

Abstract
Depolarization-induced suppression of inhibition (DSI) in central neurons is mediated by a transient reduction of γ-aminobutyric acid (GABA) release from interneurons. DSI is induced by a retrograde signal emitted from principal cells. We used electrophysiological recordings from CA1 neurons of the rat hippocampal slice to test the hypothesis that only certain classes of interneurons are susceptible to DSI.
DSI of action potential-dependent, spontaneous, inhibitory postsynaptic currents (sIPSCs) in hippocampus is facilitated by carbachol (3 μm), which increases the occurrence of large sIPSCs. Besides carbachol, noradrenaline (norepinephrine; 10 μm), or elevated extracellular potassium (8 mm), could abruptly increase the occurrence of large sIPSCs and DSI in many cases. DSI appeared and disappeared concomitantly with the onset and offset of these large sIPSCs. In contrast, application of AP-5 and CNQX often markedly increased baseline sIPSC activity without enhancing DSI.
A brief train of extracellular electrical stimulation could trigger the onset of prolonged, repetitive IPSC activity that was susceptible to DSI. The magnitude of DSI of single evoked IPSCs (eIPSCs) in a given pyramidal cell could be altered by changes in stimulus strength, but there was no simple relationship between stimulus strength and DSI.
Baclofen (0.5-5 μm) eliminated the increase in sIPSC activity and DSI induced by carbachol. A GABAB receptor antagonist, CGP 35348, reversed the effects of baclofen.
Carbachol-induced sIPSCs had relatively rapid rise and decay phases. There was no marked distinction between DSI-susceptible and non-susceptible sIPSCs. Nevertheless, two kinetically distinct components of the eIPSC could be distinguished by their decay times. DSI reduced GABAA,fast without affecting GABAA,slow. Furosemide (frusemide), which blocks only GABAA,fast, reduced the eIPSC and occluded DSI.
The data suggest that, with respect to DSI, there are at least three functionally distinct types of IPSCs. Two types (one susceptible to DSI and one not) have relatively rapid kinetics are probably made by perisomatic synapses. A third, slow IPSC, which is insensitive to DSI, may be produced by distal dendritic synapses.
After being briefly depolarized by a short train of action potentials or a voltage-clamp step, hippocampal pyramidal cells receive decreased GABAA receptor-mediated inhibitory synaptic input for ∼1 min (at 30 °C). A great deal of evidence indicates that this phenomenon, called depolarization-induced suppression of inhibition (DSI), involves a retrograde signalling process that causes a transient decrease in the release of GABA from interneurons in hippocampus (Pitler & Alger, 1992a, 1994; Ohno-Shosaku et al. 1998) and cerebellum (Llano et al. 1991; Vincent et al. 1992; Glitsch et al. 1996). Because DSI can, by reducing overlapping IPSCs, enhance EPSCs (Wagner & Alger, 1996), it could play a role in the control of neuronal excitability. In hippocampus, DSI can be induced by action potential trains (Pitler & Alger, 1992a), calcium spikes (Pitler & Alger, 1994) or low-Mg2+-induced ‘burst’ potentials (LeBeau & Alger, 1998), and therefore may be produced by physiological stimuli. Different IPSPs serve different functions in neuronal excitability control, however (Pearce, 1993; Miles et al. 1996; Kapur et al. 1997a; McMahon et al. 1998), and before DSI can be effectively incorporated into models of hippocampal functioning, it will be important to know which interneurons are affected by it.
Hippocampal interneurons are heterogeneous, with distinctions among them being made according to their morphology, location of their somata, synaptic termination zones, transmitter cofactors, synaptic inputs and presumed functions, among other features (Freund & Buzsaki, 1996). Recent evidence has disappointed simple expectations that unique, consistent groupings of these properties would readily identify orderly classes or types of interneurons. Rather, any given interneuronal feature may be randomly associated with virtually any other feature in a particular cell, so that it is difficult to make broad generalizations based on morphological, physiological or pharmacological criteria (Parra et al. 1998; McQuiston & Madison, 1999).
Nevertheless, useful classifications may still be possible on functional grounds: perisomatic GABAergic synapses tend to control pyramidal cell action potential firing and thereby to control synaptic output of the cells. Dendritic GABAergic synapses regulate dendritic integration and the expression of voltage-dependent dendritic conductances (Miles et al. 1996). Slow GABAA receptor-mediated IPSPs may be more important in the regulation of slower, NMDA-dependent EPSPs, while fast GABAA receptor-mediated IPSPs may influence fast EPSPs and postsynaptic cell firing to a greater extent (Pearce, 1993; Kapur et al. 1997b). As a step towards understanding the physiological significance of DSI, we have investigated functional properties of IPSCs to determine if systematic distinctions can be made among IPSCs with respect to their susceptibility to DSI.
Carbachol application (Pitler & Alger, 1992b; Behrends & ten Bruggencate, 1993) or release of ACh from cholinergic fibres in the slice (Pitler & Alger, 1992b) activates monosynaptic IPSCs in CA1 pyramidal cells. DSI of action potential-dependent spontaneous inhibitory postsynaptic currents (sIPSCs) seldom occurs in the absence of cholinergic stimulation (Pitler & Alger, 1992a, 1994). The increase in carbachol-enhanced sIPSC activity coincides with the onset of DSI. These carbachol effects are mediated exclusively via pirenzepine-sensitive muscarinic cholinergic receptors (mAChRs) (Martin & Alger, 1999). However, despite its pronounced facilitatory effect on DSI, mAChR activation is not mandatory for the expression of DSI, as DSI of sIPSCs (Pitler & Alger, 1994) or of monosynaptically evoked IPSCs (eIPSCs) is not blocked by atropine (Martin & Alger, 1999). This suggests that biochemical processes initiated by mAChRs are not essential for DSI.
Due to the association of increased sIPSC activity and DSI, we hypothesized that a major aspect of the effect of carbachol on DSI might be its ability to activate certain interneurons. Carbachol depolarizes some interneurons, and often raises the level of excitability in these cells to firing threshold (Reece & Schwartzkroin, 1991; Kawaguchi, 1997; McMahon et al. 1998; Parra et al. 1998; McQuiston & Madison, 1999). There are at least three ways by which this could facilitate DSI expression. DSI could be present under control conditions but not apparent because of low levels of sIPSC activity. By increasing action potential firing frequency of interneurons, carbachol could make DSI more detectable. In this case, any manipulation that increases sIPSC activity should also increase DSI expression. Alternatively, carbachol could enhance DSI by an unknown action on the pyramidal cells.
Finally, carbachol could activate a specific population of interneurons that is particularly susceptible to the DSI process. Although carbachol depolarizes some hippocampal interneurons, it hyperpolarizes others and has no effect on still others (Parra et al. 1998; McQuiston & Madison, 1999). There are evidently no morphological features or characteristic locations in the slice that identify these cells in the hippocampus (unlike the cortex, where carbachol depolarizes a subset of interneurons with distinctive neurochemical and electrophysiological properties (Kawaguchi, 1997)). We investigated DSI in the CA1 region to test the hypothesis that DSI affects functionally identifiable subsets of IPSCs. We investigated sIPSCs and eIPSCs in the presence and absence of carbachol, respectively. We conclude that DSI susceptibility distinguishes subgroups of IPSCs and that the distinctions suggest how different interneurons are affected by DSI. Some of the data presented here have previously appeared in abstract form (Martin et al. 1995; Martin & Alger, 1996).
METHODS
Preparation of slices
Hippocampal slices were obtained from young adult, male, Sprague-Dawley rats (150-250 g; 30-60 days old) using conventional techniques. All experiments were carried out in accordance with the guidelines set forth by the Institutional Animal Care and Use Committee of the University of Maryland School of Medicine. After the animals were deeply anaesthetized with halothane and decapitated, the hippocampi were removed and sectioned into slices 400 μm thick in ice-cold saline using a Vibratome (Technical Products, International, Chicago, IL, USA). The slices were maintained at room temperature in an interface holding chamber in a humidified atmosphere saturated with 95 % O2-5 % CO2. The slices were not used for at least 1 h after sectioning. The recording chamber warmed the submerged slice, and experiments were carried out at ∼30 oC (Nicoll & Alger, 1981).
Recording conditions
Whole-cell voltage-clamp recordings of CA1 pyramidal cells were done using the ‘blind’ patch method (Blanton et al. 1989) with patch electrodes (3-6 MΩ in the bath). The criteria for acceptable recordings were: input resistance > 50 MΩ, series resistance < 15 MΩ, and resting membrane potential more negative than -55 mV. Series resistances were compensated by 70-75 %. During experiments, series resistance was checked by hyperpolarizing voltage steps and adjusted if necessary. Experiments were terminated if the series resistance increased by more than 20 %. Depolarizing voltage steps (lasting 1 or 2 s) from the holding potential of -70 mV to ∼0 mV were given every 90-120 s. For DSI experiments on eIPSCs, monosynaptic IPSCs were elicited at 0.5 Hz by 50 μs extracellular stimuli delivered with concentric bipolar stimulating electrodes placed near the CA1/CA2 border in stratum (s.) oriens, s. radiatum, or s. pyramidale. The recording electrode was ∼0.5 mm away from the stimulating electrodes, which were ∼150 or ∼250 μm away from s. pyramidale in s. oriens or s. radiatum.
