

Abstract
Chronically epileptic rats, produced by prior injection of pilocarpine, were used to investigate whether changes in intrinsic neuronal excitability may contribute to the epileptogenicity of the hippocampus in experimental temporal lobe epilepsy (TLE).
Paired extra-/intracellular electrophysiological recordings were made in the CA1 pyramidal layer in acute hippocampal slices prepared from control and epileptic rats and perfused with artificial cerebrospinal fluid (ACSF). Whereas orthodromic activation of CA1 neurons evoked only a single, stimulus-graded population spike in control slices, it produced an all-or-none burst of population spikes in epileptic slices.
The intrinsic firing patterns of CA1 pyramidal cells were determined by intrasomatic positive current injection. In control slices, the vast majority (97 %) of the neurons were regular firing cells. In epileptic slices, only 53 % the pyramidal cells fired in a regular mode. The remaining 47 % of the pyramidal cells were intrinsic bursters. These neurons generated high-frequency bursts of three to six spikes in response to threshold depolarizations. A subgroup of these neurons (10.1 % of all cells) also burst fired spontaneously even after suppression of synaptic activity.
In epileptic slices, burst firing in most cases (ca 70 %) was completely blocked by adding the Ca2+ channel blocker Ni2+ (1 mm) to, or removing Ca2+ from, the ACSF, but not by intracellular application of the Ca2+ chelater 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid (BAPTA), suggesting it was driven by a Ca2+ current.
Spontaneously recurring population bursts were observed in a subset of epileptic slices. They were abolished by adding 2 μm 6-cyano-7-nitro-quinoxaline-2,3-dione (CNQX) to the ACSF, indicating that synaptic excitation is critical for the generation of these events.
All sampled pyramidal cells fired repetitively during each population burst. The firing of spontaneously active bursters anteceded the population discharge, whereas most other pyramidal cells began to fire conjointly with the first population spike. Thus, spontaneous bursters are likely to be the initiators of spontaneous population bursts in epileptic slices.
The dramatic up-regulation of intrinsic bursting in CA1 pyramidal cells, particularly the de novo appearance of Ca2+-dependent bursting, may contribute to the epileptogenicity of the hippocampus in the pilocarpine model of TLE. These findings have important implications for the pharmacological treatment of medically refractory human TLE.
Temporal lobe epilepsy (TLE) is the most common human epileptic syndrome (reviewed by Engel et al. 1997). It is characterized by recurrent complex partial seizures, which often originate in the hippocampus or in adjacent mesial temporal lobe structures (Mathern et al. 1995). It is widely believed that alterations in excitatory and/or inhibitory synaptic functions lead to the evolution of chronically epileptogenic tissue in TLE. Indeed, numerous studies in animal models of TLE have disclosed a remarkable synaptic plasticity in the hippocampus during epileptogenesis, expressed in the formation of novel recurrent excitatory synaptic connections, up-regulation of synaptic N-methyl-d-aspartate (NMDA) receptors and reduction in γ-amino-butyric acidA (GABAA) receptor-mediated inhibition (reviewed by McNamara, 1994; Dudek & Spitz, 1997). In contrast, the possibility that persistent changes in intrinsic neuronal properties may also contribute to hippocampal epileptogenesis in TLE has received less attention (e.g. Yamada & Bilkey, 1991).
In the hippocampus, the vast majority of principal cells generate only one spike in response to intracellular injection of a brief (ca 3-5 ms) depolarizing current pulse (Schwartzkroin, 1975; Jensen et al. 1994). However, the intrinsic electrical response of these neurons can change to a burst of several spikes when extracellular K+ concentration (Jensen et al. 1994) or pH (Church & Baimbridge, 1991) increases, or when extracellular Ca2+ concentration (Azouz et al. 1996) or osmotic pressure (Azouz et al. 1997) decreases. Although these alterations affect excitatory and inhibitory synaptic transmission in different ways, they all provoke the appearance of epileptiform discharges (e.g. Haas & Jefferys, 1984; Yaari et al. 1986; Aram & Lodge, 1987; Andrew et al. 1989), suggesting that intrinsic bursting may be a critical epileptogenic mechanism in acute models of hippocampal epilepsy.
Though the concept of ‘epileptic neurons’ in chronic epilepsy was entertained over three decades ago (Ward, 1969), it is not known to date whether intrinsic bursting is an important excitatory and synchronizing factor in TLE. We have now investigated the possible involvement of intrinsic bursting in TLE using the widely known pilocarpine model (Turski et al. 1983). Here we report that the development of TLE in pilocarpine-treated rats is associated with a dramatic up-regulation of intrinsic bursting in hippocampal CA1 pyramidal cells. Furthermore, we show that this novel TLE-related intrinsic bursting is driven by a Ca2+-dependent ionic mechanism and may be responsible for the initiation of epileptiform events that synchronize all CA1 pyramidal cells. These findings may open new avenues for the therapy of patients with intractable TLE.
METHODS
Preparation of pilocarpine epileptic rats
Chronically epileptic rats were prepared according to established protocols (Turski et al. 1983), which were approved by the Hebrew University Animal Care and Use Committee. Briefly, male Sabra rats (150-200 g) were injected with a single high dose of the muscarinic agonist pilocarpine (300-380 mg kg−1i.p.), which induced status epilepticus in most (≈80 %) animals. Peripheral muscarinic effects were reduced by prior administration of methyl-scopolamine (1 mg kg−1s.c.; 30 min before injecting pilocarpine). Diazepam (4 mg kg−1i.p.) was administered to all animals 2 h after the pilocarpine injection. It terminated the convulsions in the responsive rats and sedated all animals. About 5 % of the rats that experienced status epilepticus died during the acute insult or shortly thereafter. Surviving animals were closely tended, hydrated and fed in the laboratory for 48 h, after which they were monitored daily for up to 2 months for the development of spontaneous epileptic seizures. Within 2-6 weeks after this treatment, all rats that had experienced status epilepticus developed a chronic epileptic condition, expressed in two to four spontaneous ‘limbic’ seizures (characterized by chewing, head nodding, forelimb clonus, rearing and falling) per week. These animals were used as the experimental epileptic group. The other rats (comprising ≈20 % of the pilocarpine-injected animals) remained behaviourally normal and were used as the sham-control group. A group of untreated, age-matched rats was used as the naive-control group.
Preparation of hippocampal slices
Animals were decapitated under ether anaesthesia. Both hippocampi were dissected out and kept in cold (4 °C) artificial cerebrospinal fluid (ACSF). Transverse slices (400 μm thick) were cut with a vibratome and the CA3/2 region in each slice was removed. The reduced slices (comprising CA1, dentate area and subiculum) were placed on a nylon mesh support in an interface chamber at 33.5 °C and perfused from below with oxygenated (95 % O2-5 % CO2) ACSF.
