

Abstract
We have investigated the prediction of a relationship between the magnitude of activity-dependent increases in postsynaptic calcium and both the magnitude and direction of synaptic plastic change in the central nervous system. Activity-dependent increases in calcium were buffered to differing degrees using a range of concentrations of EGTA and the effects on synaptic plasticity were assessed.
Activity-dependent synaptic plasticity was induced during whole-cell recording in rat perirhinal cortex in vitro. In control conditions (0.5 mm EGTA) low frequency stimulation (LFS; 200 stimuli) delivered to neurones held at -40 or -70 mV induced long-term depression (LTD) or, at -10 mV, induced long-term potentiation (LTP).
The relationship between EGTA concentration (0.2 to 10 mm) and the magnitude of LTD was examined. This relationship described a U-shaped curve, as predicted by models of synaptic plasticity. This provides strong evidence that the magnitude of LTD is determined by the magnitude of the increase in intracellular calcium concentration.
LFS paired with depolarisation to -10 mV induced LTD, no change or LTP as activity-dependent postsynaptic calcium levels were allowed to increase progressively by the use of progressively lower concentrations of buffer (10 to 0.2 mm EGTA).
We investigated if the lack of plasticity that occurs at the transition between LTD and LTP is due to induction of both of these processes with zero net change, or is due to neither LTD nor LTP being induced. These experiments were possible as LTP but not LTD was blocked by the protein kinase inhibitor staurosporine while LTD but not LTP was blocked by the mGlu receptor antagonist MCPG. At the transition between LTD and LTP, blocking LTP mechanisms did not uncover LTD whilst blocking LTD mechanisms did not uncover LTP. This suggests that the transition between LTD and LTP is due to the lack of induction of both of these processes and also suggests that these two processes are induced independently of one another.
One of the strengths of synaptic plasticity as a model for encoding learning and memory is that synaptic transmission can be increased, as in long-term potentiation (LTP; Bliss & Collingridge, 1993), or can be decreased, as in long-term depression (LTD; Linden & Connor, 1995; Bear & Abraham, 1996). It was proposed in the Bienenstock-Cooper-Munro (BCM) model (Bienenstock et al. 1982) that the bidirectional control of synaptic strength depends on some combined function of pre- and postsynaptic activity. Thus progressively incrementing activity will result initially in LTD and then in LTP. The results of many experimental studies are consistent with the notion that there are different thresholds for the induction of LTP and LTD and it has been suggested, in the Artola-Brocher-Singer (ABS) rule (Artola & Singer, 1993), that these different thresholds are dependent on postsynaptic membrane potential. The proposal (Lisman, 1989) that the bidirectional control of synaptic plasticity relies on increases in postsynaptic calcium concentration to different levels has led to the widely held assumption that the dependence of LTP and LTD on membrane potential is directly related to some function associated with the concentration of postsynaptic calcium ions (Lisman, 1989; Artola & Singer, 1993; Yang et al. 1999). Given this assumption, an important prediction that follows from models of synaptic plasticity is that increases in postsynaptic calcium concentration beyond the initial threshold for induction of LTD should initially produce more LTD and subsequently produce less LTD, i.e. there should be a U-shaped relationship between calcium concentration and the magnitude of LTD. Still greater increases in postsynaptic calcium concentration beyond that required for LTD should then produce LTP.