Solutions
The intracellular recording solution contained (mm): 145 or 160 KCl, 1 MgCl2, 1 MgATP, 0.2 CaCl2, 2 K4-BAPTA, 10 Hepes, 0.3 Tris-GTP, and 5 QX-314. The extracellular saline included (mm): 120 NaCl, 3 KCl, 25 NaHCO3, 1 NaH2PO4, 2.5 CaCl2, 1.5 MgCl2 (or 2 MgSO4), and 10 glucose. The local anaesthetic QX-314 was included in the recording pipette to block sodium-dependent action potentials (Connors & Prince, 1982), as well as postsynaptic GABAB responses in the pyramidal cell (Nathan et al. 1990; Andrade, 1991). To isolate monosynaptic IPSCs, ionotropic glutamate receptor blockers CNQX (10 μm) and AP-5 (50 μm) (Davies et al. 1990) were present in all experiments.
Chemicals
6-Cyano-7-nitroquinoxaline-2,3-dione (CNQX), quisqualate, and baclofen were purchased from Research Biochemicals International (RBI; Natick, MA, USA), QX-314 (lidocaine (lignocaine) N-ethyl bromide) from Alomone Labs (Jerusalem, Israel) or RBI, and carbachol (carbamylcholine chloride), atropine sulphate, noradrenaline (l-arterenol bitartrate), serotonin, ascorbic acid and dl-2-amino-5-phosphonopentanoic acid (AP-5) from Sigma Chemical Co. (St Louis, MO, USA). CGP 35348 was a generous gift from Ciba-Geigy.
Data acquisition and analysis
Spontaneous IPSC data were collected using an Axopatch-1C amplifier (Axon Instruments, Foster City, CA, USA), filtered at 2 Hz and digitized at 4 kHz using a Digidata 1200 and the Clampex subroutine of pCLAMP6 software (Axon Instruments). Unfiltered data were also stored on VCR tapes using a Neuro-corder (Neuro Data Instruments, Delaware Water Gap, PA, USA). For playback, the data were fed into a computer or chart recorder after being passed through an 8-pole Bessel filter (Frequency Devices, Haverhill, MA, USA).
To quantify total sIPSC activity in some experiments (as noted), we integrated the current to measure the total charge crossing the membrane in the 8 s period before the depolarizing voltage step using the Fetchan subroutine of pCLAMP6 (Axon Instruments). We determined the baseline manually, carefully inspecting the traces to ascertain that they were free of aberrant currents that would confound the integration measure. To measure DSI, we compared the activity in this 8 s control period with the activity in an equivalent period after the step and expressed the result as a percentage of control activity. The first 2 s period after the step was disregarded to allow for maximal DSI expression (Pitler & Alger, 1994) and to exclude error introduced by any depolarization-induced afterpotentials.
Amplitude histograms were constructed by manually measuring the sIPSCs using the Clampfit subroutine of pCLAMP6. The traces were first ‘smoothed’ using a boxcar filter, which provided a running mean of five data points and minimized baseline noise. Cursors placed at the start of the sIPSC and the peak gave the amplitude, as well as a ‘time-to-peak’ measurement. All sIPSCs > 10 pA occurring 8 s prior to and 8 s after the depolarizing voltage step were measured, with the first 2 s following the step excluded, as mentioned above.
Evoked IPSC data were filtered at 2 kHz and sampled at 10 kHz. The amplitudes of the eIPSCs were measured using Clampfit. The mean of five IPSCs before and after the depolarization was compared to calculate the percentage DSI (with the first IPSC following the voltage step disregarded). The stimulation artifact was usually truncated or blanked for clarity in the illustrated examples. Spontaneous IPSCs were analysed by the time-to-peak and time-to-decay parameters of the Mini Analysis program (Synaptosoft, New York, NY, USA). The program automatically detects and measures spontaneous events. The time to peak was measured as the difference between the last point before a detected event and the event peak. The time to decay was taken as the time from the peak to a point that was 0.32 of the peak value. We verified visually that all measurements were made on IPSC waveforms.
One-way ANOVAs, followed by the Student-Newman-Keuls test for multiple comparisons, were done using Sigmastat (SPSS, Chicago, IL, USA) to determine statistical significance of the data at the level of P < 0.05. Data are presented as means ± standard error of the mean (s.e.m.). Kolmogorov-Smirnov (K-S) statistics were used to determine if there was a significant difference in the sIPSC amplitude distributions in the presence of carbachol or during the DSI period. A significance level of P < 0.001 was used to determine significant changes in distributions using the K-S test.
RESULTS
DSI-susceptible interneuronal firing can begin and end abruptly
Depolarization-induced suppression of inhibition (DSI) is a pronounced, transient suppression of IPSC activity mediated by a Ca2+-dependent retrograde signal process in hippocampus and cerebellum (see Alger, 1999, for review). As previously reported (Pitler & Alger, 1994; Martin & Alger, 1999), sIPSC activity is generally unaffected by the delivery of a depolarizing command pulse (step to 0 mV for 1 s, upward deflections in Fig. 1) to a pyramidal cell in the CA1 region of the hippocampal slice (Fig. 1Aa, left part of trace). Bath application of 3 μm carbachol often causes an abrupt increase in sIPSC activity (e.g. at arrowhead in Fig. 1Aa), and as soon as the sIPSC activity increases, the depolarizing voltage steps are followed by DSI. Thus the two events, the increase in IPSC activity and the appearance of DSI, are closely correlated in time. We have suggested that the main action of carbachol is to bring DSI-susceptible interneurons to firing threshold, and that this accounts for the connection between the sudden onset of IPSC activity and DSI (Martin & Alger, 1999). This hypothesis is compatible with the abrupt termination of DSI-susceptible sIPSC activity when muscarinic receptor antagonists, e.g. pirenzepine (Fig. 1Ab) or high concentrations of AF-DX 116 (Fig. 1Ac), are applied in the presence of carbachol. Nevertheless, the hypothesis that there are DSI-susceptible and non-DSI-susceptible interneurons has not been tested. Moreover, it was not clear whether increasing interneuron activity and DSI are unique properties of muscarinic receptor activation, or whether activation of interneurons in other ways could also induce the appearance of DSI.
Figure 1. An increase in DSI-susceptible sIPSCs could occur in an all-or-none manner.
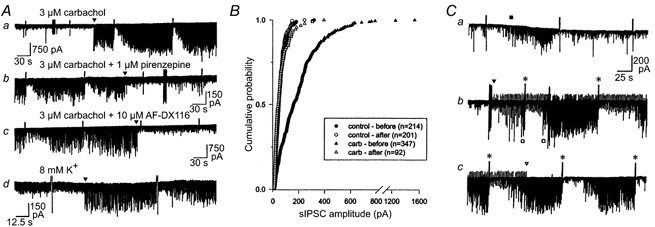
A, the traces are from four different cells that displayed a sudden onset or offset of sIPSC activity. Aa, note the abrupt onset of activity in carbachol (arrowhead). This elevated activity could be equally abruptly eliminated during perfusion with muscarinic antagonists, pirenzepine (Ab) and a high, non-specific, concentration of AF-DX 116 (Ac). This same sudden onset of relatively large amplitude sIPSCs could also be observed when 8 mm extracellular [K+] was perfused (Ad, arrowhead). There is a strong correlation between the occurrence of high-amplitude sIPSC activity and the appearance of DSI. Note that the level of sIPSC activity during maximal DSI is approximately the same as the baseline sIPSC activity prior to the onset of large-amplitude sIPSCs. B, cumulative probability distribution of the amplitudes of sIPSCs occurring 8 s prior to the depolarizing voltage step and 8 s after the step in a cell such as in A. Six cells were analysed in this manner, and all showed significant increases in amplitude of sIPSCs during carbachol perfusion and significant decreases in large sIPSCs during the DSI period (P < 0.001, K-S test). C, consecutive traces from a CA1 pyramidal cell that showed only a transient increase in sIPSCs in response to bath application of 3 μm carbachol (a, ▪), and very little DSI. Stratum (s.) radiatum was stimulated at a low frequency (0.5 Hz), beginning at the arrowhead in Cb. The eIPSCs are downward deflections. The stimulus artifacts are the regular upward deflections on the trace. Note the onset of large sIPSCs (□ beneath trace b) that obscure the eIPSCs. DSI of sIPSCs and eIPSCs occurred following the next depolarizing steps (*). Stimulation was terminated (open arrowhead, trace c), but the sIPSC activity and DSI remained in the absence of any external stimulation, and continued for the duration of the experiment (∼50 min; not shown).
To investigate the latter issue we tried other methods of activating interneurons. We first increased extracellular potassium concentration ([K+]o) in the bath perfusion from 3 mm (normal) to 8 mm (‘high [K+]’). Cells were perfused with 3 μm carbachol following return to normal-[K+] saline. In four cells, high [K+] increased sIPSC activity abruptly by inducing large-amplitude sIPSCs (e.g. Fig. 1Ad). In these cases, DSI was comparable with that observed in carbachol (53 ± 8.5 %, n = 4 in high [K+]o compared with 64.1 ± 2.2 % in carbachol, n = 32, P > 0.05) and also occurred abruptly. An example of one of these cells is illustrated in Fig. 1Ad. However, in four other cells high [K+]o failed to increase sIPSC activity or DSI, although DSI was induced when carbachol was subsequently applied.
Noradrenaline (NA) depolarizes and increases the firing frequency of several classes of interneurons located in all strata of the hippocampus (Bergles et al. 1996). We found that NA (10 μm) significantly increased sIPSC activity by nearly 2-fold over control values, and caused a small, but consistent and significant, increase in DSI (28 ± 4.5 % in NA compared with 10 ± 3.7 % in control saline for the same group of cells, n = 11; data not shown). NA increased sIPSC activity in all 11 cells and increased DSI in 10 of them. Carbachol perfusion following NA in two cells further increased DSI (to 52 ± 3.8 %).
