

Abstract
The whole-cell patch clamp technique was used to study the effect of intracellular Ca2+ on light-evoked EPSCs in on-off ganglion cells in salamander retinal slices. Both AMPA and NMDA receptors contributed to the light-evoked responses.
In the presence of strychnine and picrotoxin, ganglion cells responded to light onset and offset with transient inward currents at -70 mV. These currents were reduced by 35 ± 3 % when the light stimulus was preceded by a depolarizing step from -70 to 0 mV.
The inhibitory effect of depolarization on light-evoked EPSCs was strongly reduced in the presence of 10 mm BAPTA.
The degree of EPSC inhibition by the prepulse holding potential followed the current-voltage relationship of the Ca2+ current found in the ganglion cell.
In the presence of the NMDA receptor antagonist AP-7, glutamate-dependent current was nearly abolished when high Ca2+ was substituted for high Na+ solution.
The release of Ca2+ from internal stores by caffeine or inositol trisphosphate reduced the EPSCs by 36 ± 5 and 38 ± 11 %, respectively, and abolished the inhibitory effect of depolarization.
The inhibitory effect of depolarization on EPSCs was reduced 5-fold in the presence of AP-7, but was not reduced by the AMPA receptor antagonist CNQX.
Neither inhibition of Ca2+-calmodulin-dependent enzymes, nor inhibition of protein kinase A or C had any significant effect on the depolarization-induced inhibition of EPSCs.
Our data suggest that elevation of [Ca2+]i, through voltage-gated channels or by release from intracellular stores, reduced primarily the NMDA component of the light-evoked EPSCs.
Glutamate, the major excitatory neurotransmitter in the vertebrate retina, is released by photoreceptors and bipolar cells (reviewed in Thoreson & Witkovsky, 1999). In the inner retina of vertebrates, the amacrine and ganglion cells receive glutamatergic inputs from bipolar cells through both ionotropic receptors (iGluRs) of NMDA and AMPA subtypes (Lukasiewicz & McReynolds, 1985; Mittman et al. 1990; Diamond & Copenhagen, 1993), and a variety of metabotropic glutamate receptors (mGluRs) (Peng et al. 1995). Both iGluRs and mGluRs are linked to pathways that alter [Ca2+]i (Gilbertson et al. 1991; Linn & Christensen, 1992). Thus, light-induced depolarization increases [Ca2+]i by permitting Ca2+ influx through ionotropic glutamate-gated channels and by activating voltage-gated Ca2+ channels. In addition some mGluRs are coupled to the release of Ca2+ from intracellular stores. Finally Ca2+ entry into the neuron can stimulate Ca2+ release from intracellular stores through a process called calcium-induced Ca2+ release (Verkhratsky & Shmigol, 1996).
The possible consequences of an elevation of [Ca2+]i are a direct action of Ca2+ on the receptor-channel complex and/or initiation of second-messenger-mediated intracellular processes that modify the functioning of neurotransmitter receptors and ion channels by phosphorylating or dephosphorylating them (Huganir & Greengard, 1990). For example, in hippocampal neurons the NMDA-induced currents are modulated by internal Ca2+ (Legendre et al. 1993; Viklicky, 1993; Medina et al. 1996), and by intracellular second-messenger systems, including protein kinases and IP3 (Chen & Huang, 1992). Ca2+, acting through these pathways, serves as an important intracellular signal in the mediation of synaptic plasticity in the CNS (reviewed in Chittajallu et al. 1998). It is reasonable to suggest, therefore, that elevation of [Ca2+]i during synaptic activity might affect light-induced responses in retinal ganglion cells by modulating post-synaptic glutamate receptors. However, to our knowledge, this postulate has not been tested experimentally.
We report here that elevation of [Ca2+]i either through influx or by release from intracellular stores reduces light-evoked EPSCs in salamander retinal ganglion cells. The results indicate that NMDA receptors are the primary target of a Ca2+-induced modulation. In addition our data indicate that the inhibitory effect of Ca2+ on EPSCs is not mediated by Ca2+-calmodulin (Ca2+-CaM)-dependent processes, nor by activation of cAMP-dependent protein kinase A (PKA), or protein kinase C (PKC). Some of these results have been presented in abstract form (Akopian & Witkovsky, 2000).
METHODS
Animals
Salamanders (Ambystoma tigrinum) were obtained from a commercial supplier (Charles Sullivan, Nashville, TN, USA) and kept at 4 °C until used. The handling and maintenance of animals met the National Institutes of Health guidelines and were approved by the Animal Research committee of the New York University School of Medicine.
Slice preparation
The procedures for obtaining retinal slices were similar to those described by Lukasiewicz et al. (1995). Briefly, the animal was decapitated and double pithed. The eyes were enucleated and hemisected, and the cornea, lens and iris removed. The retina was transferred, vitreal side down, to Millipore filter paper (0.22 μm pore), then sectioned manually into 150-200 μm thick slices, which were mounted in a chamber and superfused at 2 ml min−1 during the experiment. The dissection and preparation of the slices were performed under dim red illumination and infrared illumination was used to view the slices during the experiments.
Solutions
Whole-cell voltage clamp recordings of light-evoked EPSCs were obtained with a standard solution in the patch pipette containing (mm): CsCl 100, MgCl2 2, TEA 20, CaCl2 0.2, EGTA 2, Hepes 10, ATP 2 and GTP 0.1, adjusted to pH 7.3 with CsOH. In initial experiments the bath (Ringer) solution contained (mm): NaCl 100, KCl 3, CaCl2 2, MgCl2 2 and Hepes 10, adjusted to pH 7.6 with NaOH; experiments involving the isolation of NMDA-dependent EPSCs were done in Mg2+-free Ringer solution containing 10 μm glycine, as indicated in the figure legends. Modified Ringer solution containing 20 mm TEA, 20 mm Ba2+ and 1 μm TTX was used to isolate a Ca2+ current. Two different solutions were used to test the Ca2+ permeability of AMPA receptors: a high Na+ solution, consisting of 110 mm NaCl and 5 mm Hepes; and a high Ca2+ solution consisting of 70 mm CaCl2 and 5 mm Hepes. Strychnine (20 μm) and picrotoxin (80 μm) were added to the bath solution to block glycinergic and GABAergic transmission, respectively. To isolate AMPA receptor-mediated EPSCs, NMDA receptors were blocked with d(-)-2-amino-7-phosphonoheptanoic acid (AP-7, 80 μm). The NMDA receptor-mediated EPSCs were isolated by blocking AMPA receptors with 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 2-5 μm). All drugs were obtained from Sigma-Aldrich.
Lukasiewicz & Werblin (1988) demonstrated that in the tiger salamander retina, injections of dye into the optic nerve labelled all cells in the ganglion cell layer. All of our recordings were obtained from cells located in the ganglion cell layer, thus they are presumed to be from ganglion cells.