There is indirect, but little direct, evidence that supports the changes in activity-dependent synaptic plasticity described by the BCM and ABS rules being directly dependent on the magnitude of increases in postsynaptic calcium (Artola & Singer, 1993; Yang et al. 1999; Ngezahayo et al. 2000). The most convincing evidence is that partial buffering of postsynaptic calcium by intracellular calcium chelators can result in the conversion of LTP to LTD (Kimura et al. 1990; Hansel et al. 1997). Furthermore, there is only indirect evidence for there being a U-shaped relationship between postsynaptic calcium levels and LTD. For example, afferent stimulation at different frequencies between 0.01 and 10 Hz can produce different magnitudes of LTD (Dudek & Bear, 1992; Kirkwood et al. 1993); a given stimulus protocol can produce differing magnitudes of LTD in the presence of varying concentrations of the NMDA receptor antagonist 2-amino-5-phosphonopentanoic acid (AP5; Cummings et al. 1996). In this study we have examined whether there is any relationship between the magnitude of activity-dependent LTD and the intracellular concentration of the calcium chelator EGTA. Furthermore, we have tested whether the full range of synaptic plasticity, from LTD to LTP, may be induced by a given stimulus protocol when activity-dependent increases in postsynaptic calcium are manipulated across a wide range by altering the intracellular concentration of EGTA. Finally, we have examined whether the observed transition between LTP and LTD is due to the induction of and net balance between these two processes, or reflects a lack of both LTP and LTD. These experiments have been carried out in the perirhinal cortex, a region of the brain involved in learning and memory processes (Brown & Xiang, 1998), and that displays both LTD (Cho et al. 2000) and LTP (Ziakopoulos et al. 1999). Some of these results have appeared previously in abstract format (Bashir et al. 2000).
METHODS
Slices of perirhinal cortex were prepared from adult male DA rats (150-270 g, 7-12 weeks, Bantin and Kingman, UK) as previously described (Cho et al. 2000). All efforts were made to minimise animal suffering and numbers of animals used. Animals were anaesthetised with halothane, in accordance with the UK Animals (Scientific Procedures) Act 1986, and decapitated, and the brain was rapidly removed and placed in ice-cold artificial cerebrospinal fluid (ACSF; bubbled with 95 % O2-5 % CO2) which comprised (mm): NaCl, 124; KCl, 3; NaHCO3, 26; NaH2PO4, 1.25; CaCl2, 2; MgSO4, 1; d-glucose, 10. A mid-sagittal section was made, and the rostral and caudal parts of the brain were removed by single scalpel cuts made at approximately 45 deg to the dorso-ventral axis and each half glued by its caudal end to a vibroslice stage (Campden Instruments, Sileby, UK). Slices (400 μm), which included perirhinal, entorhinal and temporal cortices were stored submerged in ACSF (20-25 °C). A single slice was placed in a submerged recording chamber (28-30 °C, flow rate ≈2 ml min−1) when required. Picrotoxin (5 μm) was present throughout the experiment. Blind whole-cell recordings were obtained from neurones in layer II-III. Pipette (4-7 MΩ) solutions (280 mosmol l−1, pH 7.2) comprised (mm): CsMeSO4, 130; NaCl, 8; Mg-ATP, 4; Na-GTP, 0.3; EGTA, 0.5; Hepes, 10; QX-314, 6. In filling solutions containing different EGTA concentrations osmolarity was maintained by substitution of the EGTA for the equivalent concentration of CsMeSO4. One stimulating electrode was placed dorso-rostrally on the temporal cortex side (area 35/36) and one ventro-caudally on the entorhinal cortex side (area 35/entorhinal cortex) of the rhinal sulcus. Stimuli (constant voltage) were delivered alternately to the two electrodes (each electrode 0.033 Hz). Neurones were voltage clamped at -70 mV unless otherwise indicated. Low frequency stimulation (LFS: 200 stimuli, 1 Hz) was delivered to one input in any one slice in order to induce LTD. LFS was delivered either at -70 mV or paired with depolarisation to -40 or -10 mV, as appropriate for different experiments. Where the membrane potential was changed this was done for the duration of LFS only. The amplitude of the evoked EPSCs was measured and expressed relative to the normalised pre-conditioning baseline. Effects of LFS were monitored for at least 15 min in all experiments and measurements for statistical analysis were made averaged over a 5 min period (between 10 and 25 min) after LFS. In experiments where LFS was delivered at -10 mV, LFS was always delivered between 5 and 6 min of obtaining whole-cell access. This ensured that these experiments did not suffer from the problem of whole-cell ‘wash-out’ of LTP. Furthermore, minimising the differences in the time between gaining whole-cell access and delivering LFS ensured that any potential differences in synaptic [EGTA] between different experiments was reduced as far as possible. If the effects of a drug were being analysed in such experiments then the drug was always present for at least 15 min prior to obtaining whole-cell access. Data were only analysed from one slice per rat (number, n). Data pooled across slices are expressed as the means ±s.e.m. and significance (P < 0.05) tested using Student's paired or unpaired t test as appropriate. Only experiments in which there was no appreciable drift (< 10 %) in the baseline were included in the pooled data. Data were recorded using an Axopatch 200 amplifier (Axon Instruments, Foster City, CA, USA), monitored and analysed on-line and re-analysed off-line (Anderson & Collingridge, 1997). MCPG (Tocris Cookson, UK) and staurosporine (Sigma) were applied by addition to the perfusate.