In some cells we observed a large increase in total sIPSC activity when AP-5 and CNQX were added to the control bathing solution (prior to carbachol application). This increase may be caused by disinhibition. Alternatively, CNQX may increase firing of a small subpopulation of CA3 s. radiatum interneurons by a direct, but unknown, mechanism (McBain et al. 1992), and if similar interneurons exist in CA1, this could account for the increase in sIPSC activity when AP-5 and CNQX were applied. The magnitude of the increase in total sIPSC activity often matched or exceeded the increase induced by carbachol. However, despite the high level of sIPSC activity in this group of cells, there was no increase in DSI (7 ± 1.8 % in AP-5 plus CNQX, n = 11) compared with cells in control solution alone, or cells in which AP-5 and CNQX did not increase baseline activity (6 ± 1.4 %, total of all control cells combined, n = 55). In these cells there was a significant increase in DSI when carbachol was subsequently applied (not shown).
Quisqualate (Poncer & Miles, 1995) and serotonin (Ropert & Guy, 1991) reportedly also increase hippocampal sIPSC activity, effects that may be mediated by selective activation of a subpopulation of interneurons. However, we found that neither 500 nm quisqualate nor 1 μm quisqualate significantly increased sIPSC activity or enhanced DSI (n = 5; data not shown). We have previously reported that, in CA1, quisqualate suppresses eIPSCs and occludes DSI (Morishita et al. 1998). Neither 10 μm nor 50 μm serotonin (5-HT) significantly increased IPSCs or DSI (n = 4). Total sIPSC activity and DSI were both greatly facilitated in all of these cells when carbachol was subsequently applied (data not shown).
In summary, there was only an imperfect correlation between increases in total sIPSC activity and the magnitude of DSI. The magnitude of DSI in NA alone was only comparable with that produced by carbachol (> 45 %) in 3 of 11 cells, and in only 4 of 8 cells in high [K+], despite approximately equivalent total levels of sIPSC activity in most cases (data not shown). Moreover, the marked increases in total sIPSC activity induced by AP-5 and CNQX were not accompanied by enhancements of DSI. Thus the increase in overall level of integrated sIPSC activity induced by carbachol cannot explain its facilitation of DSI. On the other hand, although carbachol application is the most consistently effective method we have found for inducing the appearance of DSI, it is not the only method.
Sustained DSI-susceptible sIPSC activity could be triggered by electrical stimulation
The amplitude of sIPSC activity remaining during the period of maximal sIPSC suppression was often indistinguishable from that of the baseline activity in the absence of the large, DSI-susceptible sIPSCs. Figure 1B illustrates this point quantitatively. In control conditions (AP-5 and CNQX present), sIPSCs were relatively small and infrequent. Bath application of 3 μm carbachol significantly increased the frequency and amplitude of sIPSCs, as in Fig. 1A, and a marked degree of DSI appeared. Kolmogorov-Smirnov tests indicated that the difference between the sIPSC distributions of the pre-DSI and DSI intervals in carbachol was significant (P < 0.001, n = 6 cells), but that the sIPSC amplitude distribution during DSI in carbachol did not differ from the baseline amplitude distribution. It appears that the larger sIPSCs induced by carbachol were the ones that were blocked by DSI, and that the smaller sIPSCs were unaffected by carbachol or DSI. If the large, carbachol-induced sIPSCs actually arise from the firing of one or more interneurons, then it should be possible to induce their appearance by electrical, as well as chemical, stimulation.
In approximately 30 % of the cells studied, there was very little sustained increase in sIPSC activity with carbachol, and DSI of sIPSCs could not be induced, although generally the cells were still capable of DSI of eIPSCs (Martin & Alger, 1999). In some cases, extracellular stimulation in carbachol initiated a sustained increase in sIPSC activity, and concomitantly DSI appeared. In the cell illustrated in Fig. 1C, the carbachol-induced increase in sIPSCs was small and transient (beginning at the filled square, Ca). After the onset of stimulation of s. radiatum at 0.5 Hz (regular upward deflections in Cb beginning at filled arrowhead represent stimulus artifacts) to elicit eIPSCs (regular downward deflections), there was a sudden increase in sIPSCs (Cb, open squares) that ultimately obscured the eIPSCs. In response to a DSI-inducing voltage step, there was a reversible decrease in the amplitude of both sIPSCs and eIPSCs. When extracellular stimulation was halted (Cc, open arrowhead), the elevation of sIPSCs and the ability to induce DSI remained for the duration of the experiment (∼50 min, not shown). The abrupt onset of the persistent, large-IPSC activity, and its continuation after the end of the stimulation suggested that a cell or cells began firing because of the stimulation, and continued firing after it. These data suggest that the large, carbachol-induced sIPSCs, and the eIPSCs that are susceptible to DSI (see below), could be produced by the same interneurons.
Altering the stimulus intensity affects the magnitude of DSI
In addition to finding that it is generally the larger, carbachol-induced sIPSCs that are susceptible to DSI, we have also noted many instances in which weak extracellular stimulation produced small eIPSCs that were not susceptible to DSI, but in which increases in stimulus intensity produced larger IPSCs that were susceptible to DSI (e.g. Fig. 2A). A sample of six cases of this type of result is shown in Fig. 2B. In these pyramidal cells DSI increased from 16 ± 9.3 % with ‘weak’ stimulation to 69 ± 5.8 % (n = 6) when the extracellular stimulation intensity was increased. This increase in DSI clearly represents a presynaptic phenomenon, since the activation of the postsynaptic cell was constant throughout each experiment.
Figure 2. Presynaptic stimulus intensity affects the magnitude of DSI.
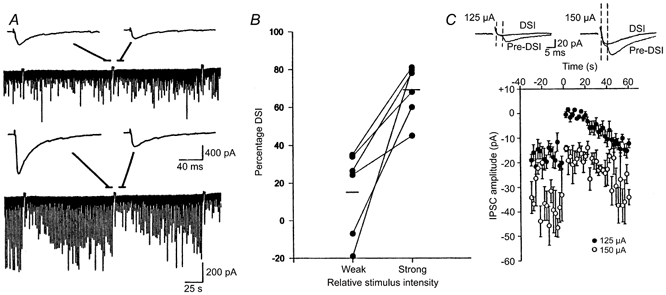
A, in this cell, with a relatively weak stimulus intensity (175 μA), the IPSCs were small, and there was little evidence of DSI following the voltage step (upper trace). The insets represent the mean of five IPSCs before and during the DSI period. When the stimulus intensity was increased (200 μA), much larger IPSCs were evoked and DSI was greatly enhanced (lower trace). B, data from six cells, showing an increase in DSI with higher stimulus intensities. Lower stimulus intensities (75-175 μA) produced 43-220 pA mean eIPSC amplitudes, whereas higher stimulus intensities (150-500 μA) elicited 215-2040 pA eIPSCs. The mean DSI values, shown by the horizontal lines, increased substantially. C, weak stimulation intensities reveal susceptible and non-susceptible eIPSCs. Stimulus intensity was adjusted to produce small, quantal-sized IPSCs and occasional failures of transmission. S. radiatum was stimulated at 0.5 Hz. At 125 μA, minimal eIPSCs were consistently elicited, as shown by the mean of 10 IPSCs occurring before the depolarizing voltage step (Pre-DSI). During the DSI period, most of the stimuli resulted in failures, as indicated by the mean of 10 sweeps shown (DSI). Increasing the stimulus intensity to 150 μA resulted in the recruitment of another component with a shorter latency that was not susceptible to DSI, although the later component still failed during DSI. Stimulus artifacts were reduced for clarity. The graph shows summary data for this cell. DSI of the peak eIPSC amplitude was apparent at the higher stimulus intensity, but could be accounted for by the reduction of the low threshold component alone.
The relationship between large IPSCs and DSI could mean that increasing stimulus intensity activated a DSI-susceptible group of interneurons, or alternatively, that the stimulation activated secondary factors, perhaps via other neuronal elements, which influenced the DSI process directly. However, the association of DSI with higher stimulation intensities was not invariant. As previously reported (Alger et al. 1996), we could occasionally induce DSI of minimal IPSCs. Minimal stimulation is defined as the lowest stimulus intensity required to elicit an eIPSC, with occasional failures of transmission occurring. Minimal stimulation is considered to activate single fibres and induce quantal-sized IPSCs (Edwards et al. 1990), as in the example of Fig. 2C. During the DSI period, the probability of failure of transmission greatly increased. Minimal stimulation is unlikely to produce extensive indirect effects that induce DSI, but this possibility is not ruled out. However, if minimal stimulation affected DSI by some indirect effect, then increasing stimulus intensity slightly beyond the minimal level should produce larger responses and more DSI. On the contrary, we found that small increases in stimulus intensity could evoke additional small eIPSCs that were not susceptible to DSI. In the example of Fig. 2C, a relatively long-latency minimal IPSC was blocked by DSI. Increasing the stimulation intensity slightly elicited a shorter latency IPSC, also of quantal size. The higher threshold component never failed in control or during DSI, even though the lower threshold component was still eliminated during DSI and, in fact, accounted for the decrease in the total IPSC amplitude during DSI. These results support the interpretation that the differences in DSI that are seen with different stimulus intensities cannot be simply explained by the production of secondary effects due to stimulation, and are compatible with the idea that the different forms of stimulation can activate different interneurons, only some of which are susceptible to DSI.