Solutions and drugs
Standard ACSF contained (mm): NaCl 124; KCl, 3.5; NaH2PO4, 1.25; MgSO4, 2; CaCl2, 2; NaHCO3, 26; and D-glucose, 10. In Ca2+-free ACSF, CaCl2 was replaced with 2 mm MnCl2. In Ni2+ ACSF, 1 mm MgCl2 was replaced with 1 mm NiCl2. In the latter two solutions, NaH2PO4 was omitted. Some ACSFs, where indicated, also contained the glutamate receptor antagonists 6-cyano-7-nitro-quinoxaline-2,3-dione (CNQX; 2 or 15 μm) and 2-amino-5-phosphono-valeric acid (APV; 50 μm) to block fast excitatory postsynaptic potentials, and the GABAA receptor antagonist bicuculline methiodide (10 μm) to block fast inhibitory postsynaptic potentials. All drugs were from Sigma (Israel), except CNQX (Tocris Cookson, UK).
Stimulation and recording
Bipolar platinum (50 μm) electrodes connected to a stimulator by an isolation unit were used for focal stimulation (1-20 V, 50-70 μs) of afferent fibres in stratum radiatum near the cut CA2/CA3 border (orthodromic stimulation) and of pyramidal cell axons in alveus (antidromic stimulation). Extracellular and intracellular recording sharp glass microelectrodes contained 1 m NaCl (5-10 MΩ) and 4 m potassium acetate (50-80 MΩ), respectively. They were inserted in the pyramidal layer with their tips separated by < 100 μm. An active bridge circuit in the amplifier (Axoprobe 2B, Axon Instruments) allowed simultaneous injection of current and measurement of membrane potential. In some experiments, 1,2-bis(o-aminophenoxy)ethane-N,N,N′, N′-tetra-acetic acid (BAPTA; 200 mm) was added to the potassium acetate solution. Negative current pulses (up to 0.5 nA; 10-30 min) were used to inject BAPTA into cells. The intracellular signals were filtered on-line at 5 kHz, digitized at a sampling rate of 10 kHz and stored by a Pentium personal computer using an acquisition system (TL-1 DMA and pCLAMP software, Axon Instruments).
Cell identification
Neurons in the pyramidal layer were identified as pyramidal cells if they responded with short latency spikes to antidromic stimulation and manifested spike frequency accommodation during sustained depolarization. All pyramidal cells accepted for this study had stable resting potential > -55 mV and overshooting action potentials.
Data analysis
Passive and active intrinsic properties were measured as described previously (Jensen et al. 1996). Briefly, small (0.1-0.5 nA), 400 ms long negative and positive current pulses were injected into the cell. The input resistance was calculated by plotting the steady-state voltages versus negative current amplitudes and measuring the slope of a linear regression of the plot. Spike threshold was defined as the membrane potential where the slope of the voltage trace increased abruptly during membrane charging induced by positive current pulses. Spike amplitude was measured as the voltage difference between the peak of the action potential and resting membrane potential (Vm). Spike width was calculated as spike duration at 50 % of the spike amplitude.
Averaged data are expressed as either means ± standard deviation (s.d.) or medians ± averaged absolute difference (a.d.). Differences between means and medians were evaluated using Student's unpaired t test and Kruskal-Wallis analysis of variance of ranks, respectively. The χ2-test was used to evaluate significance of differences between distributions. A significance level of 0.05 was used in all tests.
Terminology
In this study we defined a burst as a cluster of three or more closely spaced action potentials, riding on a distinct slow depolarizing envelope. Neurons capable of generating bursts in response to depolarizing stimuli or spontaneously are collectively termed bursters. The classification of bursters into three different subsets of increasing propensity to burst (Jensen et al. 1994) is described below.
RESULTS
Paired intra-/extracellular recordings were used to monitor simultaneously single cell and population activities of CA1 pyramidal cells in hippocampal slices obtained from epileptic rats (epileptic slices; 70 slices from 31 rats), pilocarpine-treated rats that did not develop TLE (sham-control slices; 17 slices from 6 rats), and age-matched untreated rats (naive-control slices; 27 slices from 19 rats).
Synaptic excitability in control versus epileptic slices
We first examined the responses of CA1 pyramidal cells in the three experimental groups to orthodromic stimulation. The intensity of stimulation was adjusted to vary from low intensity that evoked subliminal excitatory postsynaptic potentials (EPSPs) to high intensity that evoked maximal spike responses. Representative results are shown in Fig. 1. As expected (Schwartzkroin, 1975), the pyramidal cell responses in the naive-control slice consisted of a single population spike at the most (Fig. 1A). The intracellular correlate of this activity was a graded EPSP, which could trigger maximally a single spike, followed by an inhibitory postsynaptic potential (IPSP; Fig. 1A). Similar responses were seen in the sham-control slice (Fig. 1B), indicating that the combination of drugs used for TLE induction (methylscopolamine, pilocarpine and diazepam) per se did not have long-term effects on the synaptic excitability of the pyramidal cells. In marked contrast, the pyramidal cell responses in epileptic tissue were epileptiform in nature, comprising all-or-none bursts of population spikes, associated with large depolarization shifts and burst discharges in individual pyramidal cells (Fig. 1C). Similar differential responses were obtained in all naive- and sham-control slices versus epileptic slices.
Figure 1. Neuronal hyperexcitability and hypersynchrony in CA1 slices from epileptic rats.
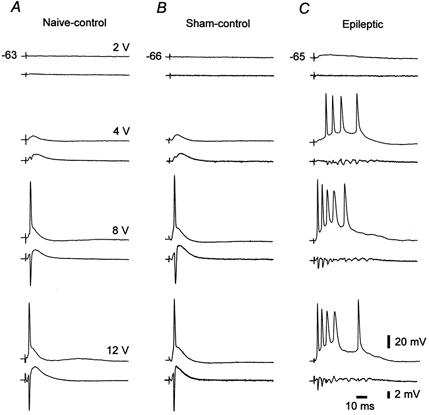
Comparative recordings were made in three CA1 slices obtained from naive-control (A), sham-control (B) and epileptic rats (C). Each pair of traces shows intra-/extracellular responses (upper/lower traces) of CA1 pyramidal cells to orthodromic stimuli of increasing intensity (from 2 to 12 V). Resting potentials (in mV) of the pyramidal cells in this and in all other figures are indicated to the left of the uppermost traces. Subthreshold stimulation evoked an EPSP-IPSP sequence in both control slices (A and B, 4 V), but a protracted EPSP in the epileptic slice (C, 2 V). Further increasing stimulus intensity evoked maximally a single spike in control tissue (as seen at both single cell and population levels; A and B, 8 and 12 V), but an all-or-none spike burst in epileptic tissue (C, 4, 8 and 12 V).
Notably, however, population spikes in epileptic slices (Fig. 1C) were considerably smaller than in naive- and sham-control tissue (Fig. 1A and B). The maximal amplitude of the first population spike in the orthodromically evoked response was 6.2 ± 2.2 mV (mean ±s.d.) in epileptic tissue compared to (and significantly different from; unpaired t test), 13.8 ± 4.4 and 12.2 ± 3.1 mV in naive- and sham-control tissue, respectively. The reduced population spikes in epileptic tissue most probably reflect the smaller neuronal density due to status epilepticus-induced death of CA1 pyramidal cells (Mello et al. 1993; Liu et al. 1994).