RESULTS
Whole-cell recordings were obtained from pyramidal neurones in layer II-III of the perirhinal cortex. Synaptic transmission was evoked by stimulation within the same layer. In the first series of experiments we examined whether there is indeed a U-shaped relationship between increases in postsynaptic calcium concentration, as determined by EGTA buffering, and the magnitude of LTD. After obtaining a stable baseline of evoked synaptic responses, low frequency stimulation (LFS; 200 stimuli, 1 Hz) was paired with depolarisation of the postsynaptic neurone to -40 mV, a protocol that results in robust LTD when 0.5 mm EGTA is included in the whole-cell pipette (Cho et al. 2000). In different experiments whole-cell solutions containing the following [EGTA] were used: 0.2, 0.5, 2, 5 and 10 mm. Under these conditions the magnitude of synaptic change was -2 ± 14, -40 ± 8, -56 ± 7, -34 ± 6 and -1 ± 5 %, respectively (n= 4, except for 10 mm EGTA where n= 3). The results of these experiments, plotted in Fig. 1, show clearly that the magnitude of LTD was dependent on the intracellular [EGTA] and described a U-shaped curve.
Figure 1. The magnitude of LTD is dependent on the activity-dependent increase in intracellular calcium.

A, LTD is not induced by pairing LFS with depolarisation to -40 mV when the filling solution contains 0.2 mm EGTA n = 4. B, however, LTD is induced with 2 mm EGTA in the filling solution n = 4. C, increasing EGTA to 5 mm reduces but does not prevent the induction of LTD n = 4. In this and subsequent figures the period of LFS is indicated by the arrows. Synaptic traces are the average of 4 consecutive responses from the time points indicated. D, graph illustrating the magnitude of LTD induced in the presence of each of the 5 different concentrations of EGTA: a U-shaped relationship exists between [EGTA] and LTD when LTD is induced at either -40 mV (○) or -70 mV (•). *Significant difference from baseline (P < 0.05).
We next repeated the above experiments but induced LTD by delivering LFS whilst the postsynaptic neurone was voltage clamped to -70 mV. This protocol results in robust LTD, but the induction mechanisms are different from those at -40 mV (Cho et al. 2000). These experiments therefore test whether the relationship between [EGTA] and LTD is similar under conditions where different induction mechanisms exist. The intracellular [EGTA] in different experiments was 0.2, 0.5, 2, 5 and 10 mm and the magnitude of synaptic change was -5 ± 5, -39 ± 8, -36 ± 10, -1 ± 4 and +4 ± 9 %, respectively (n= 4, except for 10 mm EGTA where n= 3). Thus it can be seen in Fig. 1D that the magnitude of LTD induced by LFS at -70 mV is also dependent on the intracellular [EGTA] and also describes a U-shaped relationship. These results strongly suggest that irrespective of the mode of induction there is a direct relationship between activity-dependent increases in intracellular calcium concentration and the magnitude of LTD.