IPSCs evoked from certain regions of CA1 were often more effective than those evoked from other regions
Interneurons are present throughout the hippocampus, and we wondered if large IPSCs evoked from all strata would be equally susceptible to DSI. Bath application of carbachol causes global stimulation of interneurons possessing mAChRs, but with small bipolar stimulating electrodes and studying DSI of eIPSCs, we could selectively stimulate different synaptic inputs to a pyramidal cell. By moving the stimulating electrode, two, and occasionally three, different layers of the hippocampus could be stimulated sequentially while recording from a single pyramidal cell. Stimulation intensity was adjusted to produce eIPSCs of approximately equal amplitude in each region. Substantial variability in DSI magnitude depending on the site of stimulation was observed in 12 out of 18 pyramidal cells tested (data not shown). Stimulation of s. oriens and s. radiatum was typically more effective than stimulation of s. pyramidale. Occasionally it was possible to elicit eIPSCs by stimulation in s. lacunosum-moleculare that were completely unaffected by DSI. However, in view of the extensive ramifications of the interneuronal axonal and dendritic arbors (Freund & Buzsaki, 1996), these stimulation experiments do not provide unequivocal information regarding the particular neuronal subtypes involved. Nevertheless, by revealing that DSI can differ in magnitude in a given pyramidal cell in which all conditions are identical except the presynaptic input, these results support the conclusion that variability in DSI expression is, to a large extent, attributable to activation of different interneurons.
Baclofen reversibly blocked carbachol-induced DSI of sIPSCs
Baclofen, a GABAB receptor agonist, inhibits only a subset of sIPSCs in CA3 (Lambert & Wilson, 1993), suggesting that GABAB receptors are only located on some interneurons. To determine if the DSI-susceptible interneurons that are activated by carbachol also have GABAB receptors, we used QX-314 in the recording pipette to block postsynaptic GABAB receptors and bath-applied the GABAB agonist, baclofen. Baclofen greatly reduced the carbachol-induced sIPSC activity and virtually eliminated DSI (Fig. 3). This effect could be fully reversed by addition of 1 mm CGP 35348, a GABAB receptor antagonist, demonstrating that the effect of baclofen was specific for GABAB receptors. Figure 3A and B demonstrates the reversibility of these effects. Baclofen can also reduce voltage-dependent Ca2+ currents; however, analysis of the net current during the DSI-inducing voltage step revealed no effect of baclofen or carbachol on the net current, in contrast to their marked effects on DSI (n = 6; data not shown). Grouped data in Fig. 3C show that low concentrations (0.5 or 1 μm) of baclofen decreased DSI from a mean of 63 ± 4.7 % in carbachol to 39 ± 10.5 % (n = 5), which was then further, significantly reduced to 13 ± 1.9 % (n = 4; P < 0.05) in higher baclofen concentrations (2 or 5 μm). Addition of 1 mm CGP 35348 restored DSI to 65 ± 4.9 % (n = 2). These data suggest that carbachol activates the same interneurons that are inhibited by GABAB receptor activation.
Figure 3. Baclofen blocks DSI of sIPSCs.
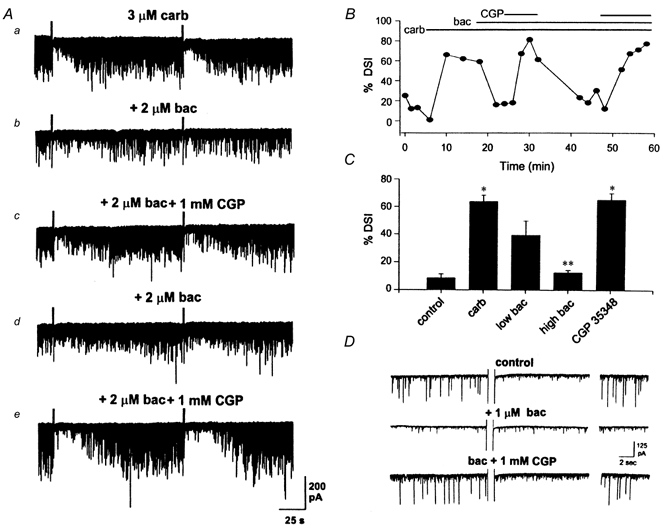
A, data traces from an experiment the time course of which is shown in B. The carbachol-induced increase in activity and DSI (a) is eliminated by the addition of 2 μm baclofen (b). Addition of 1 mm CGP 35348 to the perfusate resulted in reversal of the baclofen effect and recovery of activity and DSI (c). Baclofen and CGP 35348 applications were repeated to show reproducibility of the effect (d and e). B, time course of DSI, showing inhibition of DSI in baclofen, and recovery of DSI in CGP 35348. The bars at the top of the graph indicate the duration of drug applications. C, grouped data showing that lower concentrations (0.5 or 1 μm) of baclofen reduced DSI (n = 5) and higher concentrations (2 or 5 μm) of baclofen abolished DSI (n = 4). Addition of CGP 35348 fully recovered sIPSC activity and DSI in 2/2 of these cells (*P < 0.05 vs. control; **P < 0.05 vs. carbachol, using one-way ANOVA, followed by the Student-Newman-Keuls test) D, baclofen blocked DSI of sIPSCs in the absence of carbachol. Example from a CA1 pyramidal cell showing DSI in the absence of carbachol and presence of 1 μm atropine. Baclofen (1 μm) blocked the large sIPSCs and DSI. CGP 35348 (1 mm) recovered the large sIPSCs and DSI.
Although rare, DSI of sIPSCs does occur occasionally in the absence of cholinergic stimulation (Martin & Alger, 1999). This form of DSI in the presence of 1 μm atropine (which, by itself, had no effect in these cells (Martin & Alger, 1999)) was also eliminated by baclofen and restored by CGP 35348 (n = 2; Fig. 3D). The large sIPSCs preceding the depolarizing voltage step in control saline for the cell shown were selectively abolished during the DSI period. There was no detectable difference in the distribution or frequency of small-amplitude sIPSCs. Baclofen also eliminated most of these large events, but they returned when CGP 35348 was perfused. The reduction in large-amplitude events is similar to that observed for DSI induced by carbachol. The efficacy of baclofen in inhibiting DSI in the absence of cholinergic stimulation showed that baclofen did not directly antagonize carbachol; rather, it appeared to block the large-amplitude sIPSCs that were susceptible to DSI.
Baclofen blocks DSI of eIPSCs
DSI of eIPSCs also occurs in the presence of atropine (Martin & Alger, 1999). In these cases DSI is blocked by baclofen and is restored when CGP 35348 is added to the bath (n = 3; Fig. 4A). Control data for the cells in Fig. 4A were collected in the presence of 1 μm atropine, which had no effect on DSI of eIPSCs (Martin & Alger, 1999). Baclofen reduced the mean amplitude of the eIPSCs (Fig. 4Ab, the eIPSCs were reduced from 520 ± 87.2 pA in control to 223 ± 63.2 pA in baclofen, P > 0.05; n = 3 cells, 15-20 eIPSCs per condition per cell). This amplitude reduction is apparent in the representative cell shown in Fig. 4Aa. Baclofen could, by decreasing the eIPSC amplitude, have obscured DSI. If this were true, increasing the stimulus intensity would increase the eIPSC amplitude and DSI would again be apparent. However, this did not happen; as illustrated by the cell in Fig. 4Ac, increasing the stimulus intensity increased the mean eIPSC amplitude from 152 to 265 pA, but still there was no DSI, suggesting that baclofen blocked the portion of the eIPSC that was sensitive to DSI. Recovery of this portion of the eIPSC in CGP 35348 was sufficient to restore DSI (Fig. 4Ad).
Figure 4. Baclofen blocks DSI of eIPSCs.
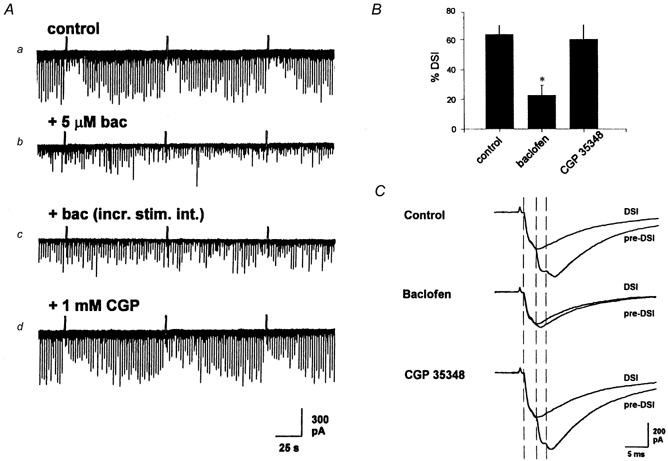
A, representative CA1 neuron in which stimulation of s. oriens at 0.5 Hz elicited eIPSCs that undergo DSI in the presence of atropine (1 μm) (a). Addition of 5 μm baclofen (b) greatly reduced the amplitude of the eIPSCs and magnitude of DSI. Increasing the stimulus intensity (c) increased the amplitude of the eIPSCs, but did not restore DSI. Addition of 1 mm CGP 35348 (d) reversed the baclofen effect, increased the amplitude of the sIPSC and restored DSI. B, group data from three cells showing a significant decrease in DSI in 5 μm baclofen, and a nearly full recovery of DSI upon addition of CGP 35348 (*P < 0.05 vs. control or CGP 35348 using one-way repeated measures ANOVA, followed by the Student-Newman-Keuls test). C, baclofen selectively eliminated the same components of eIPSCs as did DSI. A mean of 10 sweeps before the depolarizing step and 10 sweeps during the DSI period were superimposed in each perfusion medium. The dashed vertical lines indicate the time where each major eIPSC component began (see text). During the DSI period in control, there was a complete, or nearly complete, elimination of the second and third components. Perfusion with 5 μm baclofen inhibited the second and third components as did DSI, without any inhibition of the first eIPSC component. Following addition of 1 mm CGP 35348, there was recovery of all three components, and during the DSI period there was elimination again of both the second and third components. Stimulus artifacts were reduced for clarity.
