance is by comparison to the respective column of control data with no treatment (None). *P < 0.05; **P < 0.01. None, time matched controls; Tb, 200 μm tolbutamide; BIM56, 1 μm BIM-23056; L362, 1 μm L-362,855; CYN, 1 μm CYN-154806; Gly, 100 nm glibenclamide; Ba, 1 mm Ba2+.
, responses to 3rd application of SRIF-28 after washout of drug. Data are means ±s.e.m. from 4-9 cells. Statistical significance is by comparison to the respective column of control data with no treatment (None). *P < 0.05; **P < 0.01. None, time matched controls; Tb, 200 μm tolbutamide; BIM56, 1 μm BIM-23056; L362, 1 μm L-362,855; CYN, 1 μm CYN-154806; Gly, 100 nm glibenclamide; Ba, 1 mm Ba2+.
Figure 7B shows that ISRIF desensitized with successive applications of the peptide. GSRIF decreased with the 2nd and 3rd application to 61 ± 8 % (n = 9) and 32 ± 5 % (n = 7) of the control value, respectively. In another set of cells, a lower concentration (0.1 nm) of SRIF-28 activated a conductance (50 ± 46 pS, n = 7) which was < 0.5 % of that activated by 100 nm (1.2 nS). A second application of SRIF-28 at 100 nm activated a conductance (0.9 ± 0.3 nS, n = 7) comparable in magnitude to that observed when the first concentration applied was also 100 nm (0.7 ± 0.2 nS, n = 10). In five cells tested 10 pm SRIF-28 had no effect.
Receptor pharmacology of ISRIF
The nature of the somatostatin receptor subtype coupled to ISRIF was investigated. Neither BIM-23056 nor L-362,855, either at 100 nm or 1 μm, activated a current (n = 5 for each peptide at each concentration). At 1 μm however, both BIM-23056 and L-362,855 inhibited GSRIF activated by 100 nm SRIF-28 by 93 and 74 %, respectively (Fig. 7F and G). Whereas the inhibition produced by BIM-23056 was irreversible, that caused by L-362,855 was partially reversible (Fig. 7F and G). CYN-154806 (at 1 μm), a specific antagonist at the sst2 receptor (Feniuk et al. 2000), neither activated a current nor inhibited that activated by 100 nm SRIF-28 (Fig. 7G).
Identity of the conductance activated by somatostatin
To investigate the identity of the channels carrying ISRIF, a second application of SRIF-28 was made in the presence of various blockers of inwardly rectifying K+ channels. Application of 1 mm Ba2+ had no effect on the holding current, but reversibly abolished ISRIF (Fig. 7C and G). Glibenclamide (100 nm) and tolbutamide (200 μm) also had no effect on the holding current, but significantly blocked GSRIF by similar amounts: 43 % for glibenclamide and 45 % for tolbutamide (Fig. 7E and G). The effect of tolbutamide, but not glibenclamide, was reversible (Fig. 7E and G). In cells that had been pre-incubated with 100 nm glibenclamide for 2 min, GSRIF was significantly reduced by 38 % from 1.2 ± 0.1 nS (n = 67) to 0.7 ± 0.2 nS (n = 10), a decrease similar to that measured above. Glibenclamide did not affect the desensitisation of ISRIF observed with repeated applications of the peptide (Fig. 7D). These data suggest that under these conditions ≈45 % of the inwardly rectifying K+ current activated by somatostatin flows through K-ATP channels.
The observation that sulphonylurea drugs blocked only part of the current activated by somatostatin suggests that at least two types of inward rectifier K+ channels were gated by the peptide: the K-ATP channel and another of unknown identity. Application of 200 μm diazoxide, a specific opener of the K-ATP channel, activated a conductance that was several times larger and more linear than that elicited by SRIF-28 (data not shown). Since the K-ATP currents rectified less than ISRIF, the other current component of ISRIF must belong to a stronger inwardly rectifying type of K+ channel (K-SRIF).
Single-channel studies
To further characterize the properties of the channels activated by somatostatin we used cell-attached recordings. These were performed on cells whose resting potential had been clamped to 0 mV by use of a high [K+]o solution to prevent changes in Vm due to effects of glucose or somatostatin. In the absence of glucose only single-channel currents with biophysical properties consistent with activity of K-ATP channels were observed (data not shown). These were abolished shortly after the addition of 10 mm glucose to the bath (Ashcroft et al. 1988). In the presence of the sugar no single-channel currents were observed in the absence of somatostatin (n = 7).
Addition of 100 nm SRIF-14 to the bath had no effect in seven patches tested. Only when the peptide was added to the pipette solution were single-channel currents observed (Fig. 8). On the basis of single-channel current amplitude (i) and chord-conductance (γ) two types of currents were identified (Fig. 8): for one i = -5.4± 0.1 pA and γ= 77 ± 1.3 pS (n = 8), and for the other, smaller current i = -2.3± 0.1 pA and γ= 33 ± 0.9 pS (n = 14). On the basis of these values the single-channel currents are identified as flowing through ATP-sensitive K+ channels (K-ATP; Ashcroft et al. 1988; Ashcroft & Rorsman 1989) and G-protein-coupled inwardly rectifying K+ channels (GIRK; Grigg et al. 1996; Yamada et al. 1998), respectively. The sulphonylurea-insensitive component of ISRIF, K-SRIF is therefore most likely to be a member of the GIRK channel family. Out of 13 patches examined, nine exhibited activity of both types of channels whereas four patches had GIRK alone; patches never displayed the activity of just K-ATP channels. In patches that contained both types of inward rectifiers, perfusion of 100 nm glibenclamide abolished the activity of the K-ATP channels but not that associated with GIRK. In 10 out of 10 patches formed in the presence of glibenclamide (100 nm) only GIRK channels were activated by 100 nm SRIF-14. Although SRIF-14 activated K-ATP channels, openings were rare with only two to three bursts normally observed in any 60 s period.