Given that the induction of both LTP and LTD relies on increases in postsynaptic calcium concentration, then a simple prediction of the ABS and BCM rules is that a sufficient increase in calcium should result in LTP. However, LTP did not occur in either of the above series of experiments. It is most likely that, even with the lowest [EGTA], the increase in calcium concentration produced by the above protocols was not sufficient to produce LTP. To overcome this, LFS was delivered during depolarisation of the postsynaptic cell to -10 mV. In these and all subsequent experiments LFS was delivered 5-6 min after gaining whole-cell access (see Methods). The [EGTA] used in different experiments was 0.2, 0.5, 5 and 10 mm and the change in synaptic strength was +43 ± 9, +40 ± 11, -7 ± 6 and -40 ± 14 %, respectively (n= 4, except for 0.2 mm EGTA where n= 3; Fig. 2). Since LTD occurred even in the presence of 10 mm EGTA (which blocked LTD in the experiments shown in Fig. 1), the EGTA concentration was increased to 20 mm and the same induction protocol was delivered. Under these conditions no change in synaptic strength was induced (+6 ± 11 %, n= 4; Fig. 2). Taking these results together demonstrates that a given stimulus protocol can produce the full range of plasticity ranging from different magnitudes of LTD, to no effect, through to LTP.
Figure 2. Altering [EGTA] appropriately can result in LTP, no change, or LTD.
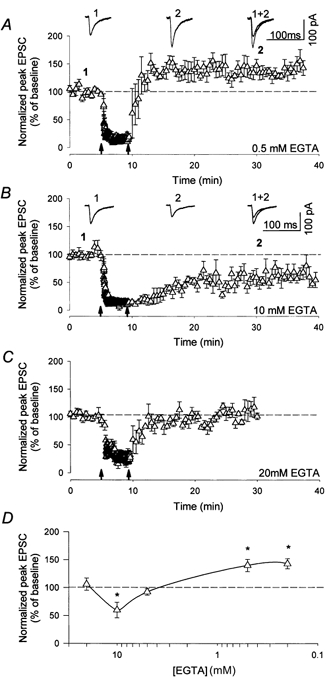
LFS delivered during depolarisation to -10 mV results in LTP in the presence of 0.5 mm EGTA n = 4 (A) but LTD in 10 mm EGTA n = 4 (B). C, changes in synaptic strength are completely prevented by the inclusion of 20 mm EGTA in the whole-cell solution n = 4. D, graph showing the effect of a range of different concentrations of EGTA on synaptic strength. *Significant difference from baseline (P < 0.05).
Following demonstration of the above results, we asked why synaptic strength does not change in the region of transition between LTD and LTP. One possible reason for the lack of plasticity is that the magnitude of the rise in calcium concentration is appropriate to induce both LTP and LTD, resulting in no net plasticity (see Fig. 3A and legend for explanation). Alternatively, the levels of calcium achieved may be inappropriate for the induction of either LTP or LTD (Fig. 3B and legend for explanation). To differentiate between these two possibilities we took advantage of the finding that LTP and LTD can rely on different induction mechanisms. Thus, in the perirhinal cortex LTP is not blocked by the broad-spectrum mGlu receptor antagonist MCPG (+40 ± 16 %, n= 3; Fig. 4A; see also Ziakopoulos et al. 1999). LTD, induced with depolarisation to -10 mV in 10 mm EGTA, is prevented by MCPG (+5 ± 7 %; n= 3, Fig. 4B; see also Cho et al. 2000). If the lack of plasticity at the cross-over between LTP and LTD is due to a balance between the two processes then the selective block of LTD by MCPG at this transition point should result in LTP. Therefore, experiments were carried out in which LFS was delivered at -10 mV with 5 mm EGTA in the whole-cell pipette since these are conditions that produce transition between LTD and LTP. However, under these conditions, neither LTP nor LTD was induced in the presence of MCPG (-4 ± 10 %, n= 3, P > 0.05, Fig. 4C and D).
Figure 3. Possible interactions between LTD and LTP in determining the outcome of synaptic strength.

