

Abstract
The mechanisms by which 5-hydroxytryptamine (5-HT) depresses transmitter release from lamprey reticulospinal axons were investigated. These axons make glutamatergic synapses onto spinal ventral horn neurons. 5-HT reduces release at these synapses, yet the mechanisms remain unclear.
Excitatory postsynaptic currents (EPSCs) evoked by stimulation of reticulospinal axons were recorded in ventral horn neurons. 5-HT depressed the EPSCs in a dose-dependent manner with an apparent Km of 2.3 μm.
To examine the presynaptic effect of 5-HT, electrophysiological and optical recordings were made from presynaptic axons. Action potentials evoked Ca2+ transients in the axons loaded with a Ca2+-sensitive dye. 5-HT slightly reduced the Ca2+ transient.
A third-power relationship between Ca2+ entry and transmitter release was determined. However, presynaptic Ca2+ currents were unaffected by 5-HT.
Further, in the presence of a K+ channel blocker, 4-aminopyridine (4-AP), 5-HT left unaltered the presynaptic Ca2+ transient, ruling out the possibility of its direct action on presynaptic Ca2+ current. 5-HT activated a 4-AP-sensitive current with a reversal potential of -95 mV in these axons.
The basal Ca2+ concentration did not affect 5-HT-mediated inhibition of release. Although 5-HT caused a subtle reduction in resting axonal [Ca2+]i, synaptic responses recorded during enhanced resting [Ca2+]i, by giving stimulus trains, were equally depressed by 5-HT.
5-HT reduced the frequency of TTX-insensitive spontaneous EPSCs at these synapses, but had no effect on their amplitude. We propose a mechanism of inhibition for transmitter release by 5-HT that is independent of presynaptic Ca2+ entry.
Insight into the mechanisms of synaptic modulation is important for understanding information processing in the central nervous system (CNS). Modulation of synaptic transmission occurs at several sites: at presynaptic terminals, by the activation of presynaptic receptors; at postsynaptic cells, by change in the postsynaptic receptor properties; and at the synaptic cleft, by change in the clearance rate of released transmitter. At the presynaptic terminal there exist at least four identifiable targets for modulation of release by receptors. (1) Changes in presynaptic resting Ca2+ concentrations, either by the opening or closing of presynaptic ligand-gated Ca2+ channels (Berretta & Jones, 1996; Schwartz & Alford, 1998; Cochilla & Alford, 1999; Glitsch & Marty, 1999) or by the regulation of Ca2+ release from internal stores (Cochilla & Alford, 1999). (2) Mechanisms that alter Ca2+ channel gating may also play a role (Dunlap & Fischbach, 1978; Takahashi et al. 1996; Wu & Saggau, 1997). (3) Changes in the properties of K+ (Ponce et al. 1996; Saugstad et al. 1996) or Na+ (Ma et al. 1997) currents will alter voltage-gated Ca2+ entry during action potentials. (4) G-protein-coupled receptor-mediated mechanisms acting on the release machinery ‘downstream’ of Ca2+ entry have been implicated in the modulation of transmitter release (Hilfiker & Augustine, 1999). These downstream mechanisms would not require any modulation of Ca2+ entry into the terminal, unlike other mechanisms suggested above (Silinsky, 1984; Scholz & Miller, 1992; Gereau & Conn, 1995; Scanziani et al. 1995). The activation of a G-protein subunit (Kowluru et al. 1996; Zhang et al. 1998) may directly alter vesicle fusion to the presynaptic membrane. Indeed Gβγ has recently been shown to bind directly to syntaxin (Jarvis et al. 2000). Alternatively, G-protein subunit activation may initiate other signal transduction cascades (e.g. by activating protein kinase C and Ca2+-calmodulin-dependent kinase II; Pasinelli et al. 1995).
5-Hydroxytryptamine (5-HT) acts throughout the CNS, at presynaptic and postsynaptic targets (Wallén et al. 1989; Hounsgaard & Kiehn, 1989; Buchanan & Grillner, 1991). Like other G-protein-coupled receptors, a number of mechanisms have been proposed for the presynaptic action of 5-HT receptors in altering transmitter release. These include the inhibition of Ca2+ channels (El Manira et al. 1997) or enhancement (Dale & Kandel, 1990; Delaney et al. 1991) or inhibition (Shupliakov et al. 1995) of release downstream of Ca2+ entry to the terminal. 5-HT1A, 5-HT1B and 5-HT1D receptors have been demonstrated to depress glutamate release in rat amyglada (Cheng et al. 1998), in the entorhinal cortex (Schmitz et al. 1998) and in the hippocampus (Schmitz et al. 1995). 5-HT-mediated depression of glutamatergic inputs to spinal motor neurons has been observed across many vertebrate groups from cyclostomes (Buchanan & Grillner, 1991) to mammals (Singer & Berger, 1996).
Access for recording techniques to the presynaptic terminal is necessary to elucidate the cellular mechanisms involved in presynaptic modulation. However, the small size of vertebrate presynaptic terminals usually prevents a direct study of these mechanisms. In the lamprey spinal cord, reticulospinal axons make en passant excitatory synapses onto motor neurons and ventral horn interneurons. The large unmyelinated axons (diameter up to 100 μm) allow an easy access for both direct electrophysiological and optical recordings. 5-HT is known to depress synaptic transmission at this synapse (Buchanan & Grillner, 1991). Using the intact lamprey spinal cord preparation, we show that 5-HT may depress synaptic transmission independently of [Ca2+]i regulation mechanisms.
METHODS
All experiments were done at physiological temperature, 8-10 °C.
Spinal cord preparation
Lamprey ammocoetes (Petromyzon marinus) were anaesthetized with tricaine methyl sulphonate (MS-222, 100 mg l−1; Sigma) and decapitated, in accordance with institutional guidelines, and sections of the spinal cord were removed. The meninx primitiva was also removed. For the electrophysiological experiments the dorsal surface of the spinal cord was sliced using a vibrating slicer (Campden Instruments). The tissue was submerged in a flowing solution (1-2 ml min−1).
Electrophysiology
Reticulospinal axons and ventral horn neurons (motor neurons or interneurons), identified by their location in the tissue and by capacity transients given by 10 mV voltage steps, were whole-cell clamped (with an Axopatch 200A amplifier; Axon Instruments) using a modified blind technique (Blanton et al. 1989; Cochilla & Alford, 1997). Patch pipettes had resistances around 5-10 MΩ. Series resistance was monitored continuously by giving a 10 mV voltage step before each episode, and if the change exceeded 15 %, the cell was discarded. Intracellular recordings were made with an electrode with a resistance of 30-60 MΩ when filled with 3 m potassium methane sulphonate, and cell types were identified by their location in the tissue and by capacity transients given by 0.1 nA current steps. The microelectrode recording technique used for paired recordings was conventional.
Imaging
Fluorescence images were recorded with a confocal microscope (Bio-Rad MRC600). Reticulospinal axons were retrogradely labelled with a dextran amine-conjugate form of the Ca2+-sensitive dye Oregon Green 488 BAPTA-1 (Molecular Probes), as described by Cochilla & Alford (1998). Briefly, immediately after the end of the spinal cord was cut, the dye was applied using a suction pipette fitted to the cut end. The tissue was then incubated overnight for the dye to be transported throughout the axons. Images were collected either at high speed by scanning a laser over a single line at 500 Hz or at lower speed by sampling two-dimensional images (170 pixels × 251 pixels) at 0.1 Hz. Imaging data were analysed using NIH Image software on a Macintosh computer. NIH Image was used to calculate the brightness value (range of possible values, 0-255 or 8 bit) for each pixel in a field of view. For each individual axon of interest, the brightness values were measured, and after background subtraction, images were normalized to the baseline level of fluorescence to give ΔF/F values, where the baseline value was 1.
Stimulation
The reticulospinal axons were stimulated at 0.05-0.1 Hz (for electrophysiology experiments) or at 50 Hz (for some imaging experiments) with a tungsten microelectrode with a tip resistance of 1-2 MΩ, as described previously (Cochilla & Alford, 1997).