Evoked IPSCs are often composed of multiple ‘components’ with reproducible latencies to onset, each probably representing the output of a different interneuron (Edwards et al. 1990; Soltesz et al. 1995). DSI often involves the selective reduction or elimination of particular components (Alger et al. 1996). This is evident in the traces in Fig. 4C, each of which represents the mean of 10 eIPSCs before and during the DSI period. The control eIPSC, in 1 μm atropine (‘pre-DSI’), is composed of four components. The three largest components, identified by the inflection points in the waveform and indicated by the dashed lines, will be discussed below. Another small component is apparent between the first and second vertical dashed lines; however, this component did not change with DSI or baclofen and so, for simplicity, has been ignored in the following. The amplitudes of the first, second and third components in the control pre-DSI trace are 311, 185 and 46 pA, respectively. The first two components are clearly large enough to be attributed to multiquantal release, because a quantal GABAA response is ∼20 pA in these cells (Edwards et al. 1990; Otis et al. 1991; Soltesz et al. 1995; Alger et al. 1996). During the DSI period, the second and third components are completely eliminated, while the initial component is unchanged (305 pA). In the same cell, perfusion with 2 μm baclofen also eliminated the third component and greatly reduced the second. The 26 pA, probably quantal, component remaining in baclofen was eliminated during the DSI period in baclofen. The initial component was essentially unchanged by baclofen or DSI. Subsequent perfusion with the GABAB antagonist CGP 35348 restored all three original components (386, 219 and 52 pA), and DSI was apparent again with the selective elimination of the second and third components. Thus the effects of both baclofen and DSI are targeted to specific multiquantal eIPSC components, an observation that is also compatible with an action of DSI on specific interneurons.
Carbachol-enhanced spontaneous IPSCs that are susceptible to DSI and those not DSI susceptible do not differ kinetically
If differences between interneurons determine DSI susceptibility, then it seemed possible that these differences might be detected in the kinetic properties of the sIPSCs. Therefore we measured the rise and decay times of sIPSCs in each of six stable, but otherwise randomly chosen, cells. For each cell, 100 sIPSCs were obtained from the 20 s interval prior to a depolarizing step and 100 from the first 20 s of the DSI periods following the steps (disregarding the first 2 s; see Methods). The measurements were made first in control saline, and then again in the presence of carbachol. Thus, there were 600 sIPSCs in each of 4 groups: pre-DSI and DSI in control saline, and pre-DSI and DSI in carbachol-containing saline. We selected isolated sIPSCs, i.e. with no evident overlap for at least 2/3 of the decay time, for this analysis. Because of the high degree of activity present in a few cells, it was necessary to accumulate the sIPSCs over two DSI trials in each condition to obtain 100 isolated ones. Measurements were made with Mini Analysis software (see Methods). There were no differences in time to peak, or time to decay, for sIPSCs in pre-DSI and DSI periods in control solution (data not shown). The mean time to peak varied from 1.2 to 2.4 ms and time to decay from 6.5 to 13.4 ms across cells (n = 6). The grouped data showed a small, though statistically significant, tendency of both the pre-DSI time-to-peak and time-to-decay measures to increase in carbachol. The group mean time to peak was 1.7 ± 0.03 ms in control and 1.9 ± 0.03 ms in carbachol; time to decay was 9.8 ± 0.16 and 10.4 ± 0.18 ms, respectively. During the DSI period in carbachol, both time to peak and time to decay significantly decreased back to control values. Nevertheless, these group effects were quantitatively small and were seen in only 3 out of 6 cells when each was examined individually. There was no correlation between initial time to peak, or time to decay, and the tendency to shift either in carbachol or during DSI. As there are no systematic differences in the absolute values of time to peak or time to decay between sIPSCs in the pre-DSI and DSI periods, DSI-susceptible sIPSCs cannot be distinguished by their kinetic properties in general.
DSI-susceptible and non-DSI-susceptible evoked IPSCs can be kinetically distinguished
Despite the lack of difference in kinetic properties between DSI-susceptible and non-susceptible sIPSCs discussed above, we noted that the eIPSC often appeared to decay more slowly during the DSI period than during the pre-DSI period. Indeed, fitting the decay phase with single exponentials showed a consistent increase in the time constant of the eIPSC decay during DSI (Fig. 5A). As this slowing of the decay phase was not seen during DSI of sIPSCs (indeed, the opposite tendency was observed in the grouped sIPSC data), it could not have been caused by postsynaptic factors.
Figure 5. DSI affects the fast but not the slow component of the eIPSC.
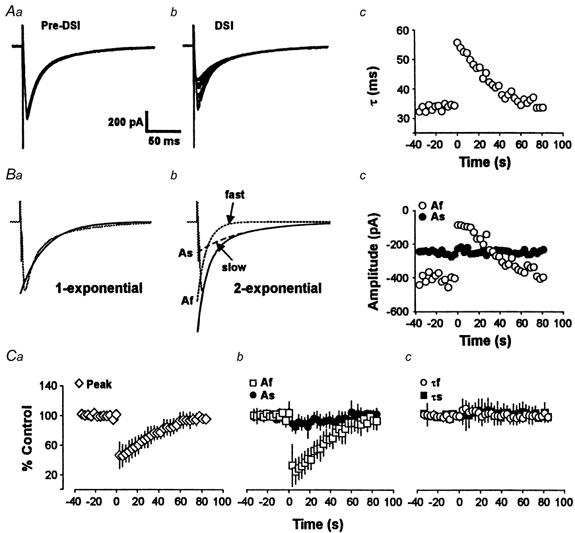
Aa, overlapped traces of the first 20 eIPSCs of a DSI trial. The DSI-inducing voltage step was given after the 20th response. Ab, overlapped traces of the last 20 eIPSCs of the same DSI trial. The eIPSCs increase gradually during recovery from DSI. Ac, plot of the time constants of decay of single exponential fits of the eIPSCs shown in Aa and Ab. The plot shows the reversible, apparent slowing of the eIPSC during DSI. Ba and Bb, the decay of the eIPSC is fitted better by the sum of two exponentials with fast and slow time constants, than by a single exponential. Af and As represent the amplitudes of the fast and slow components, respectively, at time 0. Bc, plot of Af and As for the experiment shown in A above. C, group data (n = 5) for the time course of raw peak eIPSCs (a), Af and As (b) and τf and τs(c).
Pearce and colleagues have reported the existence of two types of GABAergic eIPSCs (GABAA,fast and GABAA,slow) in CA1 cells (Pearce, 1993; Pearce et al. 1995; Banks et al. 1998) that are kinetically and pharmacologically distinguishable. We wondered if the fast and slow evoked IPSCs would be equally susceptible to DSI. We confirmed that the decay phases of eIPSCs were often better fitted by a double exponential function:
than by a single exponential (cf. Fig. 5Ba and Bb). The faster time constant, τf, was 12.1 ± 1.78 ms, while the slower one, τs, was 58.6 ± 6.17 ms. These values are close to those previously reported. Assuming the biexponential fits represent the two different classes of eIPSCs, we used the constants of the fitted equations, Af and As, as measures of the amplitudes of the two IPSCs. The results (e.g. Fig. 5Bc) were clear. During the DSI period there was a marked reversible suppression of Af, with no change in As. At the peak of DSI, Af was reduced to 32.1 ± 10.6 % of control, whereas As was 92.9 ± 5.5 % of control (difference significant, P < 0.05; n = 5). The time course of the depression of Af matched that of DSI. There was no change in τf or τs during DSI. At the peak of DSI τf and τs were 106 ± 9.4 and 101 ± 5.7 % of control values, respectively (differences not significant, n = 5). Figure 5Ca and c shows the time courses of the grouped data (n = 5) for DSI of the raw eIPSC peak (Fig. 5Ca), of the components Af and As (Fig. 5Cb), and of the time constants of decay τf and τs (Fig. 5Cc). Thus, the apparent slowing in the eIPSC during DSI can be accounted for by the selective reduction in the contribution of the fast component to the response.
Furosemide selectively reduces GABAA,fast by blocking the fast IPSC conductance without affecting the IPSC reversal potential (Pearce, 1993; Banks et al. 1998). If only GABAA,fast is DSI sensitive, then the data reported above suggest that furosemide should reduce the eIPSC amplitude and reduce DSI by blocking the fast component. In different cells from those of Fig. 5, we confirmed that bath application of 0.6 mm furosemide reduces the fast IPSC and DSI, with comparatively little effect on the slow IPSC (Fig. 6A and b). Furosemide significantly (P < 0.05) reduced the eIPSC amplitude by 55 % and DSI by 63 %. In the overlapped traces of Fig. 6B, we show the pre-DSI and DSI eIPSCs, first in raw form and then with the eIPSC during DSI scaled up to match the peak of the pre-DSI eIPSC. The slowing of the eIPSC during DSI is illustrated in the scaled traces. Furosemide reduced the eIPSC (same cell as control traces on the left), and some DSI occurred, but there was less marked slowing during DSI because furosemide had selectively reduced the fast eIPSC component. The histograms of Fig. 6C represent the data for raw eIPSC peak, the fast- and slow-amplitude components, Af and As, and time constants, τf and τs, in each condition. The data were analysed by a one-way ANOVA with repeated measures. When the fast and slow IPSC amplitudes were assessed separately it was clear that the effect of furosemide was mainly exerted on the fast IPSC, which was reduced to 28.6 ± 6.5 % of control (difference significant, P < 0.05 %, n = 4), whereas the slow IPSC was reduced only to 83.0 ± 5.5 % of control (not significant). Residual DSI in the presence of furosemide was expressed on the remaining fast eIPSCs, and not the remaining slow eIPSC. During DSI in furosemide the fast IPSC component was further reduced to 21.5 ± 7.9 % of control (difference significant, P < 0.01), but the slow IPSC component was still 83.4 ± 11.1 % of control (not significant). Thus, DSI and furosemide suppressed the same population of fast eIPSCs and spared the slow eIPSCs.