Light stimulation
Full-field, red (667 nm) light stimuli were used to evoke EPSCs and EPSPs. The intensity of the unattenuated light was 7.3 × 109 quanta cm−2 s−1, as measured by a photodiode referenced to a calibrated thermopile. The vast majority of ganglion cells recorded (> 90 %), were on-off cells that responded with transient EPSCs at light onset and offset.
Recording procedures
Whole-cell voltage and current clamp recordings were obtained in a conventional way (Hamill et al. 1981) using an Axopatch 200 amplifier (Axon Instruments). Recording pipettes were made from borosilicate glass tubing (1.2 mm o.d., 0.6 mm i.d.). Electrode resistance was typically 5-8 MΩ in the bath solution. The average input resistance, estimated from the steady-state current induced by a 10 mV voltage step from -70 mV, was 2.3 ± 0.4 GΩ (n = 14). After the seal had been ruptured the series resistance (10-15 MΩ) was compensated (70-80 %) by a standard circuit. Light-evoked whole-cell EPSCs were typically < 1 nA (usually < 600 pA), and the voltage errors resulting from inadequate compensation were estimated to be ≤ 3 mV. Currents were filtered at 1 kHz by a low-pass Bessel filter on the Axopatch 200 amplifier. Sampling frequencies were a function of episode length and varied from 0.25 to 2.5 kHz. At the lowest sampling frequency, a window, encompassing 0.125-1.0 kHz, permitted potential aliasing of the signal. We tested for aliasing by performing a Fourier analysis of the light-evoked EPSC. In the frequency band of interest, the power was reduced by more than 4 log units compared to its peak power, which occurred near 2 Hz. The data were digitized and stored on a 33 MHz 486-PC using a Digidata-1200 interface (Axon Instruments). The pCLAMP software package (Axon Instruments) was used for data acquisition and analysis. Summary data are presented as means ±s.e.m. Statistical comparison between groups was made with Student's paired t test; the corresponding P values are given in the text. In voltage clamp experiments the membrane potential usually was held at -70 mV, and a 400 ms depolarizing step from -70 to 0 mV was applied to activate voltage-gated Ca2+ channels.
RESULTS
Light-evoked EPSCs
Ganglion cell responses to subsaturating light stimuli were dependent on the duration of the light flash. Figure 1A illustrates EPSCs recorded from an on-off ganglion cell in response to light stimuli of different duration, from 100 ms to 3 s. Ganglion cells responded only to light onset (on-EPSC) when the stimulus duration was < 300 ms. When the duration of the light flash was increased to 500 ms, however, a second transient EPSC (off-EPSC) was elicited at light offset. A further increase of stimulus duration to 1-3 s resulted in an enhancement of the off-transient, whereas the response to light onset was not affected significantly. Because of the dependence of off-EPSC amplitude on stimulus duration, we conducted our analysis exclusively on on-EPSCs.
Figure 1. Light-evoked EPSCs recorded from an on-off ganglion cell with various stimulus durations.

A, with an increase in the duration of the light flash from 0.1 to 3 s the amplitude of the off-EPSCs was dramatically increased, whereas that of the on-EPSCs remained unchanged. B, the light-evoked EPSC recorded in Mg2+-free solution was reduced in the presence of CNQX (5 μm), and almost completely blocked by a combination of AP-7 (80 μm) and CNQX. The recording pipette contained standard intracellular solution (see Methods). In this and subsequent figures, horizontal hatched bars indicate the duration of the light stimuli.
In agreement with earlier studies (Lukasiewicz & McReynolds, 1985; Diamond & Copenhagen, 1993), we found that both NMDA and non-NMDA iGluRs mediated the excitatory synaptic currents of salamander retinal ganglion cells. In Mg2+-free solution, NMDA receptors have an approximately linear current-voltage relationship (Ascher et al. 1988). When CNQX (2-5 μm), an AMPA receptor antagonist, was added to the Mg2+-free bath solution, the EPSCs were reduced by 40 ± 9 % (n = 4); the remaining, CNQX-resistant component of the EPSCs was blocked by 80 μm AP-7, an NMDA receptor antagonist (Fig. 1B). Although kainate receptors have some sensitivity to CNQX, it is 3- to 10-fold lower than that of AMPA receptors (reviewed in Bleakman & Lodge, 1998). Moreover, Lukasiewicz et al. (1995) reported that salamander ganglion cells of the on-off subtype lack kainate receptors. We conclude that the AP-7-insensitive component of the EPSCs we recorded reflects current flow through AMPA receptors.
Effect of Ca2+ entry through voltage-gated channels on EPSCs
There are at least three pathways by which [Ca2+]i may be elevated as a consequence of the light-evoked synaptic activity in retinal ganglion cells: (i) by Ca2+ influx through glutamate-activated channels, (ii) by Ca2+ influx through voltage-gated Ca2+ channels whose activation level is increased by a synaptically induced depolarization, and (iii) by Ca2+ release from intracellular stores. With regard to possibility (iii) the presence of both ryanodine and IP3 receptors in turtle retinal ganglion cells has been reported previously (Akopian et al. 1998).
To study the effect of Ca2+ entry through voltage-gated channels, ganglion cells were held at -70 mV and light-evoked EPSCs were recorded before and 100 ms after the termination of a 400 ms depolarizing step from -70 to 0 mV (Fig. 2A). The data show that by comparison to the control response (continuous line, before ΔV), a depolarizing step induced a reduction in EPSC peak amplitude of 35 ± 3 % (n = 28, dotted line, after ΔV). Figure 2B illustrates an experiment in which light stimuli were presented every 30 s without or with a 400 ms depolarizing voltage step from -70 to 0 mV. It shows that the depolarization-induced suppression of the light-evoked EPSC was completely reversible. The inhibitory effect of depolarization on EPSCs was also observed when small EPSCs were elicited by lower intensity flashes (0.2-1.2 log unit attenuation; not illustrated), indicating that the depolarization-induced inhibition of EPSCs was not dependent on response saturation. Moreover, a corresponding reduction of 29 + 5 % (n = 5) was observed when the effect of depolarization was tested on EPSPs recorded in current clamp mode (Fig. 2C). The histogram in Fig. 2D summarizes these results, showing that depolarization reduced the amplitude of both EPSCs and EPSPs.
Figure 2. Depolarization-induced inhibition of light-evoked EPSCs and EPSPs.

All records were obtained in Mg2+-free Ringer solution. A, in voltage clamp mode light responses were recorded before (continuous line) and 100 ms after (dotted line) the termination of a 400 ms depolarizing step (ΔV) from -70 to 0 mV. B, light-evoked EPSCs recorded every 30 s without and with a depolarizing pre-pulse from -70 to 0 mV that induced reversible inhibition. The presentation, but not the timing, of the depolarizing pre-pulse is represented above the middle trace. C, light-evoked EPSPs were recorded in current clamp mode before (continuous line) and after a depolarizing current of +50 pA (dotted line). EPSPs were reduced following the injection of current. The resting membrane potential was -65 mV. CsCl and TEA in the pipette solution were replaced by KCl. D, histogram of the depolarization-induced inhibition (means ±s.e.m.) of EPSC and EPSP amplitudes. The number of cells studied is indicated above each bar. In this and subsequent figures, the bath solution contained 20 μm strychnine and 50 μm picrotoxin.