Intrinsic firing patterns
In hippocampal slices from normal animals, the maximal response of CA1 pyramidal cells to orthodromic stimulation is limited to a single spike due to feedforward and feedback synaptic inhibition (Alger & Nicoll, 1982). The all-or-none population burst responses in epileptic slices thus may be due to enhanced synaptic excitation, reduced synaptic inhibition, increased intrinsic excitability, or a combination of these factors. To test whether changes in the intrinsic excitability of CA1 pyramidal cells may be involved, we first determined their firing patterns in naive-control (36 cells), sham-control (38 cells) and epileptic tissue (139 cells). Long (150-200 ms) and brief (4-5 ms) rectangular depolarizing current pulses were somatically injected into each neuron. The intensity of the stimuli was increased in small steps until a spike response was obtained. The slices were perfused with ACSF containing the glutamate receptor antagonists CNQX (15 μm) and APV (50 μm). Orthodromically evoked responses were completely suppressed in this ACSF (Andreason et al. 1989).
In both naive- and sham-control groups, all but one (97.2 and 97.4 %, respectively) of the neurons were regular firing cells. That is, they generated a single spike or a short train of independent spikes as their minimal response to long stimuli and a single spike in response to brief stimuli. The remaining neuron in both naive- and sham-control groups (2.8 and 2.6 %, respectively) generated a burst in response to long, but not brief, stimuli (grade I burster, see below). The variant firing patterns of CA1 pyramidal cells in epileptic tissue are portrayed in Fig. 2. In marked contrast to both control groups, only 52.5 % of the pyramidal cells in the epileptic group fired in a regular pattern when stimulated with 1 to 2 × threshold current pulses (Fig. 2A). The remaining neurons (47.5 %) generated a burst as their minimal response to long stimuli (Fig. 2B-D). Within this group of bursters the propensity to burst fire was quite variable. Therefore, we classified them into three subgroups according to a previously suggested scheme (Jensen et al. 1994). Grade I bursters fired only one spike in response to a brief stimulus (Fig. 2Bb). Grade II bursters generated a burst also in response to a brief stimulus (Fig. 2Cb). Grade III bursters not only burst fired in response to any suprathreshold stimulus (Fig. 2Da and b), but also displayed spontaneous, rhythmic bursting (Fig. 2Dc and d), which was suppressed by hyperpolarizing the cell (see below, Fig. 6A). The overall proportion of grade I, II and III bursters in epileptic tissue was 19.4, 18.0 and 10.1 %, respectively.
Figure 2. Variant intrinsic firing patterns in CA1 pyramidal cells in epileptic tissue.
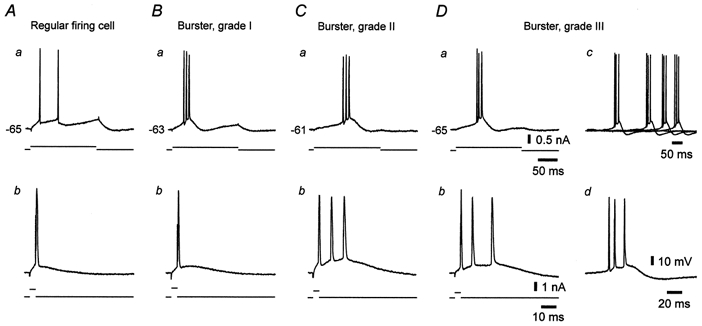
A-D, recordings from four different neurons arranged according to a gradient of increasing propensity to burst fire. The neurons were stimulated with threshold-straddling 200 ms (a) and 4 ms (b) depolarizing current pulses injected through the recording microelectrode. In each panel, upper and lower traces depict the neuronal response and the current stimulus, respectively. A, regular firing cell, responding with two separate spikes to the long stimulus (a) and with a single spike to the brief stimulus (b). B, grade I burster, responding with a burst to the long stimulus (a), but with a single spike to the brief stimulus (b). C, grade II burster, responding with bursts to both long (a) and brief stimuli (b). D, grade III burster, which in addition to bursting in response to long (a) and brief stimuli (b), also burst fired spontaneously at a frequency of 0.3 Hz (c, superposition of four sequential traces; one of the spontaneous bursts is shown on an expanded time scale in d).
Figure 6. Early recruitment of a grade III burster during spontaneous epileptiform events in epileptic tissue.
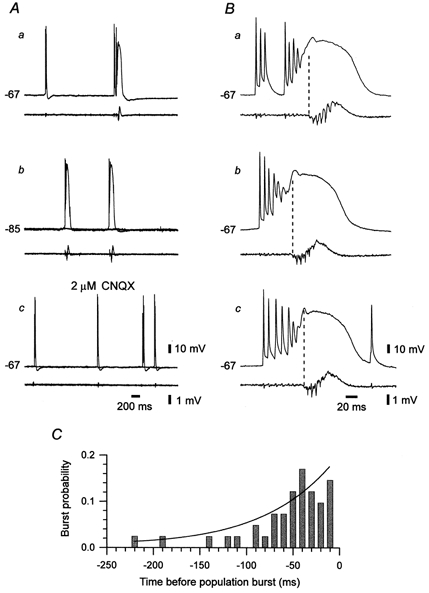
A, paired intra-/extracellular (upper/lower traces) recordings of spontaneous activity in CA1 in an epileptic slice. The intracellular recordings were obtained first at resting membrane potential (-67 mV, a), then at a hyperpolarized membrane potential (-85 mV; b) and finally at resting membrane potential again, but 30 min after addition of 2 μm CNQX to block polysynaptic activity (c). In b and c, two sequential responses are superimposed. The neuron displayed two types of spontaneous bursts, namely, intrinsic (left event in a) and network-driven (right event in a) bursts. Intrinsic bursts were not associated with a population burst (a) and were blocked by hyperpolarization (b) but not by CNQX (c). Network-driven bursts were associated with a population burst (a) and were blocked by CNQX (c) but not by hyperpolarization (b). B, temporal relation between the firing of the neuron and the population. In the three consecutive examples (a, b and c) the neuron burst fired shortly before the onset of population discharge (marked with dashed line). C, time histogram of burst probability of the neuron with respect to the onset of population discharge. Each bin depicts the probability that the neuron will start bursting during that 10 ms. The continuous line represents a single exponential function fitted to the experimental data. The time constant of exponential increase was 108.1 ms.
Pyramidal cells in CA1 are subjected to tonic GABAAergic inhibition (Alger & Nicoll, 1980), which may tonically suppress a native propensity to fire in burst mode. However, in eight regular firing CA1 pyramidal cells (in three naive-control, two sham-control, and three epileptic slices; two rats in each experimental group), adding 10 μm bicuculline to the ACSF to block GABAA receptors, had no effect on the firing mode. Thus, the appearance of intrinsic bursting in almost 50 % of the CA1 pyramidal cells in epileptic tissue (compared to ≈3 % in control tissue) is not due simply to a reduction in tonic GABAAergic inhibition (Brooks-Kayal et al. 1998). Rather, it must be due to a genuine change in the intrinsic properties of these neurons. Accordingly, we compared basic passive and active membrane properties of the pyramidal cells in the three experimental groups (Table 1). However, the three groups of pyramidal cells did not differ significantly (unpaired t test) in their resting membrane potential, input resistance, and action potential threshold, amplitude and width.
Table 1.