Figure 6. Furosemide selectively reduces the fast eIPSC component and occludes DSI.
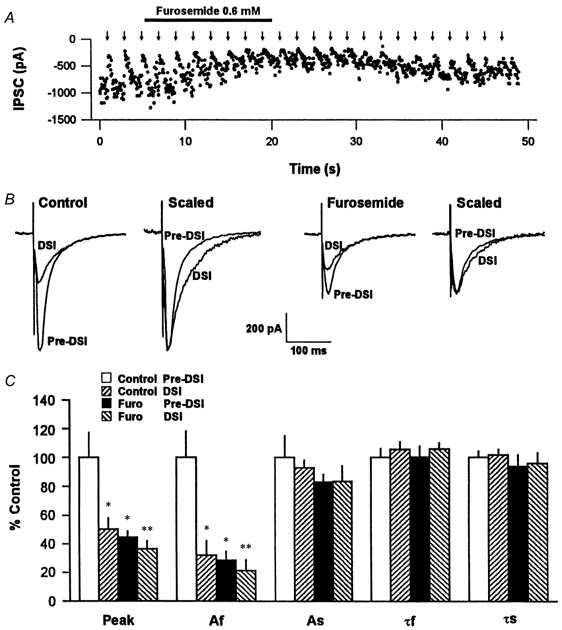
A, time course of an experiment on the effects of furosemide. Each • represents an eIPSC amplitude. Sudden decreases in eIPSC amplitudes represent DSI trials that occurred at 2 min intervals (arrows) throughout the experiment. B, representative overlapped trace means of eIPSCs in the pre-DSI (mean of 7) and DSI (mean of 5) periods, in control solution and after switching to furosemide-containing solution. In each solution, means of raw data are on the left, and the same traces scaled so the peaks match are on the right. Note the slowed time course of the eIPSC during DSI in control, and the reduction of the eIPSC and relative decrease in DSI in furosemide. C, group data for five experiments. The Peak group represents the raw eIPSCs; Af, As, τf and τs have the same significance as in Fig. 5. The data were analysed by one-way ANOVA followed by a Student-Newman-Keuls test for multiple comparisons. * Significant difference (P < 0.05) between the indicated data and the control pre-DSI data. ** Significant difference (P < 0.05) between the DSI and pre-DSI data in furosemide.
DISCUSSION
To understand the functional significance of DSI, it will be important to know which IPSCs, and by inference which interneurons, are susceptible to it. Some IPSCs are very much reduced or abolished by DSI, while others are unaffected (Alger et al. 1996). However, it was not clear from previous work if there were systematic differences between the susceptible and the non-susceptible IPSCs and, hence, whether functional classes of interneurons could be defined by DSI susceptibility. From examination of the sIPSC activity and eIPSCs in the present work, we conclude that three classes of IPSCs can be distinguished: one susceptible to DSI and two that are not susceptible. The DSI-susceptible sIPSCs are produced by GABA released by carbachol-activated and baclofen-inhibited interneurons. Their large size and rapid kinetics suggest that they are made by synapses on the pyramidal cell somata and proximal dendrites (Soltesz et al. 1995; Banks et al. 1998). Some non-DSI-susceptible sIPSCs are probably also made by synapses on the same electrotonic compartment, but in general these non-DSI-susceptible sIPSCs are small and relatively insensitive to carbachol and baclofen. The slow, furosemide- and DSI-insensitive eIPSCs make up the third functional group. The model of Fig. 7 summarizes our conclusions.
Figure 7. Interneuron model of DSI.
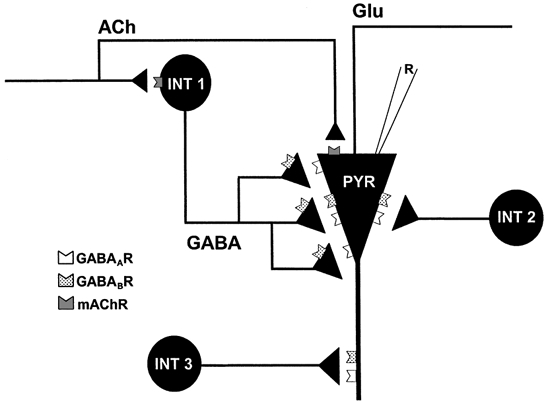
The data suggest that, with respect to DSI susceptibility, there are at least three populations of GABAergic interneurons, as summarized in the model above. Two populations make perisomatic synapses on pyramidal cells (PYR). One population (INT 1) is typically quiescent, but can be persistently activated by mAChR activation. It responds to synaptically released acetylcholine, and produces large-amplitude, DSI-susceptible sIPSCs in the pyramidal cell. The interneurons in this group can be further distinguished by their susceptibility to block by baclofen and by furosemide. The second population (INT 2) is tonically active, produces relatively small sIPSCs in the pyramidal cell, and is not susceptible to DSI. The third group of interneurons (INT 3) makes synapses in the distal dendrites of pyramidal cells, is insensitive to furosemide, and produces slowly decaying, non-DSI-susceptible IPSCs in pyramidal cells.
An unresolved issue has been the mechanism by which muscarinic agonists cause their pronounced facilitatory effects on the occurrence of DSI of sIPSCs. The agonists could affect the interneuron, the pyramidal cell, or both. Although we used bath application of carbachol, synaptically released ACh directly activates interneurons (Pitler & Alger, 1992b), and the resulting IPSCs are also susceptible to DSI (Pitler & Alger, 1994; Martin & Alger, 1999). The present studies show that the action of carbachol must to a large extent be on the interneuron. (1) In carbachol-containing solution, the occurrence of DSI was closely associated with the occurrence of large sIPSCs. (2) The abrupt onset and offset of large carbachol-induced sIPSCs can best be explained as reflecting the onset and offset of action potential firing in interneurons. (3) High K+ and NA could mimic the DSI-enhancing action of carbachol and yet would not have identical postsynaptic effects. (4) Occasionally carbachol application did not induce persistent IPSC activity, although in the presence of carbachol, lasting IPSC activity could be induced by extracellular stimulation. DSI appeared only when large sIPSCs did (Fig. 3b), despite the prior presence of carbachol. (5) Finally, baclofen occluded DSI by selectively reducing carbachol-induced, DSI-susceptible IPSCs; both the increase in sIPSC activity and DSI were abolished by baclofen, which inhibits only a subset of IPSCs (Lambert & Wilson, 1993). Baclofen also blocked the large DSI-susceptible eIPSCs that occurred in the presence of atropine, showing that it did not simply antagonize the mAChR actions pharmacologically, but rather inhibited the DSI-susceptible sIPSCs that carbachol activated. The data suggest that carbachol induces some interneurons to fire repetitively, and the output of these cells is susceptible to DSI. The data do not rule out the possibility of additional postsynaptic actions of carbachol.
In hippocampus, in normal extracellular solution DSI does not affect TTX-insensitive, i.e. action potential-independent, miniature IPSCs (mIPSCs) (Alger et al. 1996). It is only effective against action potential-dependent GABA release. Small, spontaneous IPSCs in control solution, or carbachol, are often of quantal size, thus raising the possibility that the non-DSI-susceptible IPSCs actually represent action potential-independent events. If this is the case, then the distinction between DSI-susceptible and non-DSI-susceptible IPSCs that we have been making could conceivably reflect differences between action potential-dependent and action potential-independent GABA release, rather than differences between classes of interneurons. However, we believe this interpretation is incorrect. Addition of TTX to the bathing solution markedly reduces even quantal-sized sIPSC activity, indicating that much of this activity is action potential dependent (Bergles et al. 1996), an observation we have confirmed (L. A. Martin, unpublished observation). Therefore, the small sIPSCs do not predominantly represent a DSI-insensitive mode of release. The heightened activity induced by AP-5 plus CNQX is also abolished by TTX, but is insensitive to DSI. We conclude that the simplest explanation is that the small baseline sIPSCs originate from non-DSI-susceptible interneurons.
Similarities between the DSI-susceptible sIPSCs and eIPSCs suggest that these are largely from the same, or overlapping, groups of interneurons in CA1. We infer that in the absence of carbachol these cells tend to be quiescent unless electrically stimulated. DSI-susceptible IPSCs are characterized by relatively rapid rise and decay times and sensitivity to baclofen. DSI of eIPSCs in a given pyramidal cell was also affected by factors that only affected the presynaptic interneurons, such as stimulus strength. Despite the similarities, however, it is unlikely that the DSI-sensitive sIPSCs and eIPSCs are from exactly the same interneurons in all cases. Morishita & Alger (2000) have recently found that at least some of the interneurons that are excited by carbachol differ from those that give rise to eIPSCs in CA1.