The depolarization-induced decrease of peak EPSC was not accompanied by any significant changes in the kinetics of EPSC decay. In some cells the EPSC decay was not well fitted by a sum of exponentials, because of a hump that followed its rapid onset (see Fig. 7A). Therefore, as a parameter to describe EPSC decay kinetics we used the time (D37), to reach 37 % of peak current (Higgs & Lukasiewicz, 1999). The corresponding D37 values for EPSCs were 86 ± 14 and 85 ± 13 ms (n = 10), when recorded before and after the depolarizing step, respectively.
Figure 7. Effect of depolarization-induced Ca2+ influx on NMDA- and AMPA-mediated components of EPSCs.
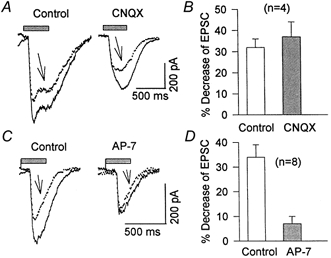
A, light-evoked EPSCs were recorded before (Control), and after (arrows) a depolarizing voltage step from -70 to 0 mV (not illustrated) in the absence (Control) and the presence of 2 μm CNQX. B, histogram summarizing the depolarization-induced inhibition of EPSCs in the absence (32 ± 2 %) and presence of 2-5 μm CNQX (37 ± 7 %). C, AMPA receptor-mediated EPSC recorded in drug-free Mg2+-free Ringer solution (left) or in the presence of 50 μm AP-7. A depolarizing step of 400 ms from -70 to 0 mV (as in A; traces indicated by arrows), reduced light-evoked EPSCs by only 7 ± 3 %, which was significantly (P < 0.05) smaller than the 34 ± 5 % inhibition seen in the absence of AP-7 (Control). D, histogram summarizing these data.
The amount of light-evoked EPSC suppression was approximately proportional to the pre-pulse potential between -50 and 0 mV. In a representative experiment (Fig. 3A), depolarizing steps from a holding potential of -70 to 0, -20 and -40 mV induced inward calcium currents (ICa) and a subsequent inhibition of EPSCs of 58, 35 and 18 %, respectively. Figure 3B provides mean data from experiments of this sort, indicating that the threshold potential to induce a reduction of EPSC was near -50 mV, and the effect was maximum at 0 mV, reducing slightly at more positive pulse potentials. The relationship between depolarizing potentials and EPSC inhibition correlated well with the I-V function of the Ca2+ current in these ganglion cells (Fig. 3C).
Figure 3. Correlation between depolarizing voltage steps and the degree of inhibition of EPSCs.

A, light-evoked EPSCs were recorded before and after depolarizing steps to 0, -20 and -40 mV from a holding potential of -70 mV. Dotted horizontal lines indicate the level of peak EPSCs recorded in the absence of a depolarizing pre-pulse. The bath solution contained 2 mm Ca2+ and 0 mm Mg2+. B, relationship between depolarizing voltage steps and EPSC inhibition. Each point represents the mean ±s.e.m. from 3-10 cells. C, I-V relationship of Ca2+ current recorded from a ganglion cell in modified Ringer solution containing TEA (20 mm), TTX (1 μm) and 20 mm Ba2+ as a charge carrier. Inset, traces of transient low (upper inset) and sustained high voltage-activated Ca2+ currents (lower inset) elicited by depolarizing steps from -90 to -40 mV, and from -60 to 0 mV, for low and high voltage-activated currents, respectively. Y-axis provides current scale for inset records.
Analysis of the dependence of the inhibitory effect of depolarizing pulses on [Ca2+]o was complicated by the fact that elevation of [Ca2+]o from 2 to 20 mm in the bath solution caused either a dramatic reduction or a complete block of light-evoked EPSCs. When [Ca2+]o was elevated to 10 mm, EPSCs were reduced to 45 ± 9 % (n = 3) of their value in normal [Ca2+]o. The Ca2+ current induced by a depolarizing step from -70 to 0 mV increased by 37 ± 10 % (n = 3) in the presence of 10 mm[Ca2+]o, and the inhibition of the EPSC induced by depolarization was enhanced from 30 ± 10 to 52 ± 14 % (n = 3, data not shown).
Effect of [Ca2+]i buffering by BAPTA
The results described above indicate that Ca2+ entry through voltage-gated channels reduces light-evoked EPSCs in salamander retinal ganglion cells. As an initial test to probe the underlying mechanism of action of Ca2+ we examined the effect of strong [Ca2+]i buffering. Accordingly we added 10 mm BAPTA to the patch pipette solution and recorded the light-evoked EPSCs in whole-cell mode every 30 s for 10-20 min. We observed that the EPSC amplitude increased significantly in a time-dependent manner during intracellular perfusion with BAPTA (Fig. 4A). The enhancement of EPSCs varied from 25 to 280 % in different cells, after 10-20 min recording with a patch pipette containing BAPTA, with a mean enhancement of 96 ± 26 % (n = 9). No enhancement of EPSCs over the same time period was observed (Fig. 4B) when the patch pipette contained the standard intracellular solution (see Methods).
Figure 4. Effect of strong intracellular Ca2+ buffering on light-evoked EPSCs.

All records were obtained in Mg2+-free Ringer solution. A, on-off ganglion cell was stimulated by 500 ms light flashes every 30 s during a 20 min whole-cell recording with 10 mm BAPTA in the patch pipette solution. B, time-dependent changes of EPSCs in the absence and presence of 10 mm BAPTA in the patch pipette (n = 10). EGTA and Ca2+ were removed from the patch pipette solution and replaced with 10 mm BAPTA.
The influence of 10 mm BAPTA on the depolarization-induced reduction of EPSCs was tested by comparing the effectiveness of a depolarizing pulse within 1 min of patch break and again after 10-15 min incubation with BAPTA. The results were that, at the beginning of the whole-cell experiment, the depolarizing pulse reduced the EPSC by 30 ± 8 % (Fig. 5A, left panel), whereas the inhibitory effect was significantly reduced to 6 ± 2 % (n = 4, P < 0.01) after 10 min incubation with BAPTA (Fig. 5A, right panel). These data, which support the idea that the inhibitory effect of depolarization on light-evoked EPSCs is mediated by an increase in [Ca2+]i, are summarized in Fig. 5B.
Figure 5. Effect of [Ca2+]i buffering on depolarization-induced inhibition of EPSCs.