Passive and active membrane properties in control versus epileptic CA1 pyramidal cells
| Naive-control | Sham-control | Epileptic | |
|---|---|---|---|
| Vm (mV) | −64.3 ± 3.1 (36) | −64.0 ± 3.9 (38) | −63.8 ± 3.8 (139) |
| Rm (MΩ) | 45.7 ± 18.3 (32) | 45.4 ± 14.3 (32) | 43.3 ± 17.1 (125) |
| AP threshold (mV) | −53.9 ± 3.8 (36) | −53.0 ± 4.2 (38) | −53.2 ± 4.5 (139) |
| AP amplitude (mV) | 90.2 ± 8.3 (36) | 89.6 ± 6.0 (38) | 89.5 ± 8.9 (139) |
| AP width (ms) | 2.0 ± 0.2 (35) | 2.1 ± 0.4 (33) | 2.0 ± 0.3 (104) |
For each of the three groups of animals examined (naive-control, sham-control and epileptic), the resting membrane potential (Vm) and input resistance (Rm), and the action potential (AP) threshold, amplitude and width are provided as means ± S.D. (n), where S.D. is the standard deviation and n is the number of observations. No significant differences in these parameters between the control groups and the epileptic group were found (Student's unpaired t test).
Effects of blocking Ca2+ currents
In normal CA1 pyramidal cells bathed in standard or in high-K+ saline, intrinsic bursting evoked by somatic depolarization is insensitive to blockage of Ca2+ currents (Azouz et al. 1996). Rather, it is driven by a persistent Na+ current that is expressed by these neurons (French et al. 1990). To test whether a similar ionic mechanism mediates intrinsic bursting in epileptic tissue, we examined the consequences of blocking Ca2+ currents on this activity. This was achieved either by adding the Ca2+ channel antagonist Ni2+ (1 mm) to the ACSF, or by removing Ca2+ from the ACSF (the total concentration of divalent cations was maintained constant; see Methods). In a series of 13 bursters examined, addition of 1 mm Ni2+ to the ACSF reversibly suppressed burst firing in 10 neurons (77 %), as illustrated in Fig. 3A. Likewise, in another series of 10 bursters, changing to nominally Ca2+-free ACSF reversibly suppressed burst firing in seven neurons, as illustrated in Fig. 3B.
Figure 3. Suppression of Ca2+ currents blocks intrinsic bursting in CA1 pyramidal cells in epileptic tissue.
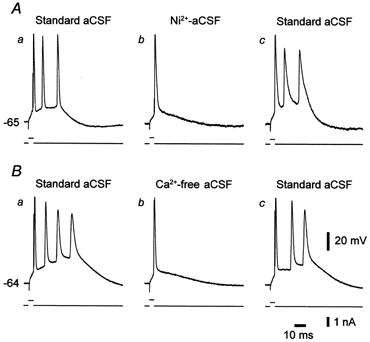
In each panel, upper and lower traces depict the neuronal response and the current stimulus, respectively. A, intrinsic bursting in a CA1 pyramidal cell perfused with standard ACSF (a) was blocked 22 min after changing to Ni2+-ACSF (b). The effect reversed after a 30 min wash with standard ACSF (c). B, intrinsic bursting in another CA1 pyramidal cell perfused with standard ACSF (a) was blocked 20 min after changing to Ca2+-free ACSF (b). The effect reversed after 30 min wash with standard ACSF (c).
Taken together, these data suggest that in epileptic tissue voltage-gated Ca2+ currents are critical for burst generation in most (ca 75 %) bursters. In the remaining bursters, persistent Na+ current may be sufficiently large to generate the burst responses independent of Ca2+ currents (Azouz et al. 1996).
Effects of chelating intracellular Ca2+
Voltage-gated Ca2+ currents may depolarize the neurons directly to produce the slow depolarization that triggers intrinsic burst discharge in epileptic tissue. Alternatively, they may secondarily activate other inward membrane currents possibly expressed in these neurons, such as the Ca2+-activated non-selective cationic current (Haj-Dahmane & Andrade, 1997). To distinguish between these two possibilities, we examined how intrinsic bursting in epileptic tissue is affected by intracellular application of the Ca2+ chelator BAPTA, previously shown to block the Ca2+-activated non-selective cationic current (Haj-Dahmane & Andrade, 1997). As illustrated in Fig. 4, BAPTA application had no effect upon intrinsic bursting (Fig. 4Aa and Ba). Nonetheless, BAPTA consistently suppressed the slow after-hyperpolarization that follows repetitive firing (Fig. 4Ab and Bb), which is generated by a Ca2+-activated K+ current (Madison & Nicoll, 1984). Similar results were obtained in all five bursters injected with BAPTA. In three cases 1 mm Ni2+ was added to the ACSF after BAPTA injection, resulting in burst suppression.
Figure 4. Intracellular BAPTA injection does not suppress intrinsic bursting.
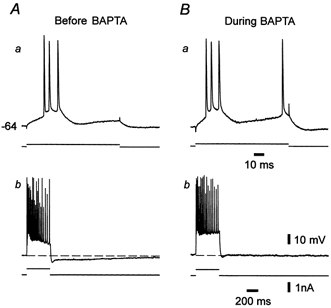
A, before injecting BAPTA the neuron burst fired when depolarized by a threshold-straddling 200 ms stimulus (a). Strong 400 ms depolarization of the neuron evoked many action potentials, and was followed by a medium and slow after-hyperpolarization (b). B, injecting BAPTA for 30 min did not affect the burst response (a), but suppressed the slow after-hyperpolarization (b).
These observations strongly suggest that a voltage-gated Ca2+ current by itself, rather than by activating another inward current, furnishes the driving force for the abnormal intrinsic bursting in epileptic tissue.
Spontaneous population bursting in epileptic slices
Spontaneous epileptiform discharges were observed in extracellular recordings in 11 of 58 (19 %) epileptic slices perfused with standard ACSF. Their frequency was quite low and variable across different slices, ranging between 0.06 and 0.1 Hz. A representative example of this epileptiform activity is shown in Fig. 5. As in other spontaneously active slices, the epileptiform discharges consisted of a positive-going field potential lasting 80-150 ms, associated with a high-frequency (120-180 Hz) cluster of five to ten population spikes (Fig. 5A and B, lower traces in a-c). This component was followed by a negative-going field potential lasting up to 200 ms. None of the naive- or sham-control slices displayed spontaneous epileptiform activity in standard ACSF.
Figure 5. Spontaneous epileptiform events in epileptic tissue.
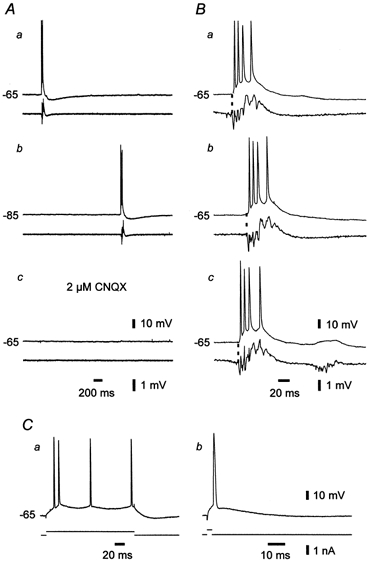
A, paired intra-/extracellular (upper/lower traces) recordings of spontaneous activity in CA1 in an epileptic slice. The intracellular recordings were obtained at resting membrane potential (-65 mV, a), at a hyperpolarized membrane potential (-85 mV; b) and at resting membrane potential 30 min after addition of 2 μm CNQX (c). The neuron displayed only network-driven burst activity (a), which was blocked by CNQX (c) but not by hyperpolarization (b). B, temporal relation between the firing of the neuron and the population. In the three sequential examples (a, b and c) the neuron fired shortly after the onset of population discharge (marked with dashed line). C, the responses of the neuron (upper traces) to brief (b) and long (a) depolarizing current pulses (lower traces) identify it as a regular firing cell.