Solutions
The patch pipette solution contained (mm): caesium methane sulphonate, 102.5; NaCl, 1; MgCl2, 1; EGTA, 5; Hepes, 5; pH adjusted to 7.2 with CsOH. The microelectrode pipette solution was either 3 m potassium methane sulphonate or 3 m potassium acetate. The external solution contained (mm): NaCl, 100; KCl, 2.1; CaCl2, 2.6; MgCl2, 1.8; NaHCO3, 26; glucose, 4; bubbled with 95 % O2-5 % CO2. For the experiments in which Ba2+ was substituted for Ca2+, a Hepes-buffered solution was used (mm): NaCl, 112; KCl, 2.1; CaCl2 or BaCl2, 2.6; MgCl2, 1.8; Hepes, 2; glucose, 4; pH adjusted to 7.4 with NaOH, bubbled with 100 % O2. For the experiments with high divalent cations a modified Hepes-buffered solution was used (mm): NaCl, 91.6; KCl, 2.1; CaCl2, 8; MgCl2, 8; Hepes, 2; glucose, 4; pH adjusted to 7.4 with NaOH, bubbled with 100 % O2. Glutamate analogues were obtained from Tocris; all other chemicals were from Sigma. Drugs were applied to the superfusate or applied over the spinal cord by pressure ejection from a fine pipette (patch pipette) with a 200 ms pulse of pressure.
Data analysis
The effects of drugs were calculated by comparing the data in the presence of a drug with the mean of the controls, before application of the drug and after washout.
Statistics
Data are given as means ±s.e.m. Student's paired two-tailed t test was used to calculate the significance of the data, unless otherwise noted.
RESULTS
5-HT depresses synaptic transmission at the reticulospinal axon to motor neuron synapse
EPSCs recorded in ventral horn motor neurons evoked by extracellular stimulation of the reticulospinal axons (Fig. 1A) are shown in Fig. 1Bi. These compound EPSCs have both an electrical and a chemical component. The chemical component is mediated by both NMDA and AMPA receptors (Buchanan et al. 1987; Brodin et al. 1988). However, electrical and chemical components of the response are not resolved when stimulated extracellularly as the inputs to the recorded cell are asynchronous due to the considerable variation in conduction velocity of axons in the lamprey. Consequently, the amplitude of the depressive effect of 5-HT on synaptic transmission is underestimated by using extracellular stimulation, although the technique is useful for determining a dose-response relationship (for the magnitude of the effect on chemical EPSC amplitude, see the paired cell recordings below). Superfused 5-HT reduced the EPSC amplitude (Fig. 1Bi and Bii). 5-HT has been shown to depress the activation of Ca2+-activated K+ currents (IK(Ca)) in lamprey spinal motor neurons. However, this postsynaptic effect of 5-HT on K+ current should be minimized by our use of Cs+ instead of K+ in the whole-cell pipette solution (Wallén et al. 1989). Indeed, in this condition, 5-HT had little effect on the holding potential (depolarized typically by 2-5 pA at a holding potential of -70 mV). 5-HT does not affect the sensitivity of the postsynaptic cell to glutamate (Buchanan & Grillner, 1991). The dose-response curve for the suppressive effect of 5-HT on the EPSCs (Fig. 1Bii) could be fitted empirically by Michaelis-Menten functions, with an apparent Km (dose producing half-maximal suppression) of 2.3 μm. The highest dose of 5-HT tested (100 μm) did not depress the peak EPSC amplitude as much as did 30 μm 5-HT, with which maximal depression to 49.6 ± 6.0 % of control was observed; this possibly reflected dose-dependent desensitization of 5-HT receptors. Therefore, in the following experiments we used 5-HT at 30 μm to depress transmitter release, unless otherwise stated.
Figure 1. Suppressive effect of 5-HT on the EPSC evoked in ventral horn neurons by stimulation of reticulospinal axons.
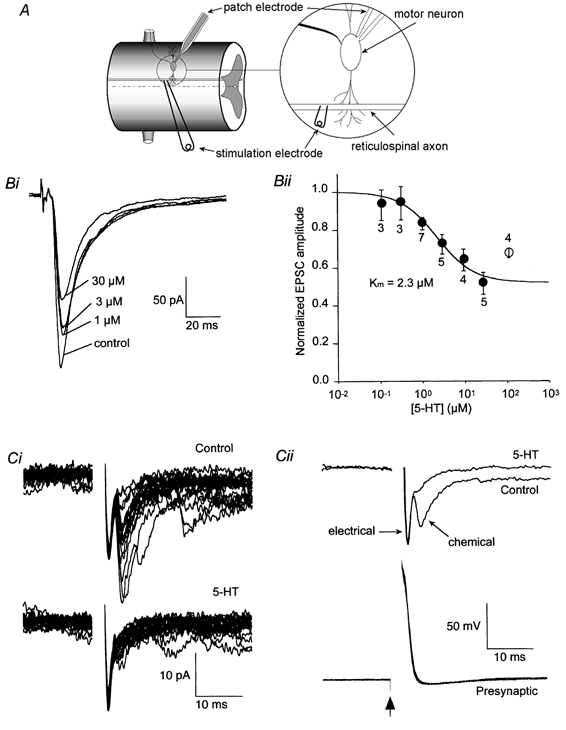
A, schematic diagram of recording configuration. Postsynaptic neurons were recorded under whole-cell voltage clamp. Presynaptic axons were stimulated either extracellularly as in B or via an intracellular recording microelectrode as in C. Bi, EPSCs recorded in control solution and with increasing doses of superfused 5-HT (1, 3 and 30 μm). The holding potential was -70 mV. Bii, mean dose-response data (±s.e.m.) obtained as in Bi. Numbers by each symbol indicate the number of cells. The continuous line is the best-fit through the points (0.1-30 μm) with the form 1 - {Fmax[5-HT]/([5-HT]+Km)}, where Fmax is the maximum fractional suppression (0.49). Ci, postsynaptic responses to action potentials evoked in a synaptically paired presynaptic giant axon. The upper record is of 20 consecutive responses in control conditions, the lower traces in the presence of 30 μm 5-HT. Note that the application of 5-HT almost completely abolished the later variable chemical component but left the early electrical component unchanged. Cii: top, the mean postsynaptic response to presynaptic action potentials (bottom) before and after application of 5-HT. Means are taken from the data in Bi. Bottom: traces showing the presynaptic action potential evoked by a 2 ms depolarizing current pulse given at the time marked by the arrow in control and in the presence of 5-HT.
To investigate the effect of 5-HT on EPSCs further, paired cell recordings were made between presynaptic reticulospinal axons and postsynaptic neurons. Presynaptic recordings were made intracellularly with microelectrodes and postsynaptic recordings with patch electrodes in the whole-cell mode. Action potentials were evoked in the axons by applying brief depolarizing current pulses through the recording microelectrode. This resulted in mixed EPSCs in the postsynaptic neurons (Fig. 1C). Separation between electrical and chemical components is clear with paired recordings (e.g. Fig. 1Cii). Application of 5-HT significantly reduced the amplitude of the chemical component of synaptic transmission (Fig. 1C) to 19.8 ± 7.5 % of control (4 pairs). However, 5-HT had no effect on the amplitude of the electrical component. This supports the contention of Buchanan & Grillner (1991) that 5-HT depresses glutamate release while having no effect on resting membrane properties of ventral horn neurons. Moreover, it also shows that the action of 5-HT does not lie in inhibition of the action potential initiation or of the action potential invasion to the presynaptic terminal. The electrical component effectively controls whether the action potential invades the presynaptic terminal, because the gap junctions that mediate the electrical component of synaptic transmission are co-localized with the vesicle release sites (Christensen, 1976; Shupliakov et al. 1996). Note also that the reduction of synaptic transmission recorded during extracellular stimulation is similar to that recorded with paired pre- and postsynaptic cells. The area under the response (total charge transfer), including both electrical and chemical components, was reduced to 43 ± 5 % of control for paired cell recordings. This is very similar to the reduction in amplitude of the extracellularly evoked responses used to construct the dose-response curves (49.6 ± 6.0 %; the reduction in area was to 48 ± 7 % of control). This implies that in both cases the 5-HT-mediated reduction in chemical neurotransmitter release (the response excluding the electrical component) was approximately 80 %.