All records were obtained in Mg2+-free Ringer solution. A, light-evoked EPSCs were recorded before and 100 ms after a 400 ms depolarizing step from -70 to 0 mV, either within 1 min of membrane rupture or after 10 min incubation with BAPTA (10 mm in the patch pipette). The presentation, but not the timing, of the depolarizing pre-pulse is represented above the traces. Dotted lines indicate the peak current observed before the depolarizing step. B, histogram of these results (n = 4, P < 0.01). C, with 10 mm caffeine in the patch pipette, the inhibitory effect of depolarization on EPSCs was recorded within 1 min of patch break and 10 min later. Caffeine by itself reduced EPSCs by 36 ± 5 % (n = 7, P < 0.01, compare first and third traces) and abolished the inhibitory effect of depolarization. D, histogram summarizing the inhibitory effect of depolarization on EPSC amplitude within 1 min of patch break, and after 10 min exposure either to caffeine (Caff, n = 5, P < 0.005) or to IP3 (50 μm, n = 4, P < 0.002).
Effect of Ca2+ released from intracellular stores on EPSCs
Effect of caffeine
Experimental evidence indicates that glutamate stimulates Ca2+ release from intracellular stores in retinal neurons (Gilbertson et al. 1991; Linn & Christensen, 1992). Increased [Ca2+]i then modulates the activity of neurotransmitter receptors in ganglion cells (Akopian et al. 1998; Shen & Slaughter, 1999). To test whether release of Ca2+ from intracellular stores inhibits the light-evoked EPSCs, we added caffeine (10 mm) to the patch pipette solution, and monitored every 30 s the EPSCs evoked by a sub-saturating light stimulus. The mean reduction of EPSCs recorded after 10-15 min incubation with caffeine was 36 ± 5 % (n = 7, see Fig. 5C). Reductions were measured in relation to the EPSCs recorded within 1 min of patch break and the difference was significant (P < 0.01). These results suggest that the elevation of [Ca2+]i caused by release from intracellular stores reduces light-evoked EPSCs, as Ca2+ influx through voltage-gated channels does.
Effect of IP3
Earlier studies indicated that retinal ganglion cells possess different classes of G protein-coupled metabotropic glutamate receptor (Hartveit et al. 1995). Activation of the mGluR1-5 subclass of receptors triggers hydrolysis of phosphoinositides, and formation of IP3, which subsequently stimulates Ca2+ release from IP3-sensitive intracellular stores (Pin & Duvoisin, 1995). We recorded from ganglion cells using a patch pipette containing 50 μm IP3 and found that peak EPSC was reduced by 38 ± 11 % (n = 4) after about 10 min incubation with IP3 (not shown), indicating that Ca2+ released from IP3-sensitive internal stores also reduces the EPSC amplitude.
Next we examined the additivity of Ca2+ released from intracellular stores and Ca2+ flux through voltage-gated channels on EPSCs. In this set of experiments the effect of depolarizing pulses on EPSCs was tested within 1 min of patch break and after 10 min incubation with caffeine. In the experiment illustrated in Fig. 5C, the EPSC amplitude was reduced by 55 % after 10 min incubation with caffeine, and depolarization-induced inhibition was abolished. The mean inhibition of EPSCs by depolarizing pulses was 40 ± 10 % when recorded at the beginning of the experiment (Fig. 5C, left panel), and was reduced to 9 ± 3 % (n = 5, P < 0.005) after a 10 min incubation with caffeine (Fig. 5C, right panel). A similar result was observed when experiments were performed with patch pipettes containing 50 μm IP3 (not illustrated). Thus, a depolarizing step reduced peak EPSC amplitude by 46 ± 5 % when recorded within 1 min of patch break, but by only 8 ± 5 % after 10 min incubation with IP3 (n = 4, P < 0.002). Figure 5D summarizes the results, which indicate that release of Ca2+ from intracellular stores reduced the light-evoked EPSCs. These data support the hypothesis that an elevation of [Ca2+]i, either by influx through voltage-gated channels or by release from internal stores, results in an inhibition of light-evoked EPSCs in salamander retinal ganglion cells.
Ca2+ permeability of AMPA receptors
Ca2+ flux through ionotropic glutamate receptors is another potential pathway through which elevation of [Ca2+]i and consequent desensitization of neighbouring NMDA receptors may occur (Kyrozis et al. 1995; Medina et al. 1996). We probed the Ca2+ permeability of AMPA receptors by recording the current-voltage relationship of glutamate-induced current, first in a high Na+ solution and then in a high Ca2+ solution (see Methods). In both solutions 50 μm AP-7 was added to block NMDA receptors. A 200 ms voltage ramp from -70 to 50 mV was used to obtain the I-V relationship in the absence and the presence of 5 mm glutamate in the bath solution. The difference between the two ramps gave a measure of the AMPA receptor-mediated glutamate current. Identical measurements were carried out in high Ca2+ solution. Figure 6 shows the data from a cell for which a substantial inward current response to glutamate was seen in high Na+ solution, but which had very little inward current in high Ca2+ solution, at all levels of membrane potential. A qualitatively similar result was obtained in four other cells.
Figure 6. Ca2+/Na+ permeability of AMPA receptors in salamander retinal on-off ganglion cells.

Current-voltage relationship of glutamate-induced current in high Na+ and high Ca2+ solutions (see Methods). A 200 ms voltage ramp from -70 to +50 mV elicited an I-V relationship in the absence and in the presence of glutamate. The difference of these two curves is shown and is taken to be the glutamate-induced current. In the presence of high Ca2+, the glutamate-evoked current was nearly abolished. External solution contained 50 μm AP-7 to block NMDA receptors. A similar result was obtained for 6 cells.
There was no change in reversal potential of the glutamate-induced current, measured by voltage ramp when the extracellular solution was switched from high Na+ to high Ca2+ solution. This is a good indication (Burnashev et al. 1992) that AMPA receptor channels in salamander retinal ganglion cells have a very low Ca2+/Na+ permeability ratio.
NMDA receptors are a target for modulation by Ca2+
It was proposed for CNS neurons that NMDA receptors are a primary target for modulation by Ca2+ (Vyclicky, 1993; Legendre et al. 1993; Kyrozis et al. 1995; Medina et al. 1996). We examined whether intracellular Ca2+ affects the NMDA receptor-mediated component of the EPSC in salamander retinal ganglion cells. In a Mg2+-free solution containing 10 μm glycine, a depolarizing step from -70 to 0 mV reduced EPSCs by 32 ± 4 %. When 2-5 μm CNQX was added to the bath solution to block AMPA receptors, the EPSCs initially were attenuated by 40 ± 9 % (n = 4), but a depolarizing step from -70 to 0 mV still reduced the CNQX-resistant component of the EPSCs by 37 ± 7 %. Sample records illustrating these effects are shown in Fig. 7A and the population data are summarized in Fig. 7B. This remaining component of the EPSC was almost completely (90 ± 3 %) blocked when 80 μm AP-7 was applied together with CNQX (not shown, but see Fig. 1B), indicating that it represented the NMDA receptor-mediated EPSC. Thus, the NMDA receptor-mediated component of the EPSC is strongly affected by a depolarizing pulse.