The intracellular correlate of the positive-going field potential in the spontaneous epileptiform event was a large depolarization shift of variable amplitude (15 to 50 mV), triggering a burst of three to eight fast action potentials (Fig. 5B, upper traces in a-c). It was followed by a slow after-hyperpolarization lasting up to 2 s (Fig. 5A, upper trace in a). Hyperpolarizing the neuron up to 20 mV did not suppress the depolarization shift and associated spike burst (Fig. 5Ab), consistent with the view that they are driven by neuronal discharge reverberating within the neuronal network (Johnston & Brown, 1981). Accordingly, they were abolished in all the 11 slices by perfusing them with ACSF containing a low concentration (2 μm) of CNQX (Fig. 5Ac), which reduces excitatory synaptic transmission in hippocampal pathways (Andreasen et al. 1989).
Recruitment of individual pyramidal cells during population bursts
All pyramidal cells monitored during spontaneous epileptiform activity (n = 22), regular firing cells and bursters alike, fired a burst of action potentials during each spontaneous population burst. Thus, the entire pyramidal cell population is recruited into the discharge zone of each epileptiform event. However, we found that the exact timing of neuronal recruitment relative to the onset of a population burst was differentially dependent on the intrinsic firing pattern of the neuron. In order to describe this observation quantitatively, we defined as ‘recruitment delay’ the time elapsing between the onset of the population burst and the peak of the first intracellular spike. For each neuron, a recruitment delay was determined by measuring and averaging the delays in a series of 15 consecutive epileptiform events. Positive values indicated that the individual neuron was recruited after the beginning of the population burst; negative values indicated that the firing of the neuron anteceded the population burst.
The pyramidal cell depicted in Fig. 5 was a regular firing cell, as deduced from its characteristic responses to long (Fig. 5Ca) and brief depolarizing current pulses (Fig. 5Cb). The temporal relation between the firing of this neuron and the synchronous discharge of the population during three sequential epileptiform events is illustrated in Fig. 5B. In all cases, the neuron began to fire concurrently with, or slightly after, the first population spike. A similar type of neuronal recruitment concurring with the first population spike was observed in all regular firing cells (n = 8) and grade I bursters (n = 4) examined in this way (altogether n = 12). The median (±a.d.) recruitment delays for the regular firing cells and grade I bursters were 1.1 ± 0.8 and 0.2 ± 0.4 ms, respectively. The difference between the medians of the two groups was not significant (Kruskal-Wallis analysis of variance of ranks). Furthermore, all these neurons were completely silent between sequential epileptiform events. Grade II bursters (n = 4) also did not fire between sequential epileptiform events. However, the recruitment of these neurons commenced slightly before the beginning of these events. The median (±a.d.) recruitment delay for grade II bursters was -1.0 ± 1.7 ms. It was significantly different from the delays of the two subsets of pyramidal cells described above (Kruskal-Wallis analysis).
The firing of grade III bursters in slices displaying spontaneous epileptiform activity consisted of two types of cellular bursts, namely network-driven bursts and intrinsic bursts. As illustrated in Fig. 6A, these two types of bursts differed in several respects. Firstly, network-driven bursts concurred with population bursts, whereas spontaneous intrinsic bursts occurred also between sequential population bursts (Fig. 6Aa). Secondly, hyperpolarizing the neuron did not affect the frequency of network-driven bursts but totally abolished the spontaneous intrinsic bursts in this neuron (Fig. 6Ab). Thirdly, changing to ACSF containing 2 μm CNQX abolished the network-driven cellular bursts and associated population bursts, without affecting the spontaneous intrinsic bursts (Fig. 6Ac).
The recruitment of grade III bursters during spontaneous population bursts markedly differed from that of the other pyramidal cells. Figure 6B describes the temporal relation between the firing of a spontaneous burster and the synchronous discharge of the population during three successive epileptiform events. In all three events, the firing of the pyramidal cell anteceded the synchronous discharge of the population. Indeed, as summarized in Fig. 6C for 40 successive epileptiform bursts, the overall probability that the neuron would fire within the 220 ms period preceding an epileptiform event was 1, and this probability increased nearly exponentially during this premonitory period. A similar early recruitment was observed in all grade III bursters examined (n = 6). The median (±a.d.) recruitment delay for this subset of neurons was -6.1 ± 18.2 ms. It was significantly different from that of the other three groups of CA1 pyramidal cells.
Because neurons in the isolated CA1 are not synchronized by exogenous inputs (e.g. from CA3), initiation of population bursts in CA1 must involve local neuronal interactions. The finding that the discharge of grade III bursters precedes that of all other neurons indicates that this spontaneously active subset of bursters may constitute the pacemaker of spontaneous epileptiform events in this chronically epileptic tissue.
DISCUSSION
In this report we describe a marked and long-lasting change in the firing behaviour of CA1 pyramidal cells associated with the development of TLE, that is the de novo appearance of intrinsic bursting in ≈50 % of hitherto regular firing cells. In most cases bursts are generated by a mechanism that critically involves the activation of voltage-gated Ca2+ channels. Spontaneously active bursters appear to be responsible for the initiation of epileptiform bursts that spread through and synchronize the entire neuronal network. Together, these findings provide a novel working hypothesis that may account for the hyperexcitability and hypersynchrony in experimental TLE.
Up-regulation of intrinsic bursting
We found about a 20-fold increase in the fraction of intrinsic bursters in epileptic versus control hippocampal tissue. This dramatic change cannot be explained simply by preferential death of regular firing because bursters are scarce in control tissue. Moreover, we show here that burst firing in epileptic tissue is Ca2+ dependent, unlike intrinsic bursting in normal pyramidal cells bathed in standard or in high-K+ ACSF, which is Ca2+ independent (Azouz et al. 1996). Thus, the development of TLE is associated with de novo induction of Ca2+-dependent intrinsic bursting in native regular firing cells.
What may be the cause of the up-regulation of intrinsic bursting following pilocarpine-induced status epilepticus? Intrinsic bursts are generated in neurons having particularly large spike after-depolarizations (Wong & Prince, 1981; Jensen et al. 1996). Slow somatic and/or dendritic inward currents that are activated by the first spike drive these after-depolarizations. An imbalance between slow inward and outward currents in favour of the former would increase the size of the spike after-depolarization and hence the propensity for burst generation. Given the Ca2+ dependency of bursting in the epileptic tissue described here, it is possible that the pilocarpine-induced status epilepticus initiates an increase in Ca2+ current density in CA1 pyramidal cells, as reportedly occurs in the electrical kindling model of TLE (Faas et al. 1996). Interestingly, hippocampal kindling is also associated with augmentation of intrinsic bursting, although the effect is less dramatic than in the present model and its ionic basis has not been determined (Yamada & Bilkey, 1991). An up-regulation of Ca2+ currents in hippocampal neurons may be mediated by neurotrophins (Levine et al. 1995), whose expression is stimulated during the pilocarpine-induced status epilepticus (Schmidt-Kastner et al. 1996).