5-HT modulates the amplitude of the presynaptic Ca2+ transient
One model for G-protein receptor-coupled presynaptic inhibition is that receptor activation inhibits presynaptic Ca2+ channels (Dunlap & Fischbach, 1978). It has been shown recently that activation of some metabotropic glutamate receptors directly leads to suppression of presynaptic Ca2+ conductance (Takahashi et al. 1996). To determine whether the observed depression of EPSCs by 5-HT was due to a direct action of the 5-HT receptor on Ca2+ influx, we used optical techniques to look at its effect on the fast Ca2+ transient triggered by presynaptic action potentials. Axons were retrogradely filled with the Ca2+-sensitive dye Oregon Green 488 BAPTA-1 (Fig. 2A), and a direct single stimulus (1 ms width) was given extracellularly onto the spinal cord to evoke an action potential in the presence of ionotropic glutamate receptor blockers, 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; 10 μm) and dl-2-amino-5-phosphonovaleric acid (AP5; 50 μm). Figure 2Bi shows a fluorescence image obtained by scanning along a fixed line (indicated by arrowheads in Fig. 2A) in the axon repetitively at 500 Hz for 512 ms (every 2 ms for 256 times). A single stimulus elicited a Ca2+ transient in the axons (Fig. 2B), as reported previously (Cochilla & Alford, 1998). The Ca2+ transient was localized to ‘hotspots’ along the membrane and then rapidly diffused into the axon at lower concentrations. 5-HT (30 μm) reduced the peak amplitude of the Ca2+ transient (n= 30, to 81.5 ± 3.9 % of control, P < 0.001; Fig. 2Bii and C). No significant difference in the effect of 5-HT on the amplitude of the evoked transient was recorded if the signal was recorded at ‘hotspots’ or elsewhere on the axons.
Figure 2. 5-HT modulates the amplitude of the presynaptic Ca2+ transient.
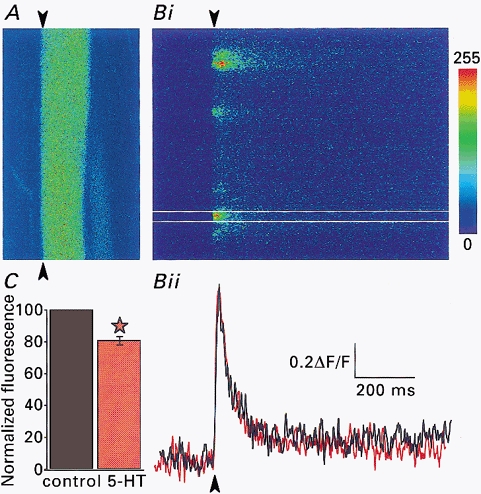
A, axon labelled with the Ca2+-sensitive dye Oregon Green 488 BAPTA-1. Bi, line scan of the axon shown in A along the line indicated by the arrowheads at 500 Hz in the presence of 50 μm AP5 and 10 μm CNQX. The laser scanned repetitively over the same line and the resultant fluorescence trace is displayed with time along the x-axis and distance along the axon on the y-axis. A single stimulus was given (arrowhead) leading to a transient increase in fluorescence level (i.e. [Ca2+]i). Bii, integrated plot of line scan from the part of Bi between the two white lines, in control (black) and in the presence of 30 μm 5-HT (red) At each time point, the fluorescence level was averaged, then normalized to the prestimulus level. C, pooled data of similar experiments to that in B (30 axons). The asterisk indicates a significant difference from control P < 0.001. ‘Box’ size in A is 150 μm × 75 μm.
The relationship between presynaptic Ca2+ entry and the release of transmitter has been shown to be non-linear. In the squid giant synapse transmitter release has been shown to demonstrate a fourth-power relationship over Ca2+ entry to the terminal (Augustine et al. 1991). A similar relationship was also found at vertebrate synapses (Schneggenburger & Neher, 2000). These results support the hypothesis that a small reduction in Ca2+ entry may lead to profound effects on synaptic transmission. Ni2+ is a potent Ca2+ channel antagonist at the lamprey giant synapse. It was, therefore, possible to investigate the relationship between the Ca2+ transient amplitude, measured optically, and the release of transmitter from these synapses. The effects of Ni2+ on Ca2+ transients (Fig. 3Ai) and on EPSCs (Fig. 3Aii) were tested similarly. The IC50 values for Ni2+ against presynaptic Ca2+ transients and EPSCs were similar, 7.0 and 2.6 μm, respectively. The relationship between Ca2+ transient amplitude and EPSC amplitude was then compared graphically (Fig. 3Bi) and was clearly supra-linear. The relationship was made linear by plotting the cube of the Ca2+ transient amplitude against EPSC amplitude. This relationship was similar to that recorded in other preparations. However, it is also clear from these graphs that to achieve an 80 % reduction in transmitter release by inhibition of Ca2+ entry, a 60 % reduction in Ca2+ transient amplitude would be required.
Figure 3. Relationship between the magnitude of presynaptic Ca2+ entry and the release of neurotransmitter.
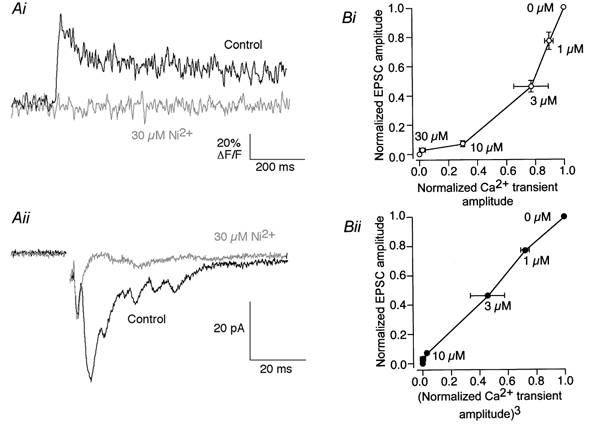
Ai, a Ca2+ transient in the reticulospinal axon (measured as in Fig. 2) was blocked by 30 μm Ni2+. Aii, an EPSC was recorded from a ventral horn neuron by extracellularly stimulating the reticulospinal axons in the ventro-medial tracts. The compound EPSC was also blocked by 30 μm Ni2+. Bi, relationship between evoked EPSC amplitude and presynaptic Ca2+ transient amplitude. Normalized EPSCs (n= 3) are plotted against normalized Ca2+ transients (n= 3) at the same Ni2+ concentration. Bii, the relationship shown in Bi became linear when the EPSC amplitude was plotted against the cube of the Ca2+ transient amplitude at the same [Ni2+]o.
To emphasize the finding that a mechanism other than an alteration of Ca2+ influx inhibits transmitter release at the giant synapse, similar graphs were plotted for the relationship between presynaptic Ca2+ transient amplitude and EPSC amplitude in the presence of 5-HT (Fig. 4). Irrespective of whether the relationship between Ca2+ entry and the EPSC amplitude was plotted linearly or as a cubic function of Ca2+ entry, the reduction in transmitter release by 5-HT cannot be accounted for by the reduction in Ca2+ entry. The graphs from Fig. 3B were superimposed on these graphs to demonstrate the difference in the relationship between presynaptic Ca2+ transients and synaptic transmission after wash-in of Ni2+, which clearly inhibits presynaptic Ca2+ entry, and 5-HT, which does so only marginally.
Figure 4. 5-HT inhibits synaptic transmission to a greater extent than Ca2+ entry.