In a parallel set of experiments, AP-7 added to the bath solution reduced EPSC amplitude (Fig. 7C, right). The inhibitory effect of a subsequent depolarizing pulse on EPSCs was approximately 5-fold smaller in the presence of AP-7 than in control solution (Fig. 7C). Thus, depolarization from -70 to 0 mV reduced peak EPSCs by only 7 ± 3 % (Fig. 7D), which was significantly less than the 34 ± 5 % (n = 8, P < 0.002) inhibition induced by the same depolarizing step in the absence of AP-7 (Fig. 7D, Control). These data support the conclusion that, in salamander retinal ganglion cells, NMDA receptors are the primary target for modulation by [Ca2+]i.
Possible intracellular mechanisms of inhibition of EPSCs by Ca2+
The next series of experiments focused on the possible mechanisms by which a Ca2+-dependent inhibition of EPSCs might occur. Studies in other preparations indicate that Ca2+ failed to inactivate NMDA receptors in inside-out patches, suggesting that the effect of Ca2+ is mediated through the Ca2+-binding protein calmodulin (Medina et al. 1996; Zhang et al. 1998; Krupp et al. 1999). We tested whether a Ca2+-CaM-dependent system is involved in the Ca2+-induced inhibition of EPSCs. Earlier studies demonstrated the presence of Ca2+-CaM-dependent phosphatases and protein kinases in retinal neurons, including ganglion cells (Cooper et al. 1985). Moreover, it has been suggested that glutamate can regulate retinal calmodulin kinase II activity through ionotropic receptors (Laabich & Cooper, 1999). Calmidazolium (50 μm), a potent inhibitor of Ca2+-CaM-dependent enzymes (Van Belle, 1981), was added to the patch pipette solution, and the effect of depolarization on EPSCs was observed after 10-15 min incubation with this drug. The mean inhibition of peak EPSC amplitude by depolarizing pulses in the presence of calmidazolium was 32 ± 5 % (n = 5), and the difference, compared with the effect obtained within 1 min of patch break, was not significant (P > 0.5; not shown). In another series of experiments done in the presence of 50 μm cyclosporin A, a selective antagonist of the Ca2+-CaM-dependent phosphatase calcineurin (Hashimoto et al. 1990), the depolarizing pulses still significantly reduced EPSCs by 29 ± 4 % (n = 3). Our results indicate that the inhibition of EPSCs induced by Ca2+ influx does not depend primarily on Ca2+-CaM-dependent enzymes. The data, however, do not exclude the possibility that calmodulin might bind to the NMDA receptor and affect its activity (Zhang et al. 1998; Krupp et al. 1999).
Recent studies in the hippocampus show that PKC enhances Ca2+-CaM-dependent inactivation of the NMDA channel (Lu et al. 2000). Accordingly we examined whether PKA, or PKC is involved in the regulation of EPSCs by Ca2+. In these experiments H-7 (20 μm), a non-specific inhibitor of PKA and PKC, was added to the patch pipette solution and the effect of depolarization was tested after 10 min incubation in the whole-cell mode. H-7 had no significant effect on the depolarization-induced inhibition of EPSCs. In sum, our data indicate that none of the second messenger-activated enzymes we tested is implicated in the inhibition of EPSCs induced by Ca2+ influx through voltage-gated channels.
Figure 8 summarizes the results obtained in our study. It documents that the depolarization-induced inhibition of EPSCs (Control) is not affected by inhibitors of Ca2+-CaM-dependent enzymes, nor by PKA and PKC, but is significantly reduced in the presence of BAPTA, and agents that stimulate Ca2+ release from intracellular stores (caffeine and IP3). The histogram also shows that the inhibitory effect of depolarization on EPSCs is largely abolished in the presence of the NMDA receptor antagonist AP-7, but not the AMPA receptor antagonist CNQX.
Figure 8. Summary of factors affecting Ca2+-mediated inhibition of EPSCs.
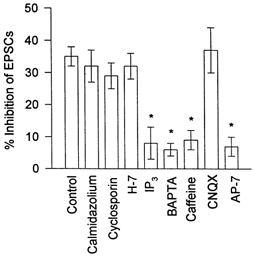
The depolarization-induced inhibition of light-evoked EPSCs in standard intracellular and bath solutions (Control) is compared with that seen in the presence of different substances. CNQX and AP-7 were added to the bath solution, other drugs were added to the patch pipette solution. (For concentrations see text.) Each bar indicates mean ±s.e.m., n = 4-10 cells. Statistical significance (*P < 0.05) was established using Student's paired t test.
DISCUSSION
The principal finding of our study is that the NMDA receptor-mediated component of the on-EPSCs of on-off retinal ganglion cells is reduced by about 35 % following a rise in [Ca2+]i. The same rise in [Ca2+]i depressed the AMPA receptor-dependent component of the same EPSC by only 7 %. Elevated [Ca2+]i, whether brought about through release from intracellular stores or caused by influx through voltage-gated channels, was equally effective in reducing the EPSC. The mechanism of Ca2+ action was shown not to depend on Ca2+-CaM-dependent protein kinases/phosphatases, or on PKA or PKC.
Our results are in broad agreement with earlier studies of hippocampal neurons indicating that neither phosphatases nor protein kinases are part of the pathway through which intracellular Ca2+ brings about an inhibition of NMDA-evoked currents (Viklicky, 1993; Legendre et al. 1993). Other than phosphorylation/ dephosphorylation, calcium could act by direct binding, either to the regulatory proteins, or to the channel. Our experiments do not directly address these possibilities. According to earlier studies (Johnson & Ascher, 1990), however, it is unlikely that Ca2+ binds to the NMDA receptors within the pore and reduces its conductance, as Mg2+ does.
In salamander retinal ganglion cells, AMPA receptors are co-localized with NMDA receptors and both these receptor classes mediate the excitatory post-synaptic responses (Lukasiewicz & McReynolds, 1985; Massey & Miller, 1990; Diamond & Copenhagen, 1993). Immunocytochemical studies indicate that these cells possess all the subunits of AMPA receptors, including those that are permeable to Ca2+ (Hamassaki-Britto et al. 1993; Brandstätter et al. 1998). In the central nervous system, including the retina, the Ca2+ permeability of AMPA receptors varies from negligible to substantial in different neurons, suggesting that some cells contain the GluR2 subunit, which imparts a low Ca2+ permeability to the glutamate receptor (Hollman & Heinemann, 1994). Ca2+ influx mediated by glutamate-gated channels during synaptic transmission may contribute to short- or long-lasting functional effects, depending on the efficiency of the Ca2+-buffering/extrusion systems operating in the cells. The data of the present study indicate that AMPA receptors in salamander retinal on-off ganglion cells have a low Ca2+ permeability. Thus, in the presence of the NMDA receptor antagonist AP-7, and when Na+ was replaced by equimolar Ca2+ in the bath solution, the glutamate-induced current was nearly abolished at all levels of membrane potential (Fig. 6). The data suggest that, consistent with their linear I-V relationship in high Na+ medium, these cells contain the GluR2 subunit, and so a significant Ca2+ influx through AMPA receptors during synaptic activity is unlikely.