An alternative to this ‘Ca2+ current increase’ hypothesis would be a ‘K+ current decrease’ hypothesis. Indeed, it has been shown recently that blockage of dendritic K+ channels leads to Ca2+-dependent intrinsic bursting in somata of CA1 pyramidal cells (Magee & Carruth, 1999), which is not seen in ordinary conditions (Azouz et al. 1996). Our present data cannot differentiate between these two hypotheses. It may be relevant, however, that no changes in K+ current density were noted in CA1 pyramidal cells in the kindling model of TLE (Vreugdenhil & Wadman, 1995), despite the reported augmentation in intrinsic bursting (Yamada & Bilkey, 1991).
Generation of spontaneous population bursts
We found that irrespective of their intrinsic firing pattern, all the CA1 pyramidal cells investigated intracellularly were transiently depolarized and fired several spikes during each spontaneous population burst. Thus, the discharge zone of this epileptiform activity comprises the entire CA1 pyramidal cell network. Two important questions regarding the generation of this activity are (i) what mechanisms initiate the excitatory process leading to a population burst and (ii) how does this process spread to all neurons in the network?
Our finding that the firing of spontaneously active bursters preceded each population burst suggests that this subset of pyramidal cells plays a critical role in the initiation of the excitatory process. Spontaneous bursters are likely candidates for initiating epileptiform bursts for two obvious reasons. Firstly, by bursting spontaneously they serve as pacemakers for their target neurons. Secondly, due to EPSP facilitation and temporal summation, a presynaptic burst will depolarize the postsynaptic neurons much more effectively than a solitary presynaptic action potential (Miles & Wong, 1986). It is also possible that synaptic excitation in bursters is stronger than in regular firing cells, as was seen in native neocortical pyramidal cells (Changac-Amitai & Connors, 1989). Together, these features predict that neuronal discharge originating in spontaneous bursters would spread to other spontaneous and conditional bursters before the remaining neuronal population would be recruited.
Intrinsic bursting was suggested previously to play a cardinal role in the initiation of epileptiform events in some acute in vitro models of hippocampal and neocortical epilepsy. Thus, spontaneous bursters were shown to be the forerunners of interictal-like and ictal-like epileptiform events in CA1 slices bathed in high-K+ ACSF (Jensen & Yaari, 1997). Likewise, in disinhibited neocortical slices, threshold stimulation of afferent fibres evoked long-latency epileptiform bursts, which originated distinctly in the cortical layers containing intrinsically bursting pyramidal cells (i.e. layers IV-V; Connors, 1984; Chagnac-Amitai & Connors, 1989).
The suppression of spontaneous population bursting in epileptic slices by CNQX indicates that recurrent excitatory transmission plays a critical role in the generation of this activity. In the CA1 field of normal rats, recurrent synapses between pyramidal cells are sparse (Deuchers & Thomson, 1996). Yet, even this loose connectivity can support the local spread of neuronal discharge once GABAAergic inhibition is reduced (Crepel et al. 1997), as may occur in CA1 in pilocarpine-treated epileptic rats (Brooks-Kayal et al. 1998). Additionally, the death of a substantial number of CA1 pyramidal cells following the pilocarpine-induced status epilepticus, is associated with local sprouting of axon collaterals and formation of novel excitatory synapses between surviving CA1 pyramidal cells (Esclapez et al. 1999). An increase in recurrent excitation due to reduced GABAAergic inhibition and/or glutamatergic synaptogenesis would provide pathways for the spread of intrinsic bursting within the neural aggregate, thus facilitating the generation of epileptiform events. Electrical interactions between neighbouring pyramidal cells mediated via gap junctions or through the extracellular space may also contribute to this spread, as well as to the spike-to-spike synchronization during each population spike (reviewed by Jefferys, 1995), but it is not known whether these mechanisms are modified in pilocarpine-treated epileptic rats.
It was recently shown that synaptically evoked bursts, but not solitary spikes, enable N-methyl-d-aspartate (NMDA) receptor-dependent long-term potentiation of EPSPs in CA1 pyramidal cells (Thomas et al. 1998; Pike et al. 1999). Presumably the depolarization associated with the burst discharge is more effective than a single spike in removing the Mg2+ block from postsynaptic NMDA receptor-channels, which is a prerequisite for the induction of long-term potentiation (Bliss & Collingridge, 1993). Therefore, it is highly likely that glutamatergic synapses in epileptic tissue would be strongly and persistently potentiated consequent to the up-regulation of intrinsic bursting. It is interesting to speculate that intrinsic bursting also may promote sprouting of axon collaterals in epileptic tissue, given that this process also is NMDA receptor dependent (Sutula et al. 1996). Through these plasticity mechanisms, intrinsic bursting may play a critical role in the progression of the epileptic condition in TLE.
Implications for human TLE
The pilocarpine rat model of TLE resembles the human disorder in several critical aspects. Not only do the animals experience several spontaneous ‘limbic’ seizures daily or weekly, but the profile of antiepileptic drug efficacy in suppressing these seizures is similar to that of human complex partial seizures (Leite & Cavalheiro, 1995). In both the animal model (Cavalheiro et al. 1991) and the human disorder (Mathern et al. 1995), the hippocampus is critically involved in seizure generation and propagation. Additionally, histological examination of pilocarpine-treated epileptic rats reveals neuronal death, gliosis and sprouting of axon collaterals in distinctive hippocampal fields (Mellow et al. 1993; Liu et al. 1994), which resemble the neuropathological changes commonly associated with human TLE (Armstrong, 1993). Therefore, the mechanisms responsible for the development and expression of TLE in pilocarpine-treated rats may be pertinent to clinical TLE.
The dramatic up-regulation of Ca2+-dependent intrinsic bursting and its active role in the generation of epileptiform events reported here might have important implications for the drug treatment of TLE. Most traditional and new drugs currently used for seizure control in TLE patients are thought to elevate seizure threshold by suppressing voltage-gated Na+ channels, enhancing GABAergic inhibition or reducing glutamatergic excitation (Macdonald & Kelly, 1995). However, seizures of temporal lobe origin are frequently resistant to these drugs (Mattson, 1992), stressing the need for drugs with novel mechanisms of action. Therefore, it would be extremely important to determine whether Ca2+-dependent intrinsic bursting occurs also in epileptogenic regions in human TLE, and which of the multiple types of Ca2+ channels expressed in forebrain neurons underlie this abnormal activity. It has been shown recently that some types of Ca2+ channels are up-regulated in hippocampal neurons isolated from TLE patients (Beck et al. 1998). If these Ca2+ channels drive intrinsic bursting, then their selective pharmacological blockage undoubtedly would reduce hippocampal hyperexcitability in TLE and may provide an effective way for controlling seizures in this devastating epileptic disorder.
Acknowledgments
We thank Dr H. Beck from critical reading of the manuscript. Supported by the German-Israel Program of BMBF and MOS and the Israel Science Foundation founded by the Israel Academy of Sciences. E.R.G.S. was supported by FAPESP and CNPq.