Figure 8. Single-channel currents activated by somatostatin.
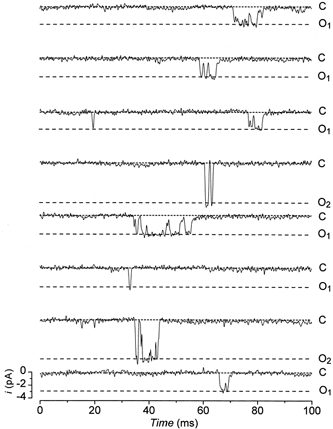
Single-channel currents activated by 100 nm SRIF-14 present in the pipette solution. Currents are recorded in the cell-attached configuration at a membrane potential of -100 mV. The baseline current (closed level, dotted lines) is indicated by C. O1 and O2 mark the single-channel current amplitude (open level, dashed lines) for GIRK and K-ATP channels, respectively. For display, currents were low pass filtered at 1 kHz (-3 dB) and sampled at 10 kHz.
Kinetic study of GIRK
The distribution of channel open times was best fitted by a single exponential component (Fig. 9A) and because of unresolved missing closures the apparent mean open time, τo, of 0.42 ± 0.06 ms is likely to be longer than actual mean open time. The closed time distributions were best fitted by the sum of three exponential components (Fig. 9B). These had mean time constants (and relative areas) of τ1= 0.32 ± 0.18 ms (40 ± 3 %), τ2= 7.6 ± 1.9 ms (39 ± 4 %) and τ3= 318 ± 183 ms (22 ± 5 %). Channel openings occurred either singly, or, more usually, were clustered together to form bursts of openings separated by brief closures (Fig. 8). Both inspection of the current traces and the presence of at least three closed time components (Fig. 9B) suggest that the bursts themselves are clustered. Bursts were defined as clusters of openings separated by closures whose length is less than a burst criterion time, τcrit (Smith et al. 1994). Since the bursts themselves appeared to be clustered, the estimation of τcrit and analyses of burst kinetics were performed twice. In the first instance, bursts were defined by a τcrit (1 ± 0.05 ms, 95 % confidence) estimated from τ1 and τ2 (short bursts). In the second case the clusters of bursts (long bursts) were defined by a τcrit (23 ± 6 ms, 95 % confidence) estimated from τ2 and τ3. The distributions for the lengths of short bursts and long bursts both appeared to consist of the sum of two exponential components (Fig. 9C and D). The two components of the short bursts had mean time constants (and relative areas) of τsb,1= 0.24 ± 0.05 ms (60 ± 7 %) and τsb,2= 2.0 ± 0.5 ms (40 ± 7 %), whereas the long bursts had mean time constants (and relative areas) of τlb,1= 0.31 ± 0.05 ms (52 ± 8 %) and τlb,2= 31 ± 6 ms (48 ± 4 %). The distributions of the number of openings within the burst for both the short bursts and long bursts were best fitted by the sum of two geometric components (Colquhoun & Sakmann 1985; Fig. 9E and F). The number of openings, r (and relative areas), for the short bursts had values of rsb,1= 1.3 ± 0.06 (70 ± 8 %) and rsb,2= 2.7 ± 0.5 (30 ± 8 %), whereas the number of openings (and relative areas) for the long bursts were rlb,1= 1.6 ± 0.16 (48 ± 8 %) and rlb,2= 14 ± 2 (52 ± 8 %). Both the mean open time and the number of openings that occurred within the short bursts were corrected for missed gaps (Colquhoun & Sigworth 1983). This process yielded a corrected rsb,2= 1.6 ± 0.1 opening per short burst with a mean open time of 0.42 ± 0.08 ms. Neither of these values is significantly different from the respective uncorrected value. Although channel activity (number of channels × probability of being open) varied widely between patches from 0.001 to 0.4, the only time constant affected by channel activity was τ3, which decreased with increased channel activity.
Figure 9. Kinetic analyses of GIRK.