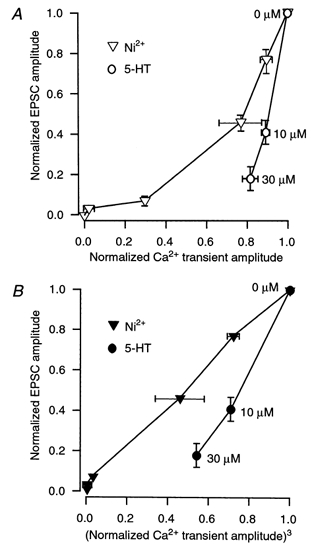
A, relationship between evoked EPSC amplitude and presynaptic Ca2+ transient amplitude. Normalized EPSCs (n= 3) are plotted against normalized Ca2+ transients (n= 3) at the same 5-HT concentrations (○) and at the same Ni2+ concentrations (▽, repeated from Fig. 3Bi). B, the relationship shown in A is plotted against the cube of the Ca2+ transient amplitude at the same concentrations of 5-HT (•), similarly to the Ca2+-EPSC amplitude plotted in the presence of Ni2+ in Fig. 3Bii, which is repeated here (▾).
To determine whether this reduction in the Ca2+ transient was due to the effect of 5-HT on presynaptic Ca2+ channels, we looked at the effect of 5-HT on axonal Ca2+ current. Using Ba2+ (2.6 mm) as a charge carrier instead of Ca2+ (and also to block Ca2+-activated K+ channels), current through Ca2+ channels in the reticulospinal axons was isolated by application of 1 μm TTX (to block Na+ current) and 1 mm 4-aminopyridine (4-AP; to block K+ current). When the presynaptic axon was whole-cell clamped in this blocker cocktail at -70 mV and depolarizing pulses (250 ms) were applied, presynaptic Ba2+ currents were evoked (Fig. 5A; Cochilla & Alford, 1998). This Ba2+ current was activated when the axon was depolarized positive to -40 mV, and peaked at around 0 mV (Fig. 5A). This current, like the Ca2+ transient, was abolished by 30 μm Ni2+ (data not shown). Superfusion of 30 μm 5-HT did not alter this current (Fig. 5B and C; n= 3), suggesting that 5-HT does not block Ca2+ channels directly. It is possible that a subgroup of channels responsible for release is affected by 5-HT; however, the nature of Ca2+ entry at ‘hotspots’ reduces the likelihood that such an effect would be significant.
Figure 5. Whole-cell axonal Ba2+ currents are not modulated by the application of 5-HT.
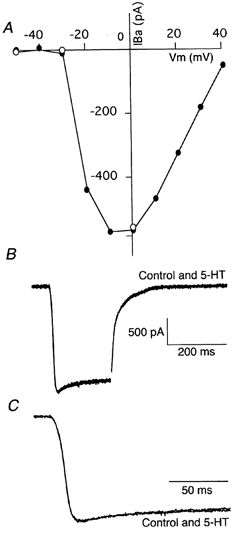
A, an axon was voltage clamped at -70 mV and 10 mV increments of test potential were applied from -70 to +40 mV to evoke a Ca2+ conductance carried by Ba2+. The peak current was measured at each test potential to generate this current-voltage plot (•). ○, peak amplitude after application of 5-HT (30 μm) to the superfusate, for 3 data points. B, raw data traces of Ba2+ current in control conditions and in the presence of 5-HT (30 μm) when the holding potential was ‘jumped’ from -70 to 0 mV. The patch solution contained Cs+ as the primary charge carrier and the external solution contained Ba2+ (2.6 mm), TTX (1 μm) and 4-AP (1 mm). C, same traces as in B but on an expanded time base.
5-HT reduces the amplitude of presynaptic action potentials by a 4-AP-sensitive mechanism
Although 5-HT reduces the amplitude of the presynaptic action potential-evoked Ca2+ transient, a direct effect on Ca2+ channel activation was not seen in voltage-clamp experiments. An alternative possibility is that 5-HT may change the amplitude of the presynaptic action potential that leads to the activation of Ca2+ channels. To test this hypothesis, action potentials were evoked in current-clamp mode using depolarizing pulses. Application of 5-HT (30 μm) reduced the amplitude of the action potential reversibly (Fig. 6A; reduced by 6 ± 3 mV or 5.0 ± 2.5 % of peak depolarization from resting membrane potential; n= 3). The rapid depolarization of action potentials in lamprey reticulospinal axons is mediated by a 4-AP-sensitive current (Fig. 6C). Addition of 4-AP (30 μm) to the superfusate depolarized the axons, broadened the action potentials and eliminated action potential after-hyperpolarizations. As shown in Fig. 6B, under these conditions, the further addition of 5-HT (30 μm) had no effect on the remaining properties of the evoked action potentials (Na+ spikes). The repolarization of the axons in the presence of 4-AP was sensitive to the replacement of extracellular Ca2+ with Ba2+ (Fig. 6Ciii). Ba2+ does not activate Ca2+-activated K+ conductances. The failure to activate these conductances leads to a significant plateau depolarization of the recorded axons following the action potential initiation. In the lamprey pre synaptic giant axons 5-HT did not affect action potential after-hyperpolarizations or repolarization in the presence of 4-AP (Fig. 5B). This is in contrast to the effect of 5-HT on post synaptic motor neurons, where 5-HT depresses the activation of IK(Ca) following action potentials in both lamprey ventral horn neurons and mammalian motor neurons.
Figure 6. The effect of 5-HT on presynaptic action potentials.
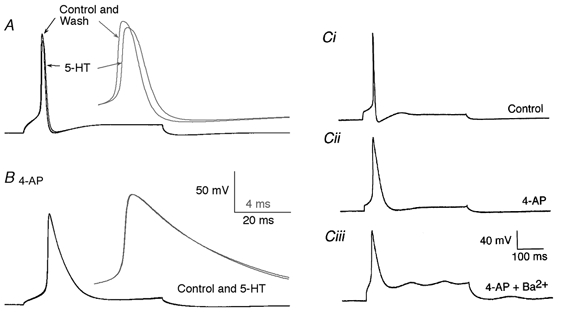
Action potentials in the reticulospinal axons were recorded with microelectrodes under current clamp. A, an action potential evoked by a depolarizing current pulse (0.5 nA). Application of 5-HT (30 μm) reduced the amplitude of the evoked action potential. The inset (grey) shows the traces on an expanded time scale. (Note that the peak of the action potential was reduced in amplitude.) B, an action potential was recorded in the same axon in the presence of 4-AP (30 μm). Application of 5-HT had no effect. The inset (grey) shows the traces on an expanded time scale as in A. Ci, another example of an action potential recorded under control conditions. An action potential was evoked in a different axon by a 1 nA depolarizing current pulse. Cii, the same axon as in Ci after the application of 4-AP (30 μm). Ciii, the same axon as in Ci after the replacement of extracellular Ca2+ with Ba2+ and in the presence of 4-AP.
To investigate the mechanism by which 5-HT modulates presynaptic action potentials, axonal currents were recorded under whole-cell voltage-clamp conditions while 5-HT (300 μm in a pressure pipette) was applied by pressure ejection (200 ms) onto the spinal cord immediately above the site of the recording. This evoked a current that lasted for more than 200 s with a reversal potential of -95 ± 5 mV (Fig. 7; n= 3). The current was blocked by 4-AP (30 μm; Fig. 7C; n= 2). The current was apparently inwardly rectified as shown by the current-voltage relationship (Fig. 7D). However, cell attached recording would be necessary to determine whether this is a direct effect on the 5-HT-evoked conductance or whether it simply represents a shunt applied by other K+ currents activated by depolarization.
Figure 7. Application of 5-HT to the spinal cord evokes a 4-AP-sensitive current in spinal axons.
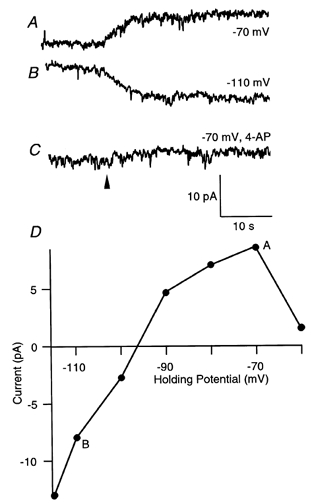
Giant axons were recorded under voltage clamp and 5-HT applied from a pressure ejection pipette placed over the spinal cord (the pipette contained 300 μm 5-HT). Pressure application (200 ms at the time indicated by the arrowhead) of 5-HT led to an outward current at -70 mV (A) and an inward current at -110 mV (B). The current was blocked by bath application of 4-AP (30 μm; C). D, plot of the amplitude of the 5-HT evoked current against holding potential.