References
- Alger BE, Nicoll RA. Spontaneous inhibitory post-synaptic potentials in hippocampus: mechanism for tonic inhibition. Brain Research. 1980;200:195–200. doi: 10.1016/0006-8993(80)91108-7. [DOI] [PubMed] [Google Scholar]
- Alger BE, Nicoll RA. Feed-forward dendritic inhibition in rat hippocampal pyramidal cells studied in vitro. Journal of Physiology. 1982;328:105–123. doi: 10.1113/jphysiol.1982.sp014255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasen M, Lambert JD, Jensen MS. Effects of new non-N-methyl-d-aspartate antagonists on synaptic transmission in the in vitro rat hippocampus. Journal of Physiology. 1989;414:317–336. doi: 10.1113/jphysiol.1989.sp017690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrew RD, Fagan M, Ballyk BA, Rosen AS. Seizure susceptibility and the osmotic state. Brain Research. 1989;498:175–180. doi: 10.1016/0006-8993(89)90417-4. [DOI] [PubMed] [Google Scholar]
- Aram JA, Lodge D. Epileptiform activity induced by alkalosis in rat neocortical slices: block by antagonists of N-methyl-d-aspartate. Neuroscience Letters. 1987;83:345–350. doi: 10.1016/0304-3940(87)90112-1. [DOI] [PubMed] [Google Scholar]
- Armstrong DD. The neuropathology of temporal lobe epilepsy. Journal of Neuropathology and Experimental Neurology. 1993;52:433–443. doi: 10.1097/00005072-199309000-00001. [DOI] [PubMed] [Google Scholar]
- Azouz R, Alroy G, Yaari Y. Modulation of endogenous firing patterns by osmolarity in rat hippocampal neurones. Journal of Physiology. 1997;502:175–187. doi: 10.1111/j.1469-7793.1997.175bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azouz R, Jensen MS, Yaari Y. Ionic basis of spike after-depolarization and burst generation in adult rat hippocampal CA1 pyramidal cells. Journal of Physiology. 1996;492:211–223. doi: 10.1113/jphysiol.1996.sp021302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck H, Steffens R, Elger CE, Heinemann U. Voltage-dependent Ca2+ currents in epilepsy. Epilepsy Research. 1998;32:321–332. doi: 10.1016/s0920-1211(98)00062-x. [DOI] [PubMed] [Google Scholar]
- Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Brooks-Kayal AR, Shumate MD, Jin H, Rikhter T, Coulter DA. Selective changes in single cell GABAA receptor subunit expression and function in temporal lobe epilepsy. Nature Medicine. 1998;4:1166–1172. doi: 10.1038/2661. [DOI] [PubMed] [Google Scholar]
- Cavalheiro EA, Leite JP, Bortolotto ZA, Turski WA, Ikonomidou C, Turski L. Long-term effects of pilocarpine in rats: structural damage of the brain triggers kindling and spontaneous recurrent seizures. Epilepsia. 1991;32:778–782. doi: 10.1111/j.1528-1157.1991.tb05533.x. [DOI] [PubMed] [Google Scholar]
- Chagnac-Amitai Y, Connors BW. Synchronized excitation and inhibition driven by intrinsically bursting neurons in neocortex. Journal of Neurophysiology. 1989;62:1149–1162. doi: 10.1152/jn.1989.62.5.1149. [DOI] [PubMed] [Google Scholar]
- Church J, Baimbridge KG. Exposure to high-pH medium increases the incidence and extent of dye coupling between rat hippocampal CA1 pyramidal neurons in vitro. Journal of Neuroscience. 1991;11:3289–3295. doi: 10.1523/JNEUROSCI.11-10-03289.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connors BW. Initiation of synchronized neuronal bursting in neocortex. Nature. 1984;310:685–687. doi: 10.1038/310685a0. [DOI] [PubMed] [Google Scholar]
- Crepel V, Khazipov R, Ben-Ari Y. Blocking GABA inhibition reveals AMPA- and NMDA-receptor-mediated polysynaptic responses in the CA1 region of the rat hippocampus. Journal of Neurophysiology. 1997;77:2071–2082. doi: 10.1152/jn.1997.77.4.2071. [DOI] [PubMed] [Google Scholar]
- Deuchars J, Thomson AM. CA1 pyramid-pyramid connections in rat hippocampus in vitro: dual intracellular recordings with biocytin filling. Neuroscience. 1996;74:1009–1018. doi: 10.1016/0306-4522(96)00251-5. [DOI] [PubMed] [Google Scholar]
- Dudek FE, Spitz M. Hypothetical mechanisms for the cellular and neurophysiologic basis of secondary epileptogenesis: proposed role of synaptic reorganization. Journal of Clinical Neurophysiology. 1997;14:90–101. doi: 10.1097/00004691-199703000-00002. [DOI] [PubMed] [Google Scholar]
- Engel J, Jr, Williamson PD, Wieser H-G. Mesial temporal lobe epilepsy. In: Engel J Jr, Pedley TA, editors. Epilepsy: A Comprehensive Textbook. Philadelphia: Lippincott-Raven Publishers; 1997. pp. 2417–2426. [Google Scholar]
- Esclapez M, Hirsch JC, Ben-Ari Y, Bernard C. Newly formed excitatory pathways provide a substrate for hyperexcitability in experimental temporal lobe epilepsy. Journal of Comparative Neurology. 1999;408:449–460. doi: 10.1002/(sici)1096-9861(19990614)408:4<449::aid-cne1>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Faas GC, Vreugdenhill M, Wadman W. Calcium currents in pyramidal CA1 neurons in vitro after kindling epileptogenesis in the hippocampus of the rat. Journal of Neuroscience. 1996;75:57–67. doi: 10.1016/0306-4522(96)00254-0. [DOI] [PubMed] [Google Scholar]
- French CR, Sah P, Buckett KJ, Gage PW. A voltage-dependent persistent sodium current in mammalian hippocampal neurons. Journal of General Physiology. 1990;95:1139–1157. doi: 10.1085/jgp.95.6.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas HL, Jefferys JG. Low-calcium field burst discharges of CA1 pyramidal neurones in rat hippocampal slices. Journal of Physiology. 1994;354:185–201. doi: 10.1113/jphysiol.1984.sp015371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haj-Dahmane S, Andrade R. Calcium-activated cation nonselective current contributes to the fast afterdepolarization in rat prefrontal cortex neurons. Journal of Neurophysiology. 1997;78:1983–1989. doi: 10.1152/jn.1997.78.4.1983. [DOI] [PubMed] [Google Scholar]
- Hirsch JC, Agassandian C, Merchan-Perez A, Ben-Ari Y, Defelipe J, Esclapez M, Bernard C. Deficit of quantal release of GABA in experimental models of temporal lobe epilepsy. Nature Neuroscience. 1999;2:499–500. doi: 10.1038/9142. [DOI] [PubMed] [Google Scholar]
- Jefferys JGR. Nonsynaptic modulation of neuronal activity in the brain: electric currents and extracellular ions. Physiological Reviews. 1995;75:689–723. doi: 10.1152/physrev.1995.75.4.689. [DOI] [PubMed] [Google Scholar]