The high Ca2+ permeability of NMDA receptors plays an important role in NMDA receptor-mediated synaptic plasticity (Mayer & Westbrook, 1987; Nicoll & Malenka, 1995). Shen & Slaughter (1998) reported that Ca2+ influx through NMDA receptors acts via Ca2+-CaM to inhibit N-type Ca2+ current in salamander retinal ganglion cells. On the other hand it is experimentally difficult to demonstrate a rise in post-synaptic Ca2+ concentration that is attributable exclusively to synaptic activation of NMDA receptors on retinal ganglion cells, excluding unequivocally the activation of voltage-gated Ca2+ channels, as was demonstrated in the hippocampus by Miyakawa et al. (1992). This would require a selective block of voltage-gated Ca2+ channels, but these same channels are utilized by pre-synaptic bipolar cell axon terminals to trigger glutamate release. Instead we examined the influence of Ca2+ influx through voltage-gated channels on light-evoked EPSCs. Ganglion cells in the salamander retina possess both transient, low voltage-activated (LVA, T-type), and sustained, high voltage-activated (HVA, L- and N-type) Ca2+ channels (Shen & Slaughter, 1998; see also Fig. 3C, inset). We found that the threshold voltage for depolarization-induced inhibition of EPSCs was near -50 mV, and that maximum inhibition occurred when cells were depolarized to 0 mV from a holding potential of -70 mV (see Fig. 3B). The degree of EPSC inhibition increased with depolarizing steps from -50 to 0 mV and then reduced at more depolarized potentials. The dependence of EPSC inhibition on pre-pulse voltage correlated well with the I-V function of HVA Ca2+ current in the same ganglion cells, consistent with the effect of depolarization being mediated by Ca2+ influx primarily through HVA Ca2+ channels.
There are controversial data in the literature regarding the effectiveness of intracellular BAPTA in reducing Ca2+-dependent inactivation of NMDA responses. In some studies BAPTA is reported to be ineffective in preventing Ca2+-dependent desensitization of NMDA responses (Vicklicky, 1993), whereas in other reports, BAPTA prevented a Ca2+-dependent inactivation of NMDA responses (Legendre et al. 1993). In our study intracellular application of BAPTA significantly reduced depolarization-induced inhibition of EPSCs. Moreover, the light-evoked EPSCs increased in a time-dependent manner during the 10-15 min whole-cell recording with 10 mm BAPTA in the patch pipette, indicating that the basal level of [Ca2+]i is sufficient to partially suppress EPSCs.
In addition to Ca2+ influx through voltage-gated and receptor-activated channels, another source of Ca2+ is by release from internal stores (Linn & Christensen, 1992). Both ryanodine- and IP3-sensitive Ca2+ stores are present in retinal ganglion cells (Akopian et al. 1998; Shen & Slaughter, 1999). In a previous study (Akopian et al. 1998), it was reported that in retinal ganglion cells, currents induced by activation of GABAA receptors were modulated by Ca2+ released from IP3- and ryanodine-sensitive intracellular stores, but not by Ca2+ influx through voltage-gated channels, presumably reflecting the compartmentalization of the cytoplasm. In contrast, in the present study Ca2+ influx through voltage-gated channels, as well as Ca2+ release from ryanodine- and IP3-sensitive stores (effects of caffeine and IP3), induced suppression of glutamate-dependent EPSCs. The inhibitory effects of Ca2+ released from intracellular stores and from voltage-gated channels on EPSCs were non-additive, indicating a common target of action for intracellular Ca2+. Probably some of the non-additivity resulted from saturation of the calcium-dependent inhibitory mechanisms.
The physiological mechanism for release of Ca2+ from stores is through a second-messenger pathway. In the retina different ganglion cells possess different mGluR subtypes including those (e.g. mGluR1) whose activation is coupled to the turnover of phosphoinositides and production of IP3 (Hartveit et al. 1995; Brandstätter et al. 1998). In CNS neurons, activation of mGluR1 results in either a reversible inhibition of NMDA-dependent responses, possibly by a membrane-delimited action, or a long-lasting potentiation, probably via activation of PKC (Anwyl, 1999). In the retina, there are reports of phosphoinositide turnover induced by light and glutamate (reviewed in Osborne & Ghazi, 1990), and the IP3 receptor has been localized in the retina (Peng et al. 1991). In the present study the peak EPSCs were reduced in a time-dependent manner following membrane rupture with an electrode containing IP3. These data suggest that activation of mGluR1 by synaptically released glutamate may contribute to the regulation of the NMDA component of post-synaptic responses. Thus the same transmitter, glutamate, initiates fast and slow processes, both of which influence the light-evoked EPSCs.
Earlier studies on CNS neurons showed that the activity of NMDA channels was unaffected by exposure to Ca2+ on the cytoplasmic side of the inside-out patches (Rosenmund & Westbrook, 1993). This finding suggested the existence of a Ca2+-dependent cytoplasmic intermediate that inactivates the NMDA receptor and washes out during the first minutes after obtaining excised patches (Medina et al. 1996). More recent studies indicate that the Ca2+-binding protein calmodulin may be the required intermediate (Ehlers et al. 1996; Zhang et al. 1998; Krupp et al. 1999). These studies demonstrated that the binding of calmodulin to the NR1 subunit inactivated the NMDA receptor, which possibly occurs by a Ca2+-CaM-dependent dissociation of the receptor complex from the neural cytoskeleton. Although our results do not clarify the mechanism through which a Ca2+-dependent inhibition of EPSCs occurs, the finding that neither Ca2+-CaM-dependent enzymes nor activation of PKA or PKC is involved in this process is consistent with the properties of the Ca2+-dependent inactivation of NMDA channels in CNS neurons (Legendre et al. 1993; Rosenmund & Westbrook, 1993; Krupp et al. 1999), possibly indicating a common mechanism.
The possible physiological significance of our study is considered briefly below. According to our data, AMPA receptors in the ganglion cells studied are impermeable to Ca2+. However, the synaptic activation of AMPA receptors and the resulting depolarization leads to an increased influx of Ca2+ through voltage-gated Ca2+ channels. The same depolarization will open Ca2+-permeable NMDA channels (Diamond & Copenhagen, 1993) by reducing the Mg2+ block of these receptors. Thus a Ca2+-induced inhibition of NMDA receptor-mediated synaptic responses is a negative feedback mechanism that helps to reduce the Ca2+ accumulation in the post-synaptic cell caused by repetitive synaptic activity. Given the large contribution of NMDA receptors to ganglion cell EPSCs (Diamond & Copenhagen, 1993), a Ca2+-mediated reduction in the EPSC will help to shape the spike pattern of the ganglion cell. A similar Ca2+-dependent inactivation of NMDA receptor-mediated monosynaptic EPSCs was reported recently in cultured hippocampal neurons (Medina et al. 1999). Although our results reflect the activity of perikaryal glutamate receptors of ganglion cells, the same mechanism may occur in neuronal dendrites which possess both voltage-gated Ca2+ channels and intracellular Ca2+ stores (Gruol et al. 1996).