5-HT modulates synaptic transmission in the presence of 4-AP
5-HT application depresses the presynaptic Ca2+ transient recorded during presynaptic action potentials. However, in the presence of 4-AP, it does not alter action potentials nor does it affect the amplitude of presynaptic Ca2+ currents. Thus if 5-HT modulates a presynaptic K+ conductance, this may have significant effects on presynaptic [Ca2+]i which would in turn account for the effect of 5-HT on synaptic transmission. To test this, Ca2+ transients were evoked in reticulospinal axons with the K+ channel blocker 4-AP (25 μm) present. 4-AP increased the peak amplitude of the Ca2+ transient (n= 14, increased to 110.0 ± 2.4 % of control), in the presence of 50 μm AP5 and 10 μm CNQX. Superfusion of 30 μm 5-HT in addition to the blocker cocktail did not alter the Ca2+ transient significantly (n= 14, 115.2 ± 6.9 % compared with 4-AP alone, P > 0.05; Fig. 8A).
Figure 8. 5-HT inhibits transmitter release independently of its action on presynaptic K+ channels.
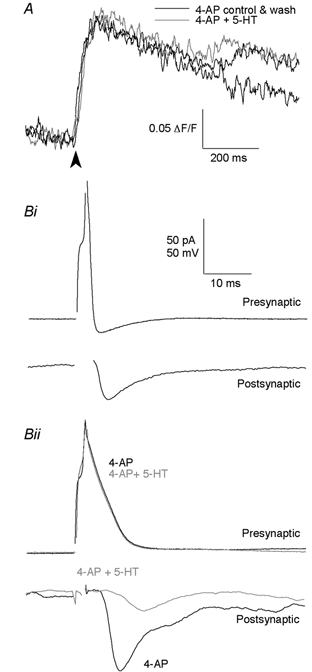
A, Ca2+ transient, extracellularly evoked at arrowhead, recorded in the presence of 50 μm AP5 and 10 μm CNQX with K+ channels blocked with 4-AP (25 μm), before (control) and after the addition of 30 μm 5-HT, and after washout of 5-HT. Bi, paired cell recording between a presynaptic reticulospinal axon and a postsynaptic ventral horn neuron. Depolarizing current steps evoked action potentials in the presynaptic reticulospinal axon, which then evoked EPSCs in the postsynaptic motor neuron. Bii, recording from the same pair as in Bi but after K+ channels were blocked with 4-AP. 5-HT (30 μm) reduced the peak amplitude of the synaptically evoked response but was without effect on the amplitude of the broadened presynaptic action potential.
To examine whether 4-AP abolished the depressant effect of 5-HT on synaptic transmission, EPSCs were recorded in the presence of 4-AP. To minimize polysynaptic transmission, we used high divalent cation external solutions with 4-AP (25 μm) and AP5 (50 μm). Experiments were performed using both paired cell recording (Fig. 8B) and extracellular stimulation (not shown). Results were similar in either case. Application of 4-AP increased the peak amplitude of EPSCs (to 165 ± 52 % of control; Fig. 8Bii) and action potential width, as expected. Superfusion of 5-HT (3-30 μm) in addition to this blocker cocktail still reduced the EPSC amplitude (to 54.7 ± 5.4 % in 3 μm 5-HT when compared with 4-AP alone, P < 0.05, n= 3, and to 25.5 %, n= 2, in 30 μm 5-HT; Fig. 8Bii). Thus we conclude that the main effect of 5-HT on depression of synaptic transmission is not on the opening of K+ channels.
5-HT does not affect Ca2+ entry upon repetitive stimulation
Presynaptic action potentials result in the opening of voltage-gated Ca2+ channels, causing Ca2+ influx. After the termination of action potentials, Ca2+ levels at the release site remain elevated for 1-2 s. This residual Ca2+ concentration is believed to play an important role in short-term synaptic plasticity (for review, see Zucker et al. 1991). Similarly, a 5-HT-dependent alteration in basal Ca2+, which is insufficient to evoke synchronous release, may still modulate action potential-induced release. Addition of 30 μm 5-HT in the presence of CNQX (10 μm) and AP5 (50 μm) reduced the basal Ca2+ level slightly (to 98.9 ± 0.005 % of control, n= 55, P < 0.001; data not shown). We wished to test whether this small alteration in basal Ca2+ concentration could alter transmitter release.
By giving five closely spaced stimuli (at 20 ms intervals, 50 Hz) to the axons, we examined the effect of 5-HT on the Ca2+ transient (Fig. 9). Superfusion of 5-HT (30 μm) decreased the peak amplitude of the Ca2+ transient (n= 25, to 84.5 ± 1.7 % of control, P < 0.001; Fig. 9A), the magnitude of reduction being similar to that after a single stimulus (Fig. 2). Note also that the Ca2+ concentration in the axon rose throughout the stimulus train.
Figure 9. The effect of 5-HT on responses to repetitive stimulation.
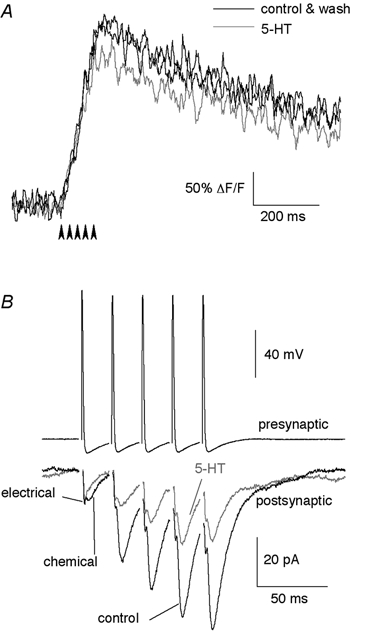
A, a Ca2+ transient evoked by a train of 5 stimuli (arrowheads) at 50 Hz, in control (with 50 μm AP5 and 10 μm CNQX), in the presence of 30 μm 5-HT and after washout of the drug. B, a paired recording was made between a reticulospinal axon and a synaptically coupled postsynaptic neuron. EPSCs (lower traces) were evoked by repetitively stimulating the presynaptic reticulospinal axon (5 stimuli at 50 Hz). 5-HT (30 μm) reduced the EPSC amplitude of each of the chemical components. Data for each trace are the average from 12 sequentially evoked responses. The holding potential was -70 mV. The upper trace is an example of one train of presynaptic action potentials.
To test the effect of 5-HT on EPSCs recorded during increased basal [Ca2+]i, EPSCs were recorded from the postsynaptic neuron in the high divalent cation external solution (to minimize polysynaptic transmission) while five consecutive stimuli were given extracellularly to the presynaptic reticulospinal axons at 20 ms intervals as for the imaging experiment above. 5-HT (30 μm) reduced the amplitude of each EPSC (n= 3; from the 1st EPSC to the 5th, amplitudes in 5-HT were: 50.7 ± 6.0, 46.9 ± 3.3, 53.7 ± 7.2, 62.9 ± 8.8 and 56.8 ± 6.2 % of control, respectively). Similarly, following paired cell recordings between synaptically coupled reticulospinal axons and whole-cell patch-clamped postsynaptic neurons, 5-HT reduced the amplitude of each EPSC in the train (Fig. 9B; n= 3; from the 1st EPSC to the 5th, amplitudes in 5-HT were: 40.0 ± 7.1, 38.9 ± 4.5, 42.0 ± 10.1, 45.1 ± 20.0 and 37.1 ± 9.1 % of control, respectively). We conclude that the reduction of transmitter release is not mediated by the effect of 5-HT on basal [Ca2+]i. Even when basal [Ca2+]i is high during a train of stimuli, 5-HT equally inhibits release.