Carbachol has heterogeneous effects on interneurons. It can depolarize, hyperpolarize, or have no effect on different groups (Kawaguchi, 1997; McMahon et al. 1998; Parra et al. 1998; McQuiston & Madison, 1999). NA also has diverse effects on different classes of interneurons (Kawaguchi & Shindou, 1998; Parra et al. 1998). Bath application of 10 μm NA significantly increased both sIPSC activity and DSI. However, DSI in NA was never as robust as that observed in carbachol, and was always increased when carbachol was subsequently applied. NA may only activate some of the DSI-susceptible interneurons. Elevating extracellular [K+] also caused DSI to occur in 50 % of cells tested. Carbachol increases sIPSC activity and DSI in ∼70 % of pyramidal cells tested (Martin & Alger, 1999). The less consistent effects of high [K+]o compared with carbachol may have resulted from a smaller sample size, or the failure of the high [K+] solution to mimic the depolarizing effects of carbachol. In any case, the large size and abrupt onset of the high [K+]-induced, DSI-susceptible IPSCs suggest they are from the same cells that are activated by carbachol. Evidently, activation of some interneurons by carbachol, K+, or NA is sufficient to induce the occurrence of DSI. In contrast, AP-5 and CNQX application (prior to carbachol application) could induce a high level of sIPSC activity without inducing DSI. AP-5 and CNQX could activate primarily non-DSI-susceptible interneurons. If only a certain population of interneurons is susceptible to DSI, then high-intensity extracellular electrical stimulation could activate DSI-susceptible as well as non-DSI-susceptible interneurons. The use of minimal stimulation (Edwards et al. 1990) permitted selective stimulation of DSI-susceptible and non-susceptible IPSCs (Alger et al. 1996) and made it clear that susceptibility to DSI was a consistent feature of certain eIPSCs and not a feature that randomly affected all of them.
Interneurons have been classified by morphological, as well as physiological, differences. Tonic inhibition represents activation of perisomatic synapses in the dentate gyrus (Soltesz et al. 1995) and CA1 (Banks et al. 1998). DSI-susceptible sIPSCs and eIPSCs had rapid rise and decay times and probably originated from perisomatic synapses. In some experiments there was a slight slowing associated with carbachol-induced, DSI-susceptible sIPSCs. As these effects were not large, or consistent across cells, we suggest that they represent cases in which cells making synapses that were slightly distal from the recording electrode site were activated by carbachol. The slowed kinetics would reflect cable filtering, which can affect IPSCs generated within 100 μm of cell somata (Soltesz et al. 1995), rather than fundamental differences in the underlying IPSCs. The similarity in the kinetic parameters of the DSI-susceptible and non-susceptible sIPSCs suggests that the two types of sIPSCs are made by synapses in approximately the same electrotonic compartment. Basket and axo-axonic cells predominantly make perisomatic synapses (Buhl et al. 1994, 1995; Freund & Buzsaki, 1996; Miles et al. 1996), and we suggest that either or both of these types of interneurons are the source of DSI-susceptible IPSCs.
Pearce and colleagues (Pearce, 1993; Pearce et al. 1995; Banks et al. 1998) have argued that the fast and slow eIPSC components represent distinct GABAA receptor-mediated IPSCs, probably induced by different types of interneurons. An apparent slowing of eIPSCs during DSI was attributable to a selective reduction in the amplitude of the fast eIPSC decay component. It should be noted that fitting exponential functions to the inhibitory currents probably systematically overestimates (Hausser & Roth, 1997) the true time courses of the underlying conductances. Nevertheless, the qualitative conclusions reported here are unlikely to change. A slow form of spontaneous mIPSCs, or sIPSCs, does exist in CA1 cells, but is rare, accounting for < 0.1 % of all sIPSCs (Banks et al. 1998). Hence, it is not surprising that, even though we analysed 2400 sIPSCs, we did not detect any slow ones. Furosemide, which blocks fast but not slow eIPSCs (Pearce, 1993), selectively antagonizes α6- and α4-subunit-containing GABAA receptors (Wafford et al. 1996), and so it is possible that the kinetic differences reflect the activation of different subtypes of GABAA receptors. Nevertheless, furosemide has numerous effects and cannot be used as a selective receptor antagonist in the hippocampus. Our conclusions depend only on the functional differentiation of fast and slow eIPSCs possible with furosemide, however, and not on its specific mechanism of action. Localized application of GABAA receptor antagonists suggested that the fast IPSC is generated by synapses on the soma and proximal dendrites, whereas the slow IPSC is generated in the distal dendrites (Banks et al. 1998). Using a loose-patch technique to stimulate individual interneurons synaptically coupled to CA1 pyramidal cells, Ouardouz & Lacaille (1997) found that lacunosum-moleculare (L-M) interneurons produced IPSCs with significantly longer decay times than did s. radiatum or s. oriens interneurons, and suggested the L-M interneurons might mediate the slow GABAA receptor-mediated IPSC. Bistratified cells also produce eIPSCs with significantly slower kinetics than those of perisomatically terminating cells (Buhl et al. 1994). Accordingly, our results imply that the L-M interneurons, or bistratified cells, or both, mediate a slow IPSC that is not susceptible to DSI. Further study of the differences among these groups of interneurons should provide insight into the functional roles of DSI.
Recently, R. Wilson and R. A. Nicoll have found that antagonists of the cannabinoid CB1 receptor inhibit DSI very significantly, and they suggest that an endocannabinoid may be the primary retrograde messenger for DSI (R. Wilson & R. A. Nicoll, personal communication). The great majority (> 90 %) of the CB1 receptor labelling in CA1 is highly specifically localized to the presynaptic terminals and preterminal axons of CCK-containing, GABAergic basket cells (Hajos et al. 2000). These findings fit together perfectly with the data reported in this paper in suggesting that DSI is selectively targeted to certain basket cells. The functional relevance of the endocannabinoid system has been suggested by the finding that an agonist of CB1 receptors suppresses hippocampal gamma rhythms (Hajos et al. 2000). The apparent association between endocannabinoids and DSI may point to a role for DSI in this important rhythmic firing behaviour.
Acknowledgments
Most of the work reported here was contained in the PhD thesis of L.A.M. The work was supported by USPHS NIH RO1 NS30219 and NS36612 to B.E.A. We thank R. I. Wilson and R. A. Nicoll for permission to cite their unpublished data.
References
- Alger BE, Pitler TA, Wagner JJ, Martin LA, Morishita W, Kirov SA, Lenz RA. Retrograde signalling in depolarization-induced suppression of inhibition in rat hippocampal CA1 cells. Journal of Physiology. 1996;496:197–209. doi: 10.1113/jphysiol.1996.sp021677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade R. Blockade of neurotransmitter-activated K+ conductance by QX-314 in the rat hippocampus. European Journal of Pharmacology. 1991;199:259–262. doi: 10.1016/0014-2999(91)90467-5. [DOI] [PubMed] [Google Scholar]
- Banks MI, Li T-B, Pearce RA. The synaptic basis of GABAA, slow. Journal of Neuroscience. 1998;18:1305–1317. doi: 10.1523/JNEUROSCI.18-04-01305.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita JC, Bruggencate tenG. Cholinergic modulation of synaptic inhibition in the guinea pig hippocampus in vitro: excitation of GABAergic interneurons and inhibition of GABA-release. Journal of Neurophysiology. 1993;69:626–629. doi: 10.1152/jn.1993.69.2.626. [DOI] [PubMed] [Google Scholar]
- Bergles DE, Doze VA, Madison DV, Smith SJ. Excitatory actions of norepinephrine on multiple classes of hippocampal CA1 interneurons. Journal of Neuroscience. 1996;16:572–585. doi: 10.1523/JNEUROSCI.16-02-00572.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanton MG, Lo Turco JJ, Kriegstein AR. Whole cell recording from neurons in slices of reptilian and mammalian cerebral cortex. Journal of Neuroscience Methods. 1989;30:203–210. doi: 10.1016/0165-0270(89)90131-3. [DOI] [PubMed] [Google Scholar]
- Buhl EH, Cobb SR, Halasy K, Somogyi P. Properties of unitary IPSPs evoked by anatomically identified basket cells in the rat hippocampus. European Journal of Neuroscience. 1995;7:1989–2004. doi: 10.1111/j.1460-9568.1995.tb00721.x. [DOI] [PubMed] [Google Scholar]
- Buhl EH, Halasy K, Somogyi P. Diverse sources of hippocampal unitary inhibitory postsynaptic potentials and the number of synaptic release sites. Nature. 1994;368:823–828. doi: 10.1038/368823a0. [DOI] [PubMed] [Google Scholar]
- Connors BW, Prince DA. Effects of local anesthetic QX-314 on the membrane properties of hippocampal pyramidal neurons. Journal of Pharmacology and Experimental Therapeutics. 1982;220:476–481. [PubMed] [Google Scholar]