A, the change in synaptic strength ( ) may be a function of the induction and expression of both LTD and LTP. The induction and expression of LTD (○) occurs at low concentrations of calcium and then reaches a plateau level. As calcium levels increase, the induction of LTP (•) also occurs and then reaches a maximum plateau level. The change in synaptic strength therefore describes the middle curve (
) may be a function of the induction and expression of both LTD and LTP. The induction and expression of LTD (○) occurs at low concentrations of calcium and then reaches a plateau level. As calcium levels increase, the induction of LTP (•) also occurs and then reaches a maximum plateau level. The change in synaptic strength therefore describes the middle curve ( ) due to both LTD and LTP and the transition between LTD and LTP is due to a sum of these two opposite processes. B, an alternative explanation is that LTD (○) and LTP (•) do not co-exist and that the change in synaptic strength (
) due to both LTD and LTP and the transition between LTD and LTP is due to a sum of these two opposite processes. B, an alternative explanation is that LTD (○) and LTP (•) do not co-exist and that the change in synaptic strength ( ) is a function of the selective presence of either LTD or LTP. Thus LTD is induced by low calcium concentrations. As the calcium concentration increases, LTD induction is prevented. As the calcium levels increase further the induction of LTP occurs. Therefore the two processes rely on different calcium concentrations and occur essentially independently of one another, and the transition between LTD and LTP is due to the lack of both LTD and LTP.
) is a function of the selective presence of either LTD or LTP. Thus LTD is induced by low calcium concentrations. As the calcium concentration increases, LTD induction is prevented. As the calcium levels increase further the induction of LTP occurs. Therefore the two processes rely on different calcium concentrations and occur essentially independently of one another, and the transition between LTD and LTP is due to the lack of both LTD and LTP.
Figure 4. MCPG, which blocks LTD but not LTP, has no effect on the position of the transition between LTP and LTD.

A, LTP, induced by depolarisation to -10 mV in the presence of 0.5 mm EGTA, is not blocked by MCPG n = 3. B, LTD, which is normally induced by depolarisation to -10 mV in the presence of 10 mm EGTA, is blocked by MCPG n = 3. C, the transition between LTP and LTD, observed at -10 mV in the presence of 5 mm EGTA, is not affected by MCPG n = 3. D, graph showing the results from the above experiments plotted together with the plasticity curve obtained from Fig. 2.
In the next series of experiments we examined the same question but used the opposite strategy. We found that the protein kinase inhibitor staurosporine prevented the induction of LTP (-7 ± 5 %, n= 3, Fig. 5A) but not LTD (-39 ± 10 %, n= 3, Fig. 5B). Therefore, experiments were carried out in which LFS was delivered at -10 mV with 5 mm EGTA in the pipette, conditions that normally produce a transition between LTP and LTD. Under these conditions, in the presence of staurosporine, there was no significant LTP or LTD induced (-11 ± 6 %, n= 5, P > 0.05 Fig. 5C and D). Therefore, staurosporine, which blocks LTP, did not uncover LTD.
Figure 5. Staurosporine, which blocks LTP but not LTD, has no effect on the transition between LTP and LTD.

A, LTP, induced by depolarisation to -10 mV in the presence of 0.5 mm EGTA is blocked by staurosporine n = 3. B, LTD, induced by depolarisation to -10 mV in the presence of 10 mm EGTA is not blocked by staurosporine n = 3. C, the transition between LTP and LTD is unaffected by staurosporine n = 5. D, graph showing the results from the above experiments plotted together with the plasticity curve obtained from Fig. 2.
Taking the staurosporine and MCPG results together suggests that the transition between LTD and LTP is not due to the induction of and balance between significant magnitudes of these two opposite forms of plasticity (see Fig. 3A and B). Furthermore, these results also suggest that once sufficient calcium concentrations are achieved for the induction of LTP, the processes of LTD are inactivated.
DISCUSSION
In this study we have tested predictions of the dependence of synaptic plasticity on the magnitude of increases in postsynaptic calcium concentration (Bienenstock et al. 1982; Lisman, 1989; Artola & Singer, 1993). The results of this study show that the magnitude of LTD is dependent upon the postsynaptic [EGTA] and describes a U-shaped relationship. Furthermore, the direction of plasticity (LTP or LTD) is also dependent on postsynaptic [EGTA]. Finally, we provide evidence suggesting that the transition in bidirectional plasticity is likely to be due to increases in calcium to levels that are inappropriate for the induction of either LTP or LTD. Since a rise in postsynaptic calcium underlies the induction of LTP and LTD in perirhinal cortex, the results of this present study are also likely to be applicable to many other brain regions.