Voltage-activated Ca2+ influx-independent suppression of glutamate release by 5-HT
By analysing the behaviour of spontaneous transmitter release, the origin of receptor-mediated modulation of synaptic responses can be examined. If the frequency of spontaneous events changes, the modulation of synaptic transmission is thought to originate from presynaptic mechanisms. On the other hand, if the amplitude of miniature events alters, postsynaptic mechanisms may be considered to be responsible for the modulation of transmission. Thus, if the depression of synaptic transmission by 5-HT observed is truly presynaptic (and Ca2+-influx independent), then the effect of 5-HT on miniature EPSCs (mEPSCs), recorded while voltage-gated channels are blocked, should be only on their frequency, and not on their amplitude. To test this hypothesis, postsynaptic motor neurons were whole-cell clamped in the presence of 1 μm TTX. Strychnine (5 μm) was also added to block glycine receptors so that only glutamatergic events were recorded as the giant axons release glutamate (Fig. 10). Note that there are no low-voltage-activated Ca2+ channels in the axons (Fig. 5). The mEPSC amplitude and frequency were 6.6 ± 0.3 pA and 11.9 ± 0.7 Hz, respectively (n= 4). Superfusion of 5-HT (30 μm) did not alter the amplitude of mEPSCs (6.8 ± 0.2 pA, n= 4, P > 0.05 in 3/4 cells, two sample Kolmogorov-Smirnov test), but reduced their frequency to 10.3 ± 1.3 Hz (P < 0.001 in 3/4 cells, two sample Kolmogorov-Smirnov test: Fig. 10C), in accordance with the presynaptic action of 5-HT. The overall effect of 5-HT on frequency was not equal to that seen for evoked release. This is probably because glutamate is released from many other terminals in the spinal cord, in addition to those of the giant axons.
Figure 10. 5-HT reduces the frequency of spontaneous EPSCs.
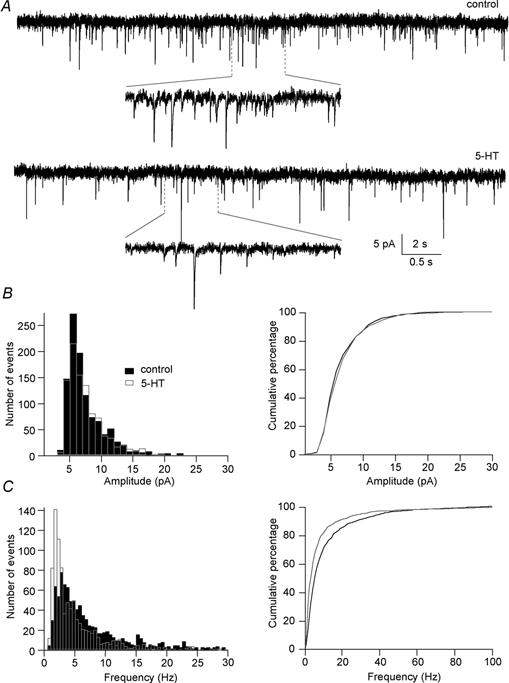
A, specimen miniature EPSCs recorded in the presence of 1 μm TTX and 5 μm strychnine (control) and with the addition of 30 μm 5-HT (5-HT). The holding potential was -70 mV. B: left, amplitude histogram of mEPSCs recorded in control (filled bars) and 5-HT (open bars) from the same cell as in A showing that 5-HT did not change the mean mEPSC amplitude (6.6 ± 0.1 pA in control and 6.7 ± 0.1 pA in 5-HT, P= 0.05, two sample Kolmogorov-Smirnov test). Right, cumulative probability plot of the amplitude of mEPSCs showing that the two distributions overlaped. C: left, frequency histogram of mEPSCs recorded in control and 5-HT from the same cell as in A showing that 5-HT significantly reduced mean mEPSC frequency (11.1 ± 0.5 Hz in control to 7.5 ± 0.4 Hz in 5-HT, P < 0.001, two sample Kolmogorov-Smirnov test). Right, cumulative probability plot of the frequency of mEPSCs showing the significant reduction in mean mEPSC frequency.
DISCUSSION
Release of transmitter follows action potential invasion of the presynaptic terminal. The resultant depolarization opens voltage-operated Ca2+ channels (Uchitel et al. 1992; Neher & Zucker, 1993; Stanley, 1993; Huston et al. 1995) causing a transient rise in presynaptic [Ca2+]i. Ca2+ then binds to a closely associated low affinity Ca2+-binding protein (Augustine et al. 1991) at concentrations of tens or hundreds of micromolar (Llinás et al. 1992a). Vesicle fusion with the terminal membrane then occurs, thus transmitter is released (Südhof, 1995). We can hypothesize that several mechanisms may be involved in the modulation of transmitter release. The degree of depolarization by the action potential and its duration will affect the amount of Ca2+ entry. Either of these processes may be altered by change in the gating properties of K+ or possibly Na+ channels in the presynaptic axon and terminal. Ca2+ entry will also be affected by the number, subtype and gating states of Ca2+ channels in the terminal (Llinás et al. 1992b). Following Ca2+ entry to the terminal, the processes activated by G-proteins or second messengers may modulate release machinery proteins. This may take the form of a change in the Ca2+-binding affinity of the proteins of the release apparatus, or an effect on the action of these proteins following Ca2+ binding.
Blockade of Ca2+ influx by 5-HT
5-HT inhibits the release of glutamate from giant axons in the lamprey spinal cord. It is difficult to relate the dose of 5-HT directly to a receptor affinity in the lamprey spinal cord because uptake mechanisms are very active in this preparation. For example, block of 5-HT uptake reveals profound physiological effects in the spinal cord (Christenson et al. 1989). Use of a specific antagonist may clarify this issue and preliminary data indicate that these receptors are 5-HT1D like (T. Blackmer & S. Alford, unpublished observation).
As hypothesized by Dunlap & Fischbach (1978) and reported by Takahashi and colleagues (1996, 1998), some G-protein-coupled receptors interact directly with voltage-gated Ca2+ channels. The activation of these receptors leads to a reduction in Ca2+ current (ICa). From our experiments on isolated IBa we conclude that this is not the case for presynaptic 5-HT receptors in the lamprey reticulospinal axon. 5-HT altered neither the amplitude nor the activation time course of ICa. However, with fast scanning of Ca2+ transients triggered by an action potential, a decrease in peak Ca2+ influx was observed. How can we explain this disagreement? This apparent discrepancy can be reconciled if 5-HT's action is not to act directly on Ca2+ channels, but rather to open K+ channels. Thus isolated ICa (with K+ channels blocked as well) remains unaffected by 5-HT. Imaging experiments revealed that 5-HT did not reduce Ca2+ influx with K+ channels blocked by 4-AP. Furthermore, 5-HT reduced action potential amplitudes, but not Na+ spike amplitudes, in the presence of 4-AP. 5-HT also activated a current in the axon that was sensitive to block with 4-AP and had a reversal potential of -95 mV. However, 5-HT still reduced the EPSCs to a similar extent in postsynaptic neurons with K+ channels blocked. Although part of the effect of 5-HT application may be on activation or potentiation of a K+ current, the contribution of this effect to the depression of synaptic transmission is minor. If we compare the effect of Ni2+ on Ca2+ entry to the presynaptic terminal with its effect on neurotransmitter release, it is clear that a much larger effect of 5-HT on Ca2+ transients would be required to account for the 80 % reduction in transmitter release that 5-HT effects. This is the case even without application of 4-AP. Therefore, it is reasonable to assume that 5-HT affects release at sites other than on Ca2+ entry. For example, this may be directly at the core complex for vesicular fusion.
5-HT application to the spinal cord leads to a very slight but significant lowering of the resting Ca2+ concentration. It seems unlikely that such a small effect would lead to a marked depression in transmitter release; however, we could control for this by measuring EPSC amplitudes before and after 5-HT application during a train of stimuli that also leads to a signficant rise in baseline Ca2+ concentrations during the train. With repetitive stimulation, we found that 5-HT reduced the peak amplitude of the Ca2+ transient by about the same magnitude as with a single stimulus. The small reduction in the Ca2+ transient is probably due to the opening of K+ channels, as was the case with a single stimulus. Similarly, synaptic transmission during repetitive stimulation, during which presynaptic Ca2+ concentrations were raised above baseline, was reduced equally throughout the stimulus train. Clearly 5-HT inhibits transmitter release even when presynaptic Ca2+ concentrations are high and we conclude that 5-HT does not mediate its inhibitory effect by altering resting Ca2+ concentrations in the axons.