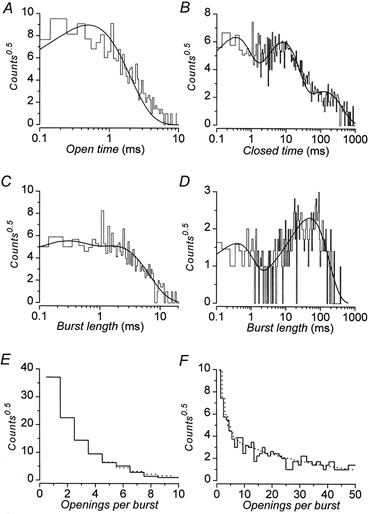
Representative distributions of kinetic parameters obtained by threshold analysis of activity of single GIRK channels. Channel activity was recorded in the cell-attached configuration. The membrane potential was -100 mV with 100 nm SRIF-14 in the pipette and 100 nm glibenclamide in the bath solution. Data are all from the same recording for which 3951 openings were detected and analysed. In all cases the bin frequency has been square root transformed (counts0.5). In A-D the ordinate is plotted on a log scale; this transformation allows identification of the number of exponential components by the number of peaks. Continuous lines are the best fits to the data obtained by maximum likelihood methods with the parameters given. A, distribution of open times; open time τo= 0.54 ms. B, distribution of closed times; time constants (and relative areas) were τ1= 0.36 ms (47 %), τ2= 8 ms (45 %) and τ3= 120 ms (8 %). C, distribution of short burst lengths; time constants (and relative areas) were τsb,1= 0.24 ms (45 %) and τsb,2= 1.8 ms (55 %). D, distribution of long burst lengths; time constants (and relative areas) were τlb,1= 0.42 ms (32 %) and τlb,2= 49 ms (68 %). E, distribution of the number of openings within short bursts; mean number of openings (and relative areas) were rsb,1= 1.4 (46 %) and rsb,2= 2 (54 %). F, distribution of the number of openings within long bursts; mean number of openings (and relative areas) were rlb,1= 2 (36 %) and rlb,2= 19 (64 %).
DISCUSSION
Receptor molecular biology
We found that the endogenous insulin-secreting cell line MIN-6 expresses all known isoforms of the somatostatin receptor family: sst1, sst2, sst3, sst4 and sst5 (Reisine & Bell 1995; Meyerhof, 1998). Assuming that these are all trafficked to the plasma membrane and are functionally coupled, this clonal cell line appears to be an excellent model in which to study the pleiotropic actions of somatostatin on β-cell function.
Potency of somatostatin
Current-clamp recording was used as a sensitive functional assay to explore the pharmacology of the inhibition of electrical activity by somatostatin in β-cells. SRIF-14 was extremely potent in its ability to inhibit the electrical activity induced by 10 mm glucose. The pEC50 of this effect (12.7) is > 100-fold that reported for the inhibition of glucose-stimulated insulin release (pEC50≈ 9-11) from a variety of β-cell preparations: 9-11 for hamster insulinoma, HIT-T15 (Ullrich et al. 1990; Thermos et al. 1990; Hsu et al. 1991); 9.8 for perfused rat pancreas (Mandarino et al. 1981); 9 for rat islets (Malm et al. 1991; Hurst & Morgan, 1990); 8.7 for isolated rat β-cells (Schuit et al. 1989). This marked divergence between the pEC50 for the inhibition of electrical activity in MIN-6 and that of insulin release by SRIF-14 may be due to a species difference since the pEC50 for the inhibition of insulin release from mouse β-cells by SRIF-14 is not yet known. Alternatively, these differences in pEC50 may be explained by the observation that the maximal inhibition of frequency of action potential firing, and presumably inhibition of Ca2+ influx and insulin secretion, produced by the peptide is incomplete at ≈50 %. Consequently, any further inhibition of insulin release by SRIF-14 must be mediated by mechanisms which have lower potencies and/or coupling efficiencies to the peptide, at sites distal to Ca2+ influx (Ullrich et al. 1990; Malm et al. 1991).
Cyclic AMP is a permissive sensitizer of insulin release in both rat and mouse (Schuit et al. 1989; Ämmäläet al. 1993) and its production is inhibited by somatostatin in both species. The pEC50 for the inhibition of insulin release by somatostatin in rat is comparable with that for reduction of cAMP levels (Schuit et al. 1989), which is ≈10-fold more potent than for the ability of the peptide to decrease Ca2+ influx within the same species (Sussman et al. 1987). Consequently, a reduction in cAMP levels alone may explain the inhibition of insulin release produced by SRIF-14 in rat (Hurst & Morgan, 1990). We found that SRIF-14 reduced the levels of forskolin-stimulated cAMP in MIN-6 cells with a pEC50 of 7.6 (P. A. Smith, A. M. Carruthers & P. P. A. Humphrey, unpublished observations). The low potency of this effect suggest that the major mechanism by which SRIF-14 inhibits insulin secretion in mouse is by a reduction in electrical activity and Ca2+ influx and not by a decrease in cAMP. Indeed in intact mouse islets, 100 nm SRIF-14 inhibits insulin secretion by 50 % (Strowski et al. 2000). Since we observed that the same concentration of peptide reduced electrical activity by ≈50 %, these data argue that, at least in mouse, the major mechanism by which somatostatin inhibits insulin secretion at concentrations ≤ 100 nm is electrophysiologically mediated. Other evidence in favour of this idea comes in a previous report (Hsu et al. 1991) in which the lowest concentration at which SRIF-14 inhibited insulin release, 0.1 pm, was the same at which the inhibition of electrical activity was first detected in the present study (Fig. 3B).