Acknowledgments
We thank Dr Piotr Bregestovski for reading the manuscript and Drs W. G. Owen and D. Tranchina for helpful comments. This work was supported by National Eye Institute grants EY-12497 to A.A. and EY-03570 to P.W., and by the Helen Hoffritz Foundation.
References
- Akopian A, Gabriel R, Witkovsky P. Calcium released from intracellular stores inhibits GABAA-mediated currents in ganglion cells of the turtle retina. Journal of Neurophysiology. 1998;80:1106–1115. doi: 10.1152/jn.1998.80.3.1105. [DOI] [PubMed] [Google Scholar]
- Akopian A, Witkovsky P. Relationship between intracellular Ca2+ and light-evoked EPSCs in retinal ganglion cells. Investigative Ophthalmology and Visual Sciences. 2000;41:S935. [Google Scholar]
- Anwyl R. Metabotropic glutamate receptors: Electrophysiological properties and role in plasticity. Brain Research Reviews. 1999;39:83–120. doi: 10.1016/s0165-0173(98)00050-2. [DOI] [PubMed] [Google Scholar]
- Ascher P, Bregestovski P, Novak L. N-Methyl-d-aspartate activated channels of mouse central neurons in magnesium-free solutions. Journal of Physiology. 1988;399:207–226. doi: 10.1113/jphysiol.1988.sp017076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleakman D, Lodge D. Neuropharmacology of AMPA and kainate receptors. Neuropharmacology. 1998;37:1187–1204. doi: 10.1016/s0028-3908(98)00139-7. [DOI] [PubMed] [Google Scholar]
- Brandstätter B, Koulen P, Wässle H. Diversity of glutamate receptors in the mammalian retina. Vision Research. 1998;38:1385–1397. doi: 10.1016/s0042-6989(97)00176-4. [DOI] [PubMed] [Google Scholar]
- Burnashev N, Monyer H, Seeburg PH, Sakmann B. Divalent ion permeability of AMPA receptor channel is dominated by the edited form of a single subunit. Neuron. 1992;8:189–198. doi: 10.1016/0896-6273(92)90120-3. [DOI] [PubMed] [Google Scholar]
- Chen L, Huang L-Y. Protein kinase C reduces Mg block of NMDA receptor channels as a mechanism of modulation. Nature. 1992;7:319–326. doi: 10.1038/356521a0. [DOI] [PubMed] [Google Scholar]
- Chittajallu R, Alford S, Collingridge G L. Ca2+ and synaptic plasticity. Cell Calcium. 1998;24:377–385. doi: 10.1016/s0143-4160(98)90061-6. [DOI] [PubMed] [Google Scholar]
- Cooper NGF, McLaughlin BJ, Tallant EA, Cheung WY. Calmodulin-dependent protein phosphatase: immunocytochemical localization in chick retina. Journal of Cell Biology. 1985;101:1212–1218. doi: 10.1083/jcb.101.4.1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS, Copenhagen DR. The contribution of NMDA and non NMDA receptors to the light evoked input-output characteristics of retinal ganglion cells. Neuron. 1993;11:725–738. doi: 10.1016/0896-6273(93)90082-3. [DOI] [PubMed] [Google Scholar]
- Ehlers MD, Zhang S, Bernhadt JP, Huganir RL. Inactivation of NMDA receptors by direct interaction of calmodulin with the NR1 subunit. Cell. 1996;84:745–755. doi: 10.1016/s0092-8674(00)81052-1. [DOI] [PubMed] [Google Scholar]
- Gilbertson TA, Scobey R, Wilson M. Permeation of calcium ions through non-NMDA glutamate channels in retinal bipolar cells. Science. 1991;251:1613–1615. doi: 10.1126/science.1849316. [DOI] [PubMed] [Google Scholar]
- Gruol DL, Netzeband JG, Parsons KL. Ca2+ signaling pathways linked to glutamate receptor activation in the somatic and dendritic regions of cultured cerebellar purkinje neurons. Journal of Neuroscience. 1996;76:3325–3340. doi: 10.1152/jn.1996.76.5.3325. [DOI] [PubMed] [Google Scholar]
- Hamassaki-Britto DE, Herman-Borgmeyer I, Heinemann S, Hughes TE. Expression of glutamate receptor genes in the mammalian retina:the localization of GluR1 through GluR7 mRNAs. Journal of Neuroscience. 1993;13:1888–1898. doi: 10.1523/JNEUROSCI.13-05-01888.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill P, Marty A, Neher E, Sakmann B, Sigworth F J. Improved patch clamp technique for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hartveit E, Brandstätter JH, Enz R, Wässle H. Expression of the mRNA of seven metabotropic glutamate receptors (mGluR1 to 7) in the rat retina. An in situ hybridization study on tissue sections and isolated cells. European Journal of Neuroscience. 1995;7:1472–1483. doi: 10.1111/j.1460-9568.1995.tb01142.x. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Perrino BA, Solderinger TR. Identification of an autoinhibitory domain in calcineurin. Journal of Biological Chemistry. 1990;265:1924–1927. [PubMed] [Google Scholar]
- Higgs MH, Lukasiewicz PD. Glutamate uptake limits synaptic excitation of retinal ganglion cells. Journal of Neuroscience. 1999;19:3691–3700. doi: 10.1523/JNEUROSCI.19-10-03691.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollman M, Heinemann S. Cloned glutamate receptors. Annual Review of Neuroscience. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- Huganir RL, Greengard P. Regulation of neurotransmitter receptor desensitization by protein phosphorylation. Neuron. 1990;5:555–567. doi: 10.1016/0896-6273(90)90211-w. [DOI] [PubMed] [Google Scholar]
- Johnson JW, Ascher P. Voltage-dependent block by intracellular Mg2+ of NMDA-activated channels. Biophysical Journal. 1990;57:1085–1090. doi: 10.1016/S0006-3495(90)82626-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupp JJ, Vissel B, Thomas CG, Heinemann S F, Westbrook G L. Interactions of calmodulin and alpha-actinin with the NR1 subunit modulate Ca2+-dependent inactivation of NMDA receptors. Journal of Neuroscience. 1999;19:1165–1178. doi: 10.1523/JNEUROSCI.19-04-01165.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyrozis A, Goldstein PA, Heath MJS, MacDermott A. Calcium entry through a subpopulation of AMPA receptors desensitized neighboring NMDA receptors in rat dorsal horn neurons. Journal of Physiology. 1995;485:373–381. doi: 10.1113/jphysiol.1995.sp020736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laabich A, Cooper NGF. Regulation of calcium calmodulin-dependent protein kinase II in the adult rat retina is mediated by ionotropic glutamate receptors. Experimental Eye Research. 1999;68:703–713. doi: 10.1006/exer.1999.0664. [DOI] [PubMed] [Google Scholar]