How does 5-HT depress transmitter release?
Our results show that 5-HT reduces transmitter release under conditions in which presynaptic [Ca2+]i is essentially unaffected (see also Robitaille et al. 1999). The observed reduction in the frequency of miniature events supports the idea that 5-HT acts independently of Ca2+ influx to reduce transmitter release. What are the possible mechanisms? There are an estimated 5000 vesicles located beneath each active zone in these axons. It seems unlikely that 5-HT is able to exhaust this vesicle pool. There is increasing evidence that G-protein-coupled receptors might affect exocytosis and transmitter release downstream of Ca2+ entry (Kowluru et al. 1996; Pinxteren et al. 1998; Zhang et al. 1998). Indeed Gβγ binds to syntaxin (Jarvis et al. 2000), one of the proteins that comprise the SNARE complex that effects exocytosis. We suggest that this is the case for the effect of 5-HT.
Acknowledgments
This work was supported by NINDS (to S.A.) and a Wellcome Prize Travelling Fellowship to M.T. We thank Marina Catsicas, Amanda Cochilla, Angus Silver and Traverse Slater for their helpful comments on the manuscript, Christophe Pouzat for some of the Igor analysis programs and Andrew Boxall for discussion.
References
- Augustine GJ, Adler EM, Charlton MP. The calcium signal for transmitter secretion from presynaptic nerve terminals. Annals of The New York Academy of Sciences. 1991;635:365–381. doi: 10.1111/j.1749-6632.1991.tb36505.x. [DOI] [PubMed] [Google Scholar]
- Berretta N, Jones RS. Tonic facilitation of glutamate release by presynaptic N-methyl-D-aspartate autoreceptors in the entorhinal cortex. Neuroscience. 1996;75:339–344. doi: 10.1016/0306-4522(96)00301-6. [DOI] [PubMed] [Google Scholar]
- Blanton MG, Lo Turco JJ, Kriegstein AR. Whole cell recording from neurons in slices of reptilian and mammalian cerebral cortex. Journal of Neuroscience Methods. 1989;30:203–210. doi: 10.1016/0165-0270(89)90131-3. [DOI] [PubMed] [Google Scholar]
- Brodin L, Grillner S, Dubuc R, Ohta Y, Kasicki S, Hokfelt T. Reticulospinal neurones in lamprey: Transmitters, synaptic interactions and their role during locomotion. Archives Italiennes de Biologie. 1988;126:317–345. [PubMed] [Google Scholar]
- Buchanan JT, Brodin L, Dale N, Grillner S. Reticulospinal neurones activate excitatory amino acid receptors. Brain Research. 1987;408:321–325. doi: 10.1016/0006-8993(87)90397-0. [DOI] [PubMed] [Google Scholar]
- Buchanan JT, Grillner S. 5-Hydroxytryptamine depresses reticulospinal excitatory post synaptic potentials in motoneurons of the lamprey. Neuroscience Letters. 1991;122:71–74. doi: 10.1016/0304-3940(91)90196-z. [DOI] [PubMed] [Google Scholar]
- Cheng LL, Wang SJ, Gean PW. Serotonin depresses excitatory synaptic transmission and depolarization-evoked Ca2+ influx in rat basolateral amygdala via 5-HT1A receptors. European Journal of Neuroscience. 1998;10:2163–2172. doi: 10.1046/j.1460-9568.1998.00229.x. [DOI] [PubMed] [Google Scholar]
- Christensen BN. Morphological correlates of synaptic transmission in lamprey spinal cord. Journal of Neurophysiology. 1976;39:197–212. doi: 10.1152/jn.1976.39.2.197. [DOI] [PubMed] [Google Scholar]
- Christenson J, Franck J, Grillner S. Increase in endogenous 5-hydroxytryptamine levels modulates the central network underlying locomotion in the lamprey spinal cord. Neuroscience Letters. 1989;100:188–192. doi: 10.1016/0304-3940(89)90682-4. [DOI] [PubMed] [Google Scholar]
- Cochilla AJ, Alford S. Glutamate receptor-mediated synaptic excitation in axons of the lamprey. Journal of Physiology. 1997;499:443–457. doi: 10.1113/jphysiol.1997.sp021940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochilla AJ, Alford S. Metabotropic glutamate receptor-mediated control of neurotransmitter release. Neuron. 1998;20:1007–1016. doi: 10.1016/s0896-6273(00)80481-x. [DOI] [PubMed] [Google Scholar]
- Cochilla AJ, Alford S. NMDA receptor-mediated control of presynaptic calcium and neurotransmitter release. Journal of Neuroscience. 1999;19:193–205. doi: 10.1523/JNEUROSCI.19-01-00193.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale N, Kandel ER. Facilitatory and inhibitory transmitters modulate spontaneous transmitter release at cultured Aplysia sensorimotor synapses. Journal of Physiology. 1990;421:203–222. doi: 10.1113/jphysiol.1990.sp017941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney K, Tank DW, Zucker RS. Presynaptic calcium and serotonin-mediated enhancement of transmitter release at crayfish neuromuscular junction. Journal of Neuroscience. 1991;11:2631–2643. doi: 10.1523/JNEUROSCI.11-09-02631.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlap K, Fischbach GD. Neurotransmitters decrease the calcium component of sensory neurone action potentials. Nature. 1978;276:837–839. doi: 10.1038/276837a0. [DOI] [PubMed] [Google Scholar]
- El Manira A, Zhang W, Svensson E, Bussieres N. 5-HT inhibits calcium current and synaptic transmission from sensory neurons in lamprey. Journal of Neuroscience. 1997;17:1786–1794. doi: 10.1523/JNEUROSCI.17-05-01786.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gereau RWIV, Conn PJ. Multiple presynaptic metabotropic glutamate receptors modulate excitatory and inhibitory synaptic transmission in hippocampal area CA1. Journal of Neuroscience. 1995;15:6879–6889. doi: 10.1523/JNEUROSCI.15-10-06879.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glitsch M, Marty A. Presynaptic effects of NMDA in cerebellar Purkinje cells and interneurons. Journal of Neuroscience. 1999;19:511–519. doi: 10.1523/JNEUROSCI.19-02-00511.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilfiker S, Augustine GJ. Regulation of synaptic vesicle fusion by protein kinase C. Journal of Physiology. 1999;515:1. doi: 10.1111/j.1469-7793.1999.001ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hounsgaard J, Kiehn O. Serotonin-induced bistability of turtle motoneurones caused by a nifedipine-sensitive calcium plateau potential. Journal of Physiology. 1989;414:265–282. doi: 10.1113/jphysiol.1989.sp017687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huston E, Cullen GP, Burley JR, Dolphin AC. The involvement of multiple calcium channel sub-types in glutamate release from cerebellar granule cells and its modulation by GABAB receptor activation. Neuroscience. 1995;68:465–478. doi: 10.1016/0306-4522(95)00172-f. [DOI] [PubMed] [Google Scholar]
- Jarvis SE, Magga JM, Beedle AM, Braun JE, Zamponi GW. G protein modulation of N-type calcium channels is facilitated by physical interactions between syntaxin 1A and Gbetagamma. Journal of Biological Chemistry. 2000;275:6388–6394. doi: 10.1074/jbc.275.9.6388. [DOI] [PubMed] [Google Scholar]
- Kowluru A, Seavey SE, Rhodes CJ, Metz SA. A novel regulatory mechanism for trimeric GTP-binding proteins in the membrane and secretory granule fractions of human and rodent beta cells. Biochemical Journal. 1996;313:97–107. doi: 10.1042/bj3130097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinás R, Sugimori M, Silver RB. Microdomains of high calcium concentration in a pre-synaptic terminal. Science. 1992a;256:677–679. doi: 10.1126/science.1350109. [DOI] [PubMed] [Google Scholar]