Since the ability of SRIF-14 to affect different aspects of the insulin secretory pathway appears to all be mediated though the sst5 receptor, there is no need for multiple receptor involvement. The actual pEC50 values reported for the inhibition of insulin release by SRIF-14 can be explained by the summation of different inhibitory mechanisms, which appear to all act through the same receptor but do so with different efficiencies of receptor-effector coupling.
Interestingly, the pEC50 values for the inhibition of insulin release by SRIF-14 are consistent with its affinity for binding to its receptor: 9 in HIT-T15 (Thermos et al. 1990), 9.6 in rat (Sussman et al. 1987) and 9.2 in MIN-6 cells (P. A. Smith, E. Jarvie & P. P. A. Humphrey, unpublished observations). The difference between the pEC50 for the inhibition of electrical activity by SRIF-14 and that obtained for its binding is probably due to the presence of an amplification cascade between the receptor and the effector ion channels that elicit the changes in electrical activity. Given the high input resistance of the β-cell, the alteration of the activity of only a few ionic channels will be sufficient to have dramatic electrophysiological consequences. A consequence of this amplification cascade mechanism is that SRIF-14 should also be extremely potent in its ability to reduce insulin secretion at concentrations ≤ 100 nm, although the maximal inhibition of secretion achieved by this mechanism will only be ≈50 %. As already discussed, other less efficiently coupled inhibitory mechanisms, which have lower potencies, must be invoked if insulin secretion is abolished at higher concentrations of SRIF-14. Indeed, the combined action of multiple inhibitory mechanisms, which have different potencies, may result in a multi-phasic relationship between the inhibition of insulin secretion and SRIF-14.
Pharmacology of the somatostatin response
The pharmacological profile that was observed for the ability of somatostatin receptor-selective analogues to affect both the glucose-induced electrical activity and ISRIF is consistent with the peptide primarily prosecuting its affects through binding to the sst5 receptor. Since the specific sst2 receptor agonist BIM-23027 did not have any effect in current clamp and the specific sst2 receptor antagonist CYN-154806 did not block ISRIF, these findings argue against an involvement of the sst2 receptor. The observation that the specific sst4 receptor agonist NNC-26,9100 had no effect rules out participation of the sst4 receptor. The observation that L-362,855, a less potent agonist at the sst3 receptor than BIM-23027 (Raynor et al. 1993a), had an effect in current clamp whereas the latter compound did not, argues against an involvement of the sst3 receptor.
The ability of the specific sst1 receptor antagonist CH-275 to partially inhibit electrical activity with an increase in Rin but without a change in Vm is consistent with inhibition of L-type Ca2+ channels. Indeed, SRIF-14 has been reported to inhibit current flowing through L-type Ca2+ channels in a variety of β-cell lines (Hsu et al. 1991; Roosterman et al. 1998; P. A. Smith & P. P. A. Humphrey, unpublished observations). In particular, in the β-cell lines RIN1046-38 and MIN-6 this inhibition appears to occur through preferential activation of sst1 receptors. Since sst1 receptors do not appear to functionally couple with G-protein coupled inwardly rectifying K+ channels, at least in recombinant systems (Kreienkamp et al. 1997), this pathway is unlikely to be involved in the effects of CH-275.
The observations that the sst5 receptor partial agonists L-362,855 and BIM-23056 mimicked the effects of SRIF-14 in current clamp and acted as antagonists of ISRIF in voltage clamp support the idea that the predominant electrophysiological actions of somatostatin are mediated by the sst5 receptor. The use of agonist concentrations several orders of magnitude higher than their reported EC50 values also adds support to the above arguments. At saturating concentrations of SRIF-14, inhibition of L-type Ca2+ channels by activation of the sst1 receptor is also expected to contribute to the reduction in electrical activity produced by the peptide. Whether the sst1 receptor is involved in the modulation of electrical activity by SRIF-14 at lower doses is unknown.
The reversal of the agonist behaviour of L-362,855 and BIM-23056 to one of antagonism on changing the recording conditions from current clamp to voltage clamp is not unexpected. This transformation probably results from changes at the cellular level, with the receptor-effector pathway shifting from an efficiently coupled system under quasi-physiological conditions of current clamp to one that is more weakly coupled (Thurlow et al. 1996; Williams et al. 1997). In voltage clamp, alterations in extracellular ionic concentrations, transmembrane ionic fluxes and intracellular ion concentrations will act to affect the receptor pathway; for example the concentration of cytoplasmic sodium is well established to affect the agonist potency and intrinsic activity of the sst5 receptor (Williams et al. 1997). Such changes will not only affect receptor- effector coupling but may also help to explain the reduced sensitivity, improved reversibility and desensitization of the somatostatin responses observed in the voltage-clamp experiments.
Identity of the conductance activated by somatostatin
We found that somatostatin activated two types of inwardly rectifying K+ channels in MIN-6 cells. On the basis of their pharmacological, biophysical and single-channel properties, one channel type is identified as the ATP-sensitive K+ channel (K-ATP; Ashcroft et al. 1988; Ashcroft & Rorsman 1989). The other channel activated by somatostatin, K-SRIF, is likely to be a member of the G-protein regulated subfamily of inward rectifier channels (GIRK, Yamada et al. 1998). The demonstration that somatostatin activates K-ATP channels in β-cells not only confirm earlier reports (De Weille et al. 1989; Ribalet & Eddlestone 1995) but now also extends this action to higher, insulinotropic glucose concentrations.