- Jensen MS, Azouz R, Yaari Y. Variant firing patterns in rat hippocampal pyramidal cells modulated by extracellular potassium. Journal of Neurophysiology. 1994;71:831–839. doi: 10.1152/jn.1994.71.3.831. [DOI] [PubMed] [Google Scholar]
- Jensen MS, Azouz R, Yaari Y. Spike after-depolarization and burst generation in adult rat hippocampal CA1 pyramidal cells. Journal of Physiology. 1996;492:199–210. doi: 10.1113/jphysiol.1996.sp021301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen MS, Yaari Y. Role of intrinsic burst-firing, potassium accumulation, and electrical coupling in the elevated potassium model of hippocampal epilepsy. Journal of Neurophysiology. 1997;77:1224–1233. doi: 10.1152/jn.1997.77.3.1224. [DOI] [PubMed] [Google Scholar]
- Johnston D, Brown TH. Giant synaptic potential hypothesis for epileptiform activity. Science. 1981;211:294–297. doi: 10.1126/science.7444469. [DOI] [PubMed] [Google Scholar]
- Leite JP, Cavalheiro EA. Effects of conventional antiepileptic drugs in a model of spontaneous recurrent seizures in rats. Epilepsy Research. 1995;20:93–104. doi: 10.1016/0920-1211(94)00070-d. [DOI] [PubMed] [Google Scholar]
- Levine ES, Dreyfus CF, Black IB, Plummer MR. Differential effects of NGF and BDNF on voltage-gated calcium currents in embryonic basal forebrain neurons. Journal of Neuroscience. 1995;15:3084–3091. doi: 10.1523/JNEUROSCI.15-04-03084.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Nagao T, Desjardins GC, Gloor P, Avoli M. Quantitative evaluation of neuronal loss in the dorsal hippocampus in rats with long-term pilocarpine seizures. Epilepsy Research. 1994;17:237–247. doi: 10.1016/0920-1211(94)90054-x. [DOI] [PubMed] [Google Scholar]
- Macdonald RL, Kelly KM. Antiepileptic drug mechanisms of action. Epilepsia. 1995;2(suppl):S2–12. doi: 10.1111/j.1528-1157.1995.tb05996.x. [DOI] [PubMed] [Google Scholar]
- McNamara JO. Cellular and molecular basis of epilepsy. Journal of Neuroscience. 1994;14:3413–3425. doi: 10.1523/JNEUROSCI.14-06-03413.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison DV, Nicoll RA. Control of the repetitive discharge of rat CA1 pyramidal neurones in vitro. Journal of Physiology. 1984;354:319–331. doi: 10.1113/jphysiol.1984.sp015378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC, Carruth M. Dendritic voltage-gated ion channels regulate the action potential firing mode of hippocampal CA1 pyramidal neurons. Journal of Neurophysiology. 1999;82:1895–1901. doi: 10.1152/jn.1999.82.4.1895. [DOI] [PubMed] [Google Scholar]
- Mathern GW, Babb TL, Pretorius JK, Melendez M, Levesque MF. The pathophysiologic relationships between lesion pathology, intracranial ictal EEG onsets, and hippocampal neuron losses in temporal lobe epilepsy. Epilepsy Research. 1995;21:133–147. doi: 10.1016/0920-1211(95)00014-2. [DOI] [PubMed] [Google Scholar]
- Mattson RH. Drug treatment of partial epilepsy. Advances in Neurology. 1992;57:643–650. [PubMed] [Google Scholar]
- Mello LE, Cavalheiro EA, Tan AM, Kupfer WR, Pretorius JK, Babb TL, Finch DM. Circuit mechanisms of seizures in the pilocarpine model of chronic epilepsy, cell loss and mossy fiber sprouting. Epilepsia. 1993;34:985–995. doi: 10.1111/j.1528-1157.1993.tb02123.x. [DOI] [PubMed] [Google Scholar]
- Miles R, Wong RKS. Excitatory synaptic interactions between CA3 neurones in the guinea-pig hippocampus. Journal of Physiology. 1986;373:397–418. doi: 10.1113/jphysiol.1986.sp016055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike FG, Meredith RM, Olding AW, Paulsen O. Postsynaptic bursting is essential for ‘Hebbian’ induction of associative long-term potentiation at excitatory synapses in rat hippocampus. Journal of Physiology. 1999;518:571–576. doi: 10.1111/j.1469-7793.1999.0571p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Kastner R, Humpel C, Wetmore C, Olson L. Cellular hybridization for BDNF, trkB, and NGF mRNAs and BDNF-immunoreactivity in rat forebrain after pilocarpine-induced status epilepticus. Experimental Brain Research. 1996;107:331–347. doi: 10.1007/BF00230416. [DOI] [PubMed] [Google Scholar]
- Schwartzkroin PA. Characteristics of CA1 neurons recorded intracellularly in the hippocampal in vitro slice preparation. Brain Research. 1975;85:423–436. doi: 10.1016/0006-8993(75)90817-3. [DOI] [PubMed] [Google Scholar]
- Sutula T, Koch J, Golarai G, Watanabe Y, McNamara JO. NMDA receptor dependence of kindling and mossy fiber sprouting: evidence that the NMDA receptor regulates patterning of hippocampal circuits in the adult brain. Journal of Neuroscience. 1996;16:7398–7406. doi: 10.1523/JNEUROSCI.16-22-07398.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MJ, Watabe AM, Moody TD, Makhinson M, O'Dell TJ. Postsynaptic complex spike bursting enables the induction of LTP by theta frequency synaptic stimulation. Journal of Neuroscience. 1998;18:7118–7126. doi: 10.1523/JNEUROSCI.18-18-07118.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turski WA, Cavalheiro EA, Schwarz M, Czuczwar SJ, Kleinrok Z, Turski L. Limbic seizures produced by pilocarpine in rats: behavioural, electroencephalographic and neuropathological study. Behavioral Brain Research. 1983;9:315–335. doi: 10.1016/0166-4328(83)90136-5. [DOI] [PubMed] [Google Scholar]
- Vreugdenhil M, Wadman WJ. Potassium currents in isolated CA1 neurons of the rat after kindling epileptogenesis. Neuroscience. 1995;66:805–813. doi: 10.1016/0306-4522(94)00587-u. [DOI] [PubMed] [Google Scholar]
- Ward AA., Jr . The epileptic neuron: chronic foci in animals and man. In: Jasper HH, Ward AA Jr, Pope A, editors. Basic Mechanisms of the Epilepsies. Boston: Little, Brown and Company; 1969. pp. 263–288. [Google Scholar]
- Wong RKS, Prince DA. Afterpotential generation in hippocampal pyramidal cells. Journal of Neurophysiology. 1981;45:86–97. doi: 10.1152/jn.1981.45.1.86. [DOI] [PubMed] [Google Scholar]
- Yaari Y, Konnerth A, Heinemann U. Non-synaptic epileptogenesis in the mammalian hippocampus in vitro. II. Role of extracellular potassium. Journal of Neurophysiology. 1986;56:424–438. doi: 10.1152/jn.1986.56.2.424. [DOI] [PubMed] [Google Scholar]
- Yamada N, Bilkey DK. Kindling-induced persistent alterations in the membrane and synaptic properties of CA1 pyramidal neurons. Brain Research. 1991;561:324–331. doi: 10.1016/0006-8993(91)91611-4. [DOI] [PubMed] [Google Scholar]