- Llinás R, Sugimori M, Silver RB. Presynaptic calcium concentration microdomains and transmitter release. Journal of Physiology (Paris) 1992b;86:135–138. doi: 10.1016/s0928-4257(05)80018-x. [DOI] [PubMed] [Google Scholar]
- Ma JY, Catterall WA, Scheuer T. Persistent sodium currents through brain sodium channels induced by G protein βγ subunits. Neuron. 1997;19:443–452. doi: 10.1016/s0896-6273(00)80952-6. [DOI] [PubMed] [Google Scholar]
- Neher E, Zucker RS. Multiple calcium-dependent processes related to secretion in bovine chromaffin cells. Neuron. 1993;10:21–30. doi: 10.1016/0896-6273(93)90238-m. [DOI] [PubMed] [Google Scholar]
- Pasinelli P, Ramakers GM, Urban IJ, Hens JJ, Oestreicher AB, De Graan PN, Gispen WH. Long-term potentiation and synaptic protein phosphorylation. Behavioural Brain Research. 1995;66:53–59. doi: 10.1016/0166-4328(94)00124-x. [DOI] [PubMed] [Google Scholar]
- Pinxteren JA, O'Sullivan AJ, Tatham PE, Gomperts BD. Regulation of exocytosis from rat peritoneal mast cells by G protein βγ-subunits. EMBO Journal. 1998;17:6210–6218. doi: 10.1093/emboj/17.21.6210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponce A, Bueno E, Kentros C, Vega-SaenzdeMiera E, Chow A, Hillman D, Chen S, Zhu L, Wu MB, Wu X, Rudy B, Thornhill WB. G-protein-gated inward rectifier K+ channel proteins (GIRK1) are present in the soma and dendrites as well as in nerve terminals of specific neurons in the brain. Journal of Neuroscience. 1996;16:1990–2001. doi: 10.1523/JNEUROSCI.16-06-01990.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robitaille R, Thomas S, Charlton MP. Effects of adenosine on Ca2+ entry in the nerve terminal of the frog neuromuscular junction. Canadian Journal of Physiology and Pharmacology. 1999;77:707–714. [PubMed] [Google Scholar]
- Saugstad J, Segerson TP, Westbrook GL. Metabotropic glutamate receptors activate G protein-coupled inwardly rectifying K+ currents in Xenopus oocytes. Journal of Neuroscience. 1996;16:5979–5985. doi: 10.1523/JNEUROSCI.16-19-05979.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanziani M, Gähwiler BH, Thompson SM. Presynaptic inhibition of excitatory synaptic transmission by muscarinic and metabotropic glutamate receptor activation in the hippocampus: are Ca2+ channels involved? Neuropharmacology. 1995;34:1549–1557. doi: 10.1016/0028-3908(95)00119-q. [DOI] [PubMed] [Google Scholar]
- Schmitz D, Empson RM, Heinemann U. Serotonin reduces inhibition via 5-HT1A receptors in area CA1 of rat hippocampal slices in vitro. Journal of Neuroscience. 1995;15:7217–7225. doi: 10.1523/JNEUROSCI.15-11-07217.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz D, Gloveli T, Empson RM, Draguhn A, Heinemann U. Serotonin reduces synaptic excitation in the superficial medial entorhinal cortex of the rat via a presynaptic mechanism. Journal of Physiology. 1998;508:119–129. doi: 10.1111/j.1469-7793.1998.119br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneggenburger R, Neher E. Intracellular calcium dependence of transmitter release rates at a fast central synapse. Nature. 2000;406:889–893. doi: 10.1038/35022702. [DOI] [PubMed] [Google Scholar]
- Scholz KP, Miller RJ. Inhibition of quantal transmitter release in the absence of calcium influx by a G protein-linked adenosine receptor at hippocampal synapses. Neuron. 1992;8:1139–1150. doi: 10.1016/0896-6273(92)90134-y. [DOI] [PubMed] [Google Scholar]
- Schwartz NE, Alford S. Modulation of pre- and postsynaptic calcium dynamics by ionotropic glutamate receptors at a plastic synapse. Journal of Neurophysiology. 1998;79:2191–2203. doi: 10.1152/jn.1998.79.4.2191. [DOI] [PubMed] [Google Scholar]
- Shupliakov O, Pieribone VA, Gad H, Brodin L. Synaptic vesicle depletion in reticulospinal axons is reduced by 5-hydroxytryptamine: direct evidence for presynaptic modulation of glutamatergic transmission. European Journal of Neuroscience. 1995;7:1111–1116. doi: 10.1111/j.1460-9568.1995.tb01099.x. [DOI] [PubMed] [Google Scholar]
- Shupliakov O, Pieribone VA, Gad H, Brodin L. Presynaptic mechanisms in central synaptic transmission: ‘biochemistry’ of an intact glutamatergic synapse. Acta Physiologica Scandinavica. 1996;157:369–379. doi: 10.1046/j.1365-201X.1996.31251000.x. [DOI] [PubMed] [Google Scholar]
- Silinsky EM. On the mechanism by which adenosine receptor activation inhibits the release of acetylcholine from motor nerve endings. Journal of Physiology. 1984;346:243–256. doi: 10.1113/jphysiol.1984.sp015019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer JH, Berger AJ. Presynaptic inhibition by serotonin: a possible mechanism for switching motor output of the hypoglossal nucleus. Sleep. 1996;19:S146–149. doi: 10.1093/sleep/19.suppl_10.146. [DOI] [PubMed] [Google Scholar]
- Stanley EF. Single calcium channels and acetylcholine release at a presynaptic nerve terminal. Neuron. 1993;11:1007–1011. doi: 10.1016/0896-6273(93)90214-c. [DOI] [PubMed] [Google Scholar]
- Südhof TC. The synaptic vesicle cycle: a cascade of protein-protein interactions. Nature. 1995;375:645–653. doi: 10.1038/375645a0. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Forsythe ID, Tsujimoto T, Barnes-Davies M, Onodera K. Presynaptic calcium current modulation by a metabotropic glutamate receptor. Science. 1996;274:594–597. doi: 10.1126/science.274.5287.594. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Kajikawa Y, Tsujimoto T. G-protein-coupled modulation of presynaptic calcium currents and transmitter release by a GABAB receptor. Journal of Neuroscience. 1998;18:3138–3146. doi: 10.1523/JNEUROSCI.18-09-03138.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchitel OD, Protti DA, Sanchez V, Cherksey BD, Sugimori M, Llinás R. P-type voltage-dependent calcium channel mediates presynaptic calcium influx and transmitter release in mammalian synapses. Proceedings of the National Academy of Sciences of the USA. 1992;89:3330–3338. doi: 10.1073/pnas.89.8.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallén P, Buchanan JT, Grillner S, Hill RH, Christenson J, Hokfelt T. Effects of 5-hydroxytryptamine on the afterhyperpolarization, spike frequency regulation, and oscillatory membrane properties in lamprey spinal cord neurons. Journal of Neurophysiology. 1989;61:759–768. doi: 10.1152/jn.1989.61.4.759. [DOI] [PubMed] [Google Scholar]
- Wu LG, Saggau P. Presynaptic inhibition of elicited neurotransmitter release. Trends in Neurosciences. 1997;20:204–212. doi: 10.1016/s0166-2236(96)01015-6. [DOI] [PubMed] [Google Scholar]
- Zhang H, Yasrebi-Nejad H, Lang J. G-protein betagamma-binding domains regulate insulin exocytosis in clonal pancreatic beta-cells. FEBS Letters. 1998;424:202–206. doi: 10.1016/s0014-5793(98)00176-8. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Delaney KR, Mulkey R, Tank DW. Presynaptic calcium in transmitter release and posttetanic potentiation. Annals of the New York Academy of Sciences. 1991;635:191–207. doi: 10.1111/j.1749-6632.1991.tb36492.x. [DOI] [PubMed] [Google Scholar]