By comparison of its properties with other G-protein-coupled inward rectifiers, K-SRIF is probably a tetramer of Kir3.x subunits (Yamada et al. 1998). In support of this notion, several studies have identified mRNA for Kir3.1, Kir3.2 and Kir3.4 in tissue from rat islets, human islets and insulinomas from a variety of different species (Stoffel et al. 1994, 1995; Ferrer et al. 1995; Tsaur et al. 1995). The use of specific antibodies has so far shown the presence of Kir3.4, but apparently not Kir3.2, in rat β-cells (Yoshimoto et al. 1999). In addition to Kir6.2 (Aguilar-Bryan et al. 1995), the only other inwardly rectifying K+ channel subunit to be reported in islet tissue to date is Kir1.1 (Yano et al. 1994).
Single-channel properties of K-SRIF
The biophysical and kinetic properties we describe for K-SRIF are almost identical to those reported for GIRK channels activated by somatostatin in rat locus coeruleus neurons under similar recording conditions (temperature of 22 °C, near-symmetrical [K+], a holding potential of -100 mV and low pass filtered at 2 kHz; Grigg et al. 1996). Immunohistochemical studies have demonstrated the presence of Kir3.1 inwardly rectifying K+ channel subunits in locus coeruleus neurons from rat (Bausch et al. 1995). This finding suggests that GIRK channels within these neurons, as is probably the case in β-cells, contain Kir3.1 subunits (Yamada et al. 1998). A similar subunit structure for GIRK channels in locus coeruleus neurons and pancreatic β-cells would explain the comparable kinetic properties for the short bursts recorded in the two cell types. Indeed the values for τo, τ1, τsb,1, τsb,2 and rsb,2 (0.54 ms, 0.42 ms, 0.32 ms, 2 ms and 2.1 openings per burst, respectively) obtained in the neurons when GIRK is activated by 10 nm SRIF-14 (Grigg et al. 1996) are similar to those measured for K-SRIF activated by 100 nm SRIF-14 in MIN-6 cells: 0.42 ms, 0.32 ms, 0.24 ms, 2 ms and 1.6 opening per burst for τo, τ1, τsb,1, τsb,2 and rsb,2, respectively.
The observation that the short bursts were clustered into longer bursts separated by long closures can be explained by a simple four-state kinetic scheme:
where G represents the activated G-protein, either α and/or βγ subunits, Kir represents closed states of the inward rectifier, Kir* is an activated closed state of the channel and Kir° is the channel open state. Transitions between Kir and GKir control the clusters of bursts, whereas transitions between GKir* and GKir control the short bursts. Transitions between GKir° and GKir* account for the openings and majority of closures (gaps) that make up the short bursts. What is unknown is the identity of the single openings that are well separated from other bursts. These may represent short bursts with unresolved gaps that occur during brief periods of G-protein activation of Kir. The finding that the mean open lifetime for the isolated openings is not significantly different from that for openings that occur within the short bursts supports this notion. Alternatively, these single openings may reflect the activity of another class of Kir channel or open state in the above kinetic scheme. Ideas akin to these have already been discussed for similar observations in locus coeruleus neurons (Grigg et al. 1996) and more generally (Colquhoun & Sakmann 1985).
The activity of Kir channels in the cell-attached patches was maintained for the life of the patch, unlike ISRIF, which exhibited desensitisation in whole-cell voltage-clamp recordings. The former observation is similar to the maintained response to SRIF-14 seen in current clamp recordings, although whether the action of SRIF-14 on single-channel activity is also irreversible cannot be readily tested in cell-attached patches. These findings further support a role for the ionic environment in the receptor-effector coupling of somatostatin.
Physiological implications
The proposal that somatostatin activates both K-ATP channels and a sulphonylurea-insensitive inwardly rectifying K+ channel, K-SRIF, which is of the GIRK moiety, explains why the membrane hyperpolarization and the inhibition of electrical activity produced by the peptide can be insensitive to sulphonylureas (Abel et al. 1996). Since at the high concentration of somatostatin (100 nm) often employed, the magnitude of the current that passes through K-SRIF would be sufficient alone to cause the reported changes in electrical behaviour (Abel et al. 1996) and effects on insulin secretion (Seaquist et al. 1992). Future study lies in the determination of the molecular identity, regulation and functional roles of K-SRIF in β-cells and, via linkage analysis, possible involvement in blood sugar disorders.
Acknowledgments
We thank A. M. Carruthers for help with the adenylate cyclase assay and E. Jarvie for performing the binding studies.