- Legendre P, Rosenmund C, Westbrook G. Inactivation of NMDA channels in cultured hippocampal neurons by intracellular calcium. Journal of Neuroscience. 1993;13:674–684. doi: 10.1523/JNEUROSCI.13-02-00674.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linn CP, Christensen BN. Excitatory amino acid regulation of intracellular Ca in isolated catfish horizontal cells measured under voltage- and concentration-clamp conditions. Journal of Neuroscience. 1992;12:2156–2164. doi: 10.1523/JNEUROSCI.12-06-02156.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu WY, Jackson MF, Bai D, Orser B A, MacDonald JF. pyramidal neurons of the hippocampus protein kinase C regulates calcium-dependent inactivation of NMDA receptors. Journal of Neuroscience. 2000;20:4452–4461. doi: 10.1523/JNEUROSCI.20-12-04452.2000. In CA1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukasiewicz PD, Lawrence JE, Valentino T L. Desensitizing glutamate receptors shape excitatory synaptic inputs to tiger salamander retinal ganglion cells. Journal of Neuroscience. 1995;15:6189–6199. doi: 10.1523/JNEUROSCI.15-09-06189.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukasiewicz PD, McReynolds JS. Synaptic transmission at NMDA receptors in the proximal retina of the mudpuppy. Journal of Physiology. 1985;367:99–115. doi: 10.1113/jphysiol.1985.sp015816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukasiewicz PD, Werblin F. A slowly inactivating potassium current truncates spike activity in ganglion cells of the tiger salamander retina. Journal of Neuroscience. 1988;12:4470–4481. doi: 10.1523/JNEUROSCI.08-12-04470.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey S, Miller RF. N-Methyl-D-aspartate receptors of ganglion cells in rabbit retina. Journal of Neurophysiology. 1990;63:16–30. doi: 10.1152/jn.1990.63.1.16. [DOI] [PubMed] [Google Scholar]
- Mayer ML, Westbrook GL. Permeation and block of NMDA receptor channels by divalent cations in mouse cultured central neurons. Journal of Physiology. 1987;394:501–527. doi: 10.1113/jphysiol.1987.sp016883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina I, Filippova N, Bakhramov A, Bregestovski P. Calcium-induced inactivation of NMDA receptor- channels evolves independently of run-down in cultured rat brain neurones. Journal of Physiology. 1996;495:411–427. doi: 10.1113/jphysiol.1996.sp021603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina I, Leinekugel X, Ben-Ari Y. Calcium-dependent inactivation of the monosynaptic NMDA EPSCs in rat hippocampal neurons in culture. European Journal of Neuroscience. 1999;11:2422–2430. doi: 10.1046/j.1460-9568.1999.00664.x. [DOI] [PubMed] [Google Scholar]
- Mittman S, Taylor WR, Copenhagen DR. Concomitant activation of two types of glutamate receptor mediates excitation of salamander retinal ganglion cells. Journal of Physiology. 1990;428:175–197. doi: 10.1113/jphysiol.1990.sp018206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyakawa H, Ross WN, Jaffe D, Callaway JC, Lasser-Ross N, Lisman J A, Johnson D. Synaptically activated increases in Ca2+ concentration in hippocampal CA1 pyramidal cells are primarily due to voltage-gated Ca2+ channels. Neuron. 1992;9:1163–1173. doi: 10.1016/0896-6273(92)90074-n. [DOI] [PubMed] [Google Scholar]
- Nicoll AE, Malenka RC. Contrasting properties of two forms of long-term potentiation in the hippocampus. Nature. 1995;377:115–118. doi: 10.1038/377115a0. [DOI] [PubMed] [Google Scholar]
- Osborne NN, Ghazi H. Agonist stimulated inositol phospholipid hydrolysis in the mammalian retina. In: Osborne NN, Chader G J, editors. Progress in Retinal Research. vol. 8. Oxford: Pergamon; 1990. pp. 221–228. [Google Scholar]
- Peng YW, Blackstone CD, Huganir RL, Yau KW. Distribution of glutamate receptor subtypes in the vertebrate retina. Neuroscience. 1995;66:483–497. doi: 10.1016/0306-4522(94)00569-q. [DOI] [PubMed] [Google Scholar]
- Peng YW, Sharp AH, Snyder SH, Yau KW. Localization of the inositol 1,4,5-trisphosphate receptor in synaptic terminals in the vertebrate retina. Neuron. 1991;6:525–531. doi: 10.1016/0896-6273(91)90055-5. [DOI] [PubMed] [Google Scholar]
- Pin JP, Duvoisin R. Neurotransmitter receptors. I. The metabotropic glutamate receptors: structure and functions. Neuropharmacology. 1995;34:1–26. doi: 10.1016/0028-3908(94)00129-g. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Westbrook G. Calcium-induced actin depolymerization reduces NMDA channel activity. Neuron. 1993;10:805–814. doi: 10.1016/0896-6273(93)90197-y. [DOI] [PubMed] [Google Scholar]
- Shen W, Slaughter MM. Metabotropic and ionotropic glutamate receptors regulate calcium channel currents in salamander retinal ganglion cells. Journal of Physiology. 1998;510:815–828. doi: 10.1111/j.1469-7793.1998.815bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Slaughter MM. Internal calcium modulates apparent affinity of metabotropic GABA receptors. Journal of Neurophysiology. 1999;82:3298–3306. doi: 10.1152/jn.1999.82.6.3298. [DOI] [PubMed] [Google Scholar]
- Thoreson W, Witkovsky P. Glutamate receptors and circuits in the vertebrate retina. Progress in Retina and Eye Research. 1999;18:765–810. doi: 10.1016/s1350-9462(98)00031-7. [DOI] [PubMed] [Google Scholar]
- Van Belle H. R24571: A potent inhibitor of calmodulin-activated enzymes. Cell Calcium. 1981;2:483. [Google Scholar]
- Verkhratsky A, Shmigol A. Calcium-induced calcium release in neurons. Cell Calcium. 1996;19:1–14. doi: 10.1016/s0143-4160(96)90009-3. [DOI] [PubMed] [Google Scholar]
- Viklicky L. Calcium-mediated modulation of NMDA responses in cultured rat hippocampal neurons. Journal of Physiology. 1993;470:575–600. doi: 10.1113/jphysiol.1993.sp019876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Ehlers MD, Bemhardt J P, Su ChT, Huganir R L. Calmodulin mediates calcium-dependent inactivation of NMDA receptors. Neuron. 1998;21:443–453. doi: 10.1016/s0896-6273(00)80553-x. [DOI] [PubMed] [Google Scholar]