Results from previous studies suggest that different levels of postsynaptic activation produce different levels of LTD. For example, the magnitude of LTD is dependent on stimulus frequency in a U-shaped manner (Dudek & Bear, 1992; Kirkwood et al. 1993). That the magnitude of LTD depends on increases in intracellular calcium to different levels was inferred from experiments in which LTD magnitude was shown to be a function of NMDA receptor activation (Cummings et al. 1996). Our current data provide further support for this assertion. Furthermore, our results suggest that there is a U-shaped relationship between calcium levels and LTD magnitude. Interestingly, we show for the first time that as the concentration of calcium buffer is reduced (from 2 mm) this also results in the induction of less LTD. This suggests that as the LFS-induced calcium rise is allowed to increase less LTD is induced and raises the possibility that either LTD is being inactivated or LTP is also being induced. Thus this experimental finding fits very well the U-shaped relationship predicted by theoretical models of synaptic plasticity (Bienenstock et al. 1982; Artola & Singer, 1993; Gold & Bear, 1994) and suggests that it is the magnitude of the postsynaptic increase in calcium concentration that directly determines the magnitude of LTD.
Under the conditions of our experiments, depolarisation to -10 mV paired with LFS successfully resulted in the induction of LTP. Manipulating the activity-dependent rise in postsynaptic calcium concentration produced the full range of synaptic plasticity from LTP, through no plasticity, to LTD and finally no plasticity again. That different thresholds for LTD and LTP may relate to different calcium concentrations has been indirectly suggested by experiments in which tetanic stimulation can produce LTP or LTD depending on extracellular calcium concentration (Mulkey & Malenka, 1992); furthermore, a given stimulus protocol can result in LTP or LTD depending on levels of NMDA receptor activation (by varying the concentration of AP5; Cummings et al. 1996). The most direct evidence to date comes from experiments in which incomplete buffering of intracellular calcium blocks LTP but allows the induction of LTD (Kimura et al. 1990; Hansel et al. 1997). Our results extend these findings by demonstrating that in the presence of appropriate [EGTA] a given stimulus protocol can induce LTP, no change or LTD, strikingly similar to the predictions of the BCM and ABS rules.
Whilst EGTA has different properties compared to, for example, BAPTA it has been shown previously that both of these buffers similarly block the induction of LTD (Egger et al. 1999). Therefore, whilst it is likely that differences in buffering properties between EGTA and BAPTA are important when studying calcium changes resulting from very brief stimuli, any differences are less likely to be influential when studying changes resulting from stimuli lasting hundreds of seconds. A potential concern when interpreting the current results is in knowing the full extent of equilibration of EGTA within the time (5-6 min) before LFS is delivered at -10 mV. However, it was necessary to perform experiments in this way in order to overcome the intracellular ‘washout’ of LTP with whole-cell recording. The exact level of equilibration of EGTA would be a concern for making accurate estimates of [Ca2+]i and, given this potential limitation, we have not attempted to correlate pipette [EGTA] directly to [Ca2+]i. Our main concern was to ensure that we could make comparisons between different experiments. Therefore, given that the diffusion constant for EGTA should not differ markedly between neurones, ensuring minimal variation in timing of LFS should ensure that equilibration of EGTA will be relatively constant between experiments.