References
- Abel KB, Lehr S, Ullrich S. Adrenaline-, not somatostatin-induced hyperpolarization is accompanied by a sustained inhibition of insulin secretion in INS-1 cells. Activation of sulphonylurea K-ATP channels is not involved. Pflügers Archiv. 1996;432:89–96. doi: 10.1007/s004240050109. [DOI] [PubMed] [Google Scholar]
- Aguilar-Bryan L, Nichols CG, Wechsler SW, Clement JP, Boyd AE, Gonzalez G, Herrera-Sosa H, Nguy K, Bryan J, Nelson DA. Cloning of the β cell high-affinity sulfonylurea receptor: a regulator of insulin secretion. Science. 1995;268:423–426. doi: 10.1126/science.7716547. [DOI] [PubMed] [Google Scholar]
- Ämmälä C, Eliasson L, Bokvist K, Larsson O, Ashcroft FM, Rorsman P. Exocytosis elicited by action potentials and voltage-clamp calcium currents in individual mouse pancreatic β-cells. Journal of Physiology. 1993;472:665–688. doi: 10.1113/jphysiol.1993.sp019966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ankersen M, Crider M, Liu S, Ho B, Andersen HS, Stidsen C. Discovery of a novel non-peptide somatostatin agonist with sst4 selectivity. Journal of the American Chemical Society. 1998;120:1368–1373. [Google Scholar]
- Ashcroft FM, Ashcroft SJH, Harrison DE. Properties of single potassium channels modulated by glucose in rat pancreatic β-cells. Journal of Physiology. 1988;400:501–527. doi: 10.1113/jphysiol.1988.sp017134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft FM, Rorsman P. Electrophysiology of the pancreatic β-cell. Progress in Biophysics and Molecular Biology. 1989;54:87–143. doi: 10.1016/0079-6107(89)90013-8. [DOI] [PubMed] [Google Scholar]
- Bausch SB, Patterson TA, Ehrengruber MU, Lester HA, Davidson N, Chavkin C. Colocalization of μ-opioid receptors with GIRK1 potassium channels in the rat brain: an immunocytochemical study. Receptors and Channels. 1995;3:221–241. [PubMed] [Google Scholar]
- Carruthers AM, Warner AJ, Michel AD, Feniuk W, Humphrey PPA. Activation of adenylate cyclase by human recombinant sst5 receptors expressed in CHO-K1 cells and involvement of Gαs proteins. British Journal of Pharmacology. 1999;126:1221–1229. doi: 10.1038/sj.bjp.0702401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun D, Sakmann B. Fast events in single-channel currents activated by acetylcholine and its analogues at the frog muscle end-plate. Journal of Physiology. 1985;369:501–557. doi: 10.1113/jphysiol.1985.sp015912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun D, Sigworth FJ. Fitting and statistical analysis of single-channel records. In: Sakmann B, Neher E, editors. Single-Channel Recording. 1. New York: Plenum Press; 1983. pp. 191–263. [Google Scholar]
- De Weille JR, Schmid-Antomarchi H, Fosset M, Lazdunski M. Regulation of ATP-sensitive K+ channels in insulinoma cells: activation by somatostatin and protein kinase C and the role of cAMP. Proceedings of the National Academy of Sciences of the USA. 1989;86:2971–2975. doi: 10.1073/pnas.86.8.2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan SP, AziADEHZZ A, Moldovan S, Ray MK, Adrian TE, Ding XZ, Coy DH, Brunicardi FC. Insulin secretion is inhibited by subtype five somatostatin receptor in the mouse. Surgery. 1998;124:254–258. [PubMed] [Google Scholar]
- Falke LC, Gillis KD, Pressel DM, Misler S. Perforated patch recording’ allows long-term monitoring of metabolite-induced electrical activity and voltage-dependent Ca2+ currents in pancreatic islet β cells. FEBS Letters. 1989;251:167–172. doi: 10.1016/0014-5793(89)81448-6. [DOI] [PubMed] [Google Scholar]
- Feniuk W, Jarvie E, Luo J, Humphrey PPA. Selective somatostatin sst2 receptor blockade with the novel cyclic octapeptide, CYN-154806. Neuropharmacology. 2000;39:1443–1450. doi: 10.1016/s0028-3908(00)00035-6. [DOI] [PubMed] [Google Scholar]
- Ferrer J, Nichols CG, Makhina EN, Salkoff L, Bernstein J, Gerhard D, Wasson J, Ramanadham S, Permutt A. Pancreatic islet cells express a family of inwardly rectifying K+ channel subunits which interact to form G-protein-activated channels. Journal of Biological Chemistry. 1995;270:26086–26091. doi: 10.1074/jbc.270.44.26086. [DOI] [PubMed] [Google Scholar]
- Grigg JJ, Kozasa T, Nakajima Y, Nakajima S. Single-channel properties of a G-protein-coupled inward rectifier potassium channel in brain neurons. Journal of Neurophysiology. 1996;75:318–328. doi: 10.1152/jn.1996.75.1.318. [DOI] [PubMed] [Google Scholar]