- Davies CH, Davies SN, Collingridge GL. Paired-pulse depression of monosynaptic GABA-mediated inhibitory postsynaptic responses in rat hippocampus. Journal of Physiology. 1990;424:513–531. doi: 10.1113/jphysiol.1990.sp018080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards FA, Konnerth A, Sakmann B. Quantal analysis of inhibitory synaptic transmission in the dentate gyrus of rat hippocampal slices: a patch-clamp study. Journal of Physiology. 1990;430:213–249. doi: 10.1113/jphysiol.1990.sp018289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, Buzsaki G. Interneurons of the hippocampus. Hippocampus. 1996;6:347–470. doi: 10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Glitsch M, Llano I, Marty A. Glutamate as a candidate retrograde messenger at interneurone-Purkinje cell synapses of rat cerebellum. Journal of Physiology. 1996;497:531–537. doi: 10.1113/jphysiol.1996.sp021786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajos N, Katona I, Naiem SS, Mackie K, Ledent C, Mody I, Freund TF. Cannabinoids inhibit hippocampal GABAergic transmission and network oscillations. European Journal of Neuroscience. 2000;12:3239–3249. doi: 10.1046/j.1460-9568.2000.00217.x. [DOI] [PubMed] [Google Scholar]
- Hausser M, Roth A. Estimating the time course of the excitatory synaptic conductance in neocortical pyramidal cells using a novel voltage jump method. Journal of Neuroscience. 1997;17:7606–7625. doi: 10.1523/JNEUROSCI.17-20-07606.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur A, Lytton WW, Ketchum KL, Haberly LB. Regulation of the NMDA component of EPSPs by different components of postsynaptic GABAergic inhibition: computer simulation analysis in piriform cortex. Journal of Neurophysiology. 1997a;78:2546–2559. doi: 10.1152/jn.1997.78.5.2546. [DOI] [PubMed] [Google Scholar]
- Kapur A, Pearce RA, Lytton WW, Haberly LB. GABAA-mediated IPSCs in piriform cortex have fast and slow components with different properties and locations on pyramidal cells. Journal of Neurophysiology. 1997b;78:2531–2545. doi: 10.1152/jn.1997.78.5.2531. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y. Selective cholinergic modulation of cortical GABAergic cell subtypes. Journal of Neurophysiology. 1997;78:1743–1747. doi: 10.1152/jn.1997.78.3.1743. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Shindou T. Noradrenergic excitation and inhibition of GABAergic cell types in rat frontal cortex. Journal of Neuroscience. 1998;18:6963–6976. doi: 10.1523/JNEUROSCI.18-17-06963.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert NA, Wilson WA. Heterogeneity in presynaptic regulation of GABA release from hippocampal inhibitory neurons. Neuron. 1993;11:1057–1067. doi: 10.1016/0896-6273(93)90219-h. [DOI] [PubMed] [Google Scholar]
- Le Beau FEN, Alger BE. Transient suppression of GABAA-receptor-mediated IPSPs after epileptiform burst discharges in CA1 pyramidal cells. Journal of Neurophysiology. 1998;79:659–669. doi: 10.1152/jn.1998.79.2.659. [DOI] [PubMed] [Google Scholar]
- Llano I, Leresche N, Marty A. Calcium entry increases the sensitivity of cerebellar Purkinje cells to applied GABA and decreases inhibitory synaptic currents. Neuron. 1991;6:565–574. doi: 10.1016/0896-6273(91)90059-9. [DOI] [PubMed] [Google Scholar]
- McBain CJ, Eaton JV, Brown T, Dingledine R. CNQX increases spontaneous inhibitory input to CA3 pyramidal neurones in neonatal rat hippocampal slices. Brain Research. 1992;592:255–260. doi: 10.1016/0006-8993(92)91683-6. [DOI] [PubMed] [Google Scholar]
- McMahon LL, Williams JH, Kauer JA. Functionally distinct groups of interneurons identified during rhythmic carbachol oscillations in hippocampus in vitro. Journal of Neuroscience. 1998;18:5640–5651. doi: 10.1523/JNEUROSCI.18-15-05640.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuiston AR, Madison DV. Muscarinic receptor activity has multiple effects on the resting membrane potentials of CA1 hippocampal interneurons. Journal of Neuroscience. 1999;19:5693–5702. doi: 10.1523/JNEUROSCI.19-14-05693.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LA, Alger BE. Muscarinic receptor activation of interneurons susceptible to depolarization-induced suppression of inhibition (DSI) Society for Neuroscience Abstracts. 1996;22:1989. [Google Scholar]
- Martin LA, Alger BE. Muscarinic facilitation of the occurrence of depolarization-induced suppression of inhibition in rat hippocampus. Neuroscience. 1999;92:61–71. doi: 10.1016/s0306-4522(98)00745-3. [DOI] [PubMed] [Google Scholar]
- Martin LA, Pitler TA, Lenz RA, Wagner JJ, Alger BE. Muscarinic enhancement of depolarization-induced suppression of inhibition in rat hippocampal pyramidal cells. Society for Neuroscience Abstracts. 1995;21:1095. [Google Scholar]
- Miles R, Toth K, Gulyas AI, Hajos N, Freund TF. Differences between somatic and dendritic inhibition in the hippocampus. Neuron. 1996;16:815–823. doi: 10.1016/s0896-6273(00)80101-4. [DOI] [PubMed] [Google Scholar]
- Morishita W, Alger BE. Differential effect of the group II mGluR agonist, DCG-IV, on depolarization-induced suppression of inhibition in hippocampal CA1 and CA3 neurons. Hippocampus. 2000;10:261–268. doi: 10.1002/1098-1063(2000)10:3<261::AID-HIPO6>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Morishita W, Kirov SA, Alger BE. Evidence for metabotropic glutamate receptor activation in the induction of depolarization-induced suppression of inhibition in hippocampal CA1. Journal of Neuroscience. 1998;18:4870–4882. doi: 10.1523/JNEUROSCI.18-13-04870.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita W, Kirov SA, Pitler TA, Martin LA, Lenz RA, Alger BE. N-Ethylmaleimide blocks depolarization-induced suppression of inhibition and enhances GABA release in the rat hippocampal slice in vitro. Journal of Neuroscience. 1997;17:941–950. doi: 10.1523/JNEUROSCI.17-03-00941.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan T, Jensen MS, Lambert JDC. The slow inhibitory postsynaptic potential in rat hippocampal CA1 neurones is blocked by intracellular injection of QX-314. Neuroscience Letters. 1990;110:309–313. doi: 10.1016/0304-3940(90)90865-7. [DOI] [PubMed] [Google Scholar]
- Nicoll RA, Alger BE. A simple chamber for recording from submerged brain slices. Journal of Neuroscience Methods. 1981;4:153–156. doi: 10.1016/0165-0270(81)90049-2. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Sawada S, Yamamoto C. Properties of depolarization-induced suppression of inhibitory transmission in cultured rat hippocampal neurons. Pflügers Archiv. 1998;435:273–279. doi: 10.1007/s004240050512. [DOI] [PubMed] [Google Scholar]
- Otis TS, Staley KJ, Mody I. Perpetual inhibitory activity in mammalian brain slices generated by spontaneous GABA release. Brain Research. 1991;545:142–150. doi: 10.1016/0006-8993(91)91280-e. [DOI] [PubMed] [Google Scholar]
- Ouardouz M, Lacaille J-C. Properties of unitary IPSCs in hippocampal pyramidal cells originating from different types of interneurons in young rats. Journal of Neurophysiology. 1997;77:1939–1949. doi: 10.1152/jn.1997.77.4.1939. [DOI] [PubMed] [Google Scholar]
- Parra P, Gulyas AI, Miles R. How many subtypes of inhibitory cells in the hippocampus? Neuron. 1998;20:983–993. doi: 10.1016/s0896-6273(00)80479-1. [DOI] [PubMed] [Google Scholar]
- Pearce RA. Physiological evidence for two distinct GABAA responses in rat hippocampus. Neuron. 1993;10:189–200. doi: 10.1016/0896-6273(93)90310-n. [DOI] [PubMed] [Google Scholar]
- Pearce RA, Grunder SD, Faucher LD. Different mechanisms for use-dependent depression of two GABAA-mediated IPSCs in rat hippocampus. Journal of Physiology. 1995;484:425–435. doi: 10.1113/jphysiol.1995.sp020675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitler TA, Alger BE. Postsynaptic spike firing reduces synaptic GABAA responses in hippocampal pyramidal cells. Journal of Neuroscience. 1992a;12:4122–4132. doi: 10.1523/JNEUROSCI.12-10-04122.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitler TA, Alger BE. Cholinergic excitation of GABAergic interneurons in the rat hippocampal slice. Journal of Physiology. 1992b;450:127–142. doi: 10.1113/jphysiol.1992.sp019119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitler TA, Alger BE. Depolarization-induced suppression of GABAergic inhibition in rat hippocampal pyramidal cells: G protein involvement in a presynaptic mechanism. Neuron. 1994;13:1447–1455. doi: 10.1016/0896-6273(94)90430-8. [DOI] [PubMed] [Google Scholar]
- Poncer J-C, Miles R. Fast and slow excitation of inhibitory cells in the CA3 region of the hippocampus. Journal of Neurobiology. 1995;26:386–395. doi: 10.1002/neu.480260310. [DOI] [PubMed] [Google Scholar]
- Reece LJ, Schwartzkroin PA. Effects of cholinergic agonists on two non-pyramidal cell types in rat hippocampal slices. Brain Research. 1991;566:115–126. doi: 10.1016/0006-8993(91)91688-w. [DOI] [PubMed] [Google Scholar]
- Ropert N, Guy N. Serotonin facilitates GABAergic transmission in the CA1 region of rat hippocampus in vitro. Journal of Physiology. 1991;441:121–136. doi: 10.1113/jphysiol.1991.sp018742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltesz I, Smetters DK, Mody I. Tonic inhibition originates from synapses close to the soma. Neuron. 1995;14:1273–1283. doi: 10.1016/0896-6273(95)90274-0. [DOI] [PubMed] [Google Scholar]
- Vincent P, Armstrong CM, Marty A. Inhibitory synaptic currents in rat cerebellar Purkinje cells: modulation by postsynaptic depolarization. Journal of Physiology. 1992;456:453–471. doi: 10.1113/jphysiol.1992.sp019346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wafford KA, Thompson SA, Thomas D, Sikela J, Wilcox AS, Whiting PJ. Functional characterization of human gamma-aminobutyric acidA receptors containing the α4 subunit. Molecular Pharmacology. 1996;50:670–678. [PubMed] [Google Scholar]
- Wagner JJ, Alger BE. Increased neuronal excitability during depolarization-induced suppression of inhibition in rat hippocampus. Journal of Physiology. 1996;495:107–112. doi: 10.1113/jphysiol.1996.sp021577. [DOI] [PMC free article] [PubMed] [Google Scholar]