A consequence of bidirectional plasticity and a prediction of the ABS and BCM rules (Bienenstock et al. 1982; Artola & Singer, 1993) is a region of transition between LTP and LTD (Dudek & Bear, 1992; Cummings et al. 1996; Hansel et al. 1997; Ngezhayo et al. 2000). One possible mechanism underlying this transition is that the levels of calcium at this point induce both LTP and LTD, producing no net change in synaptic strength (see Fig. 3A). An alternative is that the calcium concentration may be too high to induce LTD but may not be sufficiently high to induce LTP, i.e. neither LTD nor LTP is induced (see Fig. 3B). Our results suggest the latter possibility. Thus, with the appropriate [EGTA] that resulted in transition between the two forms of plasticity, the block of LTD with MCPG did not reveal LTP, and the block of LTP with staurosporine did not uncover LTD. Therefore, our interpretation is that the lack of plasticity is due to a concentration of calcium that is too high to efficiently induce LTD but too low to efficiently induce LTP. A possible mechanistic explanation for this is that the phosphatases (e.g. PP1, 2A, 2B) and kinases (e.g. PKC, CAMKII) responsible for LTD (Mulkey et al. 1994) and LTP (Bliss & Collingridge, 1993) are activated at low and high calcium concentrations, respectively (Lisman, 1989; Malenka & Nicoll, 1999; Soderling & Derkach 2000). In addition, however, our results also suggest that LTP and LTD do not co-exist once the calcium concentration increases sufficiently to induce LTP. Thus MCPG, which blocks LTD, did not enhance the magnitude of LTP. Furthermore, the block of LTP by staurosporine did not result in the uncovering of LTD. Thus our current hypothesis, in keeping with the original suggestion of Lisman (1989), requires that there is also an inactivation of LTD induction mechanisms at the calcium levels that induce LTP. Consistent with this, evidence exists that phosphorylation of Inhibitor 1 results in inhibition of the protein phosphatases (e.g. PP1) (Donella-deana et al. 1994; Strack et al. 1997) that are required for the induction of LTD (Mulkey et al. 1994). Therefore, high calcium levels can initiate phosphorylation by PKC/CAMKII to produce LTP. The same calcium levels can phosphorylate Inhibitor 1, inactivate protein phosphatases and prevent LTD (Soderling & Derkach 2000) ensuring the induction of both LTP and LTD does not occur simultaneously.
In conclusion, our results strongly suggest that the direction and the magnitude of synaptic plasticity are related to different intracellular levels of calcium. Importantly, our results also suggest that LTD and LTP are not conjointly induced at any tested calcium concentration.
Acknowledgments
This work was supported by the BBSRC and the MRC.
References
- Anderson WW, Collingridge GL. A data acquisition program for on-line analysis of long-term potentiation and long-term depression. Society for Neuroscience Abstracts. 1997;23:665. doi: 10.1016/s0165-0270(01)00374-0. [DOI] [PubMed] [Google Scholar]
- Artola A, Singer W. Long-term depression of excitatory synaptic transmission and its relationship to long-term potentiation. Trends in Neurosciences. 1993;16:480–487. doi: 10.1016/0166-2236(93)90081-v. [DOI] [PubMed] [Google Scholar]
- Bashir ZI, Aggleton JP, Brown MW, Cho K. The magnitude of cellular calcium determines the magnitude of LTD. European Journal of Neuroscience. 2000;12(suppl. 11):512. [Google Scholar]
- Bear MF, Abraham WC. Long-term depression in hippocampus. Annual Review of Neuroscience. 1996;19:437–462. doi: 10.1146/annurev.ne.19.030196.002253. [DOI] [PubMed] [Google Scholar]
- Bienenstock EL, Cooper LN, Munro PW. Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. Journal of Neuroscience. 1982;2:32–48. doi: 10.1523/JNEUROSCI.02-01-00032.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Brown MW, Xiang J-Z. Recognition memory: neuronal substrates of the judgement of prior occurrence. Progress in Neurobiology. 1998;55:149–189. doi: 10.1016/s0301-0082(98)00002-1. [DOI] [PubMed] [Google Scholar]
- Cho K, Kemp N, Noel J, Aggleton JP, Brown MW, Bashir ZI. A new form of long-term depression in the perirhinal cortex. Nature Neuroscience. 2000;3:150–156. doi: 10.1038/72093. [DOI] [PubMed] [Google Scholar]