- Hsu WH, Xiang HD, Rajan AS, Kunze DL, Boyd AE., iii Somatostatin inhibits insulin secretion by a G-protein-mediated decrease in Ca2+ entry through voltage-dependent Ca2+ channels in the beta cell. Journal of Biological Chemistry. 1991;266:837–843. [PubMed] [Google Scholar]
- Hurst RD, Morgan NG. Evidence for differential effects of noradrenaline and somatostatin on intracellular messenger systems in rat islets of Langerhans. Journal of Molecular Endocrinology. 1990;4:231–237. doi: 10.1677/jme.0.0040231. [DOI] [PubMed] [Google Scholar]
- Ishihara H, Asano T, Tsukuda K, Katagiri H, Inukai K, Anai M, Kikuchi M, Yazaki Y, Miyazaki J-I, Oka Y. Pancreatic beta cell line MIN-6 exhibits characteristics of glucose metabolism and glucose-stimulated insulin secretion similar to those of normal islets. Diabetologia. 1993;36:1139–1145. doi: 10.1007/BF00401058. [DOI] [PubMed] [Google Scholar]
- Kreienkamp HJ, Honck HH, Richter D. Coupling of rat somatostatin receptor subtypes to a G-protein gated inwardly rectifying potassium channel (GIRK1) FEBS Letters. 1997;419:92–94. doi: 10.1016/s0014-5793(97)01437-3. [DOI] [PubMed] [Google Scholar]
- Liapakis G, Hoeger C, Rivier J, Reisine T. Development of a selective agonist at the somatostatin receptor subtype sstr1. Journal of Pharmacology and Experimental Therapeutics. 1996;276:1089–1094. [PubMed] [Google Scholar]
- Malm D, Vonen B, Burhol PG, Florholmen J. The interaction between cAMP-dependent and cAMP-independent mechanisms in mediating the somatostatin inhibition of insulin secretion in isolated rat pancreatic islets. Acta Physiologica Scandinavica. 1991;143:305–310. doi: 10.1111/j.1748-1716.1991.tb09237.x. [DOI] [PubMed] [Google Scholar]
- Mandarino L, Stenner D, Blanchard W, Nissen S, Gerich J. Selective effects of somatostatin-14, -25 and -28 on in vitro insulin and glucagon secretion. Nature. 1981;291:76–77. doi: 10.1038/291076a0. [DOI] [PubMed] [Google Scholar]
- Meyerhof W. The elucidation of somatostatin receptor functions: a current view. Reviews of Physiology Biochemistry and Pharmacology. 1998;133:55–108. doi: 10.1007/BFb0000613. [DOI] [PubMed] [Google Scholar]
- Mitra SW, Mezey E, Hunyady B, Chamberlain L, Hayes E, Foor F, Wang YN. Colocalization of somatostatin receptor sst5 and insulin in rat pancreatic β-cells. Endocrinology. 1999;140:3790–3796. doi: 10.1210/endo.140.8.6937. [DOI] [PubMed] [Google Scholar]
- Miyazaki J, Araki K, Yamato E, Ikegami H, Asano T, Shibasaki Y, Oka Y, Yamamura K-I. Establishment of a pancreatic β cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology. 1990;127:126–132. doi: 10.1210/endo-127-1-126. [DOI] [PubMed] [Google Scholar]
- Nilsson T, Arkhammar P, Rorsman P, Berggren PO. Suppression of insulin release by galanin and somatostatin is mediated by a G-protein. An effect involving repolarization and reduction in cytoplasmic free Ca2+ concentration. Journal of Biological Chemistry. 1989;264:973–980. [PubMed] [Google Scholar]
- Pace CS, Tarvin JT. Somatostatin: mechanism of action in pancreatic islet β-cells. Diabetes. 1981;30:836–842. doi: 10.2337/diab.30.10.836. [DOI] [PubMed] [Google Scholar]
- Raynor K, Murphy WA, Coy DH, Taylor JE, Moreau J-P, Yasuda K, Bell GI, Reisine T. Cloned somatostatin receptors: identification of subtype-selective peptides and demonstration of high affinity binding of linear peptides. Molecular Pharmacology. 1993a;43:838–844. [PubMed] [Google Scholar]
- Raynor K, O'Carroll AM, Kong H, Yasuda K, Mahan LC, Bell GI, Reisine T. Characterization of cloned somatostatin receptors sstr4 and sstr5. Molecular Pharmacology. 1993b;44:385–392. [PubMed] [Google Scholar]
- Reisine T, Bell GI. Molecular properties of somatostatin receptors. Neuroscience. 1995;67:777–790. doi: 10.1016/0306-4522(95)00072-q. [DOI] [PubMed] [Google Scholar]
- Ribalet B, Eddlestone GT. Characterization of the G-protein coupling of a somatostatin receptor to the K-ATP channel in insulin-secreting mammalian HIT and RIN cell lines. Journal of Physiology. 1995;485:73–86. doi: 10.1113/jphysiol.1995.sp020713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roosterman D, Glassmeier G, Baumeister H, Scherubl H. A somatostatin receptor 1 selective ligand inhibits Ca2+ currents in rat insulinoma 1046–1038 cells. FEBS Letters. 1998;425:137–140. doi: 10.1016/s0014-5793(98)00221-x. [DOI] [PubMed] [Google Scholar]
- Schuit FC, Derde MP, Pipeleers DG. Sensitivity of rat pancreatic α and β cells to somatostatin. Diabetologia. 1989;32:207–212. doi: 10.1007/BF00265096. [DOI] [PubMed] [Google Scholar]