- Cummings JA, Mulkey RM, Nicoll RA, Malenka RC. Ca2+ signaling requirements for long-term depression in the hippocampus. Neuron. 1996;16:825–833. doi: 10.1016/s0896-6273(00)80102-6. [DOI] [PubMed] [Google Scholar]
- Donella-Deana MH, Krinks MH, Ruzzene M, Klee C, Pinna LA. Dephosphorylation of phosphopeptides by calcineurin (protein phosphatase 2B) European Journal of Biochemistry. 1994;219:109–117. doi: 10.1111/j.1432-1033.1994.tb19920.x. [DOI] [PubMed] [Google Scholar]
- Dudek SM, Bear MF. Homosynaptic long-term depression in area CA1 of the hippocampus and effects of N-methyl-D-aspartate receptor blockade. Proceedings of the National Academy of Sciences of the USA. 1992;89:4363–4367. doi: 10.1073/pnas.89.10.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger V, Feldmeyer D, Sakmann B. Coincidence detection and changes of synaptic efficacy in spiny stellate neurons in rat barrel cortex. Nature Neuroscience. 1999;2:1098–1105. doi: 10.1038/16026. [DOI] [PubMed] [Google Scholar]
- Gold JI, Bear MF. A model of dendritic spine Ca2+ concentration exploring possible bases for a sliding synaptic modification threshold. Proceedings of the National Academy of Sciences of the USA. 1994;91:3941–3945. doi: 10.1073/pnas.91.9.3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansel C, Artola A, Singer W. Relation between dendritic Ca2+ levels and the polarity of synaptic long-term modifications in rat visual cortex neurons. European Journal of Neuroscience. 1997;9:2309–2322. doi: 10.1111/j.1460-9568.1997.tb01648.x. [DOI] [PubMed] [Google Scholar]
- Kimura F, Tsumoto T, Niahigori A, Yoshimura Y. Long-term depression but not potentiation is induced in calcium chelated visual cortex neurones. NeuroReport. 1990;1:65–68. doi: 10.1097/00001756-199009000-00018. [DOI] [PubMed] [Google Scholar]
- Kirwood A, Dudek SM, Gold JT, Aizenman CD, Bear MF. Common forms of synaptic plasticity in the hippocampus and neocortex in vitro. Science. 1993;260:1518–1521. doi: 10.1126/science.8502997. [DOI] [PubMed] [Google Scholar]
- Linden DJ, Connor JA. Long-term synaptic depression. Annual Review of Neuroscience. 1995;18:319–357. doi: 10.1146/annurev.ne.18.030195.001535. [DOI] [PubMed] [Google Scholar]
- Lisman JA. A mechanism for the Hebb and the anti-Hebb process underlying learning and memory. Proceedings of the National Academy of Sciences of the USA. 1989;86:9574–9578. doi: 10.1073/pnas.86.23.9574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. Long-term potentiation - a decade of progress? Science. 1999;285:1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- Mulkey RM, Endo S, Shinolikar S, Malenka RC. Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature. 1994;369:486–488. doi: 10.1038/369486a0. [DOI] [PubMed] [Google Scholar]
- Mulkey RM, Malenka RC. Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron. 1992;9:967–975. doi: 10.1016/0896-6273(92)90248-c. [DOI] [PubMed] [Google Scholar]
- Ngezahayo A, Schachner M, Artola A. Synaptic activity modulates the induction of bidirectional synaptic changes in adult mouse hippocampus. Journal of Neuroscience. 2000;20:2451–2458. doi: 10.1523/JNEUROSCI.20-07-02451.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderling TS, Derkach VA. Postsynaptic protein phosphorylation and LTP. Trends in Neurosciences. 2000;23:75–80. doi: 10.1016/s0166-2236(99)01490-3. [DOI] [PubMed] [Google Scholar]
- Strack S, Barban MA, Wadzinski BE, Colbran RH. Differential inactivation of postsynaptic density-associated and soluble Ca2+/calmodulin-dependent protein kinase II by protein phosphatases 1 and 2A. Journal of Neurochemistry. 1997;68:2119–2128. doi: 10.1046/j.1471-4159.1997.68052119.x. [DOI] [PubMed] [Google Scholar]
- Yang S-N, Tang Y-G, Zucker RS. Selective induction of LTP and LTD by postsynaptic [Ca2+]i elevation. Journal of Neurophysiology. 1999;81:781–787. doi: 10.1152/jn.1999.81.2.781. [DOI] [PubMed] [Google Scholar]
- Ziakopolous Z, Tillet CW, Brown MW, Bashir ZI. Input and layer-dependent synaptic plasticity in the perirhinal cortex in vitro. Neuroscience. 1999;92:459–472. doi: 10.1016/s0306-4522(98)00764-7. [DOI] [PubMed] [Google Scholar]