- Seaquist ER, Neal AR, Shoger KD, Walseth TF, Robertson RP. G-proteins and hormonal inhibition of insulin secretion from HIT-T15 cells and isolated rat islets. Diabetes. 1992;41:1390–1399. doi: 10.2337/diab.41.11.1390. [DOI] [PubMed] [Google Scholar]
- Sellers LA, Feniuk W, Humphrey PP, Lauder H. Activated G protein-coupled receptor induces tyrosine phosphorylation of STAT3 and agonist-selective serine phosphorylation via sustained stimulation of mitogen-activated protein kinase. Resultant effects on cell proliferation. Journal of Biological Chemistry. 1999;274:16423–16430. doi: 10.1074/jbc.274.23.16423. [DOI] [PubMed] [Google Scholar]
- Smith PA, Ashcroft FM, Rorsman P. Simultaneous recordings of glucose dependent electrical activity and ATP-regulated K+-currents in isolated mouse pancreatic β-cells. FEBS Letters. 1990;261:187–190. doi: 10.1016/0014-5793(90)80667-8. [DOI] [PubMed] [Google Scholar]
- Smith PA, Williams BA, Ashcroft FM. Block of ATP-sensitive K+ channels in isolated mouse pancreatic β-cells by 2,3-butanedione monoxime. British Journal of Pharmacology. 1994;112:143–149. doi: 10.1111/j.1476-5381.1994.tb13044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoffel M, Espinosa R, iii, Powell KL, Philipson LH, Le Beau MM, Bell GI. Human G-protein-coupled inwardly rectifying potassium channel (GIRK1) gene (KCNJ3): localization to chromosome 2 and identification of a simple tandem repeat polymorphism. Genomics. 1994;21:254–256. doi: 10.1006/geno.1994.1253. [DOI] [PubMed] [Google Scholar]
- Stoffel M, Tokuyama Y, Trabb JB, German MS, Tsaar ML, Jan LY, Polonsky KS, Bell GI. Cloning of rat KATP-2 channel and decreased expression in pancreatic islets of male Zucker diabetic fatty rats. Biochemical and Biophysical Research Communications. 1995;212:894–899. doi: 10.1006/bbrc.1995.2053. [DOI] [PubMed] [Google Scholar]
- Strowski MZ, Parmar RM, Blake AD, Schaeffer JM. Somatostatin inhibits insulin and glucagon secretion via two receptor subtypes: an in vitro study of pancreatic islets from somatostatin receptor 2 knockout mice. Endocrinology. 2000;141:111–117. doi: 10.1210/endo.141.1.7263. [DOI] [PubMed] [Google Scholar]
- Sussman KE, Leitner JW, Draznin B. Cytosolic free-calcium concentrations in normal pancreatic islet cells. Effect of secretagogues and somatostatin. Diabetes. 1987;36:571–577. doi: 10.2337/diab.36.5.571. [DOI] [PubMed] [Google Scholar]
- Thermos K, Meglasson MD, Nelson J, Lounsbury KM, Reisine T. Pancreatic β-cell somatostatin receptors. American Journal of Physiology. 1990;259:E216–224. doi: 10.1152/ajpendo.1990.259.2.E216. [DOI] [PubMed] [Google Scholar]
- Thurlow RJ, Sellers LA, Coote JE, Feniuk W, Humphrey PPA. Human recombinant sst5 receptors expressed in CHO-K1 cells mediate increases in extracellular acidification by pertussis toxin-sensitive and -insensitive pathways. British Journal of Pharmacology. 1996;117:9P. [Google Scholar]
- Tsaur ML, Menzel S, Lai FP, Espinosa R, iii, Concannon P, Spielman RS, Hanis CL, Cox NJ, Le Beau MM, German MS, Jan LY, Bell JI, Stoffel M. Isolation of a cDNA clone encoding a KATP channel-like protein expressed in insulin-secreting cells, localization of the human gene to chromosome band 21q22.1. and linkage studies with NIDDM. Diabetes. 1995;44:592–596. doi: 10.2337/diab.44.5.592. [DOI] [PubMed] [Google Scholar]
- Ullrich S, Prentki M, Wollheim CB. Somatostatin inhibition of Ca2+-induced insulin secretion in permeabilized HIT-T15 cells. Biochemical Journal. 1990;270:273–276. doi: 10.1042/bj2700273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AJ, Michel AD, Feniuk W, Humphrey PP. Somatostatin5 receptor-mediated [35S]-guanosine-5'-O-(3-thio)-triphosphate binding: Agonist potencies and the influence of sodium chloride on intrinsic activity. Molecular Pharmacology. 1997;51:1060–1069. doi: 10.1124/mol.51.6.1060. [DOI] [PubMed] [Google Scholar]
- Yamada M, Inanobe A, Kurachi Y. G protein regulation of potassium ion channels. Pharmacological Reviews. 1998;50:723–760. [PubMed] [Google Scholar]
- Yano H, Philipson LH, Kugler JL, Tokuyama Y, Davis EM, Le Beau MM, Nelson DJ, Bell GI, Takeda J. Alternative splicing of human inwardly rectifying K+ channel ROMK1 mRNA. Molecular Pharmacology. 1994;45:854–860. [PubMed] [Google Scholar]
- Yoshimoto Y, Fukuyama Y, Horio Y, Inanobe A, Gotoh M. Somatostatin induces hyperpolarization in pancreatic islet α cells by activating a G protein-gated K+ channel. FEBS Letters. 1999;444:265–269. doi: 10.1016/s0014-5793(99)00076-9. [DOI] [PubMed] [Google Scholar]